

Abstract
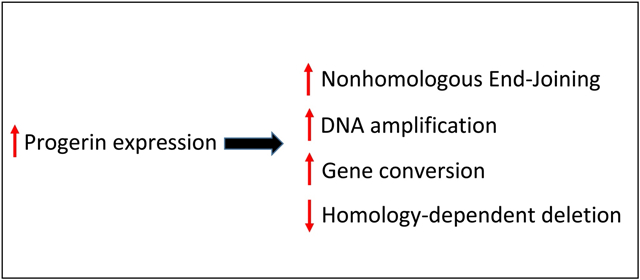
Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare autosomal, dominant genetic condition characterized by many features of accelerate aging. On average, children with HGPS live to about fourteen years of age. The syndrome is commonly caused by a point mutation in the LMNA gene which normally codes for lamin A and its splice variant lamin C, components of the nuclear lamina. The LMNA mutation alters splicing, leading to production of a truncated, farnesylated form of lamin A referred to as “progerin.” Progerin is also expressed at very low levels in healthy individuals and appears to play a role in normal aging. HGPS is associated with an accumulation of genomic DNA double-strand breaks (DSBs), suggesting corruption of DNA repair. In this work, we investigated the influence of progerin expression on DSB repair in the human genome at the nucleotide level. We used a model system that involves a reporter DNA substrate inserted in the genome of cultured human cells. A DSB could be induced within the substrate through exogenous expression of endonuclease I-SceI, and DSB repair events occurring via either homologous recombination (HR) or nonhomologous end-joining (NHEJ) were recoverable. Additionally, spontaneous HR events were recoverable in the absence of artificial DSB induction. We compared DSB repair and spontaneous HR in cells overexpressing progerin versus cells expressing no progerin. We report that overexpression of progerin correlated with an increase in DSB repair via NHEJ relative to HR, as well as an increased fraction of HR events occurring via gene conversion. Progerin also engendered an apparent increase in spontaneous HR events, with a highly significant shift toward gene conversion events, and an increase in DNA amplification events. Such influences of progerin on DNA transactions may impact genome stability and contribute to aging.
Keywords: progerin, double-strand break repair, homologous recombination, nonhomologous end-joining, genomic instability
Graphical Abstract
1. Introduction
Each day, mammalian cells must correct many forms of DNA damage in order to maintain genome stability. One type of damage that cells must contend with is the DNA double-strand break (DSB). DSBs can be generated by chemical or radiological insult. DSBs may also form spontaneously from other DNA lesions or at stalled or collapsed replication forks. It is essential that DSBs be repaired efficiently and accurately to avoid potentially deleterious chromosomal rearrangements or mutations and the consequences such events portend.
To heal DSBs, mammalian cells employ two general types of repair pathways: homologous recombination (HR) and nonhomologous end-joining (NHEJ ) [reviewed in 1–11]. Although there are various forms of HR and NHEJ, the essential difference between these two broadly defined repair schemes is that HR utilizes a template sequence to maintain or restore genetic information to the DSB site that may otherwise be lost through strand degradation, whereas NHEJ involves no template in the rejoining of DNA ends. Due to this difference in the nature of HR versus NHEJ, HR is generally considered to be accurate while NHEJ often produces deletions and, thus, is generally error-prone. Additionally, HR is largely restricted to the late S or G2 stage of the cell cycle in dividing cells, whereas NHEJ is active throughout the cell cycle and in nondividing cells.
It is clear that DNA repair processes play a critical role in the maintenance of genome integrity. With regards to DSB repair, it would seem that HR provides greater accuracy than NHEJ. Although the potential accuracy of HR should provide stability, there are certain risks associated with HR. For example, the choice of appropriate recombination partner is vital for the prevention of potential gross chromosomal rearrangements. To avoid mutation, it is important that genetic exchange be allowed to occur only between sequences that share perfect or near-perfect homology. Crossovers allowed to occur between imperfectly matched sequences can be particularly dangerous and potentially lead to translocations. Mammalian cells indeed normally exert stringent control over HR, allowing exchange to occur only between those sequences that exhibit a very high degree of similarity [12–14].
Due to a mutation of one or more important genome caretaker genes, regulation of HR and DSB repair sometimes becomes corrupted, leading to relaxed stringency or abnormally high or low rates of HR. Such breakdown in DSB repair pathways can produce “genomic instability” which can lead to mutations, aberrant chromosomal rearrangements, and loss of heterozygosity (LOH) of deleterious alleles.
The global consequences of genomic instability take a variety of forms. The association between cancer and aberrant HR or DSB repair is well-documented in the literature [15–17]. Genomic instability has also been associated with the process of aging. Increased levels of damage, mutation, and large-scale chromosomal abnormalities such as translocations, insertions and deletions have been observed with increasing age in humans, mice, and other organisms [18–31]. As the integrity of the genome is progressively compromised over time, general cellular functions would be expected to be disrupted. In addition, cell number would gradually be reduced as cells are lost due to apoptotic responses to unrepaired DNA lesions, leading to tissue depletion and loss of biological functions. Thus, the accumulation of mutation and DNA damage has come to be viewed as a possible basis for, or at least a significant contributor to, the aging process.
Evidence has been reported that the increase in genomic instability that accompanies aging correlates with a decrease in the intrinsic efficiency of a variety of DNA repair pathways as a function of age [18–31]. A dysfunction of NHEJ was recorded in rat brain during aging [24], and studies involving mice have suggested that the fidelity of DSB repair diminishes with age [25]. It has been reported that both the efficiency and fidelity of DSB repair decreases as human fibroblasts approach senescence [26]. Chromosomal DSBs accumulate in human cells approaching senescence, and it has been suggested that DSBs may be directly involved in the actual induction of senescence [18]. A marked accumulation of DSBs has also been seen in cultured cells from patients with premature aging syndromes. To summarize, there is substantial evidence indicating that impaired or altered repair of DSBs constitutes a critical component of the aging process.
Not unexpectedly, diseases that produce clinical features of premature aging (progeria) are often associated with innate DNA repair defects and associated genomic instability [19–23,27–31]. Hutchinson–Gilford Progeria Syndrome (HGPS) is one such genetic syndrome that leads to accelerated aging. The average lifespan of an individual with HGPS is about fourteen years. HGPS is most commonly caused by a point mutation in the LMNA gene which normally codes for lamin A and its splice variant lamin C. The LMNA mutation associated with HGPS leads to increased usage of a cryptic splice site which leads to the production of a truncated form of lamin A referred to as “progerin.” Significantly, it has been learned that progerin is in fact expressed at low levels in healthy individuals and appears to play a role in the normal aging process [32–35]. Unlike wild-type fully processed lamin A, progerin retains a farnesyl group at it carboxy terminus. This farnesyl group causes progerin to largely remain associated with the inner nuclear membrane rather than localize to the nuclear lamina where lamin A normally resides.
Lamin A normally serves as an important component of the nuclear lamina which plays structural as well as catalytic roles in the nucleus. In HGPS, the impact of progerin overexpression on nuclear architecture is severe. The nuclei of HGPS cells are characteristically misshapen and display blebs and invaginations. The altered nuclear structure imparts important changes to numerous nuclear functions and profoundly alters chromatin organization. Among its various roles, lamin A normally helps recruit proliferating cell nuclear antigen, DNA polymerase delta and other factors to replication forks. Progerin expression in HGPS interferes with this recruitment, leading to replication fork stalling and collapse [31,35–39]. A body of recent literature also implicates lamin A and its variants in DNA repair [40–46].
One notable consequence of progerin expression in HGPS cells is an accumulation of DSBs and increased sensitivity to DNA damaging agents [36–39,47,48]. The persistence of DSBs in HGPS cells is indicative of a general impairment of DSB repair, which likely contributes to the accelerated aging phenotype [18,39,49–51]. The high steady-state level of DSBs also leads to a persistent activation of a DNA damage response and checkpoints which may at least partly explain the reduced replicative capacity of HGPS cells. Recent work [52] shows that when the expression of a particular p53 isoform is increased in HGPS cells, DSB repair is promoted and the replicative lifespan of the HGPS cells is concomitantly increased. The general picture that has emerged, and is still developing, is that DSB repair in HGPS is delayed or, in some cases, perhaps precluded, and this corruption of DNA repair is mechanistically related to reduced lifespan of cells and individuals.
Studies have revealed that recruitment of repair proteins, most specifically those involved in HR repair (including Rad 50, Rad51, NBS1, and MRE11), to the site of a DSB is delayed in HGPS [36,48]. Consistent with such findings is work suggesting that NHEJ is enhanced while HR is concomitantly rendered defective by progerin expression [42,43,45,53].
Several recent studies have revealed that wild-type lamin A interacts with and stabilizes the protein 53BP1, a key player in NHEJ, and that depletion of lamin A leads to the degradation of 53BP1 with an attendant reduction in DSB repair via NHEJ [40,44,45]. Loss of lamin A expression has recently been shown to additionally bring about transcriptional down-regulation of the key HR proteins RAD51 and BRCA1 [41], and so lamin A deficiency compromises both HR as well as NHEJ pathways. The loss of two major DSB repair pathways is predicted to bring about profound genomic instability. The dual loss of DSB repair pathways in the absence of lamin A is notable and perhaps somewhat surprising since, as mentioned above, HGPS cells appear to exhibit an increase in NHEJ activity with concomitant reduction in HR. Thus, variations in lamin A functions and/or changes in the relative expression levels of lamin A and mutant forms of lamin A may have impacts on response to and repair of DNA damage.
Despite the unequivocal evidence that wild-type and mutant forms of lamin A such as progerin exert substantial influence on levels of DSB repair and genomic stability, there remains a surprising lack of knowledge of how progerin expression may alter the nature, at the nucleotide level, of DSB repair or spontaneous recombination events. Interestingly, a recent report [54] concludes that expression of progerin does not increase the mutation rate in a murine epithelial cell line, leading the authors to suggest that the underlying cellular defect in HGPS cells actually does not lie in defective DNA repair per se. However, neither this latter report nor any other previous report has used assays that directly examined how progerin expression influences the nature or accuracy of recombination events and other DSB repair events. In this work, we sought to explore these issues using a model experimental system employing cultured human cells containing an integrated DSB repair and HR reporter construct.
We now report that high levels of progerin in human cells correlated with an increase in DSB repair via NHEJ relative to HR, and an increased portion of HR occurring via gene conversions with no associated crossover. Progerin also engendered a significant increase in spontaneous HR events, with a highly significant shift toward noncrossover events, and an increase in DNA amplification events. Such influences of progerin may impact genome stability and contribute to the aging process.
2. Materials and Methods
2.1. General cell culture
All cell lines were derived from normal human fibroblast cell line GM637 (immortalized by SV40) which was obtained from the NIGMS. Cells were cultured in alpha-modified minimum essential medium supplemented with 10% fetal bovine serum. All cells were maintained at 37°C in a humidified atmosphere of 5% CO2.
2.2. Recombination and DSB repair reporter substate
Plasmid pLB4 (Fig. 1), used as a recombination and DSB repair reporter substrate, was described previously [55,56]. Briefly, pLB4 contains a gene comprised of herpes simplex virus type 1 thymidine kinase (tk) sequence fused to a neo gene sequence. The tk-neo fusion gene is disrupted by a 22 bp oligonucleotide containing the 18 bp recognition sequence for endonuclease I-SceI. The substrate pLB4 also contains a “donor” tk sequence that shares about 1.7 kb of homology with the tk portion of the tk-neo fusion gene. Due to several 14 scattered mismatches (Fig. 2), the donor on pLB4 displays about 1% sequence divergence with the tk portion of the tk-neo fusion gene.
Fig. 1.

Recombination and DSB repair substrate pLB4. The substrate contains a herpes tk-neo fusion gene that is disrupted by a 22-bp oligonucleotide containing the 18-bp recognition site for endonuclease I-SceI (underlined sequence). Sites of staggered cleavage by I-SceI are indicated. BamHI (B) and HindIII (H) sites are shown. Substrate pLB4 also contains a 2.5-kb HindIII fragment containing a complete herpes tk gene. The direction of transcription of both the tk gene and tk-neo fusion gene is from left to right as drawn. PCR primers used to amplify DSB repair products for subsequent sequence analysis are indicated by short horizontal arrows.
Fig. 2.

Alignment of tk-neo fusion gene sequence with donor tk sequence from pLB4. Nucleotides 308–1622, numbering according to [59], of the tk portion of the tk-neo fusion gene are aligned with the corresponding donor sequence from pLB4. The span of tk sequence shown comprises the tk portion of PCR products generated by primers AW85 and AW91. “Marker” mismatches between donor and tk-neo fusion gene sequences are highlighted. The 22 bp oligonucleotide containing the 18 bp I-SceI recognition sequence inserted in the tk-neo fusion gene (absent from donor) is depicted in bold italics, with the actual I-SceI recognition sequence in uppercase and sites of staggered cleavage indicated.
2.3. Recombination and DSB repair reporter cell lines
As previously described [57], recombination and repair substrate pLB4 (Fig. 1) had been stably integrated into the genome of human cell line GM637 cells to produce cell line pLB4/11 containing a single integrated copy of pLB4.
To produce a derivative of pLB4/11 that constitutively expresses GFP-progerin, cell line pLB4/11 was stably transfected with plasmid pBABE-puro-GFP-progerin (Addgene plasmid # 17663). This latter 8.3 kb vector, a gift of Tom Misteli from the National Cancer Institute, expresses progerin as a GFP fusion protein under the control of a Moloney murine leukemia virus LTR promoter. 5 × 106 pLB4/11 cells were electroporated with 3 ug of pBABE-puro-GFP-progerin which was first linearized by digestion with NotI. Electroporation was carried out using a Bio-Rad Gene Pulser set at 700V, 25 uF. Two days following electroporation, cells were plated at a density of 1 × 106 cells per 75cm2 flask into medium supplemented with 0.5 μg/mL puromycin to select for stable transfectants. After 14 days of selection, colonies were picked. Clones that showed GFP fluorescence were propagated further and GFP-progerin expression was confirmed by Western blot.
To produce a derivative of pLB4/11 that constitutively expresses GFP, plasmid pBABE-puro-GFP-progerin was digested with SalI and recircularized after removal of the 2.4 kb fragment containing the progerin coding sequence. The resulting 5.9 kb plasmid (pBABE-puro-GFP) was linearized with NotI and stably transfected into pLB4/11 as described above.
2.4. Western blots
Blots were performed using SDS-PAGE with 4% stacking genes and 8% separating gels. Each lane contained 30 μg of total cellular protein, and Bio-Rad Precision Plus Kaleidoscope Protein Standard (#161–0375, 5 μL) was used for molecular weight markers.
The primary antibody used was GFP (B-2): sc-9996 (mouse monoclonal, from Santa Cruz Biotechnology, Inc.) at a dilution of 1:500. Secondary antibody used was goat anti-mouse IgG-HRP: sc-2005 (Santa Cruz Biotechnology, Inc.) at a dilution of 1:1000.
Detection was accomplished using GE Healthcare Amersham ECL Select Western Blotting Detection Reagent. Imaging was conducted using a GE ImageQuant LAS 4000 courtesy of Dr. Beth Krizek, University of South Carolina.
2.5. Recovery of DSB-induced HR and NHEJ events
In order to induce a DSB at the I-SceI site within the integrated copy of substrate pLB4 in cell lines, cells were electroporated with the I-SceI expression plasmid pCMV-3xnls-I-SceI (“pSce”), generously supplied by Maria Jasin (Sloan Kettering), essentially as previously described [58]. Briefly, 5 × 106 cells were mixed with 20 μg of pSce in a volume of 800 μl of phosphate buffered saline at room temperature and electroporated in a 0.4cm gap electroporation cuvette using a Bio-Rad Gene Pulser (Bio-Rad, Hercules, CA, USA) set to 700 V and 25 μF. Following electroporation, cells were plated into growth medium under no selection and allowed to recover for 2 days. Cells were then harvested and plated into 75 cm2 flasks at a density of 1 × 106 cells per flask using medium containing 1000 ug/ml of G418 to select for cells that had undergone HR or NHEJ at the DSB site.
2.6. Determination of spontaneous intrachromosomal HR frequencies
Spontaneous recombination frequencies were determined for cell line pLB4/11 and derivatives expressing GFP-progerin or GFP by fluctuation tests. To perform such an analysis on a cell line, 10 or more subclones were first generated from the parental cell line. Subclones were propagated separately to several million cells. Cells from each subclone were then plated into several 75 cm2 flasks at a density of 1 × 106 in order to select for G418R segregants. Colony frequency was then calculated by dividing the number of G418R colonies recovered by the number of cells plated into G418. G418R colonies were harvested for further propagation and analysis. Recombination frequency was calculated by multiplying colony frequency by the percentage of analyzed events that were found to be bona fide recombination events.
2.7. PCR amplification and DNA sequence analysis
A segment of the tk-neo fusion gene spanning the I-SceI site was amplified from 500 ng of genomic DNA isolated from G418R clones using primers AW85 (5’-TAATACGACTCACTATAGGGCCAGCGTCTTGTCATTGGCG-3’) and AW91 (5’-GATTTAGGTGACACTATAGCCAAGCGGCCGGAGAACCTG-3’). AW85 is composed of nucleotides 308–327 of the coding sequence of the herpes tk gene, numbering according to [59] with a T7 forward universal primer appended to the 5’ end of the primer. AW91 is composed of 20 nucleotides from the noncoding strand of neomycin gene mapping 25 through 44 bp downstream from the neomycin start codon, with an Sp6 primer appended to the 5’ end of the primer. The position of the PCR primers are indicated in Fig. 1. PCR was carried out using Ready-To-Go PCR beads (GE Healthcare) and a “touchdown” PCR protocol as previously described [60]. PCR products were then sequenced using a T7 primer by Eton Bioscience, Inc. (Research Triangle Park, NC).
3. Results
3.1. An experimental system for monitoring intrachromosomal HR in human cells
It was our goal to assess the impact that progerin expression has on genetic recombination and DSB repair in human cells. We used our established experimental system that uses a genetic selection for HR and DSB repair events in cultured human fibroblasts. We made use of human fibroblast cell line GM637, an immortalized cell line derived from an apparently healthy individual. As previously described [57], recombination and repair substrate pLB4 (Fig. 1) had been stably integrated into the genome of GM637 cells to produce cell line pLB4/11 containing a single integrated copy of pLB4. pLB4 contains a tk-neo fusion gene disrupted by a 22 bp oligonucleotide inserted into the tk portion of the fusion gene. The 22 bp oligonucleotide contains the 18 bp recognition site for yeast endonuclease I-SceI. A complete functional tk gene is also contained in pLB4. HR between the tk gene and the tk-neo fusion gene can restore function to the fusion gene, and such an event is recoverable by selection for G418R clones. Because of the I-SceI site inserted in the tk-neo fusion gene in pLB4, a DSB can be introduced into the fusion gene by transiently transfecting cell line pLB4/11 with the I-SceI expression plasmid pSce. Cell line pLB4/11 enables the study of both spontaneous as well as DSB-induced HR. Restoration of function of the neo portion of the tk-neo fusion gene on pLB4 does not require accurate removal of the 22 bp oligonucleotide inserted into the upstream tk portion since neo expression requires only that an appropriate reading frame be restored to the fusion gene. Because of this latter feature of pLB4, recovery of error-prone NHEJ and other error-prone events in addition to accurate HR is possible when using cell line pLB4/11.
Cell line pLB4/11 does not express a detectable level of progerin (not shown). In order to study the impact of progerin on HR and DSB repair, cell line pLB4/11 was stably transfected with plasmid pBABE-puro-GFP-progerin or with pBABE-puro-GFP to produce cell lines pLB4-progerin and pLB4-GFP respectively. Cell line pLB4-progerin constitutively expresses GFP-progerin and cell line pLB4-GFP constitutively expresses GFP (Fig. 3). The level of expression of GFP-progerin in cell line pLB4-progerin was somewhat higher but comparable to the level of progerin expression in a cell line derived from an HGPS patient (Supplemental Fig. 1).
Fig. 3.

Isolation of cell lines pLB4-progerin and pLB4-GFP. (A) pLB4-progerin cells display nuclear fluorescence due to expression of GFP-progerin. (B) pLB4-GFP cells display cytoplasmic fluorescence due to expression of GFP. (C) Western blot of whole-cell extracts from cell lines pLB4, pLB4-progerin, and pLB4-GFP (lanes labeled 1,2,3 respectively). Samples are displayed in noncontiguous lanes from the same gel. An anti-GFP primary antibody was used. Cell line pLB4-progerin displays GFP-progerin at the expected mw of ~95 kD (lane 2) while cell line pLB4-GFP displays GFP at the expected mw of ~27 kD (lane 3).
3.2. Progerin expression shifts DSB repair away from HR and toward NHEJ, and shifts HR toward gene conversions
Cell lines pLB4-progerin, pLB4-GFP, and pLB4/11 were electroporated with I-SceI expression plasmid pSce to induce a DSB within the integrated pLB4 construct. Two days following electroporation, cells were plated into medium supplemented with G418 to recover clones in which a DSB repair event had restored function to the tk-neo fusion gene and thus conferred G418 resistance (Table 1).
Table 1.
Recovery of DSB repair events
| Cell line | Expt. No.a | Cells Plated (104)b | G418R Colonies | Colony Frequency (10−4)c |
|---|---|---|---|---|
| pLB4-progerin | 1 | 5.0 | 195 | 39.0 |
| 2 | 5.0 | 121 | 24.2 | |
| 3 | 4.0 | 145 | 36.3 | |
| 4 | 5.0 | 31 | 6.2 | |
| Average | 26.4 | |||
| pLB4-GFP | 1 | 5.0 | 310 | 62.0 |
| 2 | 5.0 | 401 | 80.2 | |
| Average | 71.1 | |||
| pLB4/11 | 1 | 5.0 | 238 | 46.6 |
| 2 | 5.0 | 184 | 36.8 | |
| Average | 41.7 |
Each experiment involved electroporation of 5 × 106 cells with plasmid pSce to induce a DSB.
Number of cells plated into G418 selection 48 hours after electroporation.
Number of G418R colonies divided by number of cells plated into selection.
Both HR and NHEJ events were potentially recoverable as G418R colonies from cell lines pLB4-progerin, pLB4-GFP, and pLB4/11. To determine the nature of recovered DSB repair events, genomic DNA was isolated from representative G418R clones. For each clone analyzed, a segment of DNA surrounding the DSB site was amplified by PCR, and the amplification products were sequenced. DNA sequence analysis allowed a distinction between HR and NHEJ based on the scattered “marker” nucleotide mismatches displayed by the recipient and donor sequences on integrated substrate pLB4 (Fig. 2). While these mismatches enhance sequence analysis of DSB repair products and enable an unambiguous demonstration of recombination between the tk donor sequence and the fusion gene, the mismatches are sufficiently sparse so as to preserve substantial stretches of perfect homology, including 410- and 231-bp segments of continuous homology. Critically, these stretches of continuous homology exceed the minimal amount of homology that we previously determined is required for efficient HR [13].
NHEJ events display a deletion at the I-SceI DSB site with no change to the recipient markers. HR events result in the loss of the entire 22 bp oligonucleotide insert containing the I-SceI site with a concomitant replacement of one or more recipient markers with donor markers. Each HR event could further be classified as a gene conversion versus an homology-dependent deletion (HDD)1 (Fig.4). In a gene conversion, one or several recipient markers flanking the DSB site are replaced (converted) with donor markers but there is no additional sequence rearrangement. An HDD may occur either via a crossover produced via Holliday junction intermediates, or by the nonconservative process of single-strand annealing (SSA) in which DNA end-resection uncovers complementary sequences of direct repeats [3]. In an HDD event, the donor becomes joined in-frame to the recipient, all recipient markers upstream of the DSB are expected to originate from the donor, and a variable number of markers downstream of the DSB will originate from the donor depending on the precise site of joining of donor to recipient. HDDs also result in the deletion of vector sequences between donor and recipient, including the hygromycin resistance gene. The deletion of sequences produced by an HDD allows HDDs to be distinguished easily from gene conversions on Southern blots following digestion of genomic DNA with BamHI (Fig. 4).
Fig. 4.

Analysis of HR events. On the upper panels are schematic illustrations of a gene conversion and an homology-dependent deletion (HDD) that may occur within substrate pLB4. Sequences originating from donor tk are lightly shaded to better illustrate the nature of events. BamHI (B), and HindIII (H) sites are shown. Gene conversions can be distinguished from HDDs based on distinct BamHI digest patterns revealed on Southern blots using a tk probe. The lower panel shows a representative Southern blot displaying the restriction pattern obtained for a gene conversion (lane 1) versus an HDD (lane 2).
We analyzed a total of 42 DSB repair events recovered from pLB4-progerin, 25 DSB repair events recovered from pLB4-GFP, and 22 events recovered from pLB4/11 using a combination of DNA sequencing and Southern blotting. The results of our analyses are shown in Table 2.
Table 2.
Characterization of DSB repair events
| Cell line | HR events | NHEJa | |
|---|---|---|---|
| Gene conversionb | HDD | ||
| pLB4-progerin | 13 | 14 | 15 |
| pLB4-GFP | 5 | 19 | 1 |
| pLB4/11 | 4 | 16 | 2 |
The fraction of NHEJ among DSB repair events recovered from pLB4-progerin was significantly different from the fraction of NHEJ among DSB repair events recovered from pLB4-GFP and pLB4/11 (p= 2.9 × 10−3 and p= 0.035, respectively, by two-sided Fisher exact tests).
The fraction of gene conversions among HR events recovered from pLB4-progerin was significantly different from the fraction of gene conversions among HR events recovered from pLB4-GFP and pLB4/11 (p=0.041 and p=0.047, respectively, by chi squared tests).
As presented in Table 2, we recorded an increase in DSB repair via NHEJ in pLB4-progerin cells (expressing GFP-progerin) compared with DSB occurring in either pLB4-GFP (expressing GFP only) or the parent line pLB4/11. More specifically, for pLB4-progerin cells, 15 out of 42 events were due to NHEJ whereas only 1 out of 25 and only 2 out of 22 events were due to NHEJ in pLB4-GFP or pLB4/11, respectively. The difference in NHEJ recovery from pLB4-progerin versus pLB4-GFP was highly significant (p= 2.9 × 10−3 by a two-sided Fisher exact test). The difference in NHEJ recovery from pLB4-progerin versus pLB4/11 was also significant (p= 0.035 by a two-sided Fisher exact test). Among recovered HR events, there also appeared to be a greater portion of gene conversions from pLB4-progerin compared with pLB4-GFP (p=0.041 by a chi squared test) and compared with pLB4/11 (p=0.047 by a chi squared test).
For NHEJ events recovered from all three cell lines, deletions ranged from 1 bp to as many as 1126 bp, but were primarily in the range of 1 bp to 22 bp, with each deletion restoring the correct reading frame to the fusion gene (Supplemental Fig. 2). From the data set generated, there were no remarkable differences in NHEJ events recovered from the three cell lines. Comparison with additional previously published data on NHEJ [57] also did not provide evidence for a notable impact of progerin on the nature of NHEJ events. Among gene conversions recovered from the three cell lines, all involved loss of the 22 bp oligonucleotide containing the I-SceI site. The shortest conversion tracts spanned only the first marker downstream from the DSB, while other tracts spanned the first upstream marker and as many as five downstream markers (see Fig. 2 for positions of markers). Estimated minimal conversion tract lengths thus ranged from 23 bp to 325 bp. There was no notable difference between the lengths of conversion tracts recovered from the three cell lines and no mutations were found. For all events identified as HDDs on Southern blots, sequence analysis revealed that all markers upstream from the DSB originated from the donor, as expected. The site of joining of donor to recipient downstream from the DSB was variable, with the most distal sequence junction positioned between the fifth and sixth downstream marker. All HDD events recovered from cell lines pLB4-progerin, pLB4-GFP, and pLB/11 displayed in-frame fusions of donor and recipient sequences and no mutations were found.
In summary, our results suggested that progerin expression increases the likelihood that a DSB will be repaired via NHEJ rather than HR and increases the likelihood that an HR event will occur as a gene conversion.
3.3. Progerin expression increases the frequency of spontaneous HR and shifts spontaneous HR toward gene conversions
Following the DSB repair experiments described above, we were curious to learn if progerin expression impacts spontaneous intrachromosomal HR that occurs in the absence of artificial induction of a DSB at the I-SceI site in pLB4. Such recombination may be provoked by a spontaneous DSB, a stalled or collapsed replication fork, or perhaps in the absence of any lesion or particular blockage of replication. To explore progerin’s influence on spontaneous HR, cell lines pLB4-progerin, pLB4-GFP, and pLB4/11 were subjected to fluctuation tests to measure the frequency of recovery of G418R segregants. The results of these experiments, presented in Table 3, show that the frequency of spontaneous G418R segregants recovered from pLB4-progerin was about 10-fold greater than that for pLB4-GFP and more than 30-fold greater than that for pLB4/11.
Table 3.
Recovery of spontaneous G418-resistant segregants
| Cell line | Cells Plated (106)a | G418R Coloniesb | Median No. Coloniesc | Colony Frequency (10−7)d | HR Frequency (10−7)e |
|---|---|---|---|---|---|
| pLB4-progerin | 30 (10) | 228 | 17.5 | 76 | 34.7 |
| pLB4-GFP | 30 (10) | 20 | 2 | 6.8 | 6.8 |
| pLB4/11 | 145 (30) | 29 | 0 | 2.0 | 1.8 |
For each cell line, 10 or more subclones were each propagated separately to a total of 3 to 5 million cells before being plated into medium containing G418. Total number of subclones plated is presented in parentheses.
Total number of G418R colonies recovered from all of the subclones for each cell line.
Median number of colonies recovered from the subclones plated.
Total number of colonies recovered divided by the total number of cells plated.
Calculated as colony frequency multiplied by percentage of colonies characterized as bona fide HR.
To characterize the nature of the events responsible for the production of spontaneous G418R segregants, genomic DNA was isolated from representative G418R clones from the three cell lines. The events were analyzed by a combination of DNA sequencing and Southern blots, as described above for experiments involving DSB induction, and HR events were categorized as gene conversions or HDDs. Clones for which we detected no apparent nucleotide sequence change compared with the original sequence of substrate pLB4 were categorized as “other.” The results of such analyses are presented in Table 4.
Table 4.
Characterization of spontaneous G418R segregants
| Cell Line | No. Segregants Analyzed | Gene Conversionsa | HDDs | Otherb |
|---|---|---|---|---|
| pLB4-progerin | 35 | 16 | 0 | 19 |
| pLB4-GFP | 17 | 8 | 9 | 0 |
| pLB4/11 | 24 | 5 | 16 | 3 |
The fraction of gene conversions among HR events recovered from pLB4-progerin was significantly different from the fraction of gene conversions among HR events recovered from pLB4-GFP and pLB4/11 (p = 9.3 × 10−4 and p= 1.6 × 10−6, respectively, by two-sided Fisher exact tests).
The number of “other” events recovered from pLB4-progerin was significantly different from the number of such events recovered from pLB4-GFP and pLB4/11 (p= 1.1 × 10−4 and p= 1.2 × 10−3, respectively, by two-sided Fisher exact tests).
From the data presented in Table 4, two observations were striking. First, for the pLB4-progerin cell line there was a significant shift in spontaneous HR events toward gene conversion. Among the 16 analyzed HR events that were recovered from pLB4-progerin, all were gene conversions and none were HDDs. This outcome was significantly different (p = 9.3 × 10−4 by a two-sided Fisher exact test) from the distribution of HR events recovered from pLB4-GFP in which 9 out of 17 HR events were HDDs. The distribution of events recovered from pLB4-progerin was also highly significantly different (p = 1.6 × 10−6 by a two-sided Fisher exact test) from the events recovered from parent line pLB4/11 in which 16 out of 21 HR events were HDDs. Thus, expression of progerin appeared to bring about a notable shift in spontaneous HR toward gene conversion events.
The second striking observation was the large fraction of events recovered from pLB4-progerin that displayed no change in nucleotide sequence and were placed into the “other” category (Table 4). The increased fraction “other” events recovered from pLB4-progerin versus pLB4-GFP and versus pLB4/11 was significant (p= 1.1 × 10−4 and p= 1.2 × 10−3, respectively, by a two-sided Fisher exact test). Southern blotting revealed that these clones appeared to have undergone a DNA amplification event that increased the number of copies of construct pLB4 as evidenced by an increase in hybridization signal on Southern blots (Fig. 5). HR frequencies were calculated (Table 3) by discounting the “other” colonies which did not arise from bona fide HR events. With the correction for “other” events, the HR frequency for cell line pLB4-progerin nonetheless appeared higher than than for the other two cell lines.
Fig. 5.
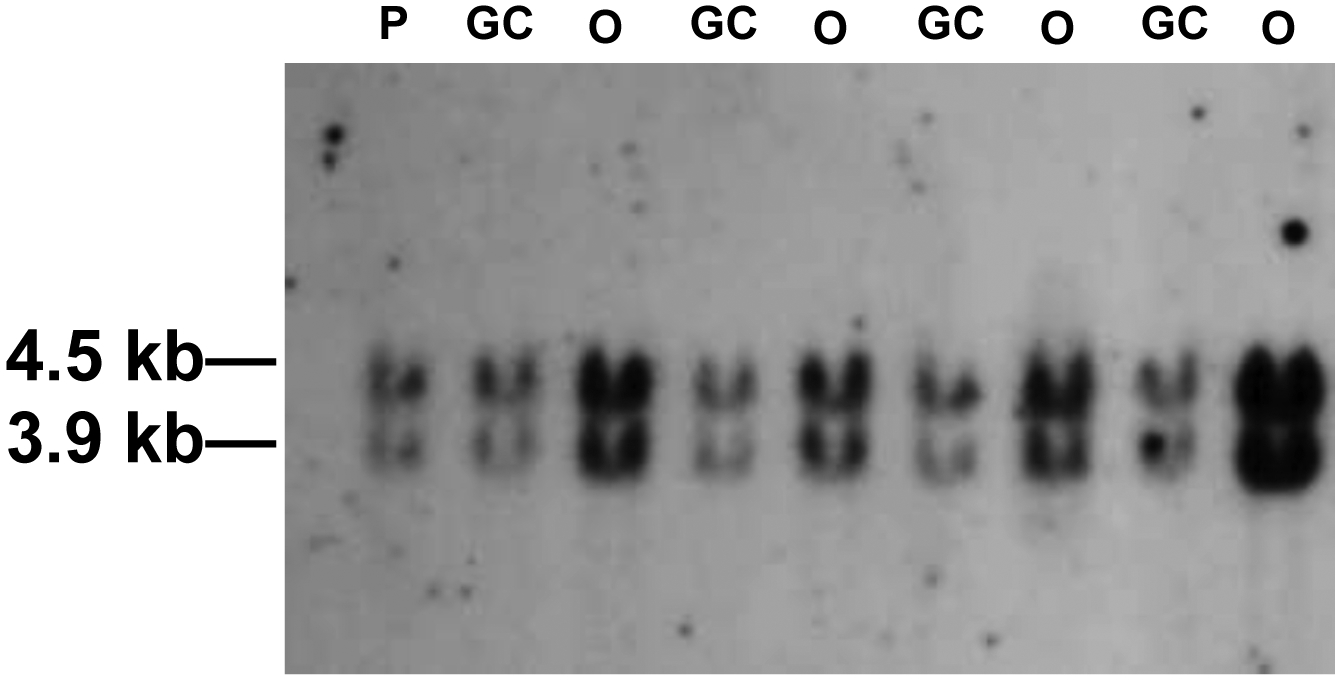
Southern blot analysis of representative G418R segregants recovered from cell line pLB4-progerin. Genomic DNA was digested with BamHI and displayed on a blot using a tk probe. DNA from cell line pLB4-progerin is presented in lane P and serves as an internal control for single-copy hybridization intensity. DNA samples from gene conversion events are presented in GC lanes, and DNA samples from “other” events are presented in O lanes. GC lanes display a hybridization intensity comparable to the P lane, while O lanes display increased hybridization intensity.
Gene conversion tracts recovered from cell lines pLB4-progerin, pLB4-GFP, and pLB4/11 displayed no notable line-to-line differences. All gene conversions displayed complete loss of the 22 bp oligonucleotide containing the I-SceI site and spanned as many as two markers upstream from the I-SceI site and as many as 7 downstream markers. No mutations were found in any of the gene conversion tracts. Similarly, all HDD events recovered from cell lines pLB4-progerin, pLB4-GFP and pLB4/11 displayed in-frame fusions of donor and recipient sequence and no mutations.
In summary, expression of progerin in cell line pLB4-progerin increased the frequency of spontaneous genetic changes that produced G418R segregants. Progerin expression appeared to provoke both an increase in HR events in the form of gene conversions as well as DNA amplification events.
4. Discussion
Lamin A is a critical component of the nuclear lamina in mammalian cells. Expression of a mutated form of lamin A known as progerin is the cause of the premature aging syndrome known as Hutchinson–Gilford Progeria Syndrome (HGPS) and has also been implicated in normal aging [32–35]. The scientific literature presents strong evidence that both lamin A and progerin exert substantial influence on DSB repair and genomic stability [19–23,27–46], and yet nucleotide-level studies on how progerin may impact characteristics of DSB repair or spontaneous recombination events have been lacking. In the current study, we sought to close this knowledge gap. Using a model system involving cultured human cells containing the integrated pLB4 DNA repair and recombination reporter construct, we report here on several impacts of progerin expression.
In experiments involving artificial induction of a genomic DSB with endonuclease I-SceI, we observed (Table 2) that progerin increased the portion of DSBs that were repaired via NHEJ rather than HR. Our results also indicated that progerin increased the portion of HR repair events that occurred by gene conversion rather than an HDD. We noted no compromise in the accuracy of HR events occurring in cells expressing progerin.
The shift in DSB repair toward NHEJ that we observed in the presence of progerin is consistent with previous reports [42,43,45,53] that NHEJ is activated in progeroid cells, apparently mediated via a progerin-associated decrease in cellular levels of PARP1 [42] which normally suppresses NHEJ. We note, however, that our work now provides direct evidence of an association between progerin expression and a shift in the pathway by which a defined DSB is repaired. The shift away from the accurate HR pathway and toward error-prone NHEJ could contribute to a compromise in genomic integrity which may play a role in the aging process.
Our data does not provide strong evidence that progerin expression reduced the overall ability of a cell to repair an induced DSB. Although colony recovery in cell line pLB4-progerin was somewhat lower than that recorded for pLB4-GFP or pLB/11 (Table 1), the relatively small decrease in recovered repair events can be explained at least in part by an increase in usage of the NHEJ pathway in cell line pLB4-progerin. Only one in three NHEJ events would be expected to re-establish an appropriate reading frame for expression of the neo coding sequence of the tk-neo gene, allowing recovery under G418 selection of only one third of NHEJ events that had actually occurred.
Our studies of spontaneous HR events that occurred in the absence of artificial DSB induction uncovered additional impacts of progerin expression. The data presented in Table 3 revealed an order of magnitude increase in spontaneous production of G418R segregants in cells expressing progerin. This increase in G418R segregants may be considered a proxy for increased genomic instability since the formation of each segregant necessarily required a genetic change. An increase in genomic DSBs and stalled or collapsed replication forks, which may be expected to occur in cells expressing progerin [18–23,26–53], could potentially provide lesions that trigger the genetic alterations needed to produce G418R segregants.
Analysis of G418R segregants also showed two conspicuous differences in the nature of events that were recovered from cells expressing progerin versus cells that did not express progerin. Unlike the clones that arose from cell lines pLB4-GFP and pLB4/11, a substantial portion of G418R clones that arose from cell line pLB4-progerin were actually not products of HR but, rather, appeared to have arisen as a result of DNA amplification events (Table 4, Fig. 5). We suggest that despite the disrupting oligonucleotide in the tk portion of the tk-neo fusion gene in construct pLB4, a low level of leaky expression from the neo coding sequence occurred. We infer that this leaky expression was at too low a level to allow growth under the concentration of G418 used in the selection medium, but increased copy number of the tk-neo gene overcame the selection due to a concomitant increase in the aggregate level of leaky expression.
Regarding recovered HR events, we observed a marked shift toward gene conversions in the absence of crossing over among spontaneous HR events in cells expressing progerin (Table 4). This outcome was similar to, if more extreme than, what we observed for DSB repair in the presence of progerin (Table 2). For our experiments using I-SceI to induce a DSB, the lesion responsible for provoking the recovered HR events was known, and each HR event directly repaired the lesion. In the case of spontaneous HR events, neither the provoking lesion nor its location was known. It is not unlikely that stalled or collapsed replication forks, rather than or in addition to classic two-ended DSBs, may contribute to some or many spontaneous events [61]. It is also possible that spontaneous events may not require any lesion to proceed. The observation that progerin facilitated gene conversions over HDDs, regardless of whether or not we introduced a strategically located DSB to trigger a repair event, is suggestive of a global cellular change in availability of one or more proteins involved in a fundamental role in HR.
How might expression of progerin selectively induce gene conversions at the expense of HDDs? A precise molecular dissection of this issue is beyond the scope of this study, but nonetheless we can speculate. A relevant issue is the question of whether HDDs are produced via crossovers or by SSA? If HDD events are largely products of SSA, it is conceivable that our results may be explained by a progerin-induced reduction of DNA end resection. Supression of end resection would likely hinder SSA which requires fairly extensive resection. Supression of end resection would also be predicted to reduce HR in favor of NHEJ. However, we believe that evidence garnered from prior work with our experimental system favors crossovers for a number of reasons, as we elaborated on previously [62]. In the discussion that follows, we will assume that the majority of HDDs occur via crossovers.
It has been shown [36,48] that recruitment of HR proteins to the site of DSBs is hampered in HGPS cells. We posit that one consequence of progerin-associated disruption of HR is a reduction in the recombination machinery’s ability to establish Holliday junction recombination intermediates at the site of HR, whether that site be a classic DSB, a stalled or collapsed replication fork, or perhaps a site of no lesion at all. The formation of a full Holliday junction or double Holliday junction is required for a crossover but is not required for a gene conversion which may occur via a synthesis dependent strand annealing mechanism [63]. This hypothesized decline in production full-fledged Holliday junction intermediates at sites at which HR is initiated could thus explain a selective reduction in crossover events with a lesser impact on gene conversions.
A general overall impairment in a cell’s ability to carry out HR combined with an activation of NHEJ [42,43,45,53] in cells expressing progerin would be expected to produce a shift toward NHEJ as a means of DSB repair, as we indeed recorded in our experiments involving DSB induction. However, our experiments involving spontaneous recovery of G418R segregants indicated an overall increase in recovery of spontaneous HR events in progerin-expressing cells, a result which may appear to counter expectations. This latter observation might be explained by a potential progerin-induced increase in stalled or collapsed replication forks, and other HR-triggering lesions or chromatin alterations. We would argue that on a per lesion basis, spontaneous HR may very well be reduced in cells expressing progerin. This latter notion suggests that, in pLB4-progerin cells, many DSBs and other lesions would be channeled into DNA transactions distinct from HR and, indeed, this may have manifested as an increased recovery of DNA amplification events seen in pLB4-progerin cells. Although mechanisms for amplification are not completely understood, most models invoke DSBs or stalled replication forks as initiating events [64–66].
One noteworthy observation reported for HGPS patients is that such individuals do not develop cancers despite the accumulation of high levels of DNA damage [67,68]. The lack of cancer in HGPS individuals also stands in contrast to the general observation of increased cancer incidence with age. The cancer suppressive phenomenon associated with HGPS was reported [68] to be largely explainable by a progerin-induced altered landscape of histone modifications that leads to a redistribution of binding sites for the bromodomain transcriptional regulator BRD4. This redistribution of BRD4 in turn impacts the cellular gene expression profile in a way that engenders an inhibition of oncogenic dedifferentiation [68].
Our current work reveals an impact of progerin that may also contribute to an anti-cancer mechanism. As discussed, our work indicates that progerin shifts DSB repair away from HR and shifts HR away from HDD which includes crossovers. It is not unreasonable to infer that such shifts in nucleic acid metabolism may result in a generalized reduction in the frequency of loss of heterozygosity (LOH) since mitotic crossing over between homologous chromosomes is likely one important mechanism for LOH. One would predict that a reduced rate of LOH of defective tumor suppressor alleles in particular would reduce the incidence of cancer. Very much related to this latter notion are the impacts of defects in the Bloom helicase (BLM) in human cells. BLM deficiency is the molecular basis of Bloom syndrome (BS), which is sometimes classified as a premature aging syndrome. We had previously shown [62] that depletion of BLM in normal cells shifts DSB repair toward HR and increases the frequency of crossovers specifically. These effects are essentially the opposite of the effects of progerin expression reported in our current work. Consistent with our findings regarding BLM depletion, the frequency of mitotic crossing-over in BS cells is estimated to be 20–50 times the frequency in normal cells and an escalation of LOH has indeed been attributed directly to BLM deficiency [69]. Of great relevance to this discussion is the observation that, in stark contrast with HGPS, BS is associated with a strong predisposition to cancer of many types [69]. A murine model has also been described in which elevated mitotic crossing-over associated with BLM deficiency drives cancer development [70]. Elevated mitotic crossing-over and LOH has additionally been reported in BLM-deficient mouse embryonic stem cells [71]. From the collective observations for impacts of BLM deficiency and our current observations for progerin expression, suppression of crossovers emerges as a potential mechanism to protect against carcinogenesis in humans and other mammals.
Finally, we note that for DSB-induced HR as well as spontaneous HR events, we found that accuracy of HR in cells expressing progerin was not compromised as we recovered no mutations. Our finding is consistent with a previous report [54] in which no increase in mutation rate was found in a murine epithelial cell line engineered to express progerin. However, our studies lead us to differ with the general assessment expressed in the previous work that the cellular defect in HGPS cells does not lie in the repair of DNA damage per se. We must be cognizant that the findings that are obtainable in any study are a function of the assay(s) used. In our current work, we obtained data indicating that progerin expression may shift the pathway of DSB repair away from HR, shift spontaneous and DSB-induced HR away from HDDs, and increase gene amplification events. None of these alterations would have been observable in the assay used previously [54], and each of these alterations may be a reflection of one or more defects in DNA repair.
Alterations in DNA transactions are associated with both HGPS and normal aging. Proper maintenance of the genome is undeniably critical for the health and well-being of a living organism, and changes in genome maintenance schemes are likely inextricably linked to the biology of aging. As diverse experimental approaches shed more light on how DNA maintenance pathways change with age, we hope to better understand the complex process of aging and to become better positioned to further ameliorate some of the consequences of accelerated and normal aging.
Supplementary Material
Highlights.
Progerin increases nonhomologous end-joining relative to homologous recombination
Progerin promotes homologous recombination via gene conversion
Progerin expression increases the frequency of genomic DNA amplification
Acknowledgements
This work was supported by the National Institute on Aging [grant R03AG064525] and the University of South Carolina [ASPIRE Proposal 13010-19-50807].
Gratitude is expressed toward the University of South Carolina for strongly encouraging undergraduate research. C.J.K., A.O.G., S.R.C., T.L.T., E.A.O., A.V.H. and S.C. all contributed to this research as undergraduates.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement
The authors declare that there are no conflicts of interest.
Abbreviation: HDD, homology-dependent deletion
References
- [1].Sonoda E, Hochegger H, Saberi A, Taniguchi Y, Takeda S, Differential usage of non-homologous end-joining and homologous recombination in double-strand break repair, DNA Repair (Amst.) 5 (2006) 1021–1029. [DOI] [PubMed] [Google Scholar]
- [2].San Filippo J, Sung P, Klein H, Mechanism of eukaryotic homologous recombination, Annu. Rev. Biochem 77 (2008) 229–257. [DOI] [PubMed] [Google Scholar]
- [3].Hartlerode AJ, Scully R, Mechanisms of double-strand break repair in somatic mammalian cells, Biochem. J 423 (2009) 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pardo B, Gómez-González B, Aguilera A, DNA double strand break repair: how to fix a broken relationship, Cell Mol. Life Sci 66 (2009) 1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shrivastav M, De Haro LP, Nickoloff JA, Regulation of DNA double-strand break repair pathway choice, Cell Res. 18 (2009) 134–147. [DOI] [PubMed] [Google Scholar]
- [6].Heyer WD, Ehmsen KT, Liu J, Regulation of homologous recombination in eukaryotes, Annu. Rev. Genet 44 (2010) 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kass EM, Jasin M, Collaboration and competition between DNA double-strand break repair pathways, FEBS Lett. 584 (2010) 3703–3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chapman JR, Taylor MR, Boulton SJ, Playing the end game: DNA double-strand break repair pathway choice, Mol. Cell 47 (2012) 497–510, 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- [9].Goodarzi AA, Jeggo PA, The repair and signaling responses to DNA double-strand breaks, Adv. Genet 82 (2013) 1–45, 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- [10].Iyama T, Wilson III DM, DNA repair mechanisms in dividing and non-dividing cells, DNA Repair (Amst.) 12 (2013) 620–636, 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Scully R, Panday A, Elango R, Willis NA, DNA double-strand break repair-pathway choice in somatic mammalian cells, Nat. Rev. Mol. Cell Biol 20 (2019) 698–714, 10.1038/s41580-019-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Waldman AS, Liskay RM, Differential effects of base-pair mismatch on intrachromosomal versus extrachromosomal recombination in mouse cells, Proc. Natl. Acad. Sci. USA 84 (1987) 5340–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Waldman AS, Liskay R.M RM, Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology, Mol. Cell. Biol 8 (1988) 5350–5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lukacsovich T, Waldman AS, Suppression of intrachromosomal gene conversion in mammalian cells by small degrees of sequence divergence, Genetics 151 (1999) 1559–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bishop AJ, Schiestl RH, Role of homologous recombination in carcinogenesis, Exp. Mol. Pathol 74 (2003) 94–105. [DOI] [PubMed] [Google Scholar]
- [16].Eyfjord JE, Bodvarsdottir SK, Genomic instability and cancer: networks involved in response to DNA damage, Mutat. Res 592 (2005) 18–28. [DOI] [PubMed] [Google Scholar]
- [17].Kennedy RD, D’Andrea AD, DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes, J Clin. Oncol 24 (2006) 3799–3808. [DOI] [PubMed] [Google Scholar]
- [18].Gorbunova V, Seluanov A, Making A ends meet in old age: DSB repair and aging, Mech. Ageing Dev 126 (2005) 621–628. [DOI] [PubMed] [Google Scholar]
- [19].Kyng KJ, Bohr VA, Gene expression and DNA repair in progeroid syndromes and human aging, Ageing Res. Rev 4 (2005) 570–602. [DOI] [PubMed] [Google Scholar]
- [20].Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW, DNA repair, genome stability, and aging, Cell 120 (2005) 497–512. [DOI] [PubMed] [Google Scholar]
- [21].Gorbunova V, Seluanov A, Mao Z, Hine C, Changes in DNA repair during aging, Nucleic Acids Res 35 (2007) 7466–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Freitas AA, de Magalhães JP, A review and appraisal of the DNA damage theory of ageing, Mutat. Res 728 (2011) 12–22. [DOI] [PubMed] [Google Scholar]
- [23].Tiwari V, Wilson DM 3rd, DNA damage and associated DNA repair defects in disease and premature Aging, Am. J. Hum. Genet 105 (2019) 237–257. doi: 10.1016/j.ajhg.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ren K, de Ortiz SP, Non-homologous DNA end joining in the mature rat brain, J Neurochem. 80 (2002) 949–59. [DOI] [PubMed] [Google Scholar]
- [25].Vijg J, Dolle MET, Large genome rearrangements as a primary cause of aging, Mech. Ageing Dev 123 (2002) 907–915. [DOI] [PubMed] [Google Scholar]
- [26].Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, Gorbunova V, DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc. Natl. Acad. Sci. U S A 101 (2004) 7624–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bachrati CZ, Hickson ID, RecQ helicases: suppressors of tumorigenesis and premature aging, Biochem. J 374 (2003) 577–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].O’Driscoll M, Jeggo PA, The role of double-strand break repair - insights from human genetics, Nat. Rev. Genet 7 (2006) 45–54. [DOI] [PubMed] [Google Scholar]
- [29].Brosh RM Jr., Bohr VA, Human premature aging, DNA repair and RecQ helicases, Nucleic Acids Res. 35 (2007) 7527–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G, The hallmarks of aging, Cell 153 (2013) 1194–1217, doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Burla R, La Torre M, Merigliano C, Vernì F, Saggio I, Genomic instability and DNA replication defects in progeroid syndromes, Nucleus, 9 (2018) 368–379, doi: 10.1080/19491034.2018.1476793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS, Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts, J. Clin. Invest 121 (2011) 2833–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, S Collins F, Djabali K, The mutant form of lamin A that causes Hutchinson-Gilford Progeria is a biomarker of cellular aging in human skin, PLoS ONE 2 (2007) e1269. doi: 10.1371/journal.pone.0001269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Scaffidi P, Misteli T, Lamin A-dependent nuclear defects in human aging, Science 312 (2006) 1059–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ashapkin VV, Kutueva LI, Kurchashova SY, Kireev II, Are there common mechanisms between the Hutchinson-Gilford Progeria Syndrome and Natural Aging?, Front. Genet 10 (2019) 455. doi: 10.3389/fgene.2019.00455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, Zou Y, Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A, FASEB J. 22 (2008) 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Musich PR, Zou Y, Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A, Aging 1 (2009) 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Serio RN, Unraveling the mysteries of aging through a Hutchinson–Gilford progeria syndrome model, Rejuvenation Res. 14 (2011) 133–141. [DOI] [PubMed] [Google Scholar]
- [39].Musich PR, Zou Y, DNA-damage accumulation and replicative arrest in Hutchinson–Gilford progeria syndrome, Biochemical Soc. Trans 39 (2011) 1764–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gonzalez-Suarez I, Redwood AB, Grotsky DA, Neumann MA, Cheng EH, Stewart CL, Dusso A, Gonzalo S, A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair, EMBO J. 30 (2011) 3383–3396. doi: 10.1038/emboj.2011.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage J, Roti-Roti JL, Stewart CL, Zhang J, Gonzalo S, A dual role for A-type lamins in DNA double-strand break repair, Cell Cycle 10 (2011) 2549–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang H, Xiong ZM, Cao K, Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1, Proc. Natl. Acad. Sci. USA 111 (2014) E2261–E2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gonzalo S, DNA damage and lamins, Adv. Exp. Med. Biol 773 (2014) 377–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gibbs-Seymour I, Markiewicz E, Bekker-Jensen S, Mailand N, Hutchison CJ, Lamin A/C-dependent interaction with 53BP1 promotes cellular responses to DNA damage, Aging Cell 14 (2015) 162–169. doi: 10.1111/acel.12258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gonzalo S, Kreienkamp R, DNA repair defects and genome instability in Hutchinson-Gilford Progeria Syndrome, Curr. Opin. Cell. Biol 34 (2015) 75–83. doi: 10.1016/j.ceb.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Huang X, Pan Y, Cao D, Fang S, Huang K, Chen J, Chen A, UVA-induced upregulation of progerin suppresses 53BP1‑mediated NHEJ DSB repair in human keratinocytes via progerin-lamin A complex formation, Oncol. Rep 37 (2017) 3617–3624. doi: 10.3892/or.2017.5603. [DOI] [PubMed] [Google Scholar]
- [47].Ragnauth CD, Warren DT, Liu Y, McNair R,Tajsic T, Figg N, Shroff R, Skepper J, Shanahan CM, Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging, Circulation 121(2010) 2200–2210. [DOI] [PubMed] [Google Scholar]
- [48].Constantinescu D, Csoka AB, Navara CS, Schatten GP, Defective DSB repair correlates with abnormal nuclear morphology and is improved with FTI treatment in Hutchinson-Gilford progeria syndrome fibroblasts, Exp. Cell Res 316 (2010) 2747–2759. [DOI] [PubMed] [Google Scholar]
- [49].Richards SA, Muter J, Ritchie P, Lattanzi G, Hutchison CJ, The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine, Hum. Mol. Genet 20 (2011) 3997–4004. [DOI] [PubMed] [Google Scholar]
- [50].Hutchison CJ, The role of DNA damage in laminopathy progeroid syndromes, Biochem. Soc. Trans 39 (2011) 1715–1718. [DOI] [PubMed] [Google Scholar]
- [51].Gonzalez-Suarez I, Gonzalo S, Nurturing the genome: A-type lamins preserve genomic stability, Nucleus 1 (2010) 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].von Muhlinen N, Horikawa I, Alam F, Isogaya K, Lissa D, Vojtesek B, Lane DP, Harris CC, p53 isoforms regulate premature aging in human cells, Oncogene 37 (2018)2379–2393. doi: 10.1038/s41388-017-0101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadinanos J, Lopez-Otin C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z, Genomic instability in laminopathy-based premature aging, Nat. Med 11 (2005) 780–785. [DOI] [PubMed] [Google Scholar]
- [54].Deniaud E, Lemaître C, Boyle S, Bickmore WA, Expression of progerin does not result in an increased mutation rate, Chromosome Res. 25 (2017) 227–239. doi: 10.1007/s10577-017-9556-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Smith JA, Bannister LA, Bhattacharjee V, Wang Y, Waldman BC, Waldman AS, Accurate homologous recombination is a prominent double-strand break repair pathway in mammalian chromosomes and is modulated by mismatch repair protein Msh2, Mol. Cell. Biol 27 (2007) 7816–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bannister LA, Waldman BC, Waldman AS, Modulation of error-prone double-strand break repair in mammalian chromosomes by DNA mismatch repair protein Mlh1, DNA Repair 3 (2004) 465–474. [DOI] [PubMed] [Google Scholar]
- [57].Waldman BC, Wang Y, Kilaru K, Yang Z, Bhasin A, Wyatt MD, Waldman AS, Induction of intrachromosomal homologous recombination in human cells by raltitrexed, an inhibitor of thymidylate synthase, DNA Repair 7 (2008) 1624–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wang Y, Li S , Smith K , Waldman BC, Waldman AS, Intrachromosomal recombination between highly diverged DNA sequences is enabled in human cells deficient in Bloom helicase, DNA Repair 41 (2016) 73–84. doi: 10.1016/j.dnarep.2016.03.005. [DOI] [PubMed] [Google Scholar]
- [59].Wagner MJ, Sharp JA, Summers WC, Nucleotide sequence of the thymidine kinase of herpes simplex virus type 1, Proc. Natl. Acad. Sci. U.S.A 78 (1981) 1441–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chapman KM, Wilkey MM, Potter KE, Waldman BC, Waldman AS, High homology is not required at the site of strand invasion during recombinational double-strand break repair in mammalian chromosomes, DNA Repair (Amst) 60 (2017)1–8. doi: 10.1016/j.dnarep.2017.10.006. [DOI] [PubMed] [Google Scholar]
- [61].Helleday T, Pathways for mitotic homologous recombination in mammalian cells, Mutat. Res 532 (2003) 103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- [62].Wang Y, Smith K, Waldman BC, Waldman AS, Depletion of the Bloom syndrome helicase stimulates homology-dependent repair at double-strand breaks in human chromosomes, DNA Repair 10 (2011) 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Morrical SW, DNA-pairing and annealing processes in homologous recombination and homology-directed repair, Cold Spring Harb. Perspect. Biol 7 (2015) :a016444. doi: 10.1101/cshperspect.a016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lee JA, Carvalho CM JR. Lupski JR, A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders, Cell 131 (2007) 1235–1247. [DOI] [PubMed] [Google Scholar]
- [65].Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD SD, Lupski JR, The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans, Nat. Genet 41 (2009): 849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mondello C, Smirnova A, Giulotto E, Gene amplification, radiation sensitivity and DNA double-strand breaks, Mutat Res 704 (2010) 29–37. [DOI] [PubMed] [Google Scholar]
- [67].Gordon LB, Rothman FG, López-Otín C, Misteli T, Progeria: a paradigm for translational medicine, Cell 156 (2014) 400–407. doi: 10.1016/j.cell.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Fernandez P, Scaffidi P, Markert E, Lee JH, Rane S, Misteli T, Transformation resistance in a premature aging disorder identifies a tumor-protective function of BRD4, Cell Rep. 9 (2014) 248–260. doi: 10.1016/j.celrep.2014.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].de Renty C, Ellis NA, Bloom’s syndrome: Why not premature aging?: A comparison of the BLM and WRN helicases, Ageing Res Rev. 33 (2017) :36–51. doi: 10.1016/j.arr.2016.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A,. Cancer predisposition caused by elevated mitotic recombination in Bloom mice, Nat. Genet 26 (2000) 424–429. [DOI] [PubMed] [Google Scholar]
- [71].LaRocque JR, Stark JM, Oh J, Bojilova E, Yusa K, Horie K, Takeda J, Jasin M, Interhomolog recombination and loss of heterozygosity in wild-type and Bloom syndrome helicase (BLM)-deficient mammalian cells, Proc. Natl. Acad. Sci. U S A 108 (2011) 11971–11976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.