

Abstract
Intestinal organoids have proven to be useful in vitro models for many basic and translational studies aimed at understanding and treating disease. However, their routine culture relies on animal derived matrices that limit their utility for clinical applications. In fact, there are few fully defined, synthetic hydrogel systems that allow for the development and expansion of intestinal organoids. Here, we present an allyl sulfide photodegradable hydrogel, achieving rapid degradation through radical addition-fragmentation chain transfer reactions, to support the routine passaging of intestinal organoids. We use shear rheology to first characterize the effect of soluble thiol and allyl sulfide crosslink structures on degradation kinetics. Irradiation with 365nm light (5 mW cm−2) in the presence of a soluble thiol (glutathione at 15 mM), and a photoinitiator (lithium phenyl-2,4,6-trimethylbenzoylphosphinate at 1mM), led to complete hydrogel degradation in less than 15 seconds. Allyl sulfide hydrogels were used to support the formation of epithelial colonies from single intestinal stem cells, and rapid photodegradation was used to achieve repetitive passaging of stem cell colonies without loss in morphology or organoid formation potential. This platform could be used to support long term culture of intestinal organoids, potentially replacing the need for animal derived matrices, while also allowing systematic variations to the hydrogel properties tailored for the organoid of interest.
Under appropriate culture conditions, many stem cells have the remarkable capacity to proliferate and self-organize into organ-specific three-dimensional constructs called organoids. These miniature tissues possess hallmarks of the parent tissue, including stem cell compartmentalization and rudimentary function, which enables investigations into organ development[1,2], disease progression[3,4], and opens new avenues for drug discovery and screening.[5–7] Moreover, organoids represent a potentially scalable source of transplantable tissue.[8–10] The broad applicability of organoid cultures has created an explosion in organoid research involving a growing number of different tissues, including brain, lung, and kidney.[11,12] Among epithelial tissues, intestinal organoids have grown to be one of the most widely used and studied model systems, after Clevers and coworkers first reported the growth of intestinal organoids with crypt-villus architecture from single stem cells isolated from intestinal crypts.[13] Despite these advancements, translation to a clinical setting has yet to come to fruition. This is at least partially due to a lack of appropriate cell culture platforms. Current organoid culture techniques rely almost exclusively on basement membrane extracts, such as Matrigel. While supporting cellular growth, these animal-derived matrices are an ill-defined mixture of proteins and growth factors that present problems towards achieving reproducible, scalable, and transplantable tissues.[14] As such, there is a need to develop alternatives to Matrigel, based on well-defined material chemistries for the growth and expansion of intestinal organoids.
Synthetic matrices provide a “blank slate” environment in which the minimal matrix requirements for organoid growth can be determined using a bottom-up approach. As an example of using this approach, Lutolf et al. developed a synthetic poly(ethylene glycol) (PEG) based hydrogel to support intestinal organoid development and determined the dependence of stem cell colony formation and organoid patterning on matrix elastic modulus and adhesive ligand presentation.[15] The proliferation of single encapsulated stem cells requires sufficient mechanical activation through a matrix stiffness of approximately 1.3 kPa. Progression from the symmetric spheroid to the crypt-villus architecture, however, was found to rely on matrix softening – in this case introduced through hydrolysis of network ester groups – to relieve the compressive forces built up during proliferation. Similarly, degradation permits the in vitro expansion of organoids, which requires successive rounds of passaging and growth in supportive, 3D environments. Through judicious selection of synthetic hydrogel chemistries, conditions can be identified where organoid formation precede complete gel erosion; however, the predefined degradation kinetics and lack of spatial control ultimately limits the versatility of this system. This motivated our interest in exploiting photocontrolled degradation mechanisms[16,17] that when integrated into organoid culture scaffolds would offer spatial, temporal, and rate control by manipulating the location, timing, and intensity of applied light. Photodegradation has been used extensively to modulate hydrogel mechanical properties[18–20], allowing for investigations into cellular mechanosensation mechanisms[21,22] and cell spreading and migration.[23–25]
While photochemistry offers certain benefits over other degradation mechanisms, many photodegradable hydrogels utilize inefficient cleavage mechanisms or suffer from light attenuation in thick samples; thus necessitating long exposure times, high light intensities, or thin films that are not always suitable for 3D cell culture. In response, allyl sulfide hydrogels were developed, and shown to degrade rapidly under mild light intensity and photoinitiator concentrations by an amplified chemical process.[16,17] Specifically, a photoinitiator is used to generate free thiyl radicals in solution, which cleave allyl sulfide crosslinks through a reversible addition-fragmentation chain transfer (RAFT) reaction. The allyl sulfide is regenerated along with a new thiyl radical that can initiate further cycles of exchange through chain transfer to soluble thiols (Scheme 1).[16,26] The rate of thiol-ene addition has been extensively studied[27,28] and shown to depend on the protonation state of the thiol in aqueous solutions.[29] Recently, the formation of disulfide radical anions (DRAs) by the recombination of a thiolate anion and a thiyl radical, was shown to sequester catalytic radical species and slow thiol-ene conversion rates.[30] These results suggest that the protonation state of thiols involved in allyl sulfide exchange, either as soluble monofunctional thiols or network thiols released from allyl sulfides, could influence the rate of photodegradation. Here, we present a new allyl sulfide crosslinker that undergoes degradation more rapidly than previously reported kinetics.[16] This platform is then used as a reversible scaffold to promote colony growth from single intestinal stem cells (ISCs), maintaining organoid formation potential through multiple rounds of photodegradation-mediated passaging.
Scheme 1.
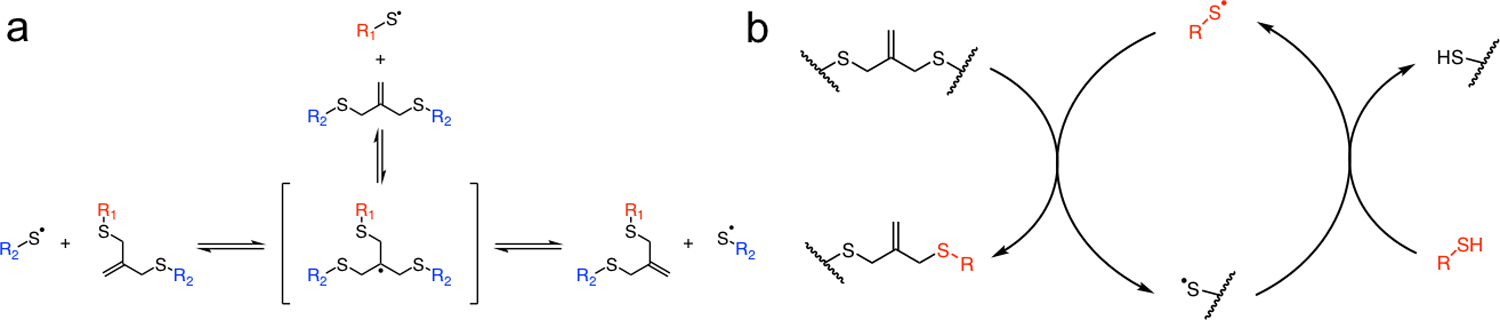
Light initiated radical network degradation. a Thiyl radicals react with an allyl sulfide to form a radical intermediate that subsequently undergoes beta scission, resulting in the fragmentation of crosslinks and the regeneration of a thiyl radical and allyl sulfide. b A soluble thiol species (red) exchanges with the allyl sulfide, resulting in a cleaved crosslink and the generation of a network tethered thiyl radical. The released network thiyl radical undergoes chain transfer to a soluble thiol species, which can initiate further exchange reactions, amplifying the degradation process.
Results:
Crosslinked hydrogels were formed through a strain promoted azide alkyne (SPAAC) reaction with tetrafunctional poly(ethylene glycol) dibenzylcyclooctyne (PEG-4DBCO) and an allyl sulfide bis(PEG3-azide) (Figure 1a). This bio-orthogonal click reaction proceeds rapidly in solution at physiological conditions and has previously been used as a cytocompatible crosslinking strategy (Figure 1b).[24,31] Three different diazide-functionalized allyl sulfide (AS) crosslinkers were synthesized, with either acetamide (AS-AA), propionamide (AS-PA), or phenyl acetamide (AS-PhAA) groups adjacent to the allyl sulfide functionality. These functionalities were chosen to study the effect of allyl sulfide crosslinker structure on degradation kinetics. Hydrogels formed with 6 wt% PEG-4DBCO (12 × 10−3 m DBCO) and either AS-AA, AS-PA, and AS-PhAA (14.4 × 10−3 m azide) crosslinkers reached the gel point (crossover of G’ and G”) in <30s and achieved the final storage modulus before 600 seconds, which remained unchanged upon plateauing (Figure 1c and Figure S1, Supporting Information). Hydrogels achieved equilibrated storage moduli of 1400 ± 270, 1200 ± 200, and 1100 ± 180 Pa, respectively. The differences in storage modulus are not statistically significant (Figure S2, Supporting Information). This hydrogel formulation was chosen based on the targeted range of stiffness for optimal ISC culture in synthetic hydrogels, previously shown to be 1.3 kPa.[15] A frequency sweep revealed that the storage and loss moduli display no frequency dependence, indicating elastic behavior in the absence of radical generation (Figure 1d).
Figure 1.
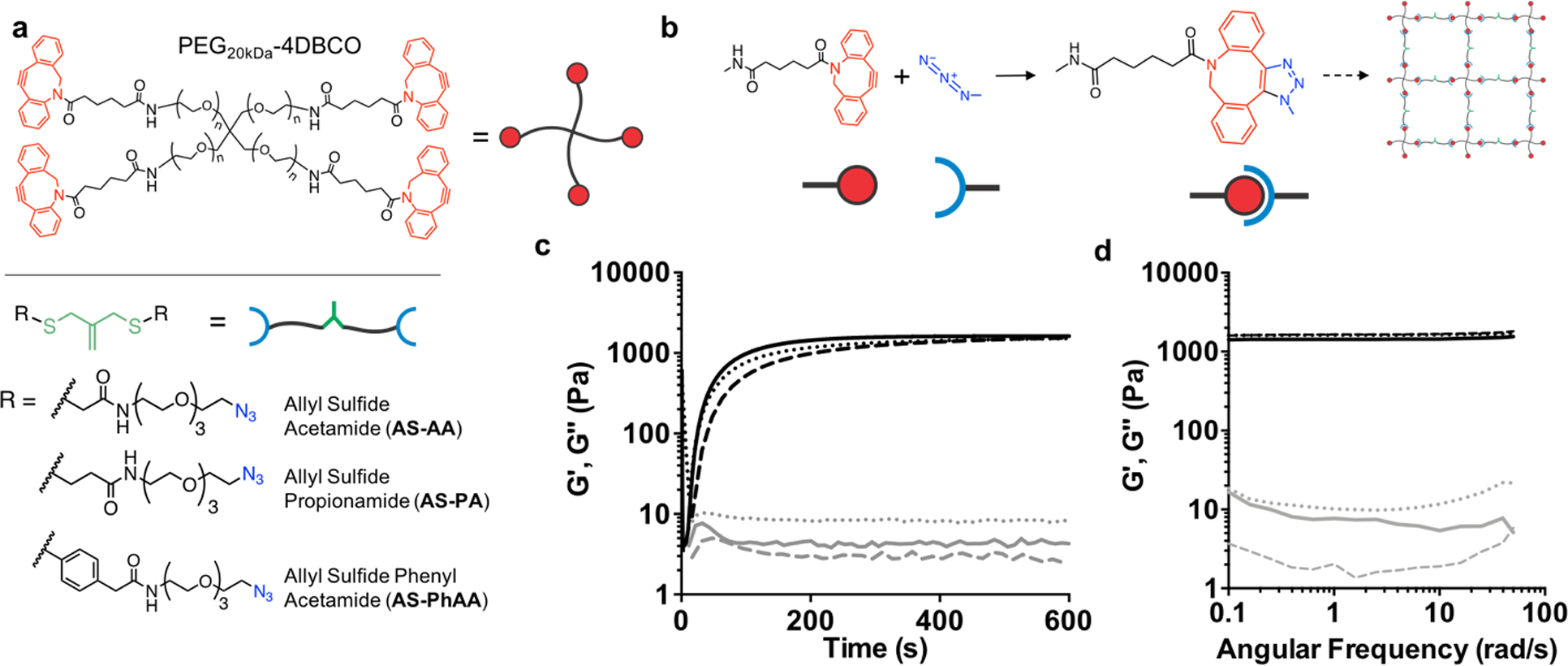
Network structure of AFCT-based photodegradable hydrogels. a Structures of the cyclooctyne functionalized PEG macromer, PEG-4DBCO, and the azide functionalized crosslinkers, AS-AA, AS-PA, AS-PhAA containing the allyl sulfide functionality. b Upon mixing, the strained octyne reacts with the azide to form a bond through strain-promoted azide alkyne cycloaddition (SPAAC), resulting in the formation of a hydrogel network incorporating the allyl sulfide functionalities. c The evolution of shear storage (black) and loss (gray) moduli are monitored with shear rheology for AS-PA (solid line), AS-AA (dotted), and AS-PhAA (dashed) crosslinkers. Hydrogels reach a similar shear modulus after 10 minutes. d A frequency sweep of the shear storage (black) and loss (gray) moduli for AS-PA (solid line), AS-AA (dotted), and AS-PhAA (dashed) crosslinked hydrogels shows a lack of frequency dependence, indicative of elastic networks.
To determine the effect of the crosslinker structure on degradation kinetics, hydrogels were formed using AS-PA, AS-AA, and AS-PhAA crosslinkers and placed in aqueous solutions containing 3-methyl mercaptopropionate (3MMP, pKa = 10.4[32]) at 15 × 10−3 m and the photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) at 1 × 10−3 m. A small amount of tri(ethylene glycol) monoazide (PEG3-N3) was also included to cap any unreacted DBCO functionalities, which have been shown to undergo side reactions in the presence of photoinitiator.[33] The addition of PEG3-N3 shows no effect on the equilibrated modulus over time (Figure S3, Supporting Information). In the presence of a soluble thiol, AS-PA, AS-AA, and AS-PhAA cleavage products present thiols of decreasing pKa values (Figure 2a). Crosslink cleavage was characterized using a rheometer equipped with a light curing accessory to track the change in shear storage (G’) modulus during irradiation (365 nm, 5 mW cm−2). As seen in Figure 2b, the AS-PA and AS-AA achieved reverse gelation, with AS-PA being more rapid, while AS-PhAA did not (i.e., remained a percolating 3D network). The degradation of AS-PA, AS-AA, and AS-PhAA hydrogels with 3MMP corresponds to rate constants of kapp / I0 × 10−4 of 310 ± 50, 190 ± 60, and 70 ± 10 cm2 mW−1 s−1, respectively (Figure 1c). This trend follows the pKa of the cleavage product, in which the propylamide thiol has the highest pKa (10.4)[32], while the phenyl thiol has the lowest pKa (6.6)[34]. As thiol pKa decreases, increased thiolate concentration in the presence of thiyl radicals leads to increased formation of DRAs, which sequester radicals and inhibit the degradation process.[30] Previous work with AS hydrogels used the AS-AA crosslinker.[16] These results show that the use of the AS-PA crosslinker, which yields a cleavage product with a higher pKa, results in more rapid degradation, ultimately allowing for lower light exposure and decreased radical generation, potentially improving the cytocompatibilty of the degradation process. The AS-PA crosslinker was therefore chosen to conduct all cell studies.
Figure 2.
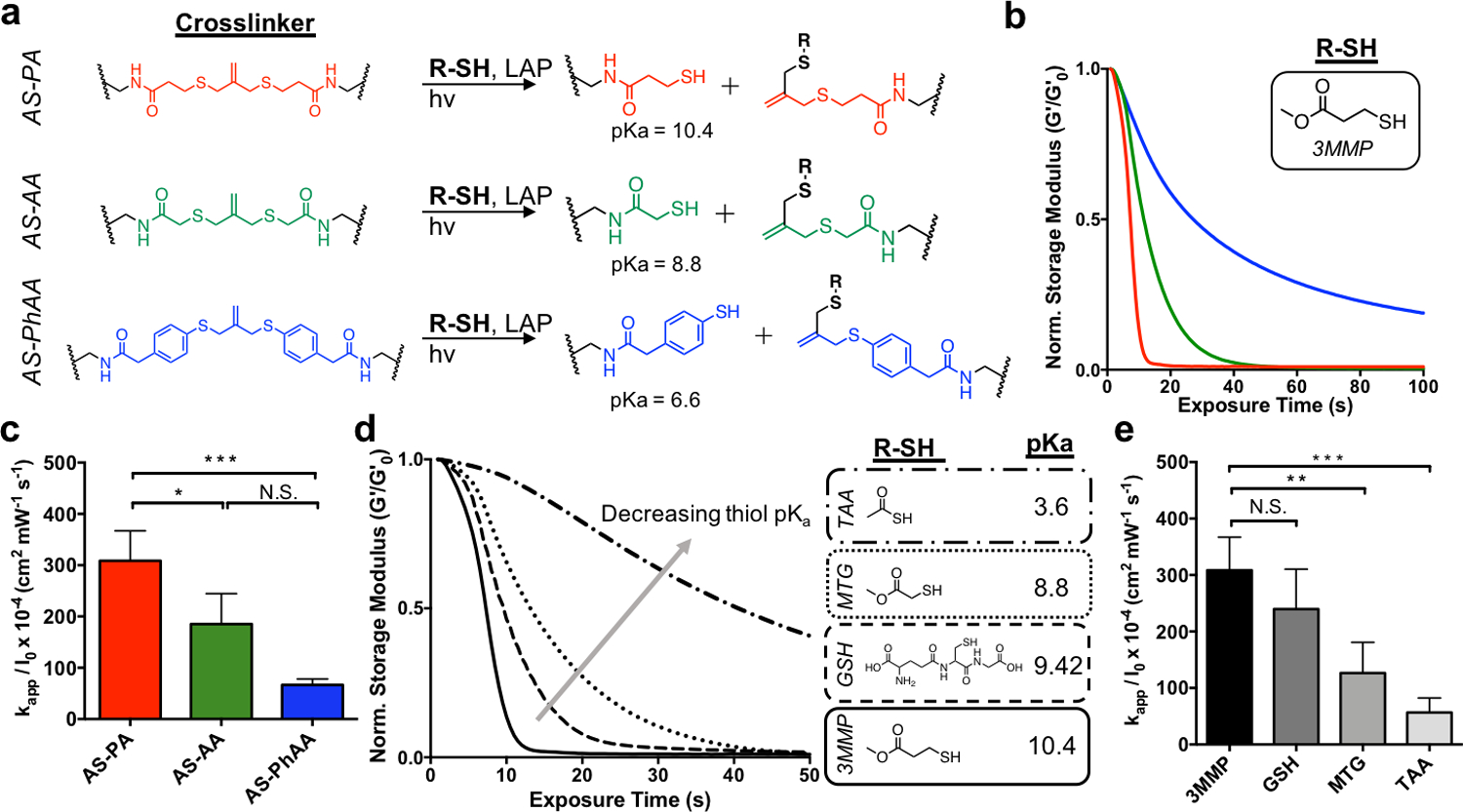
The effect of thiol structure on allyl sulfide hydrogel degradation kinetics. a Reaction mechanisms for AS-PA, AS-AA, and AS-PhAA crosslinkers, resulting in the generation of network tethered thiol species with pKa values of 10.4, 8.8, and 6.6, respectively. b AS-PA (red) and AS-AA (green) hydrogels undergo reverse gelation when degraded with the thiol, 3MMP. AS-PhAA crosslinked hydrogels do not achieve reverse gelation. c Apparent rate constants for photodegradation of crosslinkers AS-PA, AS-AA, and AS-PhAA using 3MMP are shown. Significance determined by one-way ANOVA, N=3, mean ± s.d., *p < 0.05, ***p < 0.001. d. The extent of degradation of AS-PA hydrogels decreases when degraded with thiols of decreasing pKa. The thiols, 3MMP (solid), glutathione (GSH, dash), MTG (dot), and TAA (dot-dash), have pKa values of 10.4, 9.42, 8.8, and 3.6, respectively. e Apparent rate constants for photodegradation of AS-PA crosslinking using 3MMP, GSH, MTG, and TAA are shown. Significance determined by one-way ANOVA, N=3, mean ± s.d., **p < 0.01, ***p < 0.001. Hydrogels were equilibrated with 15 mM soluble thiol and 1 mM lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) at pH 7.4 and exposed to light (365 nm, 5 mW cm−2) at t = 0. All rheological plots show a representative trace from N=3.
To determine the effect of free thiol protonation state on allyl sulfide degradation, hydrogels crosslinked with the AS-PA were then equilibrated with soluble thiols of various pKa, along with LAP, and degraded using similar conditions as before. The thiol pKa was chosen as a control handle to change protonation state of the thiol, rather than solution pH, as mammalian cells only survive within a narrow range of pH values. The chosen thiols, 3MMP, reduced glutathione (GSH), methyl thioglycolate (MTG) and thioacetic acid (TAA), which have pKa values of 10.4[32], 9.42[35], 8.8[32], and 3.6[36], respectively, were dissolved in a pH 7.4 buffer. Using the Henderson-Hasselbalch equation, the ratio of deprotonated to protonated thiols for 3MMP, GSH, MTG, and TAA at pH 7.4 are approximately 0.001, 0.009, 0.04, and 6300, respectively. Again, degradation was monitored on the rheometer while irradiating with 365nm light at 5 mW cm−2. The AS-PA hydrogels showed rapid degradation using 3MMP, followed by increasingly slower rates of degradation with GSH, MTG and TAA, respectively (Figure 2d). The degradation of AS-PA hydrogels with 3MMP, GSH, MTG, and TAA corresponds to rate constants of kapp / I0 × 10−4 of 310 ± 50, 240 ± 70, 130 ± 60 and 56 ± 25 cm2 mW−1 s−1, respectively (Figure 1e). These results show a decreasing rate of degradation with increasing thiol deprotonation. Under conditions of increasing thiol deprotonation, more thiolate anions are present to participate in radical sequestration, resulting in a decrease in degradation rate. The rate of degradation using MTG at pH 3 (more protonated) is increased (although not significant) compared to the rate at pH 7.4, while the rate of degradation using MTG at pH 9 (more deprotonated) is significantly decreased (Figure S4, Supporting Information). The rate of degradation shows a clear dependence on the protonation state of the thiol, which can be tuned using thiol pKa or solution pH. These results present the use of thiol pKa to tune hydrogel mechanical properties through the rate of degradation.
Interestingly, the trend in thiol protonation state was not seen when hydrogels were degraded with a soluble, aromatic thiol, 4-mercaptophenylacetic acid (4MPAA). Hydrogels crosslinked with AS-PA, AS-AA, and AS-PhAA and equilibrated with 4MPAA were degraded using similar conditions as above (Figure 3a). Contrary to degradation in the presence of an alkyl thiol, the AS-PA and AS-AA crosslinked hydrogels showed little degradation when irradiated in the presence of an aromatic thiol. Meanwhile, the AS-PhAA crosslinked hydrogel exhibited rapid degradation. The degradation of AS-PA, AS-AA, and AS-PhAA hydrogels with 4MPAA corresponds to rate constants of kapp / I0 × 10−4 of 2.1 ± 0.5, 1.1 ± 0.3, and 270 ± 130 cm2 mW−1 s−1, respectively (Figure 2b). The thiol pKa of 4MPAA is 6.6[34], corresponding to a ratio of deprotonated to protonated thiols of approximately 6.3, and so degradation is expected to behave according to the trend of thiol pKa and protonation state described previously. The lack in degradation seen is likely due to the increased radical stability of the aromatic thiol. The lower S-H bond dissociation energy (BDE) of aromatic thiols make them more reactive towards initiating radicals than alkyl thiols.[37] Following thiol-ene addition onto the allyl sulfide and formation of a radical intermediate, reformation of the aromatic thiyl radical is the favored cleavage product (Figure S5a, Supporting Information). In the case of the alkyl thiol crosslinkers, AS-PA and AS-AA, this decomposition inhibits cleavage of the allyl sulfide crosslinker and negates any degradation. These hydrogels show moderate degradation only in the presence of photoinitiator (no thiol), so lack of degradation following addition of the aromatic thiol species 4MPAA indicates active inhibition of the exchange mechanism (Figure S6, Supporting Information). Conversely, the symmetrical aromatic intermediate formed between AS-PhAA and 4MPAA has no favored degradation product, in terms of radical stability of the generated aromatic thiyl radicals, affording rapid degradation (Figure S5b, Supporting Information). While the effects of radical stability are apparent, the effect of thiol protonation state is still present in the case of aromatic thiols. Hydrogels crosslinked with AS-PhAA degrade more rapidly at pH 5, compared to pH 7.4 where more deprotonated thiols are present (Figure 3c). The degradation of AS-PhAA hydrogels with 4MPAA at pH 5 and pH 7.4 corresponds to rate constants of kapp / I0 × 10−4 of 570 ± 120, and 270 ± 130 cm2 mW−1 s−1, respectively (Figure 3d). While it is clear that protonation state and radical stability are both contributing factors for aryl thiols, the effect of radical stability is less practically relevant, as most biologically relevant thiols are alkyl.
Figure 3.
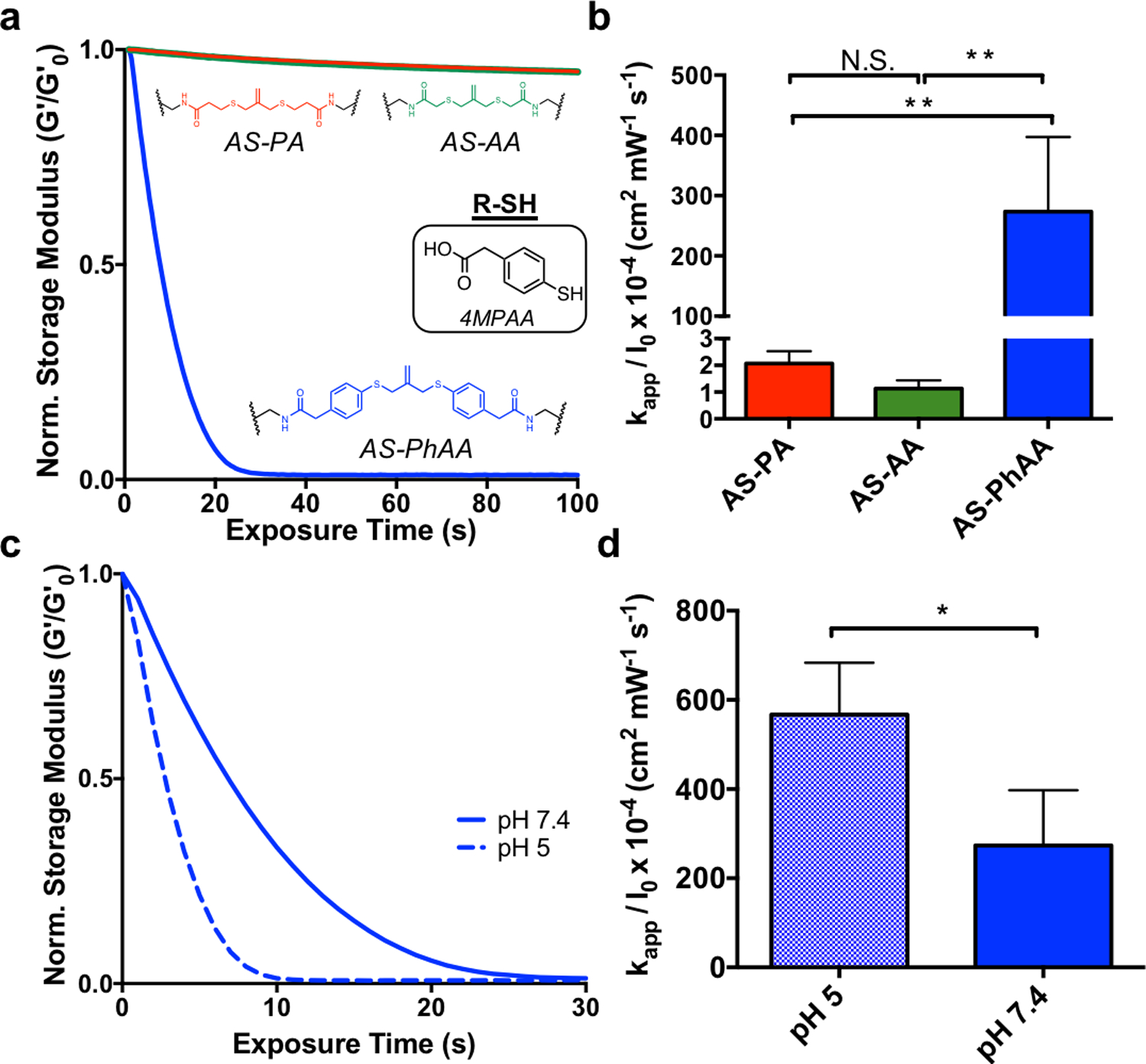
The effect of aromatic thiols on allyl sulfide hydrogel degradation kinetics. a. When equilibrated with the aromatic thiol, 4MPAA, AS-PhAA (blue) hydrogels degrade rapidly, while AS-PA (red) and AS-AA (green) hydrogels experience limited degradation. b Apparent rate constants for photodegradation of crosslinkers AS-PA, AS-AA, and AS-PhAA using 4MPAA are shown. Significance determined by one way ANOVA, N=3, mean ± s.d., **p < 0.01. c AS-PhAA crosslinked hydrogels equilibrated with 4MPAA degrade more rapidly at pH 5 (dash) than at pH 7.4 (solid). d Apparent rate constants for photodegradation of AS-PhAA using 4MPAA at pH 5 and pH 7.4 are shown. Significance determined by t test, N=3, mean ± s.d., **p < 0.01. Hydrogels equilibrated with 15 mM soluble thiol and 1 mM LAP at pH 7.4 (or pH 5) were exposed to light (365 nm, 5 mW cm−2) at t = 0. All rheological plots show a representative trace from N=3.
Using our understanding of the exchange reaction mechanism, we can begin to tailor the experimental conditions to suit stem cell expansion as related to a key stage of organoid derivation. Previous work with allyl sulfide degradable hydrogels used PEG-SH as the soluble monofunctional thiol species.[16] However, it is desirable to extend the applicability of photodegradation to biologically relevant thiols, such as cysteine or glutathione. Glutathione (pKa = 9.42) was ultimately chosen over cysteine (pKa = 8.6[38]) due to the elevated pKa, indicating more thiol protonation at physiological pH and thus rapid degradation kinetics. Also, the concentration of glutathione is 5−10 × 10−3 m within cells, which approaches conditions necessary for degradation.[39] Additionally, glutathione acts as an antioxidant to protect against radical damage from reactive oxygen species[40] and ultraviolet light[41], and could therefore be beneficial during photodegradation. However, as the extracellular environment is generally viewed as oxidizing, it may be advantageous to avoid exposing encapsulated cells to elevated levels of reduced thiols.[42] Disulfide bonds have a lower BDE compared to S-H bonds in thiols and are also reactive in radical reactions.[37,43] AS-PA hydrogels were equilibrated with 15 × 10−3 m solutions of both oxidized (GSSG) and reduced (GSH) glutathione, along with 1 × 10−3 m LAP, and subsequently exposed to 365nm light at 5 mW cm−2 (Figure 4a). The degradation of AS-PA hydrogels with GSH and GSSG corresponds to rate constants of kapp / I0 × 10−4 of 240 ± 70, and 150 ± 70 cm2 mW−1 s−1, respectively (Figure 4b). Rates of degradation for GSSG and GSH were statistically similar, and equally rapid compared to degradation with thiols of similar protonation states. These results indicate that glutathione is a biologically relevant alternative, that still allows for rapid degradation.
Figure 4.
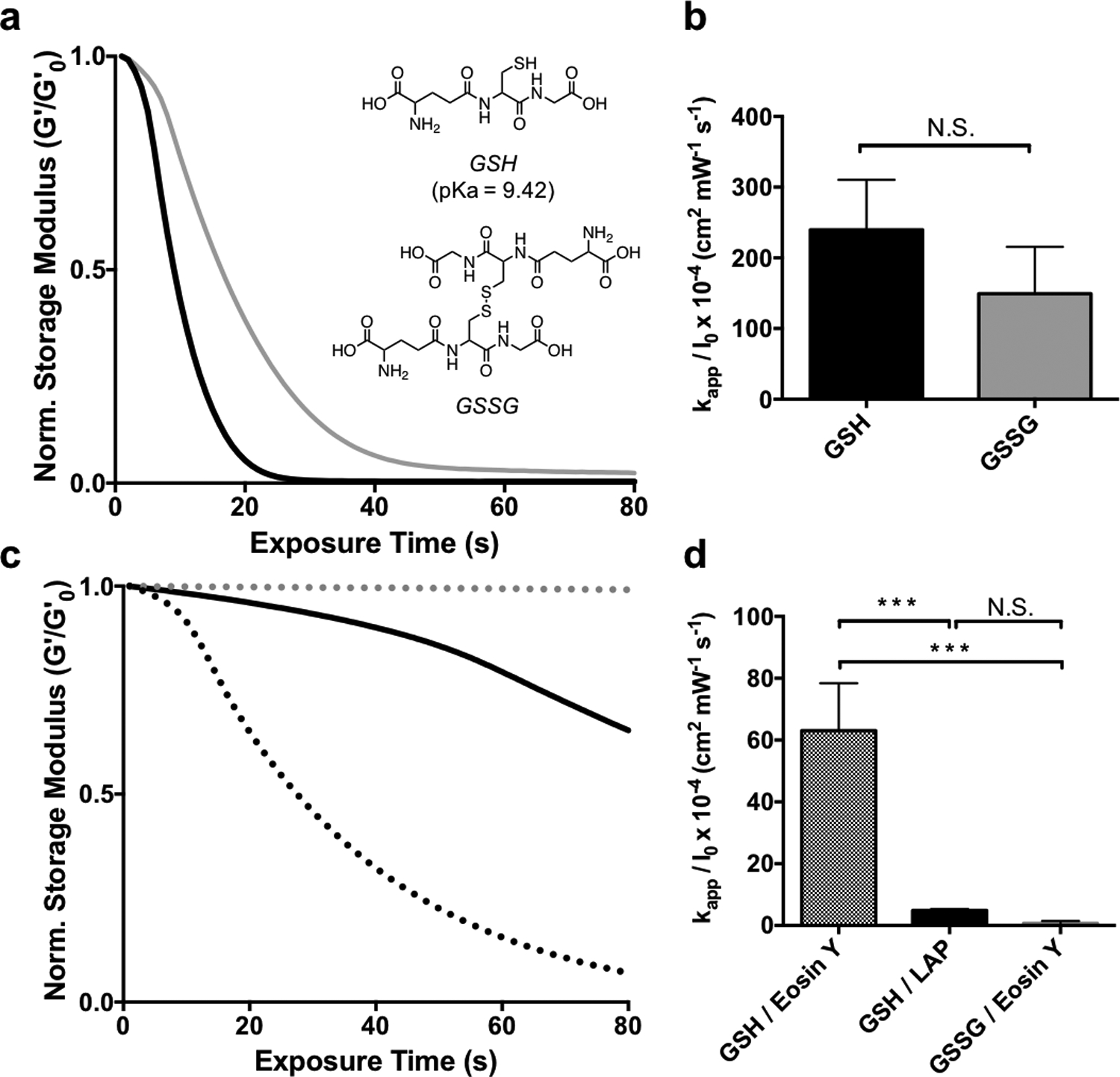
The effect of thiol oxidation state and initiator type on allyl sulfide hydrogel degradation kinetics. a AS-PA hydrogels equilibrated with glutathione (GSH, black) and glutathione disulfide (GSSG, grey) display similar degradation kinetics when degraded using 1mM LAP and 365nm light (5 mW cm−2). b Apparent rate constants for photodegradation of AS-PA using GSH and GSSG are shown. Significance determined by t test, N=3, mean ± s.d.. c Degradation is slower using GSH and LAP at higher wavelengths, such as 405 nm light (solid black). However, using 405 nm light and the photoinitiator eosin Y, degradation is more rapid with GSH (black dot), while GSSG (grey dot) exhibits little degradation. d Apparent rate constants for photodegradation of AS-PA using thiols, GSH and GSSG, and photoinitiators, LAP and eosin y, are shown. Significance determined by one way ANOVA, N=3, mean ± s.d., ***p < 0.001. Hydrogels equilibrated with 15 mM soluble thiol and 1 mM photoinitiator at pH 7.4 were exposed to light (365 nm or 405/436, 5 mW cm−2) at t = 0. All rheological plots show a representative trace from N=3.
In addition to the rapid and versatile nature of the thiol-ene reaction, it has the ability to be photoinitiated by a variety of strategies, including visible light and/or type II photoinitiators. Eosin Y is a water-soluble type II photoinitiator that has found utility as a visible light photoinitiator for 3D cell encapsulation.[44,45] AS-PA hydrogels equilibrated with 15 × 10−3 m GSH and 1 × 10−3 m eosin Y degraded more rapidly at 405/436 nm (5 mW cm−2) than hydrogels initiated with 1 × 10−3 m LAP at the same wavelength (Figure 4c), due to the larger molar absorptivity of eosin Y. As expected, hydrogels equilibrated with LAP degraded more slowly when irradiated with 405/436 nm light compared to 365nm light, due to the lower molar absorptivity of LAP at longer wavelengths.[46] Additionally, hydrogels equilibrated with 15 × 10−3 m GSSG and 1 × 10−3 m eosin Y showed no degradation (Figure 4b). As a type II initiator, eosin Y requires abstraction of a hydrogen from a co-initiating species.[45] Such a role can be fulfilled by the sulfhydryl group of GSH, however, GSSG lacks a reduced thiol to facilitate generation of a radical by type II photoinitiation. The degradation of AS-PA hydrogels with GSH/eosin Y, GSH/LAP, and GSSG/eosin Y using 405/436 nm (5 mW cm−2) light corresponds to rate constants of kapp / I0 × 10−4 of 63 ± 15, 4.8 ± 0.5, 0.7 ± 0.7 cm2 mW−1 s−1, respectively (Figure 4e) The use of visible light photoinitiators extends the application of allyl sulfide degradable hydrogels, as visible light allows for deeper tissue penetration than UV light.[47]
The current study reveals that cleavage of the AS-PA crosslinker with glutathione allows for more biologically relevant, and rapid degradation than previously reported.[16] These advancements allow for lower UV light doses and lower concentrations of photoinitiating species, which extend the utility of this photodegradation strategy for use with sensitive cell types. In combination with the bioorthogonal SPAAC click reaction for gel formation, allyl sulfide degradable hydrogels provide a practical reversible scaffold for intestinal organoids. Additionally, the dimensions of hydrogel pucks used for rheology are similar to the dimensions of cell-laden matrices used for routine organoid passaging, ensuring translation to in vitro studies. Murine Lgr5-GFP intestinal organoids (maintained in Matrigel) were isolated, dissociated into single cells, and encapsulated into allyl sulfide containing hydrogels supplemented with the integrin-binding ligand RGD (Arg-Gly-Asp) to promote cell survival and proliferation.[15,48] Importantly, hydrogels were not supplemented with laminin-111, a component of the basement membrane necessary for intestinal crypt homeostasis[49,50] that is included in formulations of other synthetic hydrogel platforms for intestinal organoid development.[15,48] The formation of colonies from single ISCs encapsulated into synthetic hydrogels is mechanosensitive and depends on the hydrogel storage modulus.[15] Therefore, hydrogels were formulated with increasing concentrations of PEG macromer, providing increasing modulus values (Figure 5a). After four days, single cells had grown into spherical colonies of Lgr5+ stem cells, where the colony forming efficiency (CFE) is defined as the fraction of single cells that grows into an epithelial cyst (Figure 5b). The CFE is lowest at low modulus values, reaches a plateau around 1500 Pa, and then starts to decline at higher modulus values, a trend that has been seen in other synthetic hydrogel platforms.[15,48] The colonies polarized to form epithelium and central lumen, a key morphological feature of intestinal organoids (Figure 5c). Gene expression determined by quantitative real-time PCR (qPCR) of stem (Lgr5) and differentiation markers (Lct, Lyz1, Muc2, Chga) was similar in organoids grown in allyl sulfide hydrogels compared to organoids grown in Matrigel under stem-inducing conditions (Figure 5d), further indicating the maintenance of the stem population in allyl sulfide photodegradable hydrogels. The expression of Dclk1 was significantly elevated compared to the Matrigel control, indicating the increased presence of Tuft cells. However, this difference is not expected to be significant to the cellular organization of intestinal organoids, considering Tuft cells account for only 0.4% of cells in the intestinal epithelium.[51]
Figure 5.
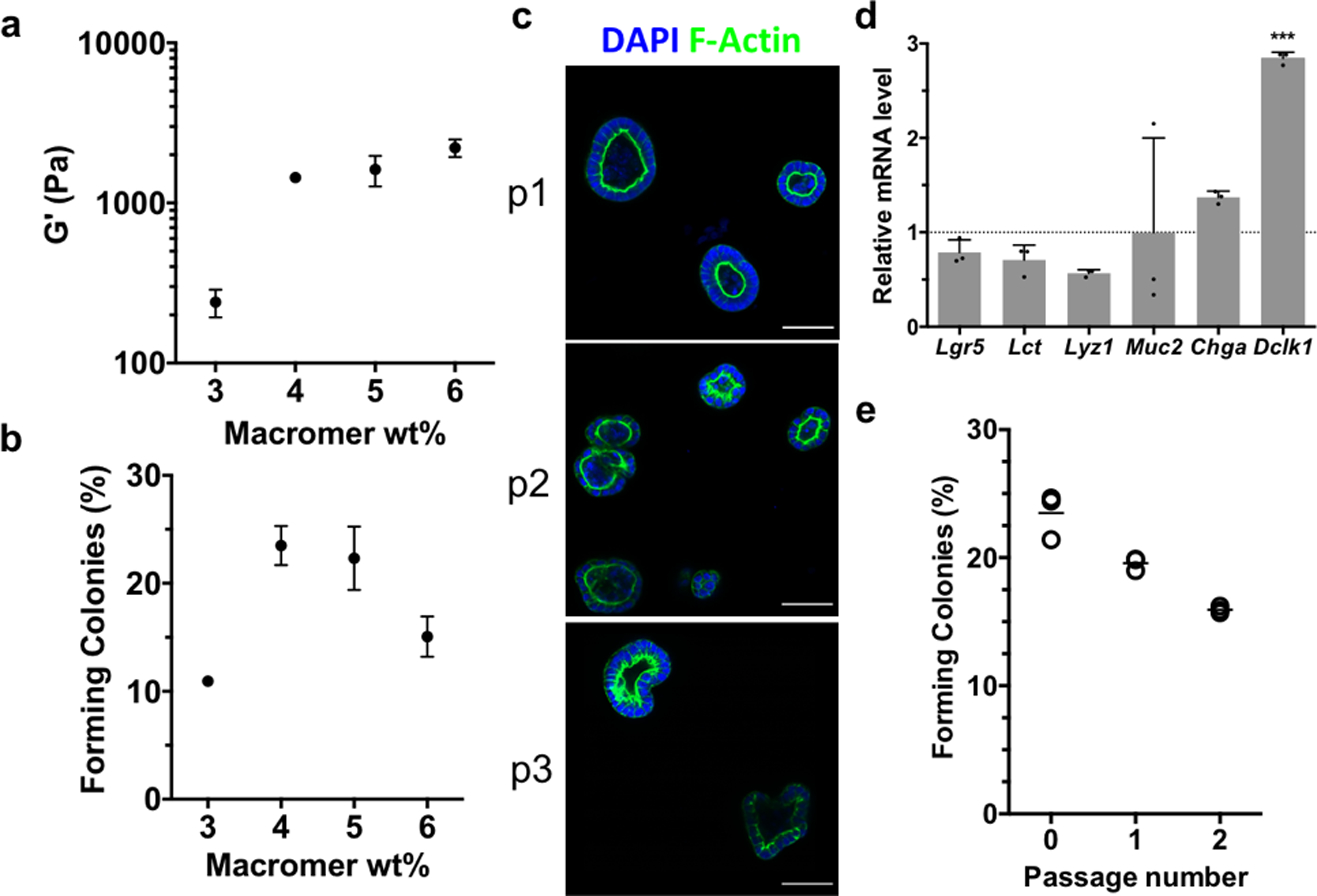
Allyl sulfide photodegradable hydrogels support the expansion and repetitive passaging of intestinal organoids. a Shear rheology of swollen allyl sulfide hydrogels shows a correlation between macromer concentration and shear storage modulus. b The efficiency of colony formation from single encapsulated ISCs shows a dependence on the macromer concentration. c Allyl sulfide photodegradation was used to repetitively passage ISC colonies (p1, p2, p3), which maintained polarized epithelium, as seen by DAPI (blue) and f-actin (green) staining. Scale bars 50μm. d Relative mRNA expression for intestinal genes was similar in organoids grown in allyl sulfide photodegradable hydrogels compared to Matrigel controls. Significance determined by one-way ANOVA, N = 3, mean ± s.d., ***p < 0.001. e Colonies were formed at a slightly decreasing rate upon repetitive passaging. Open circles represent average measurements from one gel replicate, while bars represent the average of the three gel replicates.
The utility of this system as a reversible scaffold is dependent on the ability to repetitively passage ISC colonies. AS-PA hydrogels containing colonies grown from single encapsulated cells were equilibrated with GSH (15 × 10−3 m) and LAP (1 × 10−3 m), and exposed to 365 nm light (5 mW cm−2), leading to complete network degradation. The colonies were collected, dissociated into single cells and encapsulated into allyl sulfide hydrogels as “p1”. After 4 days, colonies had formed from the encapsulated stem cells and the passaging process was again repeated on these colony laden gels to form “p2”. Single cells encapsulated as p1 and p2 formed colonies with slightly decreasing efficiency, possibly due to a decrease in the encapsulated cell density following each passaging step (Figure 5e). However, the values for CFE are still similar to other reported values.[15] Colonies of p2 and p3 also formed polarized epithelium similar to their p1 counterparts (Figure 5c). These results indicate that allyl sulfide hydrogels can support the growth and expansion of intestinal organoids.
To enhance utility as a reversible scaffold, the degradation reactions must proceed in the presence of cells without compromising viability or stemness. AS-PA hydrogels laden with ISC colonies were equilibrated with GSH (15 × 10−3 m) and LAP (1 × 10−3 m), and exposed to 365 nm light (5 mW/cm2), leading to complete network degradation. Temporary subjection of intestinal organoids to these concentrations of GSH and LAP do not result in any loss of cell viability (Figure S7, Supporting Information). The colonies were collected, reseeded into Matrigel, and cultured in differentiation medium lacking exogeneous CHIR 99021 and valproic acid to promote crypt formation. After 3 days, visible crypts were noticed budding from the encapsulated organoids. Immunostaining confirmed the presence of lysozyme, a marker for the differentiated Paneth cell, a key cell type of the intestinal crypt (Figure 6a). Additionally, 5-ethynyl-2’-deoxyuridine (EdU, 24 h pulse) staining was restricted to the crypt (Figure 6b). Crypts staining positive for lysozyme and EdU were found in 22.5 ± 1.0 % and 27.5 ± 0.9 % of transplanted organoids, respectively (Figure 6c). The cohabitation of Paneth cells and proliferative stem cells in the formed crypts indicate the formation of morphologically accurate crypts that display correct compartmentalization of intestinal epithelial cell types. Allyl sulfide photodegradable hydrogels are able to maintain cell viability and multipotency during the photodegradation process.
Figure 6.
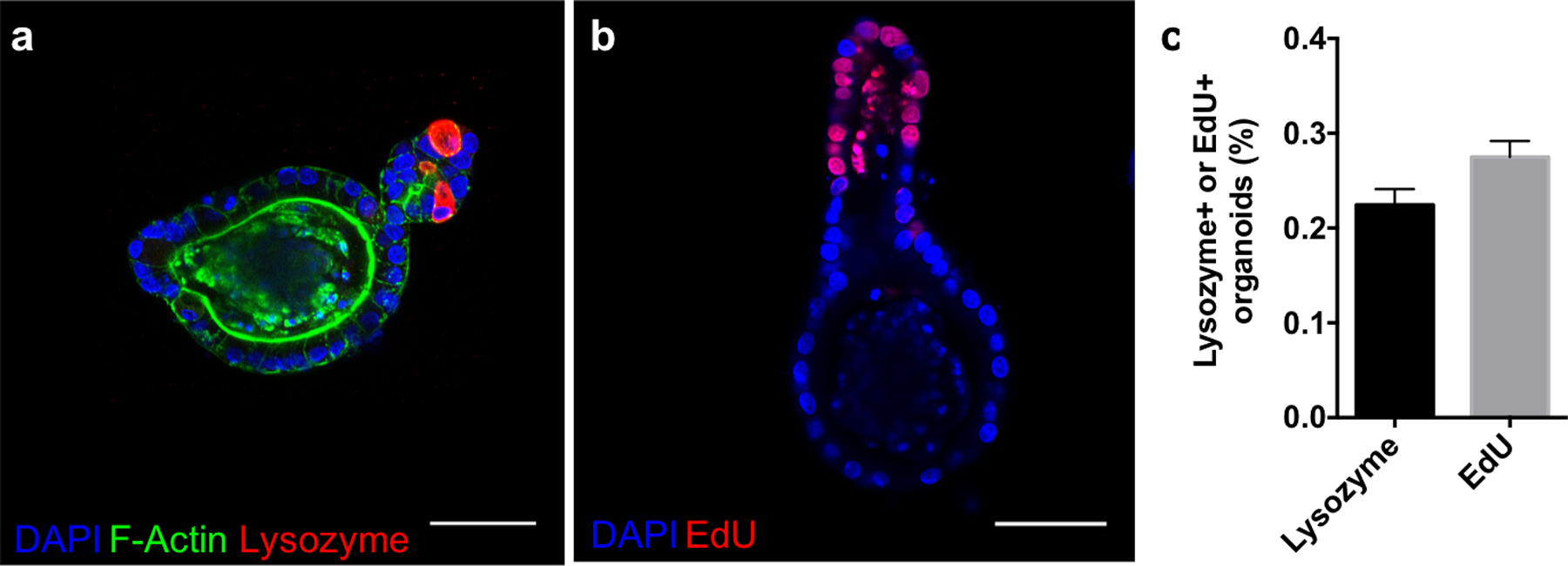
Allyl sulfide photodegradation maintains stem cell differentiation potential. Allyl sulfide hydrogels laden with colonies were degraded to release the encapsulated colonies, which were encapsulated into Matrigel and cultured with differentiation media (lacking CHIR99021 and valproic acid). a After 3 days, buds had protruded off of the colonies to form crypts, which immunostained for lysozyme (red), an indicator of Paneth cells and a mature crypt, as well as DAPI (blue) and f-actin (green). b Staining for EdU (red, 24 hour pulse) and DAPI (blue) showed proliferative cells residing in the crypt ends. Scale bars 50μm. c A low percentage of organoids formed from ISC colonies expanded in allyl sulfide gels and transplanted into Matrigel formed crypts containing lysozyme (22.5 ± 1.0 %, N = 3) and EdU positive cells (27.5 ± 0.9 %, N = 4). Mean ± s.d..
Current culture platforms rely on Matrigel, a murine-derived mixture of proteins and growth factors notorious for its undefined and xenogeneic composition and batch-to-batch variation.[14] These limitations can confound interpretation of mechanistic studies of organoids, and prevents the application of intestinal organoids for clinical use. While synthetic hydrogel platforms are beginning to be developed that support the growth and expansion of intestinal organoids[15,48,52–54], these platforms rely on predefined degradation kinetics, lacking spatial and temporal control over material properties. However, the use of allyl sulfide photodegradable hydrogels allows for the spontaneous release and capture of encapsulated cells, potentially providing a replacement for Matrigel and other animal derived scaffolds. Passaging in animal derived matrices is often a time-consuming and rate limiting step in the use of organoids that introduces variability, creating problems for applications such as drug screening. The ability to degrade rapidly using light provides a standardized means to release organoids without compromising function, thus improving quality and yield of cultures. Additionally, allyl sulfide hydrogels do not require supplementation with tissue derived proteins, such as laminin, for the growth of ISC cultures. These hydrogels utilize only RGD to facilitate cell-matrix interactions, therefore providing a completely defined and synthetic system to support ISC development. While this system supports the growth of ISC cultures, the absence of laminin prevents applications with differentiated cultures, which is a necessary component of intestinal organoid differentiation.[15,48] The utility of allyl sulfide photodegradable hydrogels is therefore restricted to the expansion and culture of ISC cultures.
In addition to the use of this platform as a means to propagate ISC colonies with the potential to form organoids, allyl sulfide hydrogels can also be used to investigate cell-matrix interactions that govern intestinal organoid development. While intestinal development has been linked to changes in cell shape following localized tissue remodeling, the exact bio-mechanical pathway involved in intestinal organoid development is unknown.[55,56] Allyl sulfide degradable hydrogels could provide a controlled environment to study the biomechanical pathways involved in mechanosensation, allowing on-demand control of the scaffold properties, which should be a useful tool for research aimed at understanding intestinal development.
Allyl sulfide moieties have been previously used to facilitate photo-induced plasticity in crosslinked polymer networks[26,57,58] and to reversibly photopattern biomolecules.[59,60] However, their use in modulating hydrogel mechanical properties has been significantly limited, in part, due to the lack of understanding of the exchange mechanism. In this work, we report the thiol pKa as the determining factor mediating the kinetics of allyl sulfide exchange. This understanding was leveraged to improve the rate of degradation by selecting crosslinker and soluble thiols that present high pKa values. The use of the AS-PA crosslinker and glutathione (15 × 10−3 m) allowed for complete hydrogel degradation in under 15 seconds of irradiation, using LAP (1 × 10−3 m) and 5 mW cm−2 of 365 nm light, which improves on previously reported results.[16] Ultimately, this advance allows for reduced light and radical exposure, as well as degradation of thicker samples. This scaffold was then shown to support the expansion of ISC colonies from single encapsulated stem cells, using only RGD adhesive peptides, with similar efficiencies to previously reported results. Rapid photodegradation allowed for repetitive passaging, which preserved the potential of intestinal stem cells to form colonies and fully developed organoids bearing crypt-villus domains. The use of this platform could provide a replacement for Matrigel used in routine organoid culture, or can be used to investigate the mechanobiological processes involved in organ development.
Methods and Materials
Synthesis of PEG-4DBCO.
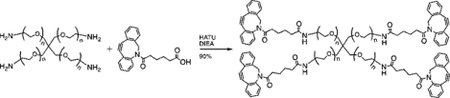
Four-arm, 20 kDa PEG-dibenzylcyclooctyne (PEG-4DBCO) macromers were synthesized by coupling DBCO-C6 acid (100mg, 0.3 mmol, Click Chemistry Tools) to 4arm PEG amine (1g, 0.05 mmol, Mw = 20,000, JenKem USA) using (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU, 114mg, 0.3 mmol, ChemPep) as an activator and N,N-diisopropylethylamine as a base (0.139 mL 0.8 mmol, Aldrich). The reaction was allowed to proceed overnight, after which the reaction mixture was precipitated in cold diethyl ether (x3) to yield an off white solid. The product was collected by centrifugation, dissolved in water, dialyzed (8kDa MW cutoff, Spectrum Chemical) for 48 hours against deionized (DI) water, frozen, and lyophilized to yield the final product (90% yield, 85% functionalization by 1H NMR).
Synthesis of allyl sulfide crosslinker, AS-AA.
The compound AS-AA was synthesized as previously described[16].
Synthesis of allyl sulfide crosslinker, AS-PA
Synthetic Scheme for AS-PA
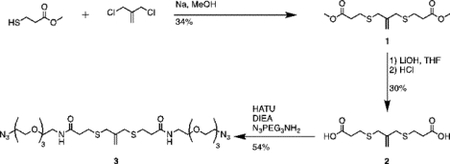
Synthesis of compound 1 [allyl sulfide bis(methyl propionate ester)]:
Sodium methoxide (2.26mL, 24 mmol) and methyl 3-mercaptopropionate (2.43mL, 22 mmol) were added to a flame dried flask charged with 50mL anhydrous methanol. The mixture was heated to reflux under argon atmosphere for 20 minutes. 3-chloro-2-chloromethyl-1-propene (1.15mL, 10 mmol) dissolved in 10mL anhydrous methanol was then added dropwise to the reaction vessel over 20 minutes. The reaction mixture was purged with argon and stirred for 20h at 60°C. The resulting mixture was filtered and concentrated through rotary evaporation to yield a pale-yellow crude oil with a white precipitate. The mixture was taken up in 50mL water (Milli-Q, EMD Millipore), acidified to pH 1 with HCl and extracted 4x with 50mL diethyl ether. The combined organics phase was washed with 100mL saturated sodium chloride (brine), dried over sodium sulfate and concentrated by rotary evaporation to yield a pale-yellow oil. The crude product was purified by column chromatography (Biotage Isolera) using a gradient of 0% to 20% EtOAc in hexanes. Two products were collected and TLC in 20:80 ethyl acetate:hexanes resulted in Rf values of 0.4 and 0.6 under UV and KMnO4 stain. The product at Rf = 0.4 was collected and concentrated by rotary evaporation to yield a colorless oil (1g, 34%). 1H NMR (400 MHz, Methanol-d4) δ 5.08 (s, 2H), 3.70 (s, 6H), 3.35 (d, J – 0.75 Hz 4H), 2.66 (m, 8H).
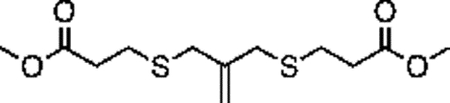
Synthesis of compound 2 [allyl sulfide bis(propionic acid)]:
40mL of 1M LiOH was added to a solution of 1 (1g, 3.42 mmol) in 40mL THF on ice. The turbid solution was stirred for 5 h on ice, after which the solution was acidified (to pH = 0) by addition of ~50mL 2M HCl. 20 mL of brine was added and the solution was extracted with EtOAc. The combined organics were dried over sodium sulfate and concentrated to yield a white solid. The product was purified by column chromatography using a gradient of up to 10% MeOH in DCM with 1% acetic acid. TLC in 5% MeOH:95% DCM with 1% acetic acid yielded a fraction with Rf = 0.3. This fraction was collected and concentrated by rotary evaporation to yield a white solid (300mg, 30%). 1H NMR (400 MHz, Methanol-d4) δ 5.09 (t, J – 0.69Hz, 2H), 3.37 (t, J – 0.65 Hz, 4H), 2.63 (m, 8H).
Synthesis of compound 3 [allyl sulfide bis(propionamide PEG3-azide]:
![]()
A flame dried RBF was charged with 2 (300mg, 1.13mmol), diisopropylethylamine (DIEA, Sigma) (1.96mL, 11.3 mmol), O-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU, Chem-Impex Int’l) (0.859g, 2.26 mmol), and 125 mL of ethyl acetate. The resulting slurry was purged with argon and reacted for 1hr at room temperature. 11-Azido- 3,6,9-trioxaundecan-1-amine (TCI America) (0.537 mL, 2.712 mmol) was added to the flask and the mixture was stirred overnight at room temperature under argon atmosphere. The resulting mixture was vacuum filtered and washed with 1N HCl, saturated aq. sodium bicarbonate, water, and brine. The organic phase was dried over sodium sulfate and concentrated by rotary evaporation to yield a yellow oil (406mg, 54%). 1H NMR (400 MHz, Methanol-d4) δ 5.09 (s, 2H), 3.72–3.62 (m, 20H), 3.57 (t, J = 5.44 Hz, 4H), 3.39 (q, J = 5.31Hz, 8H), 3.36 (s, 4H), 2.70 (t, J = 7.3Hz, 4H), 2.49 (t, J = 7.6Hz, 4H)
Synthesis of allyl sulfide crosslinker, AS-PhAA
Synthetic Scheme for AS-PhAA
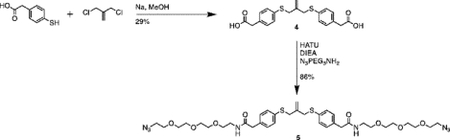
Synthesis of compound 4 [allyl sulfide bis(phenyl acetic acid)]:
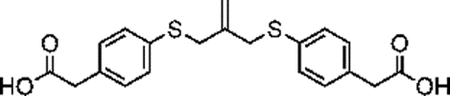
Sodium methoxide solution (0.8mL, 12 mmol) and 4 mercaptophenylacetic acid (0.925g, 5.5 mmol) were added to a flame dried flask charged with 30mL anhydrous methanol. The mixture was heated to reflux under argon atmosphere for 20 minutes. 3-chloro-2-chloromethyl-1-propene (0.289mL, 2.5 mmol) dissolved in 10mL anhydrous methanol was then added dropwise to the reaction vessel over 20 minutes. The reaction mixture was purged with argon and stirred for 20h at 60°C. The resulting mixture was filtered and concentrated through rotary evaporation to yield a pale-red crude oil with a pink precipitate. The mixture was taken up in 25mL of 1N HCl and extracted 4x with 50mL ethyl acetate. The combined organics phase was washed with 100mL brine, dried over sodium sulfate and concentrated by rotary evaporation to yield a pale-yellow oil. The crude product was purified by column chromatography (Biotage Isolera) using a gradient of 0% to 10% MeOH in DCM with 1% acetic acid. The fractions were analyzed by TLC using 5% MeOH in DCM and 1% acetic acid. Fractions with Rf values of 0.36 were collected and concentrated by rotary evaporation to yield a pale yellow solid (280mg, 29%). 1H NMR (400 MHz, CDCl3) δ 7.27–7.14 (m, 8H), 5.01 (p, J = 0.81 Hz, 2H), 3.71 (t, J = 0.88 Hz, 4H), 3.61 (s, 4H).
Synthesis of compound 5 [allyl sulfide bis(phenylacetamide PEG3-azide)]:
![]()
A flame dried RBF was charged with 4 (280mg, 0.72mmol), diisopropylethylamine (DIEA, Sigma) (1.25mL, 7.2 mmol), O-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU, Chem-Impex Int’l) (0.574g, 1.512 mmol), and 100 mL of ethyl acetate. The resulting slurry was purged with argon and reacted for 1hr at room temperature. 11-Azido- 3,6,9-trioxaundecan-1-amine (TCI America) (0.371 mL, 1.872 mmol) was added to the flask and the mixture was stirred overnight at room temperature under argon atmosphere. The resulting mixture was vacuum filtered and washed with 1N HCl, saturated aq. Sodium bicarbonate, water, and brine. The organic phase was dried over sodium sulfate and concentrated by rotary evaporation to yield a yellow oil (488mg, 86%). 1H NMR (400 MHz, CDCl3) δ 7.31 – 7.13 (m, 8H), 4.96 (m, 2H), 3.75–3.64 (m, 18H), 3.6 (m, 8H), 3.53 (m, 8H), 3.42 (m, 8H).
Characterization of SPAAC hydrogel degradation.
Hydrogels containing allyl sulfide crosslinks were formed by strain promoted azide-alkyne cycloaddition (SPAAC) by mixing stock solutions of 20wt% PEG-4DBCO in phosphate buffered saline (PBS) and 50 × 10−3 m allyl sulfide crosslinkers in DMSO to a final concentration of 6 wt% PEG-4DBCO and 5 × 10−3 m allyl sulfide crosslinker in PBS. The precursor solutions were vortexed for ~5 seconds and placed as 20 μL drops between two glass slides treated with Sigmacote, which were separated by two 0.5 mm spacers. The glass slides were clamped together and set aside to gel for 30 minutes. The fully formed gels were then placed in 500 μL baths containing a monothiol (15 × 10−3 m) in buffers of various pH, LAP (1 × 10−3 m), and a monofunctional PEG3-azide (0.92 × 10−3 m) for 30 minutes to reach a uniform concentration profile. The monothiols used were glutathione (GSH, Sigma), methyl 3-mercaptopropionic acid (3MMP, Sigma), methyl thioglycolate (MTG, Sigma), thioacetic acid (TAA, Sigma), and 4-mercaptophenylacetic acid (4MPAA, Sigma). Buffers were created at pH 3 (100 × 10−3 m citrate), pH 5 (50 × 10−3 m citrate), pH 7.4 (100 × 10−3 m 3-(N-morpholino)propanesulfonic acid (MOPS)), and pH 9 (200mM tris(hydroxymethyl)aminomethane (Tris)). Oscillatory rheology was performed on the gels using a TA Instruments DHR-3 rheometer with an 8 mm parallel plate geometry. The rheometer was fitted with an adaptor to allow for light exposure from a mercury arc lamp (Omnicure) fitted with a 365 nm or 400–500 nm bandpass filter. The storage and loss moduli were recorded using a strain of 5% and a frequency of 1 Hz, while the sample was irradiated with 365 nm light at 5 mW cm−2 or 400–500 nm light at 5 mW cm−2.
Crypt Isolation and organoid culture.
Murine small intestinal crypts were extracted from Lgr5-eGFP-IRES-CreERT2 mice as previously described[15]. The crypts were cultured as organoids by embedding in reduced growth factor Matrigel (Corning). The organoids were maintained using Advanced DMEM-F12 (Invitrogen), containing N2 and B27 Supplements (Thermo Fischer Scientific), Glutamax (Gibco), HEPES, penicillin-streptomycin, and supplemented with epidermal growth factor (50 ng/mL, R&D Systems), Noggin (100 ng/mL, PeproTech), R-Spondin conditioned media (5% v/v)[61], CHIR99021 (3uM, Selleckchem) and valproic acid (1 × 10−3 m, Sigma-Aldrich). The medium was changed every 2 days, and organoids were passaged every 4 days.
Encapsulation of single cells into photodegradable gels.
Hydrogel precursor solutions were prepared by diluting 4-arm PEG-DBCO in Advanced DMEM-F12 media supplemented with Glutamax, HEPES, and penicillin-streptomycin to varying concentrations. Azide functionalized RGD (0.8 × 10−3 m) was added to the hydrogel precursor solutions, which were then kept on ice until the addition of cells. Organoids were released from Matrigel using cold media followed by mechanical stimulation from a pipette tip. The organoids were enzymatically dissociated into single cells by incubation at 37°C for 8 minutes in 1 mL TrypLE (Life Technologies), supplemented with DNAseI (~10mg, Sigma-Aldrich), 1 × 10−6 m N-acetylcysteine (Sigma-Aldrich), and 10 × 10−6 m Y27632 (Stemgent). Following incubation, the single cell suspension was diluted with 1 mL FBS and 8 mL Advanced DMEM/F12, and passed through a 40μm filter to remove multicellular aggregates. The single cell suspension was then centrifuged (1200 RPM, 4 minutes) and the pellet resuspended in media which was added to the hydrogel precursor solutions on ice. Gelation was initiated by the addition of AS-PA, added such that the allyl sulfide azide groups were on stoichiometry with total DBCO groups. Immediately after the allyl sulfide crosslinker was added, the solutions were vortexed for 5 seconds and added as 20μL drops to thiol-functionalized coverslips to gel. The gels were left at room temperature for 10 minutes, then cultured in media with the appropriate supplements as described above, along with 2.5 × 10−6 m of thiazovinin (Stemgent). The medium was replaced without thiazovivin after 2 days.
Quantification of colony forming efficiency.
Colonies growing in allyl sulfide degradable hydrogels were fixed with 4% paraformaldehyde following 4 days of growth. A laser scanning confocal microscope (Zeiss LSM 710) with a 488nm laser was used to collect stacks spanning 500μm of the thickness of the cell laden hydrogels (three stacks per gel). The Cell Counter plugin in ImageJ (NIH) was used to manually count the fraction of encapsulated cells that formed colonies (three gels per condition).
Complete photodegradation of cell laden hydrogels.
On day 3 post encapsulation, a small, monofunctional PEG-azide was added to the media in a final concentration of 0.76 × 10−3 m, in order to cap any unreacted DBCO groups. On day 4 post-encapsulation, the culture medium was removed and replaced with 500μL of FluoroBrite medium (Thermo Fischer Scientific), supplemented with N2 and B27 supplements (Thermo Fischer Scientific), 15 × 10−3 m reduced glutathione (Sigma), and 1 × 10−3 m LAP. The cell laden gels were allowed to equilibrate for 30 minutes at 37°C, after which the medium was removed and the gels exposed to 365nm light at 5mW cm−2 for 30s to ensure complete degradation. The degraded gels were collected by washing with Advanced DMEM/F12 medium and passed through a ~1mm filter to remove any undegraded gel. The resulting suspension of cells was centrifuged (900 RPM, 4 minutes) and either resuspended into Matrigel or dissociation media (described above). Colonies encapsulated into Matrigel were cultured in differentiation medium (lacking CHIR99021 and valproic acid)[15], while cells in dissociation media were then re-encapsulated into allyl sulfide hydrogels following the above protocol.
Immunofluorescence analysis.
ISC colonies growing in Matrigel were fixed using 4% paraformaldehyde in PBS (30 minutes, room temperature). The fixation process led to the complete dissolution of the Matrgiel (Corning), leaving behind a suspension of cells. Cells were collected by centrifugation (900 RPM, 4 minutes), washed with deionized (DI) water, and pelleted. The resulting pellet was resuspended in DI water and encapsulated into a hydrogel formed by a thiol-ene click reaction of 1% 8 arm PEG thiol (MW 20kDa, Jenkem Technology USA), 1% 8 arm PEG-norbornene (MW 20kDa, synthesized as described elsewhere[62], and 0.5% LAP. Encapsulation into a thiol-ene hydrogel allows for staining and preservation of the ISC colonies. ISC colonies were then solubilized using 0.2% Triton X-100 (Sigma) in PBS (1 hour at room temperature) and blocked using 10% horse serum (Gibco) and 0.01% Triton X-100 in PBS (overnight, 4°C). The samples were then incubated overnight at 4°C with a rabbit primary antibody against lysozyme (1:50, Invitrogen PA1–29680) diluted in blocking buffer. After washing with fresh PBS every hour for 5 hours to remove residual primary antibody, the samples were incubated with rhodamine phalloidin (1 U/mL, Invitrogen), DAPI (1.5μM), and an Alexa-647 goat-α-rabbit (1:1000, Invitrogen) secondary antibody in blocking buffer. After washing with PBS remove any residual antibody, the fluorescently labeled colonies were imaged using confocal microscopy (Zeiss LSM 710).
Imaging of EdU expression.
Colonies growing in Matrigel were pulsed with 5-ethynyl-2’-deoxyuridine (EdU, 10 × 10−6 m) 24 hours prior to fixation. Here, half of the media was removed and replaced by a 2X solution of differentiation media containing EdU. The colonies were then fixed, solubilized, and blocked as described above. Samples were then incubated overnight at 4°C with 4 × 10−3CuSO4, 2 × 10−6 m Sulfo-Cyanine3 azide (Lumiprobe), and 100 × 10−3 m sodium ascorbate in Tris buffered saline (pH 7.4, 100 × 10−3 m). Following incubation, the samples were washed with PBS to remove reagents and imaged using confocal microscopy (Zeiss LSM 710).
Quantitative Real-Time PCR (qPCR).
ISC colonies or organoids grown in Matrigel or hydrogels were dissociated as described above, and RNA was extracted using an RNeasy Mini Kit (Qiagen). cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR was carried out using the SYBR Select Master Mix (Applied Biosystems) and the primers listed in Extended Data Table 1.
Supplementary Material
Acknowledgments
F.M.Y and T.E.B contributed equally to this work. This work was supported in part by grants from the NIH (R01 DK120921 and T32 GM-065103). F.M.Y and E.A.H acknowledge DoEd GAANN fellowship for funding. T.E.B acknowledges NSF GRFP for funding.
References:
- [1].Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y, Nature 2011, 472, 51. [DOI] [PubMed] [Google Scholar]
- [2].McCracken KW, Catá EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, Tsai YH, Mayhew CN, Spence JR, Zavros Y, Wells JM, Nature 2014, 516, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Xia Y, Nivet E, Sancho-Martinez I, Gallegos T, Suzuki K, Okamura D, Wu MZ, Dubova I, Esteban CR, Montserrat N, Campistol JM, Izpisua Belmonte JC, Nat. Cell Biol 2013, 15, 1507. [DOI] [PubMed] [Google Scholar]
- [4].Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA, Nature 2013, 501, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, Yoon KJ, Jeang W, Lin L, Li Y, Thakor J, Berg DA, Zhang C, Kang E, Chickering M, Nauen D, Ho CY, Wen Z, Christian KM, Shi PY, Maher BJ, Wu H, Jin P, Tang H, Song H, Ming GL, Cell 2016, 165, 1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Takasato M, Er PX, Chiu HS, Maier B, Baillie GJ, Ferguson C, Parton RG, Wolvetang EJ, Roost MS, De Sousa Lopes SMC, Little MH, Nature 2015, 526, 564. [DOI] [PubMed] [Google Scholar]
- [7].Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C, Drowley L, Plowright AT, Needham EJ, Wang Q-D, Gregorevic P, Xin M, Thomas WG, Parton RG, Nielsen LK, Launikonis BS, James DE, Elliott DA, Porrello ER, Hudson JE, Proc. Natl. Acad. Sci 2017, 114, E8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yui S, Nakamura T, Sato T, Nemoto Y, Mizutani T, Zheng X, Ichinose S, Nagaishi T, Okamoto R, Tsuchiya K, Clevers H, Watanabe M, Nat. Med 2012, 18, 618. [DOI] [PubMed] [Google Scholar]
- [9].Fordham RP, Yui S, Hannan NRF, Soendergaard C, Madgwick A, Schweiger PJ, Nielsen OH, Vallier L, Pedersen RA, Nakamura T, Watanabe M, Jensen KB, Cell Stem Cell 2013, 13, 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang RR, Ueno Y, Zheng YW, Koike N, Aoyama S, Adachi Y, Taniguchi H, Nature 2013, 499, 481. [DOI] [PubMed] [Google Scholar]
- [11].Lancaster MA, Knoblich JA, Science 2014, 345, DOI 10.1126/science.1247125. [DOI] [PubMed] [Google Scholar]
- [12].Clevers H, Cell 2016, 165, 1586. [DOI] [PubMed] [Google Scholar]
- [13].Sato T, Vries RG, Snippert HJ, Van De Wetering M, Barker N, Stange DE, Van Es JH, Abo A, Kujala P, Peters PJ, Clevers H, Nature 2009, 459, 262. [DOI] [PubMed] [Google Scholar]
- [14].Hughes CS, Postovit LM, Lajoie GA, Proteomics 2010, 10, 1886. [DOI] [PubMed] [Google Scholar]
- [15].Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordóñez-Morán P, Clevers H, Lutolf MP, Nature 2016, 539, 560. [DOI] [PubMed] [Google Scholar]
- [16].Brown TE, Marozas IA, Anseth KS, Adv. Mater 2017, 29, DOI 10.1002/adma.201605001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Killaars AR, Grim JC, Walker CJ, Hushka EA, Brown TE, Anseth KS, Adv. Sci 2018, 1801483, 1801483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tsang KMC, Annabi N, Ercole F, Zhou K, Karst DJ, Li F, Haynes JM, Evans RA, Thissen H, Khademhosseini A, Forsythe JS, Adv. Funct. Mater 2015, 25, 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kloxin AM, Kasko AM, Salinas CN, Anseth KS, Science 2009, 324, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].He M, Li J, Tan S, Wang R, Zhang Y, J. Am. Chem. Soc 2013, 135, 18718. [DOI] [PubMed] [Google Scholar]
- [21].Yang C, Tibbitt MW, Basta L, Anseth KS, Nat. Mater 2014, 13, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rosales AM, Vega SL, DelRio FW, Burdick JA, Anseth KS, Angew. Chemie - Int. Ed 2017, 56, 12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].McKinnon DD, Brown TE, Kyburz KA, Kiyotake E, Anseth KS, Biomacromolecules 2014, 15, 2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].DeForest CA, Anseth KS, Nat. Chem 2011, 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kloxin AM, Tibbitt MW, Kasko AM, Fairbairn JA, Anseth KS, Adv. Mater 2010, 22, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Scott TF, Schneider AD, Cook WD, Bowman CN, Science 2005, 308, 1615. [DOI] [PubMed] [Google Scholar]
- [27].Hoyle CE, Lee TY, Roper T, J. Polym. Sci. Part A Polym. Chem 2004, 42, 5301. [Google Scholar]
- [28].Hoyle CE, Bowman CN, Angew. Chem. Int. Ed. Engl 2010, 49, 1540. [DOI] [PubMed] [Google Scholar]
- [29].Colak B, Soares TA, Gautrot JE, 2016, DOI 10.1021/acs.bioconjchem.6b00349. [DOI]
- [30].Love DM, Kim K, Goodrich JT, Fairbanks BD, Worrell BT, Stoykovich MP, Musgrave CB, Bowman CN, J. Org. Chem 2018, 83, 2912. [DOI] [PubMed] [Google Scholar]
- [31].DeForest CA, Polizzotti BD, Anseth KS, Nat. Mater 2009, 8, 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bernasconi CF, Killion RB, J. Am. Chem. Soc 1988, 110, 7506. [Google Scholar]
- [33].Brown TE, Silver JS, Worrell BT, Marozas IA, Yavitt FM, Günay KA, Bowman CN, Anseth KS, J. Am. Chem. Soc 2018, 140, 11585. [DOI] [PubMed] [Google Scholar]
- [34].Moyal T, Hemantha HP, Siman P, Refua M, Brik A, Chem. Sci 2013, 4, 2496. [Google Scholar]
- [35].Tajc SG, Tolbert BS, Basavappa R, Miller BL, J. Am. Chem. Soc 2004, 126, 10508. [DOI] [PubMed] [Google Scholar]
- [36].Irving RJ, Nelander L, Wadso I, Acta Chem. Scand 1964, 18, 769. [Google Scholar]
- [37].Dénès F, Pichowicz M, Povie G, Renaud P, Chem. Rev 2014, 114, 2587. [DOI] [PubMed] [Google Scholar]
- [38].Awoonor-Williams E, Rowley CN, Chem J. Theory Comput. 2016, 12, 4662. [DOI] [PubMed] [Google Scholar]
- [39].Griffith OW, Free Radic. Biol. Med 1999, 27, 922. [DOI] [PubMed] [Google Scholar]
- [40].Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ, Vousden KH, Dev. Cell 2013, 25, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Biaglow JE, Varnes ME, Clack EP, Epp ER, Radiat. Res 1983, 95, 437. [PubMed] [Google Scholar]
- [42].Bakaic E, Smeets NMB, Hoare T, RSC Adv. 2015, 5, 35469. [Google Scholar]
- [43].Fairbanks BD, Singh SP, Bowman CN, Anseth KS, Macromolecules 2011, 44, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Halstenberg S, Panitch A, Rizzi S, Hall H, Hubbell JA, Biomacromolecules 2002, 3, 710. [DOI] [PubMed] [Google Scholar]
- [45].Cruise GM, Hegre OD, Scharp DS, Hubbell JA, Biotechnol. Bioeng 1998, 57, 655. [DOI] [PubMed] [Google Scholar]
- [46].Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS, Biomaterials 2009, 30, 6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ash C, Dubec M, Donne K, Bashford T, Lasers Med. Sci 2017, 32, 1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Cruz-Acuña R, Quirós M, Farkas AE, Dedhia PH, Huang S, Siuda D, García-Hernández V, Miller AJ, Spence JR, Nusrat A, García AJ, Nat. Cell Biol 2017, 19, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Simon‐Assmann P, Orian‐Rousseau V, Arnold C, Kedinger M, Duclos B, Mathelin C, Engvall E, Dev. Dyn 1994, 201, 71. [DOI] [PubMed] [Google Scholar]
- [50].Benoit YD, Groulx J-F, Gagné D, Beaulieu J-F, Signal Transduct J 2012, 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gerbe F, Legraverend C, Jay P, Cell. Mol. Life Sci 2012, 69, 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang Y, Gunasekara DB, Reed MI, DiSalvo M, Bultman SJ, Sims CE, Magness ST, Allbritton NL, Biomaterials 2017, 128, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tong Z, Martyn K, Yang A, Yin X, Mead BE, Joshi N, Sherman NE, Langer RS, Karp JM, Biomaterials 2018, 154, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Broguiere N, Isenmann L, Hirt C, Ringel T, Placzek S, Cavalli E, Ringnalda F, Villiger L, Züllig R, Lehmann R, Rogler G, Heim MH, Schüler J, Zenobi-Wong M, Schwank G, Adv. Mater 2018, 1801621, DOI 10.1002/adma.201801621. [DOI] [PubMed] [Google Scholar]
- [55].Walton KD, Wang S, Gumucio DL, Signals and Forces Shaping Organogenesis of the Small Intestine, Elsevier Inc., 2019. [DOI] [PubMed] [Google Scholar]
- [56].Bonnans C, Chou J, Werb Z, Nat. Rev. Mol. Cell Biol 2014, 15, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Scott TF, Draughon RB, Bowman CN, Adv. Mater 2006, 18, 2128. [Google Scholar]
- [58].Kloxin CJ, Scott TF, Bowman CN, Macromolecules 2009, 42, 2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gandavarapu NR, Azagarsamy MA, Anseth KS, Adv. Mater 2014, 26, 2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Grim JC, Brown TE, Aguado BA, Chapnick DA, Viert AL, Liu X, Anseth KS, ACS Cent. Sci 2018, 4, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Jones EJ, Matthews ZJ, Gul L, Sudhakar P, Treveil A, Divekar D, Buck J, Wrzesinski T, Jefferson M, Armstrong SD, Hall LJ, Watson AJM, Carding SR, Haerty W, Di Palma F, Mayer U, Powell PP, Hautefort I, Wileman T, Korcsmaros T, Dis. Model. Mech 2019, 12, dmm037069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Roberts JJ, Bryant SJ, Biomaterials 2013, 34, 9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.