

SUMMARY
Ataxia Telangiectasia and Rad3-Related kinase (ATR) is a master regulator of genome maintenance, and participates in DNA replication and various DNA repair pathways. In a genome-wide screen for ATR-dependent fitness genes, we identified a previously uncharacterized gene, C17orf53, whose loss led to hypersensitivity to ATR inhibition. C17orf53 is conserved in vertebrates and is required for efficient cell proliferation. Loss of C17orf53 slowed down DNA replication and led to pronounced interstrand crosslink (ICL) repair defect. We showed that C17orf53 is a ssDNA- and RPA-binding protein and both characteristics are important for its functions in the cell. In addition, using multiple omics methods, we found that C17orf53 works with MCM8/9 to promote cell survival in response to ICL lesions. Taken together, our data suggest that C17orf53 is a novel component involved in ICL repair pathway.
Keywords: Genome-wide screen, C17orf53, DNA replication, ICL repair, ssDNA-binding, RPA-binding, HROB, MCM8IP
In Brief
We identified a previously uncharacterized protein C17orf53 that promotes cell survival in response to ATR inhibition. C17orf53 is a conserved protein in vertebrates, and its expression is associated with cell proliferation. We further demonstrated that C17orf53 loss leads to defective interstrand crosslink (ICL) repair. We propose that C17orf53 functions with MCM8/9 and promotes ICL repair.
INTRODUCTION
The Ataxia Telangiectasia mutated and Rad3-related (ATR) protein kinase is a master operator in genome maintenance through coordinating DNA replication, DNA repair, and cell cycle transition [1–3]. Single-stranded DNA (ssDNA) produced at sites of stressed or stalled replication forks is bound by the replication protein A (RPA) complex, which activates ATR with the help of ATR interacting protein (ATRIP) [4–6]. The activated ATR further initiates a series of cellular processes including replication stress responses and DNA repair. Among these cellular processes, one critical role of ATR is control of the cell cycle checkpoint. ATR activates its downstream CHK1 kinase, which further suppresses cyclin-dependent kinases (CDKs) to prevent cells with DNA lesions from entering mitosis [7–9]. Another important function of ATR is preventing replication catastrophe by stabilizing the stressed replication fork [2, 10–12]. Moreover, ATR activity is important for various DNA damage repair pathways, such as homologous recombination repair (HR) pathway and interstrand crosslink (ICL) repair pathway [13–17].
The multiple functions of ATR in coordinating the responses to replication stress and DNA damage make it a promising target for cancer therapy [18, 19]. Tumors often harbor high levels of replication stress due to escalated proliferation, which may increase their need for ATR activity. Similarly, due to the roles of ATR in various DNA damage repair pathways, combination with ATR inhibitors may enhance the efficacy of chemotherapy and radiation therapy that directly or indirectly induce DNA lesions.
DNA ICL is considered as one of the most toxic DNA lesions because DNA double helix is covalently linked, causing an obstacle to replication and transcription [20–24]. Repair of ICL is a complex process, which requires the coordination of several distinct DNA repair pathways, including Fanconi anemia (FA) pathway, Homologous Recombination (HR) pathway and translesion synthesis (TLS) pathway [25–30]. FANCD2 and FANCI proteins are the core components of FA pathway, which acts in part by initiating ICL repair [31]. Monoubiqitination of the FANCI-D2 complex promotes “unhooking” of ICL, which further generates DNA strand breaks at stalled replication forks. HR machinery acts at stalled replication forks to complete DNA repair. HR may also act with TLS to bypass remaining DNA lesions and start DNA replication [32–36]. ATR plays an important role in ICL repair. ATR is activated by ICL-induced stalled replication forks and then in turn phosphorylates numerous downstream effectors, including FA proteins [37, 38]. Since many of these effectors are regulated by ATR kinase, ATR inhibition sensitizes cells to a variety of DNA damage-inducing agents including cisplatin and mitomycin C (MMC) that induce ICLs.
To understand the various cellular processes that require ATR activity in humans, several large-scale genetic screens have been conducted in human cells. Previous studies with RNA interference identified many genes that when lost, confer heightened sensitivity to ATR inhibition [19, 39, 40]. The advance of CRISPR/Cas9-based screens further prompted us to perform genome-wide profiling of vulnerabilities to ATR inhibition [41]. The unbiased profiling of genes whose loss may affect sensitivity to ATR inhibition is useful for at least two purposes. First, these studies allow the identification of new genetic alternations and biomarkers that endow hypersensitivity of tumors to ATR inhibition, which can be exploited in the clinic. Second, these studies reveal novel genes that have functional and/or genetic interaction with ATR pathway.
In this study, we focused on C17orf53 whose loss led to increased sensitivity to ATR inhibition in our previous screen [41]. Human C17orf53 was reported in a few studies and the functions of C17orf53 remain largely unknown until very recently. A previous study indicated a potential relationship between C17orf53 single nucleotide polymorphisms (SNPs) and bone mineral density disorder [42], and another study suggested that C17orf53 is a G2/M specific gene [43].
Here, we showed that C17orf53 is conserved in vertebrates. The expression of C17orf53 is associated with cell proliferation and loss of C17orf53 reduced the rate of DNA replication. Loss of C17orf53 also resulted in pronounced ICL repair defect. We showed that C17orf53 is ssDNA- and RPA-binding protein and both characteristics are essential for its function. Moreover, using multiple omics approaches, we demonstrated that C17orf53 cooperates with MCM8/9 to mediate ICL repair. Collectively, our data suggest that C17orf53 is a new repair protein that promotes ICL repair.
RESULTS
C17orf53 Loss Sensitizes Cells to ATR Inhibition
We recently reported genome-wide CRISPR screens for profiling genes that affect cellular responses to ATR inhibition [41] (Figure 1A). In this study, many genes involved in DNA replication or replication-related DNA repair pathways were enriched, including FA pathway, HR pathway and nucleotide excision repair (NER) pathway (Figure 1B). We found 24 candidate genes correlated robustly with response to ATR inhibition, with 18 genes whose loss induced hypersensitivity to ATRi and 6 genes whose loss induced resistance to ATRi (Figure 1C). Some of these candidate genes have already been reported by us and others [41, 44–46]. We then focused on an uncharacterized gene, C17orf53, the loss of which induced hypersensitivity to ATR inhibition, since significant dropouts of sgRNAs targeting this gene were noted in the ATR inhibitor treated groups (Figure 1D).
Figure 1. C17orf53 was Identified as a Determinant of ATRi Sensitivity.
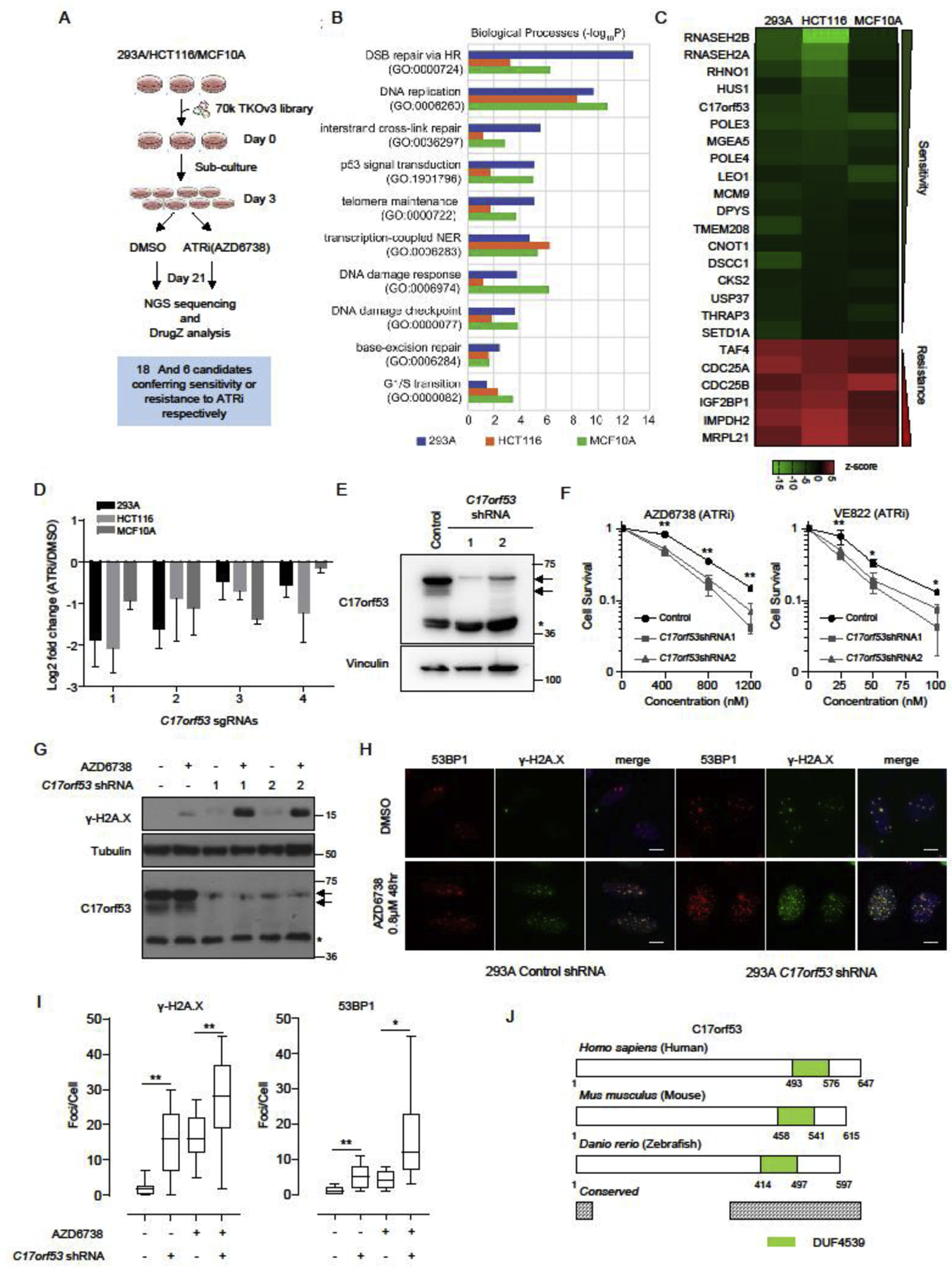
(A) Schematic representation of the workflow for CRISPR screens performed in 293A, HCT116, and MCF10A cells. (B) Gene ontology (GO) analysis of the synthetic lethality genes identified in the CRISPR screens. The top10 GO terms significantly enriched are listed here. Different color represents different cell line. Blue: 293A, Orange: HCT116, Green: MCF10A. (C) Visualization of the high-confidence genes in these screens. The drugZ scores are displayed as a heatmap summarizing the common genes uncovered in all three screens (FDR<0.01) which show synthetic lethality (score<0) or synthetic survival (score>0) with ATR inhibition. (D) Fold change ratios of sgRNAs targeting C17orf53 in 3 cell lines comparing ATRi treated group with DMSO treated group. Mean±SD, n=3. (E) Immunoblot of wild-type and C17orf53 knock-down 293A cells. Arrows mark the C17orf53 protein, asterisk marks a non-specific band. (F) Clonogenic survival assay of cells treated with ATR inhibitors AZD6738 or VE-822. Mean±SD, n=3, *p<0.05, **p<0.01, student t-test. (G) Wild-type and C17orf53 knock-down 293A cells were treated with AZD6738 (0.8 μM, 48hr). Cells were harvested and lysates were analyzed by immunoblotting with indicated antibodies. Arrows mark the C17orf53 protein, asterisk marks a non-specific band. (H) Wild-type and C17orf53 knock-down 293A cells were treated with DMSO or AZD6738 (0.8 μM, 48hr). Cells were fixed and processed for γ-H2A.X and 53BP1 immunofluorescence staining. The scale bar represents 1 μm. (I) Quantification of γ-H2A.X and 53BP1 foci in different groups in H). n=100 cells, *P<0.05, **P<0.01, student t-test. (J) Schematic diagram of C17orf53 proteins from human to zebrafish. Green color marks a conserved domain which was named as Domain with Unknown Function 4539 (DUF4539). The protein regions with >80% sequence similarity from zebrafish to human were marked as “conserved” (Grey).
We first confirmed this finding with a different gene depletion method. By using shRNAs, we efficiently knocked down C17orf53 in 293A cells, which were validated in both mRNA and protein levels (Figure 1E and Figure S1A). Cell survival assays showed that the C17orf53 knock-down cells were sensitive to treatments with two different ATR inhibitors (Figure 1F and Figure S1B), further confirming that C17orf53 is a determinant of ATRi sensitivity.
On molecular levels, ATR inhibition induced dramatic increase of γ-H2A.X in C17orf53 knock-down cells compared with WT cells (Figure 1G). Moreover, γ-H2A.X and 53BP1 foci were also significantly increased in C17orf53 knock-down cells upon ATR inhibitor treatment (Figures 1H and 1I), indicating that more double strand DNA breaks (DSBs) were generated due to depletion of C17orf53. All these results suggest a possible function of C17orf53 in genome maintenance. Furthermore, as genes co-essential with ATR inhibition usually participate in DNA replication or repair [1, 18, 41], our data further indicate that C17orf53 may function in these processes.
C17orf53 Is a Conserved Gene Involved in Cell Proliferation
C17orf53 is highly conserved in vertebrates, which is schematically depicted in Figure 1J (please also see Figure S1C for protein sequence alignment). While the function of C17orf53 remains unknown, a previous study indicated that C17orf53 mutations are associated with bone mineral density disorder [42].
We first noticed that C17orf53 expression levels were significantly higher in various cancer types than its expression levels in the matched normal tissues in TCGA (The Cancer Genome Atlas) database (Figure 2A and Figure S2A). Moreover, higher expression levels of C17orf53 were associated with poor prognosis (Figure 2B). We also found that C17orf53 expression level was higher in proliferating cells than that in quiescent cells (Figure 2C). These observations suggest that C17orf53 may have a function in cell proliferation. Whereas a recent study indicated that C17orf53 is a highly expressed gene in S/G2 cell cycle phases [43], the C17orf53 protein level did not change significantly during the cell cycle in our experiment (Figure S2B). These findings suggest that C17orf53 is a cell proliferation related gene but its protein level is not significantly regulated during cell cycle.
Figure 2. C17orf53 is Necessary for Optimal Cell Proliferation and DNA Replication.
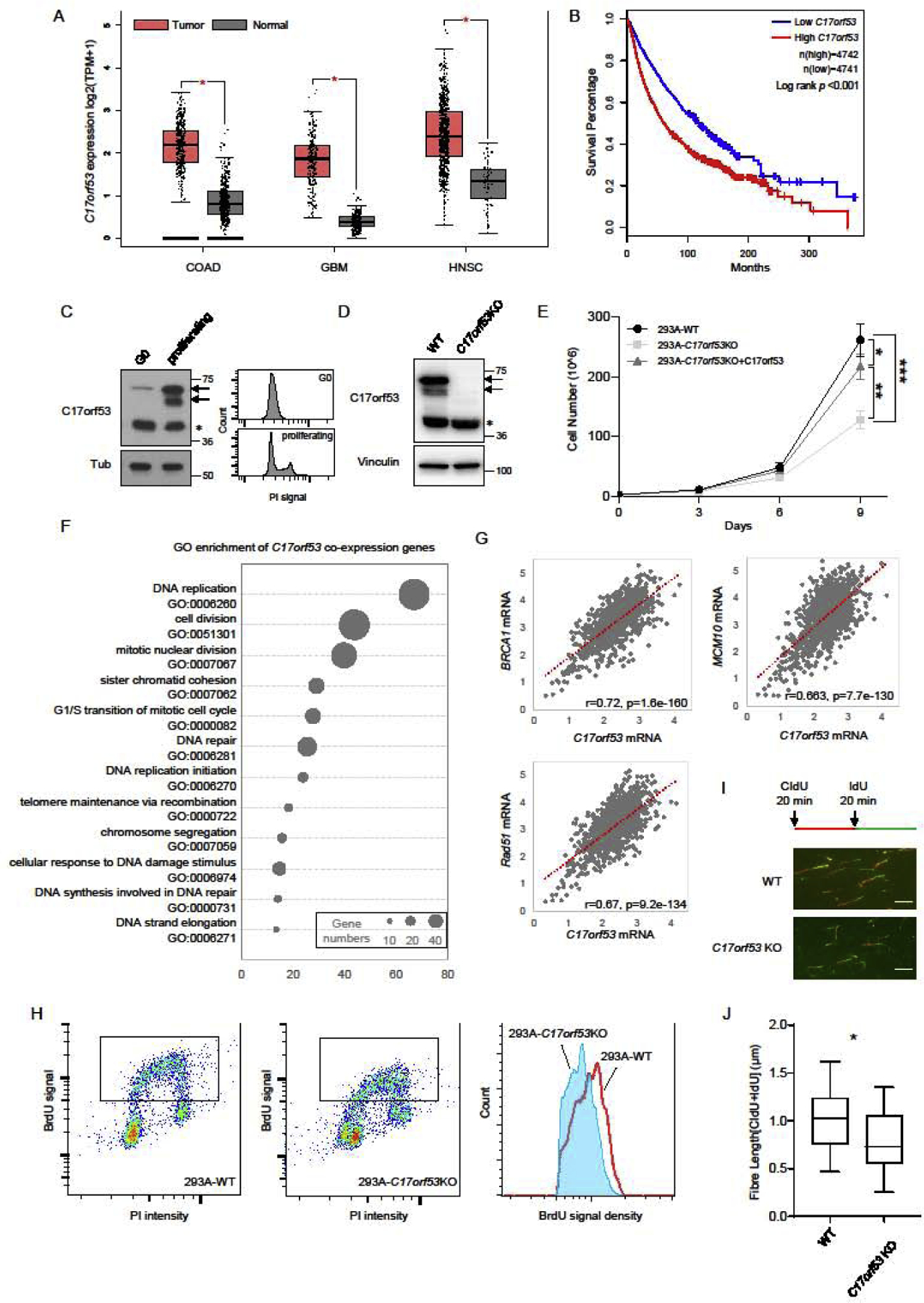
(A) C17orf53 expression is upregulated in various types of tumors (data from TCGA). Analysis of C17orf53 expression (log2 [TPM+1]) in TCGA tumors and the matched normal tissues. COAD: Colon adenocarcinoma; GBM: Glioblastoma; HNSC: Head and Neck squamous cell carcinoma. *p<0.01, Wilcoxon test. (B) Kaplan-Meier curves of overall survival for the overall population (more than 9000, data from TCGA) according to the expression levels of C17orf53. Statistical significance was assessed by the log-rank test and presented. (C) 293A cells were contact inhibited (G0) then released for 12 hrs (proliferating). (Left panel) Total cell lysates were prepared from each group and immunoblotted with the indicated antibodies. Asterisk marks the non-specific band. (Right Panel) The same samples were fixed, stained with Propidium iodide (PI) and analyzed by fluorescence-activated cell sorting (FACS). (D) Immunoblot analysis of 293A wild-type and C17orf53 KO cells. Arrows mark the C17orf53 protein, asterisk marks non-specific band. (E) Growth curves of indicated cells. Different cells were plated in 6-well plates, passaged and counted every 3 days. *p<0.05, **p<0.01, ***p<0.001, ANOVA test. (F) Gene ontology (GO) terms significant enriched among the genes co-expressed with C17orf53 (Gene list is available in Table S1). (G) Expression level of C17orf53 correlates with expression levels of BRCA1, RAD51 and MCM10 in 1019 cell lines in CCLE. Expression level was calculated by log2.FPKM+1. The correlation scores were measured by Pearson correlation. (H) BrdU incorporation assay. Growing wild-type and C17orf53KO 293A cells were incubated in medium containing BrdU (10 μM) for 30 mins, fixed, denatured with 2N HCl and stained with BrdU antibody and propidium iodide (PI). Cells with BrdU incorporation are marked with frame and histograms of BrdU intensity are shown in the right panel. (I) Quantitation of the flow cytometry data shown in (H), presented as the mean BrdU fluorescence intensity. (±SD, n=3, *p<0.05). (J) 293A WT and C17orf53 KO were treated with CIdU (25 μM) for 20 mins and then treated with IdU (250 μM) for 20 mins. DNA fibers from these cells are indicated. The scale bar represents 1 μm. (K) Quantification of DNA fiber lengths in J). Both CIdU and IdU fibers were counted. The box plots show twenty-fifth to seventy-fifth percentiles with lines indicating the median. n =100, *p<0.05, student t-test.
We then generated C17orf53 knock-out (KO) 293A cells using CRISPR/Cas9 gene editing technology, which were validated by immunoblotting and genome sequencing (Figure 2D and Figure S2C). Consistent with the C17orf53 knock-down cells, the C17orf53-KO cells also showed slow cell growth which could be rescued by reconstruction with ectopically expressed C17orf53 (Figure 2E). We also generated C17orf53 KO U2OS cells and confirmed the retarded cell growth phenotypes in these KO cells (Figure S2D). We further checked apoptosis among different groups using Annexin-V staining. As shown in Figure S2E, there was no significant difference in apoptotic cell population among different groups, indicating that the slow growth phenotype is not due to cell death. All these results suggest that C17orf53 is necessary for sufficient cell proliferation at least in the cell lines we tested.
To gain further insight into the possible functions of C17orf53, we performed coexpression analysis of C17orf53 using the gene expression datasets available from Genomics of Drug Sensitivity in Cancer (GDSC) and Cancer Cell Line Encyclopedia (CCLE). We calculated the Pearson correlation factors and false discover rates (FDR) between the expression levels of C17orf53 and the expression levels of other genes in hundreds of cancer cell lines (Table S1). Using a stringent cutoff of FDR<10^−40, we found 230 genes whose expression levels are robustly co-related with that of C17orf53 (Figure S2F and Table S1). GO enrichment analysis showed that most of these genes are involved in DNA replication and replication-associated DNA repair processes such as HR and FA pathways (Figure 2F and Table S1). For example, in cancer cell lines, the expression levels of C17orf53 are closely correlated with the expression levels of BRCA1, RAD51 and MCM10 (Figure 2G), which was further confirmed by the TCGA data (Figure S2G). These data further implicated a possible relationship between C17orf53 expression and cell proliferation.
The nice correlation of C17orf53 with numerous genes required for faithful DNA replication (Figure 2F and Table S1) also implied a similar role for C17orf53. Thus, we first checked the DNA replication in C17orf53 KO cells. Although C17orf53 KO cells showed cell cycle distribution similar to that of WT cells (Figure S2H and S2I), the BrdU incorporation was reduced in C17orf53 KO cells (Figures 2H and 2I). The DNA fiber assay further showed slower replication fork progression in C17orf53 KO cells (Figures 2J and 2K). These findings suggest that loss of C17orf53 may affect DNA replication, which may be one of the reasons for that C17orf53-depleted cells showed cell proliferation defects and were sensitive to ATR inhibition.
C17orf53 Depleted Cells Show Profound Defect In ICL Response
To gain more insight into the functions of C17orf53, we tested the sensitivity of 293A C17orf53 KO cells to treatments with a variety of DNA-damaging agents. Besides ATR inhibitor, we found that 293A C17orf53 KO cells were sensitive to ICL-inducing agents mitomycin C (MMC) and cisplatin, but were not consistently sensitive to hydroxyurea (HU), camptothecin (CPT), irradiation (IR), or UV (Figure 3A and Figure S3A). Moreover, compared with WT cells, 293A C17orf53 KO cells showed significant G2/M arrest upon MMC treatment (Figures 3B and 3C). Furthermore, we assayed C17orf53 KO cells for sensitivity to psoralen or isopsoralen plus whole cell UVA irradiation (365nm). The products of activated psoralen are predominantly ICLs, whereas the activated isopsoralen forms monoadducts [22, 47]. As shown in Figure S3B, C17orf53 KO cells were sensitive to psoralen+UVA but not sensitive to isopsoralen+UVA. All these results suggest a function for C17orf53 in the processing of ICL. It is also of note that C17orf53 KO cells were sensitive to etoposide treatment (Figure S3A), implying possible additional functions for C17orf53 in other repair pathways.
Figure 3. C17orf53 Deficient Cells Show Pronounced ICL Repair Response Defects.
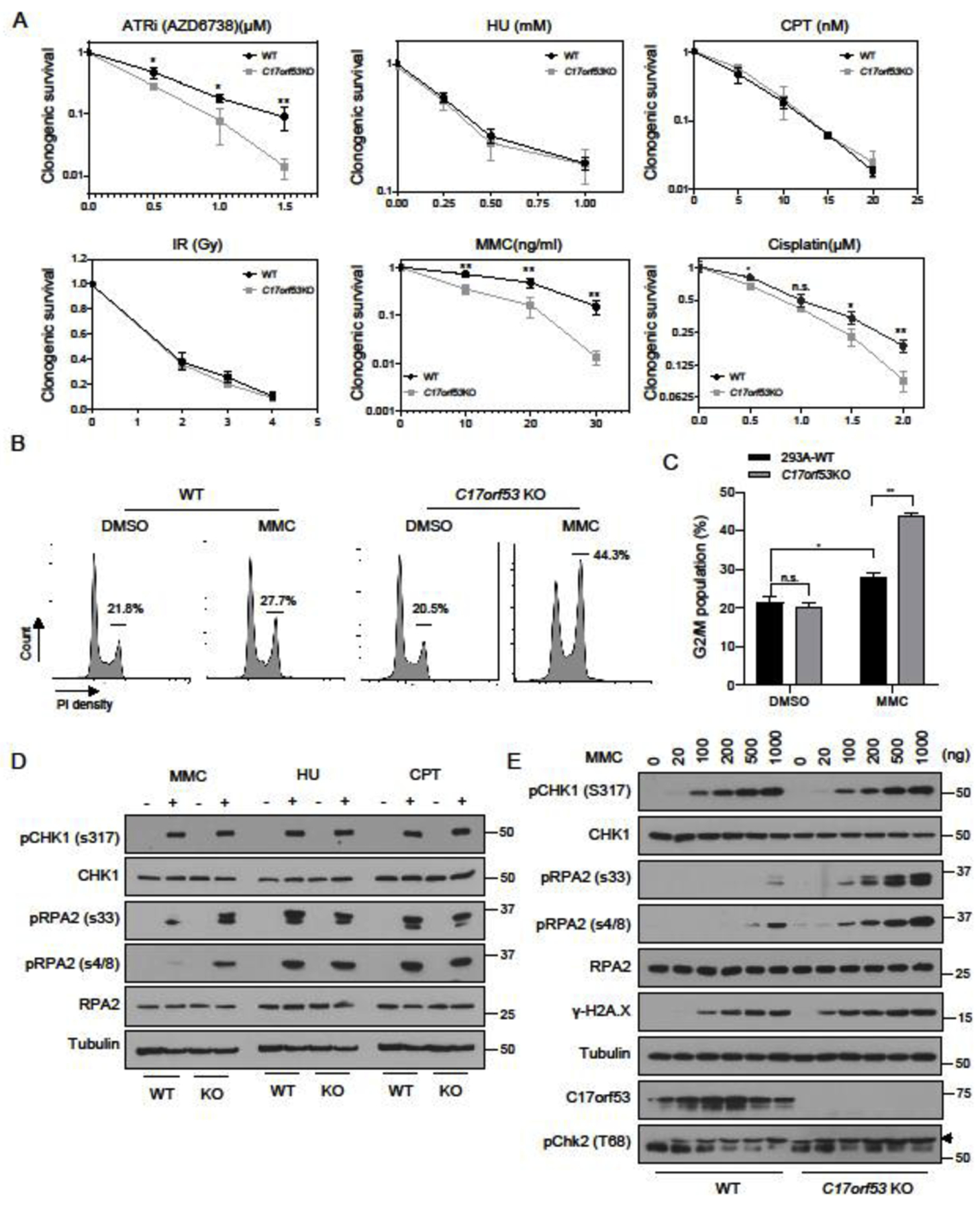
(A) Clonogenic survival assay for WT and C17orf53KO 293A cells exposed to ATRi (AZD6738), HU, CPT, IR, MMC and Cisplatin. Mean±SD, n=3, *p<0.05, **p<0.01, student t-test. (B) 293A WT or C17orf53 KO cells were treated with DMSO or MMC (10 ng/ml) for 48 hr before ethanol fixation, propidium iodide (PI) staining, and fluorescence-activated cell sorting (FACS) analysis. Cells in G2-M cell cycle stages were marked and measured. Representative data are shown. (C) Quantification of the experiments shown in B). Mean±SD, n=3, *p<0.05, **p<0.01, student t-test. (D) WT or C17orf53KO 293A cells were treated with MMC (500 ng/ml), HU (4 mM) or CPT (200 nM) for 8 hours and then lysed. The total cell lysates were immunoblotted with the indicated antibodies. (E) WT or C17orf53KO 293A cells were treated with MMC of different doses for 8 hours and then lysed. The total cell lysates were immunoblotted with the indicated antibodies.
Compared with WT cells, MMC treatment induced exacerbated checkpoint activation as indicated by the dramatic increase of pRPA2 in C17orf53 KO cells, whereas neither HU nor CPT treatment could induce such dramatic differences between C17orf53 KO and WT cells (Figure 3D). This result was further confirmed in cells under treatments of different doses of MMC. As shown in Figure 3E, pRPA2 and γH2A.X levels were significantly increased in C17orf53 KO cells more than WT cells upon MMC treatment. Although treatments with different doses of HU or CPT could also lead to significant increase in pRPA2 levels in cells, we did not observe any obvious difference of pRPA2 levels between WT and C17orf53 KO cells (Figure S3C). In addition, consistent to C17orf53 KO 293A cells, MMC treatment could also induce significant increase in pRPA2 level (Figure S3D) and dramatic G2/M arrest (Figures S3E and S3F) in C17orf53 depleted U2OS cells more than in WT U2OS cells. These results further confirmed that loss of C17orf53 appears to induce ICL repair defects in cells and suggest a function for C17orf53 in the processing and/or repair of ICL lesions.
C17orf53 Functions in the Late Stage of ICL Repair
ICL repair is a complex process that requires the action of multiple DNA repair pathways [22, 24, 48, 49] . To address the function of C17orf53 in this process, we treated 293A WT or 293A C17orf53 KO cells with MMC (1000 ng/ml) for 1 hour, released them from MMC treatment, and then monitored DNA repair and DNA damage markers in these cells at different time points (i.e. 8, 24 and 48 hr). We found that the pRPA2 level was significantly increased in C17orf53 KO cells more than WT cells at each checked time point (Figure 4A). The increased pRPA2 level was probably not induced by long stretches of single strand DNA. As shown in Figures S4A and S4B, while native BrdU signals could be detected in cells treated with HU when long stretches of single strand DNA were generated, we could hardly detect any native BrdU signal in either WT or C17orf53 KO cells treated with MMC, which is consistent with a previous report [48]. We also noticed that γH2A.X level was higher in C17orf53 KO cells, especially at late time points (Figure 4A), indicating an accumulation of DNA damage, which agrees with the observations above that showed ICL repair defects caused by C17orf53 depletion.
Figure 4. C17orf53 participates in ICL repair through affecting HR repair.
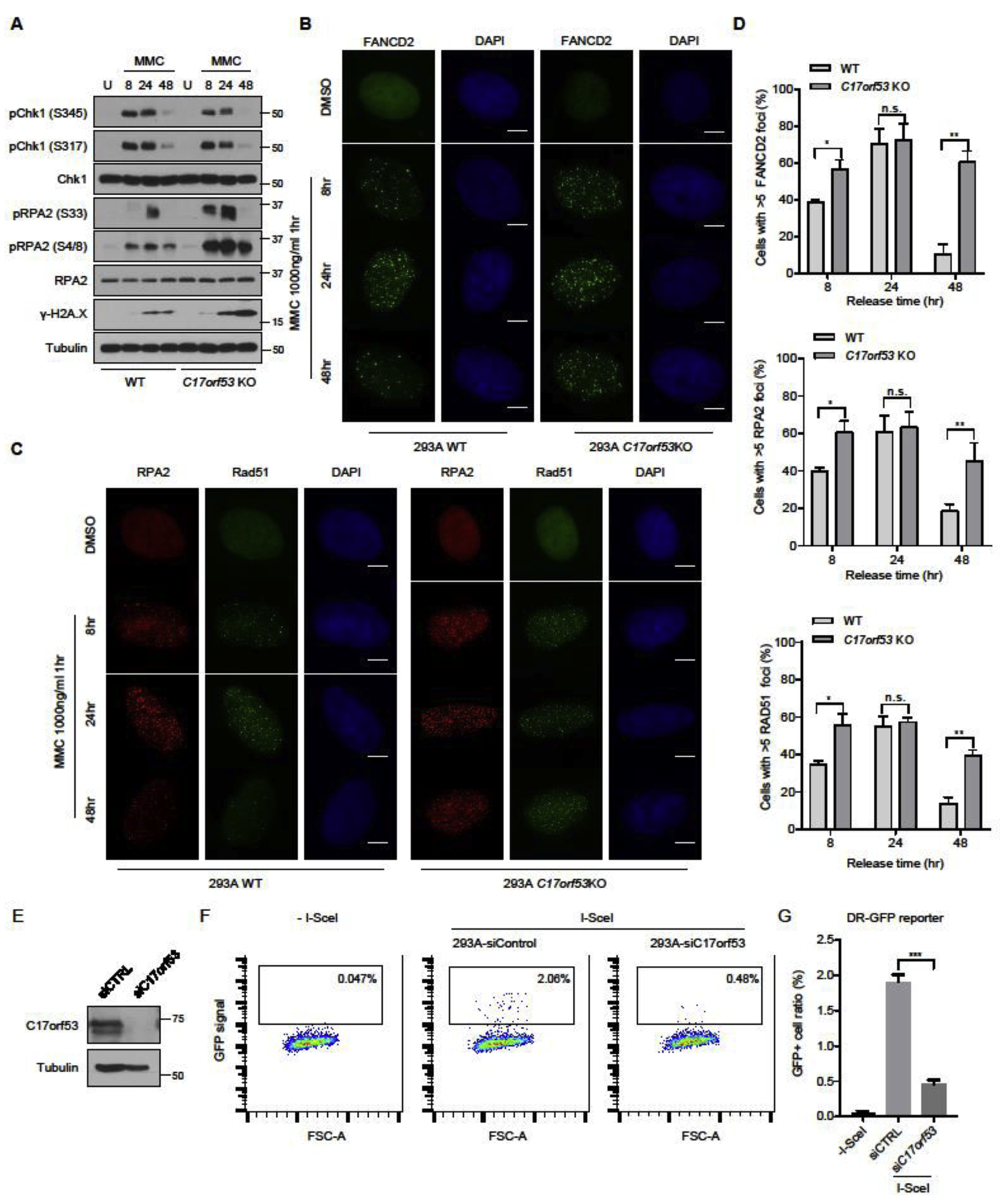
(A) WT cells and C17orf53 KO cells were treated with DMSO or MMC (1000 ng/ml, 1 hr). The cells were harvested at different time points after treatment. The total cell lysates were immunoblotted with the indicated antibodies. (B) MMC-induced FANCD2 foci formation in WT and C17orf53 KO 293A cells. The indicated cells were left untreated or treated with MMC (1000 ng/ml, 1hrs). At different time points after treatment, the cells were fixed and immunostained with FANCD2 antibody. The scale bar represents 1 μm. (C) MMC-induced RPA2 and RAD51 foci in WT and C17orf53KO 293A cells. The same cells in B) were fixed and immunostained with RPA2 and RAD51 antibodies. The scale bar represents 1 μm. (D) Quantification of FANCD2, RPA2 and RAD51 FANCD2 foci in B) and C). n=3 biological independent experiments. *p<0.05, **p<0.01, student t-test. (E) U2OS cells were transfected with control siRNA or C17orf53 siRNA. Cell lysates were immunoblotted with indicated antibodies. (F) Loss of C17orf53 results in decreased HR-directed DNA repair. U2OS-DR-GFP were transfected with control siRNA or C17orf53 siRNA, followed by transfection of I-SceI. 48hr after transfection, cells were harvested and assayed for GFP expression by FACS. Representative data are shown. (G) Quantification of the experiments shown in F). Mean±SD, n=3. ***p<0.001, student t-test.
It is speculated that ICL lesions induced by MMC treatment may be sensed and processed by FA, NER, and other pathways, which are further repaired by multiple subsequent repair pathways including HR [50, 51]. To assess where along the ICL repair process that C17orf53 functions, we monitored the foci formations of several essential proteins in these pathways, which include FANCD2, RPA2 and RAD51. We first monitored the foci formation of FANCD2, a core component of the FA pathway which is important for the initiation of the ICL repair. As shown in Figures 4B and 4D, in 293A C17orf53 KO cells, MMC treatment induced more FANCD2 foci at the early time point (8 hrs), which persisted much longer (48 hrs) than those in WT cells, suggesting that while the recruitment of FANCD2 was not affected by C17orf53 depletion, the ICL repair capacity was impaired. Consistently, under MMC treatment, the monoubiquitination changes of FANCD2 and FANCI were unaffected in C17orf53 KO cells comparing to those in WT cells (Figure S4C). These results suggest that C17orf53 is not likely involved in the initial stage of ICL repair. We also monitored the foci formation of RPA2 and RAD51. Both RPA and RAD51 are known to participate in replication fork protection, which may be involved in the early stage of ICL processing. Moreover, RPA and RAD51 are key factors required for HR repair, which is known to act at the late stage of ICL repair. As shown in Figures 4C and 4D, RPA2 and RAD51 foci increased significantly at early time point (8 hrs) and persisted much longer (48 hrs) in 293A C17orf53 KO cells than those in WT cells. The persistence of RPA2 and RAD51 foci further suggest that C17orf53 may have a unique role in ICL repair, i.e. it may function after or in parallel to RAD51 recruitment.
We then used well-established reporter assays to monitor HR capacity in 293A C17orf53 KO cells. We knocked down C17orf53 using siRNA in U2OS cells (Figure 4E) and found that C17orf53 depletion led to a pronounced reduction in HR repair efficiency (Figures 4F and 4G). Meanwhile, we tested NHEJ capacity in 293A C17orf53 knock-down cells. As shown in Figures S4D and S4E, the NHEJ repair efficiency was only modestly impaired in C17orf53 knock-down cells. These results suggest that defective ICL repair observed in C17orf53 depleted cells may be at least in part due to defective HR repair measured by these reporter assays and further implied that C17orf53 may function in HR induced by ICL.
Both RPA Binding Motifs and ssDNA Binding Domain-DUF4539 are Essential for the Function of C17orf53 in ICL Repair
To get further insight into the C17orf53 protein function, we performed mass spectrometry analysis following Tandem affinity purification (TAP-MS) or GFP-pull down to search for C17orf53-binding partners. As shown in Figure 5A and Table S2, the C17orf53 interactome revealed enrichment of the RPA complex. Moreover, we also found C17orf53 in the TAP-MS results of RPA1 (Figure 5A and Table S2). Indeed, co-immunoprecipitation (Co-IP) experiments showed that C17orf53 could bind strongly to RPA1 and RPA2 (Figure 5B) and the in vitro pull-down assays demonstrated that RPA1 and RPA2 could bind directly to C17orf53 (Figure 5C). We further mapped the binding between C17orf53 and RPA1/RPA2 (Figure 5D and 5E) and showed that residues 1–100 (F1) and residues 433–492 (F5) of C17orf53 mediate its binding to RPA1, while residues 433–592 (F5) mediates its binding to RPA2. Through alignment with other RPA binding proteins (ETAA1, ATRIP, XPA, NBS1, SMARCAL1), we identified two RPA binding motifs (RBM1: residues 10–23 and RBM2: residues 479–492) in C17orf53 (Figure 5F), which were further confirmed by Co-IP experiments using deletion variants of C17orf53 (Figure 5G).
Figure 5. C17orf53 is a RPA-binding Protein and Contains a ssDNA-binding Domain DUF4539.
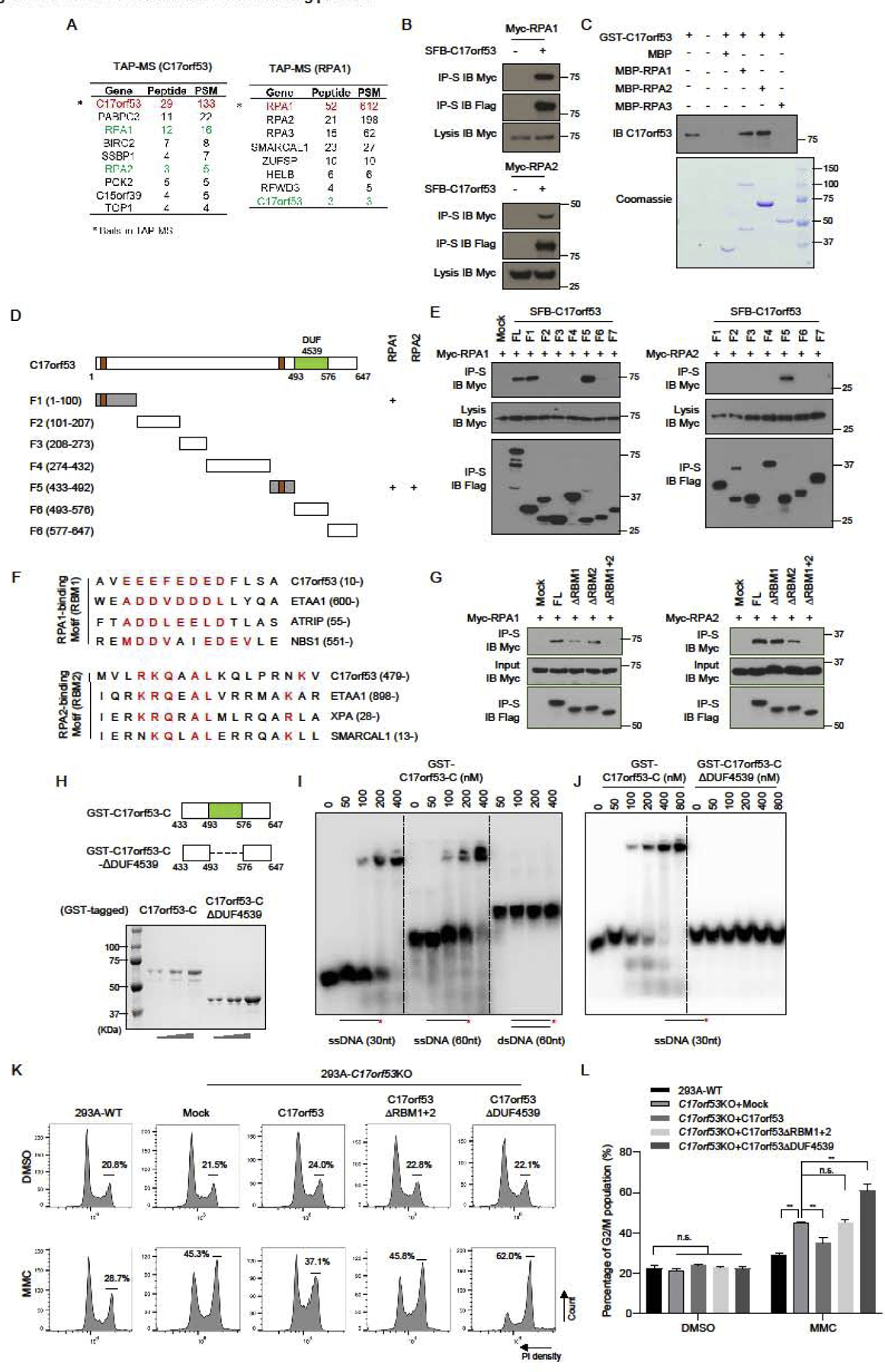
(A) 293T cells stably expressing SFB-C17orf53 or SFB-RPA1 were used for TAP. Tables are representative proteins identified by mass spectrometry analysis. Letters in Red indicate the bait proteins. Letters in Green indicate the candidate proteins. (B) C17orf53 interacts with RPA1 and RPA2. 293T cells were transiently transfected with indicated plasmids. Cell lysates were immunoprecipitated with S beads, and immunoblot was performed with the indicated antibodies. (C) Direct binding between recombinant GST-C17orf53 and MBP-tagged RPA1 and RPA2. Upper panel: C17orf53 was detected by immunoblotting. Lower panel: purified proteins were visualized by Commassie staining. Due to its low concentration, GST-C17orf53 protein could only be detected by immunoblotting. (D) Schematic diagram of C17orf53 indicating the truncation fragments tested in the following experiments. + represents the positive binding. (E) 293T cells were transiently transfected with indicated plasmids. Cell lysates were immunoprecipitated with S beads, and immunoblot was performed with the indicated antibodies. (F) The protein sequence of C17orf53 was compared with the sequences of known RPA-binding proteins and the similar motifs were listed above. They were named as RBM1 (RPA1-binding Motif) and RBM2 (RPA2-binding Motif). (G) 293T cells were transiently transfected with indicated plasmids. Cell lysates were immunoprecipitated with S beads, and immunoblot was performed with the indicated antibodies. (H) Schematic diagram of C17orf53 indicating the location of DUF4539 domain and the truncation fragments tested in the following experiments. (I) The radioactive labeled ssDNA (30 nt), ssDNA (60 nt) and dsDNA (60 nt) were incubated with indicated doses of purified MBP-C17orf53-C (residues 433–647) protein. Electrophoretic mobility shift was used to assess DNA-binding activity. (J) The radioactive labeled ssDNA (30 nt) were incubated with indicated doses of purified MBP-C17orf53-C (residues 433–647 aa) protein or MBP-C17orf53-ΔDUF4539 protein. Electrophoretic mobility shift was used to assess DNA-binding activity. (K) 293A WT cells, 293A C17orf53 KO cells or 293A C17orf53 KO cells with ectopic expression of indicated constructs were treated with 10 ng/ml MMC for 48 hours before ethanol fixation, PI staining, and FACS analysis. Cells in G2-M cell cycle stages were marked and measured. (L) Quantification of the experiments shown in K) Mean±SD, n=3, *p<0.05, **p<0.01, student t-test.
Moreover, we performed alignment of the most conserved domain of C17orf53, i.e. DUF4539 (Domain with Unknown Function 4539), with other known protein domains in the Pfam database (http://pfam.xfam.org/) (Figure S1C). The alignment analysis revealed a putative oligonucleotide/oligosaccharide-binding (OB) fold within DUF4539, suggesting that this domain may be able to bind to nucleotides (Figures S5A and S5B). We then conducted electrophoretic mobility shift assay (EMSA) assays using purified Maltose-binding protein (MBP) proteins fused with the C-terminus of C17orf53 (residues 433–647) containing the DUF4539 domain (Figure 5H). As shown in Figure 5I, the C-terminus of C17orf53 could preferentially bind to ssDNA, but not to dsDNA. Moreover, deletion of DUF4539 abolished the ssDNA-binding ability of the C-terminus of C17orf53 (Figures 5H and 5J). These results demonstrate that C17orf53 could bind to ssDNA via its DUF4539 domain.
To validate whether RPA-binding motifs and the ssDNA-binding domain of C17orf53 are important for its function in ICL repair, we performed reconstruction experiments by ectopic expression of C17orf53, C17orf53-ΔRBM1+2, and C17orf53-ΔDUF4539 separately in 293A C17orf53 KO cells. As shown in Figures 5K and 5L, while the ectopic expression of C17orf53 could partially rescue the G2/M arrest upon MMC treatment in 293A C17orf53 KO cells, neither the ectopic expression of C17orf53-ΔRBM1+2 nor the ectopic expression of C17orf53-ΔDUF4539 could lead to consistent rescue. It is also of note that the ectopic expression of C17orf53-ΔDUF4539 induced more severe G2/M arrest in C17orf53 KO cells upon MMC treatment.
These findings above suggest that both RPA binding and ssDNA binding are important for the function of C17orf53 in ICL repair, possibly by recruiting C17orf53 to the DNA damage sites. To test this possibility, we used both immunostaining and chromatin loading approaches. Although we observed co-localization of C17orf53 and RPA2 at DNA damage sites induced by UV micro-irradiation, we were not able to show convincing foci formation of C17orf53 following MMC treatment (Figure S5C). This result may due to the limited specificity and/or affinity of endogenous C17orf53 antibody that fails to detect damage-induced C17orf53 foci formation by immunostaining. Another possibility is that C17orf53 may not need to form visible foci when it functions. As shown in Figure S5D, we were able to detect MMC-induced chromatin enrichment of WT C17orf53, whereas this damage-induced chromatin loading was significantly decreased for C17orf53-ΔRBM1+2 or C17orf53-ΔDUF4539 variants. These results suggest that in response to MMC treatment, C17orf53 may be recruited to chromatin through its binding to RPA and ssDNA, and therefore play a role in ICL repair.
C17orf53 acts with MCM8/9 in DNA repair
To gain more insight into how C17orf53 functions in ICL repair, we carried out CRISPR screens to search for co-functional genes of C17orf53. As shown in Figure 6A, we performed two parallel CRISPR screens in 293A WT and 293A C17orf53 KO cells upon MMC treatment. In brief, 293A WT and C17orf53 KO cells were infected with TKOv3 sgRNA library and were then treated with appropriate doses of MMC (20 ng/ml for 293A WT cells and 5 ng/ml for 293A C17orf53 KO cells) which could lead to 20% killing. Cells were harvested after 18 days of MMC treatment. Total genomic DNA were extracted, labeled with barcodes and sequenced as previously described [41]. Using drugZ [52], we calculated the Z-scores of each gene by comparing the final sgRNAs counts in MMC-treated WT cells with those in MMC-treated C17orf53 KO cells (Figure 6B and Table S3). In this assay, genes with negative Z-scores means that they are more essential for MMC resistance in WT cells. As expected, we found that C17orf53 is critical for MMC resistance in WT cells, compared with C17orf53 KO cells (rank 12 for C17orf53). That is because of that sgRNAs against C17orf53 could not lead to MMC sensitivity in cells already lack of C17orf53. Of note, we found MCM8 and its partner MCM9 at the top of the essential gene list (rank 1 for MCM8 and rank 19 for MCM9), suggesting that they may function together with C17orf53 in MMC resistance. Meanwhile, we searched the DepMap database (https://depmap.org/portal/) for C17orf53 co-functional genes [53]. Based on the gene effect scores derived from the CRISPR screens in 625 cancer cell lines in DepMap database [53], we calculated the Pearson correlation scores between C17orf53 and other genes (Table S4), with the assumption that genes with higher correlation scores are more likely to function together with C17orf53. As shown in Figure 6C and Table S4, both MCM8 and MCM9 were found to be top genes that likely share functional similarity with C17orf53 (rank 3 for MCM8 and rank 8 for MCM9). Since MCM8 and MCM9 also function in HR repair induced by ICL [54], it is highly likely that C17orf53 cooperates with MCM8-MCM9 complex. We thus validated the genetic interaction between C17orf53 and MCM8 under MMC treatment. In cell survival assays, while both C17orf53 KO and MCM8 depletion induced by MCM8-siRNA sensitized cells to MMC treatment, depletion of MCM8 in C17orf53 KO cells did not show additive MMC sensitivity (Figures 6D and 6E), suggesting they probably function in the same pathway. Moreover, we found MCM8 and MCM9 in the GFP-C17orf53 binding protein list derived from GFP-Pull down MS experiment that we described previously (Table S2 and Figure 6F), suggesting that they may directly interact with each other. Indeed, our follow-up CoIP experiments demonstrated that C17orf53 could interact with MCM8 and MCM9 (Figure 6G). All these results suggest that C17orf53 may act with MCM8 and MCM9 to function in HR repair induced by ICL (Figure 6H).
Figure 6. C17orf53 Cooperates with MCM8-MCM9 in ICL Repair.
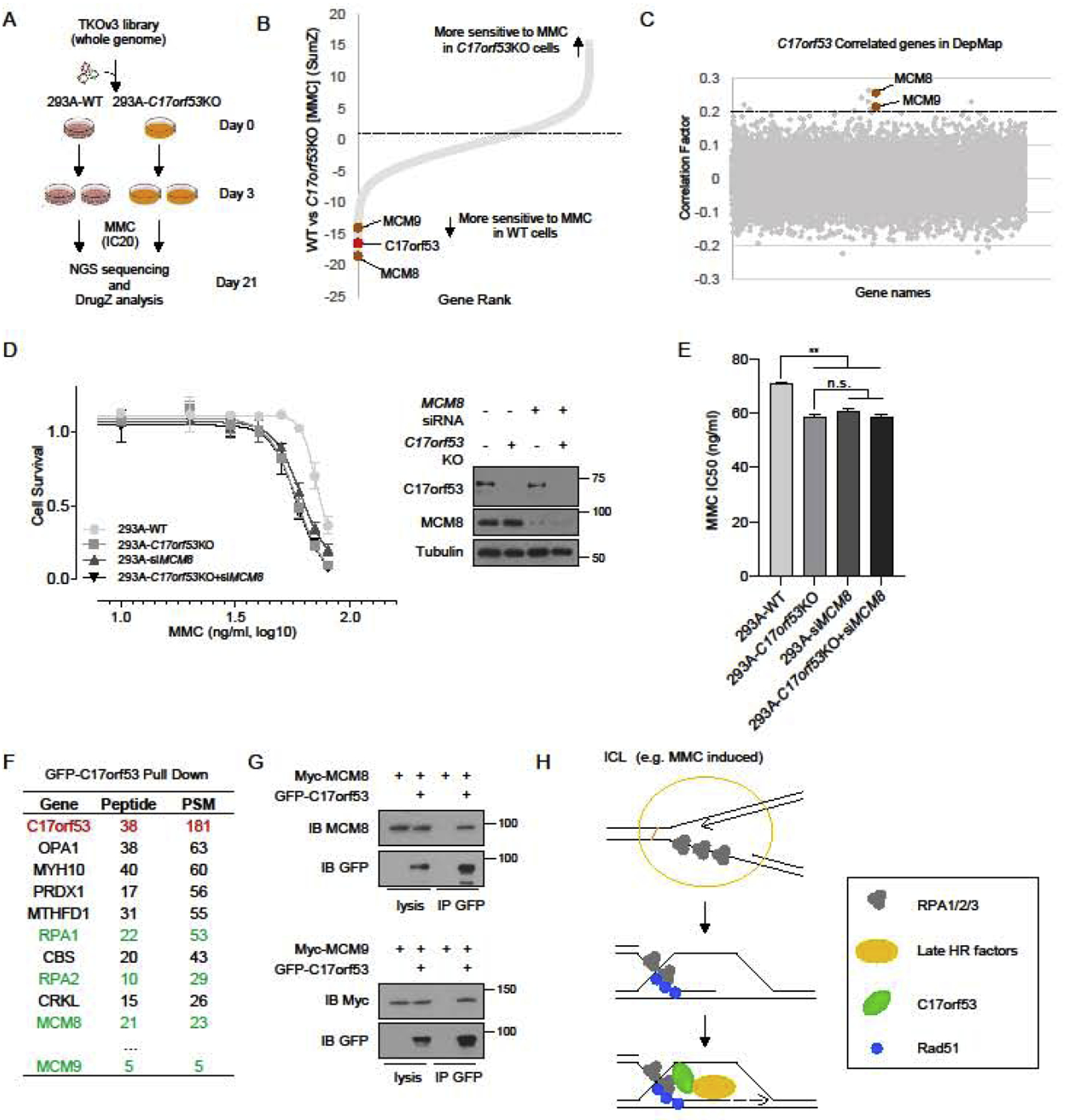
(A) Schematic representation of the workflow for CRISPR screens performed in WT and C17orf53 KO 293A cells with TKOv3 whole-genome library under MMC treatment. (B) DrugZ analysis of the CRISPR screens in A). DrugZ scores were calculated by comparing the sgRNAs counts from MMC-treated WT 293A cells with those from MMC-treated C17orf53 KO cells. Lower scores indicate higher MMC sensitivity in WT cells than that in C17orf53 KO cells. C17orf53 is marked as red. Candidate genes are marked as brown. (C) Based on the gene effect scores from DepMap CRISPR screen datasets, Pearson correlation scores between C17orf53 and other genes were calculated and presented. Genes with higher correlation scores were more likely to function together with C17orf53. The candidate genes are marked in brown. (D) 293A WT cells or 293A C17orf53 KO cells were transfected with control siRNA or MCM8 siRNA. 24 hr later, the cells were treated with different doses of MMC for 72 hr. Cell survival was determined by cell titer Glo assay as described in Methods. Half of the same cells were harvested, lysed and immunoblotted with the indicated antibodies. (E) The IC50 of MMC sensitivity for each group in D) were calculated. Mean±SD, n=3, *p<0.05, **p<0.01, student t-test. (F) 293T cells were transfected with constructs encoding GFP-C17orf53 and treated with MMC (1000 ng/ml, 8 hr). The cells were harvested, lysed and pull down with GFP antibody. The pull-down samples were analyzed by MS. The representative gene list is shown. (G) 293T cells were transiently transfected with indicated plasmids. Cell lysates were immunoprecipitated with GFP antibody, and immunoblot was performed with the indicated antibodies. (H) Model of that C17orf53 functions in HR induced by ICL. Following MMC treatment, C17orf53 is recruited by RPA and ssDNA to sites of DNA damage and further recruits MCM8-MCM9 to facilitate the loading of other repair factors.
DISCUSSION
In this study, we focused on C17orf53, which we identified as promoting viability in cells treated with ATR inhibitor. We validated that the loss of C17orf53 sensitized cells to ATR inhibition. We showed that C17orf53 expression correlates with cell proliferation. Moreover, we found C17orf53 depleted cells showed profound ICL repair defect. Further experiments revealed that C17orf53 functions at the late stage of ICL repair. Mechanically, we found that C17orf53 binds to RPA1/RPA2 through its RBM motifs and binds to ssDNA through its DUF4539 domain. Both the RBM motifs and the DUF4539 domain are important for the chromatin loading of C17orf53 when cells are treated with MMC. With use of functional screening, we found that C17orf53 cooperates with MCM8/9. In sum, our results suggest that C17orf53 functions in homologous recombination induced by ICL.
While our manuscript was in submission, a study reported by Dr. Durocher and colleagues used RPE-hTERT TP53KO cells and identified C17orf53 as a protein involved in HR repair through loading MCM8-MCM9 helicase complex [55]. They renamed this protein as HROB (Homologous Recombination OB-fold). Their results agree with the findings reported in this paper. Using a HR-GFP reporter assay, we showed that C17orf53 depletion led to significant reduction of HR repair efficiency, indicating that C17orf53 is involved in HR repair measured by this reporter assay. We would like to point out that although our experiments showed that C17orf53 facilitates the late HR steps induced by ICL, C17orf53 does not behave like a canonical HR repair protein. For example, C17orf53 KO 293A cells are sensitive to ICL-induced agents such as MMC, cisplatin and psoralen+UVA, but they are not sensitive to a wide range of other DNA damaging agents, such as IR, CPT, HU and isopsoralen+UVA. Thus, the function of C17orf53 is likely dependent on specific DNA lesions, which needs further clarification. Additionally, C17orf53 KO cells are sensitive to etoposide, implying that it may also function in other repair pathways.
Most recently, Dr. Ciccia and colleagues identified that C17orf53 binds to MCM8 and facilitates the MCM8–9 dependent DNA synthesis during DNA recombination and replication [56]. They renamed this protein as MCM8IP. Using CRISPR screens following MMC treatment, we independently identified MCM8 and MCM9 as genes that operate with C17orf53 in MMC resistance, which is similar to the findings reported recently [55, 56]. Moreover, we analyzed genes that may share functions with C17orf53 using the CRISPR screen data available from DepMap, and uncovered again MCM8 and MCM9 as genes with functional correlation with C17orf53. Furthermore, we demonstrated that C17orf53 is a RPA-binding protein and contains a ssDNA-binding domain DUF4539. We mapped the interaction between C17orf53 and RPA, and uncovered two RPA-binding motifs in C17orf53. Both the RPA-binding motifs and the ssDNA-binding domain are necessary for the function of C17orf53 in ICL repair. Based on these observations, we propose that C17orf53 is recruited by RPA and ssDNA to sites of DNA damage and carry out its functions in ICL repair, probably by facilitating efficient chromatin loading of MCM8-MCM9 and other late DNA repair factors. Further mechanistic investigations are needed to define the precise roles of C17or53 in DNA repair.
In addition, we showed that C17orf53 is required for optimal DNA replication, efficient cell proliferation and may be a reliable marker to discriminate pre-cancerous cells from normal cells. The precise function of C17orf53 in DNA replication remains unclear. We would like to determine whether or not this role of C17orf53 in DNA replication is also linked with its association and function with MCM8/9. It is also of note that C17orf53 is a conserved protein, but seems to appear exclusively in vertebrates. It is possible that increased genome complexity may require more sophisticated regulations and thus necessitate the functions of C17orf53 and others during DNA replication and DNA repair, which warrant further investigation.
METHODS
Key resources table
| Reagents or Resources | Source | Identifier | ||
|---|---|---|---|---|
| Antibodies | ||||
| C17orf53 | SIGMA | Cat. HPA023393 | ||
| RPA2 | Cell Signaling | Cat. 2208 | ||
| pRPA2(s33) | Bethyl | Cat. A300–246A | ||
| pRPA2(s4/8) | Bethyl | Cat. A300–245A | ||
| FANCD2 | Santa Cruz | Cat. sc-20022 | ||
| FANCD2 | Novus Biologicals | Cat. NB-100182 | ||
| FANCI | Santa Cruz | Cat. sc-271316 | ||
| CHK1 | Cell Signaling | Cat. 2360 | ||
| pCHK1(s317) | Cell Signaling | Cat. 2344 | ||
| pCHK1(s345) | Cell Signaling | Cat. 2348 | ||
| CHK2 | Cell Signaling | Cat. 6334 | ||
| pCHK2(T68) | Cell Signaling | Cat. 2197 | ||
| rH2A.x | Biolegend | Cat. 613401 | ||
| rH2A.x | Millipore | Cat. 05–636 | ||
| MCM8 | OriGene Technologies | TA322142 | ||
| RAD54 | Abcam | ab224763 | ||
| REV1 | Santa Cruz | sc-393022 | ||
| POLH (eta) | Cell Signaling | 13848S | ||
| shRNAs and esiRNA | ||||
| shRNA1 (C17orf53) | Dharmcon | V3LHS_325872 | ||
| shRNA2 (C17orf53) | Dharmcon | V3LHS_403475 | ||
| esiRNA (C17orf53) | Sigma | EHU090691 | ||
| esiRNA (MCM8) | Sigma | EHU132941 | ||
| sgRNAs | ||||
| C17orf53 sgRNA1 | GGACCTAATTCCTTACCCGT | exon6 | ||
| C17orf53 sgRNA2 | GCGCGGCATTACCATAGCGA | exon1 | ||
| C17orf53 sgRNA3 | TCTCTCATCCAGGCCTAGCG | exon5 | ||
| Oligonucleotides (for Gel-shift Assay) | ||||
| 60nt-f | GACGCTGCCGAATTCTACCAGTGCCTTGCTGGACATCTTTGCCCACCTGCAGGTTCACCC | |||
| 60nt-r | GGGTGAACCTGCAGGTGGGCAAAGATGTCCAGCAAGGCACTGGTAGAATTCGGCAGCGTC | |||
| 30nt-f | GACGCTGCCGAATTCTACCAGTGCCTTGCT | |||
| Database and Tools Used | ||||
| CCLE(Cancer Cell Line Encyclopedia) | https://portals.broadinstitute.org/ccle | |||
| GDSC(Genomics of Drug Sensitivity in Cancer) | https://www.cancerrxgene.org/ | |||
| GEPIA(Gene Expression Profiling Interactive Analysis) | http://gepia.cancer-pku.cn/ | |||
| CellMinerCDB | https://discover.nci.nih.gov/cellminercdb/ | |||
| TCGA(The Cancer Genome Atlas Program) | http://www.cbioportal.org/ | |||
| DepMap portal | https://depmap.org/portal/ | |||
| Pfam database | http://pfam.iam.org/ | |||
EXPERIMENTAL PROCEDURES
Cell Culture
293A, HCT116, MCF10A, and U2OS cells were obtained from ATCC. Dulbecco’s modified Eagle medium (DMEM) with 10% fetal calf serum was used to culture 293A, HCT116, and U2OS cells. MCF10A cells were cultured in DMEM/F12 Ham’s mixture supplemented with 5% equine serum (Gemini Bio), 20 ng/mL epidermal growth factor (Sigma), 10 μg/mL insulin (Sigma), 0.5 mg/mL hydrocortisone (Sigma), 100 ng/mL cholera toxin (Sigma), 100 units/mL penicillin, and 100 μg/mL streptomycin.
Generation of Knock-out Cells
pLentiCRISPRv2 was used to generate KO cells. Cells were transiently transfected with the indicated plasmids and selected using puromycin (2 mg/mL). Single cells were then plated into 96-well plates. After 10 days, clones were picked and checked by Western blotting.
Viability Assay
For cell growth assays, cells were seeded in 6-well plates (104 per well) and passaged every 3 days until cell numbers were determined. For clonogenic assays, cells were seeded in 6-well plates (200 cells per well for WT cells and 400 cells per well for C17orf53 KO cells), exposed to chemicals treatment (HU, CPT, MMC, Cisplatin, etoposide) for 24hr, and cultured for 14 days. For ATR inhibitors treatment (VE822, AZD6738), the cells were continuously exposed to chemicals for 14 days. Cells were stained with crystal violet solution (Sigma-Aldrich). Colonies were counted manually. All cell survival assays were performed at least in triplicate. For cell survival assays of psoralen or isopsoralen plus UVA, cells were seeded in 6-well plates (3000 per well) and treated with psoralen (10μg/ml) or isopsoralen (10μg/ml) for 30mins before UVA irradiation using a Stratalinker (Stratagene, La Jolla, CA). On day 8, the cells were trypsinized and viable cells were counted using Bio-rad TC20 cell counter (Bio-Rad).
BrdU Incorporation and Cell Cycle Analysis by Flow Cytometry
Cells were incubated with 10 μM 5-bromo-2-deoxyuridin (BrdU) for 30 mins before fixation with 70% ethanol. BrdU was detected with BrdU antibody (BDB347583, Fisher Scientific). Propidium iodide (PI) was used to measure DNA content. Data were collected with BD C6 (Becton Dickinson) and analyzed with Flowjo (Becton, Dickinson & Company).
Native and Denatured BrdU staining
Cells were incubated with 20 μM BrdU for 24hr. After the indicated treatments, cells were fixed in 4% paraformaldehyde and permeablized with phosphate-buffered saline (PBS) with 0.5% Triton X-100. Then, cells were incubated with primary antibodies diluted in PBS with 0.05% Triton X-100 and 1% BSA (PBST-BSA) for 1h at room temperature. After 3 washes with PBS, fluorescently labeled secondary antibodies in PBST-BSA were added for 1 h. Cells were then washed in PBS with Hoechst stain (1:10,000). Slides were imaged at 40× on a Leica microscope. For denatured BrdU staining, cells were denatured using 2N HCl for 30min at room temperature after the fixation and permeablization.
Analysis of Protein Localization to UV Laser Induced Sites of DNA Damage
In brief, cells were seeded into 35-mm, glass-bottomed dishes. The next day, UV laser-induced damage of cells was conducted under a Nikon (Tokyo, Japan) TE200 inverted microscope coupled with a 365-nm UV laser MicroPoint system. After irradiation, cells were immediately fixed with 3% paraformaldehyde and processed for immunostaining.
Immunofluorescence Staining Analysis
Cells were grown on coverslips for 24 h before treatment. After the indicated treatment, cells were fixed in 4% paraformaldehyde and permeablized with phosphate-buffered saline (PBS) with 0.5% Triton X-100. Then, cells were incubated with primary antibodies diluted in PBS with 0.05% Triton X-100 and 1% BSA (PBST-BSA) for 1h at room temperature. After 3 washes with PBS, fluorescently labeled secondary antibodies in PBST-BSA were added for 1 h. Cells were then washed in PBS with Hoechst stain (1:10,000). Slides were imaged at 40× on a Leica microscope.
Assessment of apoptosis with Annexin-V Staining
The proportion of cells undergoing apoptosis (annexin positive) or death (annexin and propidium iodide dual positive; propidium iodide positive) was determined using a standard flow cytometry Annexin-VFITC binding assay according to the manufacturer’s instructions (Bio-RAD ANNEX300F).
DNA fiber Assay
Cells were labeled with 30 μM CIdU for 30 mins, washed quickly twice with PBS and exposed to 250 μM IdU for another 30 mins. Cells were harvested and resuspended in PBS. Cells were then lysed with lysis buffer (200 mM TrisHCL pH7.4, 50 mM EDTA, 0.5% SDS), and DNA fibers stretched onto glass slides. The fibers were then denatured with 2.5 M HCL for 1 hr, washed with PBS and blocked with 2% BSA in PBST for 30 mins. The fibers were stained with anti-BrdU antibodies recognizing CIdU (Rat anti-BrdU, AbD Serotec OBT0030) and IdU (Mouse anti-BrdU, BD 347580). Slides were imaged at 40× on a Leica microscope. The sum of IdU and CldU length were calculated to measure the DNA fiber length.
siRNA Transfection
C17orf53 esiRNA and control siRNA were products of Sigma. 10 nM of each siRNA was transfected to cells with Lipfectamin RNAiMAX Reagent (13778, Invitrogen) according to the manufacturer’s instructions. Culture medium was changed 12 hrs after the transfection.
RNA extraction, reverse transcription and real-time PCR
RNA samples were extracted with TRIZOL reagent (Invitrogen). Reverse transcription assay was performed by using the iScript cDNA Synthesis Kit (BioRad, Hercules, CA, USA) according to the manufacturer’s instructions. Real-time PCR was performed by using Power SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA). For quantification of gene expression, the 2 − ΔΔCt method was used. Actin expression was used for normalization.
Immunoprecipitation (IP) and Mass Spectrometry
Expression plasmids were transfected to HEK293 suspension cells with polyethyleneimine. Cells were harvested 64 hrs after transfection and pellets were directly lysed with NETN buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 10% glycerol, 0.5% NP40, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg ml−1 leupeptin, 1 μg ml−1 aprotinin). The lysates were ultra-centrifuged at 15,000 rpm for 15 min and then the supernatant was incubated with Streptavidin beads for 3–4 h at 4 °C. The beads were washed four times with NETN buffer and eluted with NETN containing 2 mg/ml biotin. The elution was then incubated with S-protein beads for 2 hrs. Subsequently, S-protein beads were washed and the complexes were eluted, and analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and mass spectrometry.
For mapping the interactions between RPA1/2 and C17orf53, modified NETN buffer (20 mM Tris-HCl [pH 7.5], 250 mM NaCl, 10% glycerol, 0.5% NP40, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml leupeptin, 1 μg/ml aprotinin, 25 U/mL Benzonase and 100 μg/mL RNase A) were used to eliminate any possible chromatin-mediated interactions.
Whole-cell Extract and Chromatin Extract Preparation
For preparation of whole-cell extract, cells were collected, washed with PBS, and boiled at 95°C for 10 min in 1× Laemmli buffer. For preparation of chromatin extract, cells were lysed with ice-cold NETN lysis buffer (50 mM Tris–HCl, pH 7.4, 100 mM NaCl, 0.4% NP-40, 1 mM EDTA) and protease inhibitor cocktail for 10 min on ice. Cell lysates were centrifuged at 13 200 rpm for 10 min in a cold room. Pellets were washed with NETN lysis buffer two more times and boiled at 95°C for 10 min in 1× Laemmli buffer.
Western Blotting
Cells were lysed in NET-N buffer (20 mM Tris [pH 7.6], 1 mM ethylenediaminetetraacetic acid, 1% NP40, 150 mM NaCl) supplemented with protease inhibitor cocktail tablets (Roche). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to membranes, and immunoblotted with antibodies as indicated.
Gel-shift Assay
The DNA substrates were made by annealing oligos 60nt-f and 60nt-r, 30nt-f and 60nt-r, respectively. The 5ʹ ends of the oligo 60nt-f and 30nt-f were labeled with 32P using T4 polynucleotide kinase before annealing. In all, 5 nM 32P -labeled DNA subtracts and the indicated amount of proteins were incubated at 25 °C in 10 μl reaction buffer (20 mM HEPES at pH 7.5, 5 mM MgCl2, 100 mM KCl, 1 mM DTT, 0.05% Triton X-100, 100 μg ml−1 BSA and 5% glycerol) for 15 min. Reaction mixture was loaded and resolved on a 5% Tris-borate-EDTA (TBE) gel.
sgRNA screening
The TKOv3 library was a gift from Traver Hart lab, MD Anderson Cancer Center. The CRISPR screen was as described before. In brief, 120 million 293A or C17orf53KO 293A cells were infected with the TKOv3 library lentiviruses or DDR library lentivirus at a low MOI (< 0.3). Twenty-four hours after infection, the medium was replaced with fresh medium containing puromycin (2 μg/mL). After selection, cells were split into 2 replicates containing ~20 million cells each, passaged every 3 days, and maintained at 200-fold coverage. At day 0 and every 3 days from day 6 to day 21, 25 million cells (> 300-fold coverage) were collected for genomic DNA extraction. Genomic DNA was extracted from cell pellets using the QIAamp Blood Maxi Kit (Qiagen), precipitated using ethanol and sodium chloride, and resuspended in Buffer EB (10 mM Tris-HCl, pH 7.5). gRNA inserts were amplified via PCR using primers harboring Illumina TruSeq adapters with i5 and i7 barcodes as previously reported [57], and the resulting libraries were sequenced on an Illumina HiSeq 2500 system. The sequencing results were used for further analysis. MAGecK and DrugZ analysis were used to calculate the difference between the DMSO- and MMC-treated groups.
HR-GFP and NHEJ-GFP reporter Assays
U2OS cells stably expressing HR reporter DR- GFP or NHEJ-GFP (EJ5-GFP) reporter were gifts from Albert C. Koong lab in MD Anderson Cancer Center. 1 × 106 U2OS DR- GFP or NHEJ-GFP cells were transfected with 50 nmol control siRNA or C17orf53 siRNA with RNAimax. 24 hrs later, 2 μg of pCBASce plasmid (an I- SceI expression vector) were transfected into the cells with Lipo2000. Cells were cultured for another 48 hrs and subjected to flow cytometry analysis to determine percentages of GFP- positive cells, which result from HR repair or NHEJ repair induced by DNA DSBs. Means were obtained from 3 independent experiments.
Bioinformatics Analysis
For gene expression analysis of C17orf53, TCGA datasets were analyzed with GEPIA [58] and GDSC/CCLE datasets were analyzed with CellMinerCDB [59]. The overall survival analysis of patients with tumors were done with GEPIA. For gene co-function analysis, gene effect data were downloaded from DepMap (https://depmap.org/portal/) and the Pearson correlation factors were calculated between C17orf53 and other genes using Python 3.7.1.
Statistics
Statistical analyses were performed using GraphPad Prism software version 8.0. All of the statistical methods used are described in the main text. Each experiment was repeated three times or more. Differences between groups were analyzed by the Student t-test, unless otherwise noted. A P-value <0.05 was considered statistically significant.
Supplementary Material
Highlights.
C17orf53 promotes cell survival in response to ATR inhibition
C17orf53-deficient cells show profound defect in response to ICL
C17orf53 is a ssDNA-binding protein and directly interacts with RPA
C17orf53 acts with MCM8/9 in DNA repair
ACKNOWLEDGEMENT
We thank the members of Dr. Junjie Chen’s lab for their kind help. We also thank Tamara K. Locke in the Department of Scientific Publications from The University of Texas MD Anderson for help in the scientific editing of the manuscript. U2OS DR-GFP and NHEJ-GFP (EJ5-GFP) cells were kindly provided by Dr. Albert C. Koong at the University of Texas MD Anderson Cancer Center. This work was supported in part by CPRIT (RP160667) and NIH grants (CA193124, CA210929, CA216911, and CA216437) to J.C. and MD Anderson’s NIH Cancer Center Support Grant (CA016672).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
The authors declare no competing financial interests.
Reference
- [1].Saldivar JC, Cortez D, and Cimprich KA, The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol, 2017. 18(10): p. 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hekmat-Nejad M, et al. , Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol, 2000. 10(24): p. 1565–73. [DOI] [PubMed] [Google Scholar]
- [3].de Klein A, et al. , Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol, 2000. 10(8): p. 479–82. [DOI] [PubMed] [Google Scholar]
- [4].Cortez D, et al. , ATR and ATRIP: partners in checkpoint signaling. Science, 2001. 294(5547): p. 1713–6. [DOI] [PubMed] [Google Scholar]
- [5].Bomgarden RD, et al. , A novel protein activity mediates DNA binding of an ATR-ATRIP complex. J Biol Chem, 2004. 279(14): p. 13346–53. [DOI] [PubMed] [Google Scholar]
- [6].Zou L and Elledge SJ, Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science, 2003. 300(5625): p. 1542–8. [DOI] [PubMed] [Google Scholar]
- [7].Sorensen CS and Syljuasen RG, Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res, 2012. 40(2): p. 477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu Q, et al. , Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev, 2000. 14(12): p. 1448–59. [PMC free article] [PubMed] [Google Scholar]
- [9].Abraham RT, Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev, 2001. 15(17): p. 2177–96. [DOI] [PubMed] [Google Scholar]
- [10].Cha RS and Kleckner N, ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science, 2002. 297(5581): p. 602–6. [DOI] [PubMed] [Google Scholar]
- [11].Costanzo V, et al. , An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell, 2003. 11(1): p. 203–13. [DOI] [PubMed] [Google Scholar]
- [12].Luciani MG, Oehlmann M, and Blow JJ, Characterization of a novel ATR-dependent, Chk1-independent, intra-S-phase checkpoint that suppresses initiation of replication in Xenopus. J Cell Sci, 2004. 117(Pt 25): p. 6019–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang H, et al. , ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res, 2004. 64(19): p. 7139–43. [DOI] [PubMed] [Google Scholar]
- [14].Fasullo M and Sun M, UV but not X rays stimulate homologous recombination between sister chromatids and homologs in a Saccharomyces cerevisiae mec1 (ATR) hypomorphic mutant. Mutat Res, 2008. 648(1–2): p. 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Buisson R, et al. , Coupling of Homologous Recombination and the Checkpoint by ATR. Mol Cell, 2017. 65(2): p. 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhu W and Dutta A, An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol Cell Biol, 2006. 26(12): p. 4601–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Collis SJ, et al. , FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell, 2008. 32(3): p. 313–24. [DOI] [PubMed] [Google Scholar]
- [18].Rundle S, et al. , Targeting the ATR-CHK1 Axis in Cancer Therapy. Cancers, 2017. 9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Williamson CT, et al. , ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nature Communications, 2016. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Knipscheer P, et al. , The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science, 2009. 326(5960): p. 1698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huang H, et al. , Solution structure of a cisplatin-induced DNA interstrand cross-link. Science, 1995. 270(5243): p. 1842–5. [DOI] [PubMed] [Google Scholar]
- [22].Huang J, et al. , The DNA Translocase FANCM/MHF Promotes Replication Traverse of DNA Interstrand Crosslinks. Molecular Cell, 2013. 52(3): p. 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rohleder F, et al. , FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Research, 2016. 44(7): p. 3219–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Huang J, et al. , Remodeling of Interstrand Crosslink Proximal Replisomes Is Dependent on ATR, FANCM, and FANCD2. Cell Reports, 2019. 27(6): p. 1794−+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kruyt FA, et al. , Fanconi anemia genes act to suppress a cross-linker-inducible p53-independent apoptosis pathway in lymphoblastoid cell lines. Blood, 1996. 87(3): p. 938–48. [PubMed] [Google Scholar]
- [26].D’Andrea AD, Cellular function of the Fanconi anemia pathway. Nat Med, 2001. 7(12): p. 1259–60. [DOI] [PubMed] [Google Scholar]
- [27].Rosselli F, Briot D, and Pichierri P, The Fanconi anemia pathway and the DNA interstrand cross-links repair. Biochimie, 2003. 85(11): p. 1175–84. [DOI] [PubMed] [Google Scholar]
- [28].Shen X, et al. , Recruitment of fanconi anemia and breast cancer proteins to DNA damage sites is differentially governed by replication. Mol Cell, 2009. 35(5): p. 716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Huang Y, et al. , Modularized functions of the Fanconi anemia core complex. Cell Rep, 2014. 7(6): p. 1849–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Williams HL, Gottesman ME, and Gautier J, The differences between ICL repair during and outside of S phase. Trends Biochem Sci, 2013. 38(8): p. 386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Joo W, et al. , Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science, 2011. 333(6040): p. 312–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shukla P, et al. , DNA interstrand cross-link repair: understanding role of Fanconi anemia pathway and therapeutic implications. Eur J Haematol, 2013. 91(5): p. 381–93. [DOI] [PubMed] [Google Scholar]
- [33].Haynes B, et al. , Crosstalk between translesion synthesis, Fanconi anemia network, and homologous recombination repair pathways in interstrand DNA crosslink repair and development of chemoresistance. Mutat Res Rev Mutat Res, 2015. 763: p. 258–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Roy U and Scharer OD, Involvement of translesion synthesis DNA polymerases in DNA interstrand crosslink repair. DNA Repair (Amst), 2016. 44: p. 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sasaki MS, et al. , Recombination repair pathway in the maintenance of chromosomal integrity against DNA interstrand crosslinks. Cytogenet Genome Res, 2004. 104(1–4): p. 28–34. [DOI] [PubMed] [Google Scholar]
- [36].Hinz JM, Role of homologous recombination in DNA interstrand crosslink repair. Environ Mol Mutagen, 2010. 51(6): p. 582–603. [DOI] [PubMed] [Google Scholar]
- [37].Shigechi T, et al. , ATR-ATRIP kinase complex triggers activation of the Fanconi anemia DNA repair pathway. Cancer Res, 2012. 72(5): p. 1149–56. [DOI] [PubMed] [Google Scholar]
- [38].Tomida J, et al. , A novel interplay between the Fanconi anemia core complex and ATR-ATRIP kinase during DNA cross-link repair. Nucleic Acids Res, 2013. 41(14): p. 6930–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kwok M, Davies N, and Agathanggelou A, ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells (vol 127, pg 582, 2016). Blood, 2016. 127(21): p. 2647–2647. [DOI] [PubMed] [Google Scholar]
- [40].Mohni KN, Kavanaugh GM, and Cortez D, ATR Pathway Inhibition Is Synthetically Lethal in Cancer Cells with ERCC1 Deficiency. Cancer Research, 2014. 74(10): p. 2835–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang C, et al. , Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene, 2019. 38(14): p. 2451–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Styrkarsdottir U, et al. , New sequence variants associated with bone mineral density. Nat Genet, 2009. 41(1): p. 15–7. [DOI] [PubMed] [Google Scholar]
- [43].Giotti B, et al. , Assembly of a Parts List of the Human Mitotic Cell Cycle Machinery. J Mol Cell Biol, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ruiz S, et al. , A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol Cell, 2016. 62(2): p. 307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee J, Kumagai A, and Dunphy WG, The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem, 2007. 282(38): p. 28036–44. [DOI] [PubMed] [Google Scholar]
- [46].Cotta-Ramusino C, et al. , A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science, 2011. 332(6035): p. 1313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Duquette ML, et al. , CtIP Is Required to Initiate Replication-Dependent Interstrand Crosslink Repair. Plos Genetics, 2012. 8(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Huang M, et al. , The FANCM/FAAP24 Complex Is Required for the DNA Interstrand Crosslink-Induced Checkpoint Response. Molecular Cell, 2010. 39(2): p. 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang H, et al. , SLX4IP acts with SLX4 and XPF-ERCC1 to promote interstrand crosslink repair. Nucleic Acids Res, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang LC and Gautier J, The Fanconi anemia pathway and ICL repair: implications for cancer therapy. Critical Reviews in Biochemistry and Molecular Biology, 2010. 45(5): p. 424–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sengerova B, Wang AT, and McHugh PJ, Orchestrating the nucleases involved in DNA interstrand cross-link (ICL) repair. Cell Cycle, 2011. 10(23): p. 3999–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Colic M, et al. , Identifying chemogenetic interactions from CRISPR screens with drugZ. Genome Medicine, 2019. 11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Meyers RM, et al. , Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nature Genetics, 2017. 49(12): p. 1779−+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nishimura K, et al. , Mcm8 and Mcm9 Form a Complex that Functions in Homologous Recombination Repair Induced by DNA Interstrand Cross links. Molecular Cell, 2012. 47(4): p. 511–522. [DOI] [PubMed] [Google Scholar]
- [55].Hustedt N, et al. , Control of homologous recombination by the HROB-MCM8-MCM9 pathway. Genes & Development, 2019. 33(19–20): p. 1397–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Huang JW, et al. , MCM8IP activates the MCM8–9 helicase to promote DNA synthesis and homologous recombination upon DNA damage. Nat Commun, 2020. 11(1): p. 2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hart T, et al. , Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 (Bethesda), 2017. 7(8): p. 2719–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tang ZF, et al. , GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Research, 2017. 45(W1): p. W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Reinhold WC, et al. , CellMinerCDB and CellMiner web-applications for genomics and pharmacogenomics analyses of cancer cell lines. Cancer Research, 2019. 79(13). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.