

Abstract
Immunohistochemistry (IHC) can provide detailed information about protein expression within the cell microenvironment and is one of the most common techniques in biology and medicine due to the broad availability of highly specific antibodies and well-established bioconjugation methods for modification of these antibodies with chromogens and fluorophores. Despite recent advances in this field, it remains challenging to simultaneously achieve high multiplexing, sensitivity, and throughput in single-cell profiling experiments. Here, we report the combination of two powerful technologies, quantum dot and signal amplification by exchange reaction (QD-SABER), for sensitive and multiplexed imaging of endogenous proteins. Compared to the conventional IHC process using dye-labeled secondary antibodies (which already has a built-in signal amplification mechanism), QD-SABER provides an additional 7.6-fold signal amplification. In addition, the DNA hybridization-based IHC can be rapidly removed to regenerate the sample for subsequent cycles of immune-staining (>10 cycles), greatly expanding the multiplexing capability.
Keywords: quantum dots, DNA nanotechnology, single-cell, imaging, multiplexing
Graphical Abstract
Highly multiplexed and high-sensitivity immunohistochemistry imaging was achieved by combining the Qdot nanotechnology with DNA nanotechnology. Qdots’ unique optical properties and Primer Exchange Reaction’s programmability allow rapid, sensitive, and specific immunostaining in single cells.
Recent advances in molecular cell analysis have generated enormous insights into the complex biological systems. For instance, advanced sequencing technologies are capable of analyzing the genome and transcriptome of single cells, linking abnormal genes and gene expressions with various diseases.[1–3] Mapping the genomic and transcriptomic information to the corresponding proteomic phenotypes in a highly multiplexed and quantitative fashion, however, remains to be one of the long-standing challenges in biology and medicine. Although cells in a human body share a nearly identical genome, the proteomic phenotypes are often different to realize the diverse biological functions and can be highly dynamic in response to stimuli (e.g., therapies) and changes in the cellular microenvironments. Increasing evidence suggests that cell phenotypes are heterogeneous, even in cultured cells from the same origin,[4–7] and this heterogeneity becomes especially important in the era of immunotherapy where the immune contexture often decides the treatment outcome and disease prognosis. [8–10] Unfortunately, conventional proteomic analysis techniques such as protein chips, electrophoresis, and mass spectrometry using homogenized solutions cannot retain spatial information about this kind of heterogeneous sample. In contrast, microscopy approaches such as IHC (either based on chromogens or fluorophores) allow biomarker interrogation and cell typing within the context of cellular microenvironment with high imaging resolution. Comprehensive immunoprofiling of single cells, however, is currently limited by multiple factors, in particular, the unfavorable spectral properties of organic dyes.
Currently, two general approaches derived from the conventional IHC offer important advantages and are highly suitable for in situ single-cell immunoprofiling. The first approach uses mass spectrometry as the detection readout.[11–14] Angelo et al. reported a method that uses secondary ion mass spectrometry (SIMS) to image antibodies tagged with isotopically pure elemental metal reporters.[15] The so-called multiplexed ion beam imaging (MIBI) technique is capable of analyzing multiple targets in adherent cells or clinical tissue sections with a large detection dynamic range. Remarkably, using small laser spots and step sizes, MIBI under scanning mode can create cell staining images with resolution comparable to optical imaging. More recently Giesen et al. have shown the imaging of 32 proteins and protein modifications at subcellular resolution.[16] The advantages of mass spectrometry-based readout are the high multiplexing capability and short immunostaining time (simultaneous incubation of all primary antibodies). The downside includes the tradeoff between high-resolution scanning and long scanning time, the large metal-chelator polymers tethered to antibodies that limits tissue penetration during staining, and the high cost of mass spectrometers.
The second category of immunoprofiling technologies was developed based on the concept of multicycle IHC, keeping the conventional fluorescence microscopy as the readout mechanism. The general process includes multiple steps: i) label a panel of antibodies with spectrally distinguishable fluorophores; ii) stain cultured cells or tissue sections using the antibody-fluorophore conjugates for fluorescence imaging; iii) regenerate the samples by removing the fluorescent stains (also known as de-staining) for additional rounds of IHC using a different subset of antibodies. In another word, the high-level multiplexing is gained at the cost of prolonged experiment time, because IHC is repeated multiple times on the same sample. Multiple innovative designs have been demonstrated recently including chemical quenching or removal of fluorescent reporters and DNA-encoding of antibodies. [17–26] For example, we have developed a QD-adaptor protein platform for easy preparation of large QD-antibody libraries and antibody stripping,[18, 19] whereas other groups have reported highly innovative chemical conjugation methods to link biomolecules including both antibodies and oligonucleotides to QDs with controlled orientations.[27–29] In parallel, owing to the unmatched programmability of DNA, a number of enabling technologies have been developed for biological imaging and sensing.[30–33] For example, recently Kishi et al. reported a highly innovative DNA nanotechnology termed primer exchange reaction (PER)[34] and the associated technique termed signal amplification by exchange reaction (SABER), in which the controlled growth of long DNA concatemers from a short primer serves as an efficient substrate for multiplexed and amplified signal detection in cells via the recruitment of fluorescently labeled detection oligos to the specific nucleic acid or protein targets. [25, 26]
Here, we report the combination of QD nanotechnology and DNA nanotechnology to simultaneously take advantage of the unique optical properties of QDs (such as higher level of brightness, photostability, and multiplexing) with the flexibility and programmability of DNA nanotechnology (such as signal amplification, and reduced antibody incubation cycles). The overall experiment flow is schematically illustrated in Scheme 1. Primary antibodies barcoded with unique oligonucleotide sequences (bridge oligos) are applied to cells in parallel. The bridge oligos barcode each antibody and serve as an anchor point for immobilization of orthogonal ssDNA concatemers, which are pre-synthesized in vitro using PER. In parallel, fluorescent imagers for hybridization with the long concatemers are made by simple mixing of biotinylated oligos with QD-streptavidin. In each hybridization cycle, 5–10 colors of QDs can be applied simultaneously for rapid, sensitive, and specific immunostaining. Because QDs are linked to the target antigens through DNA-barcoding, there is no need to remove the primary antibodies, a process that requires harsh treatments such as low pH as we demonstrated previously.[18, 19] A gentle stripping step using formamide in combination with pH 5.5 buffer allows complete removal of QD fluorescence, restoring the sample for additional rounds of imager hybridization.
Scheme 1. Schematic diagrams of QD-SABER for multicolor multicycle IHC.

Key steps in QD-SABER: 1) Bridge oligo-barcoded antibodies (antibody-oligo) are used to simultaneously stain multiple targets in cells. 2) Concatemers hybridize to the corresponding bridge oligos. 3) QD-imagers hybridize to the long concatemer for fluorescence microscopy. Note that the QD-imager sequences are designed to hybridize with two copies of the extended primer sequences in the concatemer as described previously.[25, 26] 4) De-hybridization of the QD-imagers for subsequent rounds of QD-imager hybridization.
Because linking antibodies with the bridge oligos involves chemical modifications of the antibodies, we first characterized the structure and function of the bioconjugates (Supplementary Figure S1). PAGE and single-color cell staining confirmed the conjugation between the partially reduced antibodies and bridge oligo (Supplementary Figure S1b), and preserved antigen-recognition specificity compared to the positive control (conventional two-step immunofluorescence staining with primary antibody and Alexa Fluor 555-labeled secondary antibody). The microtubule protein β-tubulin showed the characteristic fibrous structures inside cells (Supplementary Figure S1c–e). In parallel, we also characterized the PER concatemer, whose length determines the signal amplification level (Supplementary Figure S1g). It is worth mentioning that in contrast to the previously reported DNA nanotechnology approaches that amplify signals in situ (generally slow processes due to diffusion limitation of the reagents or difficulty to control the degree of extension), [24,35,36] PER concatemer extensions are pre-made in solution, eliminating the concern of slow or hard-to-control reaction kinetics. [25, 26]
To evaluate the staining fluorescence intensity and imaging sensitivity, a head-to-head comparison was made by staining five biomarkers (HSP90, Ki-67, Lamin A, Calnexin, and β-tubulin) in HeLa cells using four different IHC approaches: i) conventional two-step staining using a primary antibody (1’Ab) and dye-labeled secondary antibody (2’Ab), ii) conventional two-step staining using 1’Ab and QD-labeled 2’Ab, iii) SABER using dye-labeled imager strands, and iv) SABER signal amplification using QD-labeled imager strands. Representative images are shown in Figure 1 for the five targets. All four staining methods showed consistent staining patterns, confirming specificity cross the board. Quantitative analysis using equal exposure times indicates that compared against the conventional 2’Ab-dye (which already has a built-in signal amplification mechanism), the 2’Ab-QD, organic dye-based SABER, and QD-SABER on average further enhanced the signal strength by another 3.0-, 3.0-, and 7.6-fold, respectively. Intuitively, these values match expectations (Figure 1e). Although single QDs have been reported to be 10–20 times brighter than single dye molecules due to their large molar extinction coefficients,[37–40] considering each 2’Ab can accommodate multiple dye molecules where QDs on average have 2–3 copies of 2’Ab on their surface due to QDs’ large size, the steric hindrance effect reduces the number of QDs that can bind with each antigen, resulting in modest signal enhancement. In contrast, the long PER concatemer extends out from the primary antibody, allowing a large number of dye- or QD-labeled DNA imager strands to hybridize for improved detection sensitivity. Comparing the results using dye- or QD-labeled DNA imager strands, the QD-imager is approximately 2.5 times brighter than the dye-imager. Considering single QDs are 10–20 times brighter than single dye molecules, the number of QDs hybridized onto the same concatemer is less than that of the dyes, likely due to the steric hindrance caused by QDs’ size. Although it is difficult to measure the actual number of QDs hybridized onto each concatemer, the above fluorescence intensity measurements indicate QD-imager strands on average can tile 1/6 of the total binding sites. This low tiling density can be potentially addressed using concatemers with the monomers spaced out farther from each other.
Figure 1. Comparison of IHC using various types of IHC.

a) Conventional two-step IHC using organic dye-labeled 2’Ab, b) conventional two-step IHC using QD-labeled 2’Ab, c) SABER technology using organic dye-labeled imager strands, and d) SABER technology using QD-labeled imager strands. The imaging protocols are described in Supplementary Information. Five model targets representing antigens in both the cytoplasm and nucleus are HSP90, Ki-67, Lamin A, Calnexin, and β-tubulin, from left to right. The organic dye is Alexa Fluor 555 emitting at 580 nm, and the QDs emit at 585 nm. The staining across the four types of IHC showed similar patterns, confirming the staining specificity. For quantitative comparison, images were captured with a 100x objective and a Qcolor5 CCD with constant exposure times, and the results were analyzed with Image J. The images were false-colored and presented in a LUT scaled from 0–255. e) Schematic illustration of how methods b), c), and d) improve staining brightness over method a), the conventional two-step staining using organic dye-labeled 2’Ab (which already has an amplification mechanism because multiple 2’Abs can bind to each 1’Ab and the 2’Abs often have multiple fluorophores), by 3.0, 3.0, and 7.6 folds, respectively. The fluorescence intensities from randomly selected cells (>35 for all samples) were measured. At the camera exposure times optimal for visual presentation in a), b), and c), the fluorescence intensity of QD-SABER in d) was too bright and saturated the detector (top panels). The bottom panels show the same images obtained with the exposure time shortened by 6 times. Scale bar, 50 μm. Control experiments without the primary antibodies were also performed to confirm the QD-SABER staining specificity (Supplementary Figure S2). Non-specific binding from QD-oligo was largely negligible. Since the high-magnification images shown here have a limited number of cells, additional views are also provided in Supplementary Figure S2.
It is worth mentioning that the QD-imager oligo conjugates were prepared via a simple incubation of QD-streptavidin and biotinylated oligos (Supplementary Figure S1h) instead of covalent crosslinking to avoid complex chemical reactions and purification steps that often reduce the yield of the products substantially. This strategy requires complete (or nearly complete) capture of the biotinylated oligo onto the QD surface because free oligo would compete for hybridization sites in the concatemer, thus reducing fluorescence signal strength. To optimize the ratio of QD-streptavidin and biotinylated imager oligo, a series of molar ratios ranging from 1:1 to 1:7 was tested in QD-SABER using β-tubulin as the target. A decrease of fluorescence intensity was observed with increasing concentrations of the biotinylated imager (Supplementary Figure S3). QDs’ large surface can have multiple copies of streptavidin per particle.[41] Although each streptavidin, in theory, can bind with up to four biotins, the IHC staining results suggests that not every binding site in streptavidin is accessible when they are immobilized on QD surface.
Because all the imager strands are linked to QDs via the same interaction (streptavidin-biotin binding), it is crucial to confirm the probe stability to avoid potential cross-reactivity (also known as crosstalk). The streptavidin-biotin binding is among the strongest in biological interactions with a dissociation constant of 10−14 M, but it is still a non-covalent interaction, leaving biotinylated strands exchanging between particles possible. To test crosstalk, a simple verification using dual-color QD-imager probes (emission 525 and 585 nm) was implemented in three separate experiments (Figure 2). In these experiments, cells sequentially labeled with the 1’Ab-bridge oligo and the long concatemer were incubated with (1) preassembled QD525-imager mixed with QD585-streptavidin, (2) preassembled QD585-imager mixed with QD525-streptavidin, or (3) QD525-streptavidin and QD585-streptavidin simultaneously added to biotinylated imager oligo (control experiment). If crosstalk existed due to the exchange of the biotinylated imager strands between different QDs, fluorescence would be expected in both the 525 nm and 585 nm channels in the first two experiments. However, fluorescence was only observed in the preassembled QD-imager channel but not the free QD-streptavidin channel (Figure 2a–b), proving the absence of imager oligo hopping (dissociate/re-associate) between different QDs and consequently crosstalk. In parallel, QD525-streptavidin and QD585-streptavidin simultaneously mixed with biotinylated imager oligo produced specific antigen staining in both channels with a nearly equal contribution (Figure 2c–d). These experiments prove the feasibility of parallel multicolor QD-SABER for IHC.
Figure 2. Characterization of QD-SABER potential crosstalk during staining.

a) Pre-assembled QD525-imager mixed with QD585-Streptavidin for Lamin A staining, and b) pre-assembled QD585-imager mixed with QD525-Streptavidin for Lamin A staining. Specific nuclear envelope staining was only observed in the fluorescence channel of the pre-assembled QD-imagers, confirming the absence of imager strands dissociate with the original QD and re-associate with a different QD. c) QD585-streptavidin and QD525-streptavidin of equal molar concentration were mixed with the biotinylated imager probe and applied to cells for Lamin A imaging, signals of similar strength were observed in both fluorescence channels. d) Quantitative bar plots of the cell fluorescence intensity in a), b), and c). HeLa cells were used in this study. Dual-color images were obtained on a microscope equipped with a 100x objective and an HSI camera. Scale bars, 50 μm.
To demonstrate QD-SABER for multiplexed staining, the five biomarkers representing target antigens in both the cytoplasm and nucleus (HSP90, Ki67, Lamin A, Calnexin, and β-tubulin) were labeled in parallel using five sets of QD-SABER (unique bridge, concatemer, and imager sequences combined with five-color QDs). Hyperspectral imaging (HSI) was used for fluorescence image capture and quantitative analysis because of its capability in removing autofluorescence and resolving fluorescent components that spectrally overlap.[38] The biomarker intracellular distribution patterns obtained through parallel staining (Figure 3a–b) were consistent with those obtained with the single-color IHC (Figure 1 and Figure 3c). Remarkably, quantitative analysis revealed that the expression profiles of individual biomarkers stained in the multiplexed QD-SABER were similar to the one used obtained from the single color QD-SABER (Figure 3d), showing the absence of interferences during the multiplexed SABER hybridization step. In this multicolor staining experiments, the incubation time was kept consistent for QDs of different colors, because the overall hydrodynamic sizes of the QD bioconjugates are largely determined by the surface coating layers (amphiphilic polymer, polyethylene glycol, and biomolecules) rather than the core particle sizes, as shown in previous studies.[18] Indeed, when QDs of different colors were used in QD-PER, no major difference was observed in hybridization kinetics (Supplementary Figure S4). The different colored QDs, however, have different brightness due to variance in molar extinction coefficient (in general, larger QDs have much higher molar extinction coefficient) and quantum yield, the brightness across different colors cannot be directly compared. To account for this photophysical bias, the brightness of individual QD colors was calibrated by measuring the fluorescence intensity of QD solutions of a fixed concentration using the same microscope setup.[18, 19]
Figure 3. Multiplexed QD-SABER staining of HSP90, Ki-67, Lamin A, Calnexin, and β-tubulin.

a) A reconstructed false-color composite image from parallel multiplexed staining (see Panel b). b) False-colored individual channels obtained with HSI for clear visualization of target intracellular location and distribution (from left to right, QD emitting fluorescence peaked at 525, 565, 585, 605 and 655 nm). The staining patterns and intensities are consistent with c) single-color staining conducted with color-matched QD-imager probes. These single-color images were obtained with a true-color CCD. d) Bar plots of the fluorescence intensity of the stained biomarkers after applying the QD brightness correction factors. The error bars represent s.d. of the average staining intensity between three different fields of view on the same specimen. The Nuance image analysis software was employed to measure the fluorescence intensity of individual QD signal intensity. Scale bar, 50 μm.
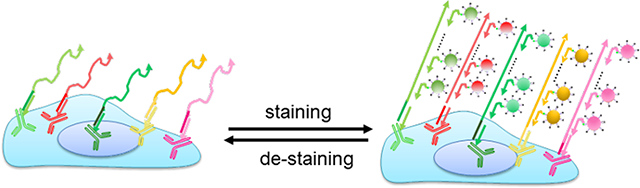
After demonstrating improved signal strength and the absence of crosstalk during immuno-staining and fluorescence imaging, we investigated the compatibility of QD-SABER with cyclic staining for expanded multiplexing capability. Previously, using protein A as the adaptor protein to link QDs with primary antibodies, we have shown that cell specimens immunostained with QDs can be de-stained and regenerated for additional rounds of staining (cyclic staining).[18, 19] If a panel of N biomarkers can be stained in parallel, staining the same samples for M times of different biomarker subsets would allow N x M biomarkers to be probed in the same sample, making IHC a content-rich technique. Because all color QDs are linked to 1’Abs via the same type of biomolecular interaction (Ab-protein A binding), the 1’Abs binding to the sample after each round of staining would have to be removed to avoid crosstalk in subsequent rounds of immunostaining. This requirement has two consequences: 1) the primary antibodies are applied to the specimen one subset at a time rather than all together, resulting in longer assay time (cell incubation with antibodies is a slow process) and laborious manual handling (blocking, mixing, washing, etc.); and 2) breaking protein-protein interactions in the de-staining step requires harsh chemical treatments. For QD-SABER combining QD optical properties with DNA nanotechnology, both issues can be addressed because 1’Abs encoded by unique DNA sequences can be applied to the samples all at once while the conditions for DNA de-hybridization are mild.
For cyclic staining, two requirements should be met. First, the QD fluorescence after each cycle of staining and imaging must be completely removed to avoid signal carry-over into the subsequent cycles. Second, the oligo-encoded 1’Abs applied to the sample all at once and the long concatemers should be stably immobilized on the sample throughout the cycles (free of dissociation). To meet these requirements, the 1’Abs were covalently linked to cell endogenous proteins with an amine-to-amine homobifunctional crosslinker, bis(sulfosuccinimidyl)suberate (BS(PEG)5), and the binding affinity between the bridge oligo and concatemer was designed to be higher (higher melting temperature Tm) than that between the imager oligo and concatemer. For DNA de-hybridization, the addition of formamide has been reported as a highly effective approach. More importantly, by controlling the concentration of formamide, DNA duplexes with lower Tm can be selectively broken apart without affecting DNA duplex with higher Tm. With optimized conditions, we observed that the QD-imager oligo was readily removed with greater than 90% of QDs lifted, and the minute amount of residual QDs was completely quenched with a sodium acetate buffer of pH 5.5, rendering the sample ready for subsequent staining cycles. A simple illustration of the clean de-staining and re-staining was illustrated in Figure 4a, where Calnexin (a cytoplasmic target) and Lamin A (a nuclear target) were initially stained with QD565 imager and QD585 imager. Upon complete de-staining, the two QD imagers were swapped in the second-round QD-SABER for the two targets. Identical staining patterns between the two cycles for both targets were observed. To quantitatively evaluate the antigenicity, we tested 10 cycles of de-staining and re-staining processes. As shown in Figure 4b, after each cycle of de-staining, re-application of the QD-imager completely restored the same target staining patterns, proving that only the QD-imager oligo was washed away during the de-staining process but not the long concatemers. Quantitative analysis showed that the 10 (likely more cycles can be done) staining and de-staining cycles were achieved without affecting the cell morphology, antigenicity, hybridization specificity, or resulting in a signal loss (Figure 4c).
Figure 4. Cyclic staining via QD-SABER.

a) Dual-color cell labeling enabled a complete exchange of QD-imagers between Lamin A and Calnexin in two cycles (staining-destaining-staining). The two colors were swapped in the second round of staining. The fluorescence micrographs were collected with the HSI camera. b&c) To quantitatively assess of repeated rapid sample regeneration. 10 staining and de-staining cycles were conducted using QD-SABER for Lamin A. No change in fluorescence staining pattern or intensity was detected, demonstrating excellent preservation of specimen antigenicity. The images were obtained with a Qcolor5 camera and a 40X objective. Constant exposure time was used for direct comparison of the staining intensity using Image J. The error bars represent s.d. of the average fluorescence intensity between different fields of view on the same specimen. Scale bar, 50 μm.
Besides these cycling performance characterizations, assay time is another factor that is important for future practical applications in biology and medicine. As aforementioned, cyclic staining offers excellent multiplexing capability, but at the cost of assay time since the same protocols are repeated on the sample samples multiple times. For example, among the key steps in SABER, immuno-recognition, where antibodies diffuse inside the cells to find the corresponding antigens, is a slow step that often takes 1–2 hours to achieve decent results and overnight incubation to reach antibody-antigen binding equilibrium. Fortunately, the unique design of SABER enables all antibodies to be incubated with the sample in a single step and the long concatemer incubation in a single step. The cycling process occurs in the hybridization step between the QD-imagers and the concatemers, a relatively fast process. In the current study, we fixed the hybridization time at 1h, which can be potentially shortened as previously shown using dye-imagers.[26] The current protocol can also be extended to formalin-fixed paraffin-embedded (FFPE) tissue specimens with minor changes. Figure S5 shows an example of QD-SABER staining of clinical prostate tissue sections. Compare with the conventional IHC method, QD-SABER shows significantly enhanced staining intensity for improved visualization.
In conclusion, we have developed a simple and powerful platform, QD-SABER, for sensitive and multiplexed protein analysis at the single-cell level. The key features of this technology including multicolor staining, antibody encoding by oligonucleotide, fluorescence signal enhancement, and efficient de-staining are enabled by the unique optical properties of QDs and the flexibility and programmability of DNA nanotechnology. Compared to conventional IHC using dye-labeled 2’Abs where only 2–3 targets can be imaged, 5–10 QD colors can be simultaneously, and the labeling of subsets of target antigens can be repeated for at least 10 cycles without affecting sample antigenicity. In the current study that focuses on technology development, model antigens with relatively high antigen expression levels are used. For future applications, we expect QD-SABER to become an advanced imaging tool for biomedical research and clinical diagnostics. In particular, it could have an impact on high-sensitivity imaging where the target is of low abundance or when the background signal is high (such as autofluorescence), and on high-content imaging where a large number of targets need to be evaluated in the same samples.
Supplementary Material
Acknowledgments
This work was supported in part by NIH (R21CA192985), the Department of Bioengineering at the University of Washington, and a Damon Runyon Dale Frey Innovator Award (BJB). We also thank Dr. Pavel Zrazhevskiy for his advice on the experimental design, and Professor Larry True for the de-identified prostate tissue sections.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Wen Zhou, Department of Bioengineering, University of Washington, Seattle, WA 98195, USA.
Yan Han, Department of Bioengineering, University of Washington, Seattle, WA 98195, USA.
Brian J. Beliveau, Department of Genome Sciences, University of Washington, Seattle, WA 98195, USA
Xiaohu Gao, Department of Bioengineering, University of Washington, Seattle, WA 98195, USA.
References
- [1].Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, Goolam M, Saurat N, Coupland P, Shirley LM, Smith M, Van der Aa N, Banerjee R, Ellis PD, Quail MA, Swerdlow HP, Zernicka-Goetz M, Livesey FJ, Ponting CP, Voet T, Nat. Methods 2015, 12, 519. [DOI] [PubMed] [Google Scholar]
- [2].Gawad C, Koh W, Quake SR, Nat. Rev. Genet. 2016, 17, 175. [DOI] [PubMed] [Google Scholar]
- [3].Saikia M, Burnham P, Keshavjee SH, Wang MFZ, Heyang M, Moral-Lopez P, Hinchman MM, Danko CG, Parker JSL, De Vlaminck I, Nat. Methods 2019, 16, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Altschuler SJ, Wu LF, Cell 2010, 141, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, Strathdee CA, Dempster J, Lyons NJ, Burns R, Nag A, Kugener G, Cimini B, Tsvetkov P, Maruvka YE, O’Rourke R, Garrity A, Tubelli AA, Bandopadhayay P, Tsherniak A, Vazquez F, Wong B, Birger C, Ghandi M, Thorner AR, Bittker JA, Meyerson M, Getz G, Beroukhim R, Golub TR, Nature 2018, 560, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Turajlic S, Sottoriva A, Graham T, Swanton C, Nat. Rev. Genet. 2019, 20, 404. [DOI] [PubMed] [Google Scholar]
- [7].Rohban MH, Abbasi HS, Singh S, Carpenter AE, Nat. Commun. 2019, 10, 2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoué F, Bruneval P, Cugnenc P-H, Trajanoski Z, Fridman W-H, Pagès F, Science 2006, 313, 1960. [DOI] [PubMed] [Google Scholar]
- [9].Fridman WH, Pages F, Sautes-Fridman C, Galon J, Nat. Rev. Cancer 2012, 12, 298. [DOI] [PubMed] [Google Scholar]
- [10].Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G, Nat. Rev. Clin. Oncol. 2017, 14, 717. [DOI] [PubMed] [Google Scholar]
- [11].Bendall SC, Simonds EF, Qiu P, Amir E.-a. D., Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP, Science 2011, 332, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bendall SC, Nolan GP, Nat. Biotechnol. 2012, 30, 639. [DOI] [PubMed] [Google Scholar]
- [13].Frei AP, Bava FA, Zunder ER, Hsieh EW, Chen SY, Nolan GP, Gherardini PF, Nat. Methods 2016, 13, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Han G, Spitzer MH, Bendall SC, Fantl WJ, Nolan GP, Nat. Protoc. 2018, 13, 2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S, Natkunam Y, Nolan GP, Nat. Med. 2014, 20, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schüffler PJ, Grolimund D, Buhmann JM, Brandt S, Varga Z, Wild PJ, Günther D, Bodenmiller B, Nat. Methods 2014, 11, 417. [DOI] [PubMed] [Google Scholar]
- [17].Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, Hollman D, Kamath V, Kaanumalle S, Kenny K, Larsen M, Lazare M, Li Q, Lowes C, McCulloch CC, McDonough E, Montalto MC, Pang Z, Rittscher J, Santamaria-Pang A, Sarachan BD, Seel ML, Seppo A, Shaikh K, Sui Y, Zhang J, Ginty F, Proc. Natl. Acad. Sci. USA 2013, 110, 11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zrazhevskiy P, Gao X, Nat. Commun. 2013, 4, 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zrazhevskiy P, True LD, Gao X, Nat. Protoc. 2013, 8, 1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, Black S, Nolan GP, Cell 2018, 174, 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L, Nat. Methods 2014, 11, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shah S, Takei Y, Zhou W, Lubeck E, Yun J, Eng C-HL, Koulena N, Cronin C, Karp C, Liaw EJ, Amin M, Cai L, Cell 2018, 174, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shah S, Lubeck E, Zhou W, Cai L, Neuron 2016, 92, 342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nagendran M, Riordan DP, Harbury PB, Desai TJ, Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kishi JY, Lapan SW, Beliveau BJ, West ER, Zhu A, Sasaki HM, Saka SK, Wang Y, Cepko CL, Yin P, Nat. Methods 2019, 16, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Saka SK, Wang Y, Kishi JY, Zhu A, Zeng Y, Xie W, Kirli K, Yapp C, Cicconet M, Beliveau BJ, Lapan SW, Yin S, Lin M, Boyden ES, Kaeser PS, Pihan G, Church GM, Yin P, Nat. Biotechnol. 2019, 37, 1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hildebrandt N, Spillmann CM, Algar WR, Pons T, Stewart MH, Oh E, Susumu K, Díaz SA, Delehanty JB, Medintz IL, Chem. Rev. 2017, 117, 536. [DOI] [PubMed] [Google Scholar]
- [28].Qiu X, Hildebrandt N, ACS Nano 2015, 9, 8449. [DOI] [PubMed] [Google Scholar]
- [29].Jennings TL, Becker-Catania SG, Triulzi RC, Tao G, Scott B, Sapsford KE, Spindel S, Oh E, Jain V, Delehanty JB, Prasuhn DE, Boeneman K, Algar WR, Medintz IL, ACS Nano 2011, 5, 5579. [DOI] [PubMed] [Google Scholar]
- [30].Samanta D, Ebrahimi SB, Mirkin CA, Adv. Mater. 2019, Doi: 10.1002/adma.201901743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Paige JS, Nguyen-Duc T, Song W, Jaffrey SR, Science 2012, 335, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Thekkan S, Jani MS, Cui C, Dan K, Zhou G, Becker L, Krishnan Y, Nat. Chem. Biol. 2019, 15, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zrazhevskiy P, Akilesh S, Tai W, Queitsch K, True LD, Fromm J, Wu D, Nelson P, Stamatoyannopoulos JA, Gao X, Angew. Chem. Int. Ed. 2016, 55, 8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kishi JY, Schaus TE, Gopalkrishnan N, Xuan F, Yin P, Nat. Chem. 2018, 10, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lin R, Feng Q, Li P, Zhou P, Wang R, Liu Z, Wang Z, Qi X, Tang N, Shao F, Luo M, Nat. Methods 2018, 15, 275. [DOI] [PubMed] [Google Scholar]
- [36].Choi HMT, Beck VA, Pierce NA, ACS Nano 2014, 8, 4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gao X, Yang L, Petros JA, Marshall FF, Simons JW, Nie S, Curr. Opin. Biotechnol. 2005, 16, 63. [DOI] [PubMed] [Google Scholar]
- [38].True LD, Gao X, J. Mol. Diagn. 2007, 9, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Medintz IL, Uyeda HT, Goldman ER, Mattoussi H, Nat. Mater. 2005, 4, 435. [DOI] [PubMed] [Google Scholar]
- [40].Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li J, Sundaresan G, Wu A, Gambhir SS, Weiss S, Science, 2005, 307, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rauf S, Glidle A, Cooper JM, Adv. Mater. 2009, 21, 4020. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.