

Abstract
Oncogene-induced metabolic reprogramming is a hallmark of pancreatic cancer (PDAC), yet the metabolic drivers of metastasis are unclear. In PDAC, obesity and excess fatty acids accelerate tumor growth and increase metastasis. Here, we report that excess lipids, stored in organelles called lipid droplets (LD), are a key resource to fuel the energy-intensive process of metastasis. The oncogene KRAS controlled the storage and utilization of LD through regulation of hormone sensitive lipase (HSL), which was downregulated in human PDAC. Disruption of the KRAS-HSL axis reduced lipid storage, reprogrammed tumor cell metabolism, and inhibited invasive migration in vitro and metastasis in vivo. Finally, microscopy-based metabolic analysis revealed that migratory cells selectively utilize oxidative metabolism during the process of migration to metabolize stored lipids and fuel invasive migration. Taken together, these results reveal a mechanism that can be targeted to attenuate PDAC metastasis.
Keywords: Metastasis, pancreatic cancer, lipid droplets, hormone-sensitive lipase
Introduction
Metastases are responsible for 90% of cancer-related deaths and represent a critical challenge for cancer therapy (1,2). Because metastasis is a dynamic process (3), the mechanisms cancer cells utilize during dissemination must be adaptable for successful invasion, migration, and establishment of a secondary tumor. Metastatic invasion is a highly energy-intensive process, and the metabolic processes used by invading tumor cells are poorly defined. While proliferative primary tumors rely heavily on glycolysis (4), metastatic tumor cells have drastically different metabolic requirements (5) including reliance on mitochondrial integrity and increased oxidative phosphorylation, likely to generate ATP for migration (6–8). The metabolic changes that occur during tumor cell invasion may represent a novel therapeutic target to reduce metastasis and improve outcomes for patients with metastatic cancer.
Pancreatic ductal adenocarcinoma (PDAC) ranks among the deadliest cancers, due in part to a high incidence of metastasis at diagnosis. One of the few known risk factors for pancreatic cancer is obesity, which correlates with worse prognosis (9,10). Corroborating these epidemiological studies, high fat diets accelerate tumorigenesis and increase metastasis in mouse models of PDAC (11,12). Additionally, increased intratumoral fatty acids (FAs) may result from elevated de novo synthesis (9,13–15). However, the mechanisms exploited by PDAC cells to store and utilize fat to promote cancer progression and metastasis are not well defined.
Excess FAs are stored as triglycerides within organelles called lipid droplets (LDs). These stored neutral lipids can be catabolized by lipolysis, which uses the sequential activity of lipases to liberate free FAs (16). Best studied in adipocytes, adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL) sequentially catabolize triacylglycerol to glycerol and free FAs (17). Lipolysis, thus, mediates the release of bioavailable FAs from inert storage within LDs. Therefore, dysregulation of lipolysis, or any of the crucial enzymes mediating this process, may be a novel mechanism by which cancer cells fuel tumor progression and metastasis.
The GTPase KRAS is activated in over 90% of PDAC, and is a driver of tumorigenesis and metabolic reprogramming (18–20). Oncogenic KRAS mutations instill a greater dependency on glycolysis and amino acid metabolism, promoting biomass synthesis to support tumor growth (21–24). How KRAS-mediated metabolic regulation is altered during metastasis, and the mechanisms involved in this transient process, are not well understood.
Here, we describe a dynamic mechanism in which oncogenic KRAS regulates HSL to control metabolism in metastatic pancreatic cancer cells by regulating lipid storage and utilization. Oncogenic KRAS suppresses HSL expression as part of a metabolic shift away from FA oxidation in pancreatic cancer cells, leading to LD accumulation and priming tumor cells for invasion. Live cell imaging revealed that these stored lipid droplets are then catabolized during invasion by the action of lipases, including residual HSL, resulting in increased FA oxidation and oxidative metabolism that drives tumor cell invasion. These findings reveal a novel mechanism by which KRAS regulates tumor cell metabolism and controls metastatic invasion in pancreatic cancer.
Materials and Methods
Cell culture
Panc-1, MiaPaCa-2, CFPAC, and mKPC cells were cultured in DMEM containing 10% FBS and Pen/Strep. BxPC3 cells were cultured in RPMI-1640 containing 10% FBS and Pen/Strep. iKRAS cells were cultured in RPMI-1640 containing 10% FBS, Pen/Strep, and 1μg/mL doxycycline hyclate (Sigma, D9891). HPDE cells were cultured in keratinocyte-SFM containing epidermal growth factor, bovine pituitary extract, and Pen/Strep. DMEM and RPMI-1640 were from Corning, and FBS was from Sigma. Cell lines were mycoplasma tested using PCR (Southern Biotech), DAPI staining, or by IDEXX BioAnalytics. Atglistatin (15284) and CAY10499 (10007875) were from Cayman Chemical. U0126 (662005) was from Calbiochem, H89 (B1427) and inhibitors to DGAT1 (PZ0207) and DGAT2 (PZ0233) were from Sigma-Aldrich. Cell lines were from ATCC or were provided by the laboratory of Dr. David Tuveson (mKPC T4-2D, Cold Spring Harbor Laboratory), Dr. Marina Pasca di Magliano (iKRAS 4292, University of Michigan), or Dr. Daniel Billadeau (HPDE, CFPAC, Mayo Clinic).
Transfections and establishment of stable cell lines
Lipofectamine 2000 (11668-027, Invitrogen) was used for plasmid transfection. The murine HSL-GFP plasmid was a gift from J. Liu (Mayo Clinic). HSL was cloned into the pLenti6.3 lentiviral vector by the Mayo Clinic C-SiG Gene Editing and Epigenomics Core. To generate lentivirus, the HSL plasmid, or a pLenti6.3.FLAG control vector, was cotransfected into HEK 293T cells with VSV-G-pseudotyped viral packaging plasmids (provided by Dr. Y. Ikeda, Mayo Clinic). After 72 hours, supernatants were collected and passed through 0.45 μm filters to collect virus. mKPC cells were transduced with virus for 72 hours with 10 μg/mL polybrene and selected with 5 μg/mL Blasticidin for 14 days to obtain cells stably overexpressing control vector or HSL. Murine siGENOME SMARTpools (Dharmacon) for LIPE (HSL) and CPT1A were used for knockdowns, with Lipofectamine RNAiMAX (13778-150).
Immunoblotting
For western blotting, cells were lysed in NP-40 lysis buffer (20 mM Tris-Cl, pH 7.4, 137 mM NaCl, 10% glycerol, 1% NP-40 (IGEPAL), and 2 mM EDTA, pH 8.0) with cOmplete Protease Inhibitor Cocktail (11873580001, Roche). Protein levels were quantified by BCA assay (23225, Pierce), and equal amounts were resolved on SDS-PAGE gels and transferred to PVDF for probing. Primary antibodies were: HSL (4107, Cell Signaling), phospho-HSL (4139, Cell Signaling), ATGL (2439, Cell Signaling), MGL (sc-398942, Santa Cruz), phospho-ERK (4377, Cell Signaling), ERK (9102, Cell Signaling), Vimentin (5741, Cell Signaling), Claudin-1 (13255, Cell Signaling), ZO-1 (8193, Cell Signaling), Snail (3879, Cell Signaling), E-Cadherin (610182, BD Biosciences), N-Cadherin (610921, BD Bioscience), CPT1 (12252, Cell Signaling), PLIN-2 (3121, LSBio), PLIN-3 (10694, Proteintech), PLIN-5 (46215, Invitrogen), Actin (A2066, Sigma), and GAPDH (5174, Cell Signaling). Secondary antibodies were conjugated to horseradish peroxidase (Biosource International) and immunoreactive signals were detected using SuperSignal West Pico or Femto substrates (Thermo Fisher Scientific) and HyBlot CL film (Denville Scientific), and developed using an X-Omat film processor. Densitometry values were quantified using ImageJ
Immunohistochemistry (IHC)
For human samples, 30 histological samples from 10 de-identified patients were obtained from the Mayo Clinic SPORE in Pancreatic Cancer. Patients provided written informed consent and approval of the Institutional Review Board was granted prior to investigation. From each patient, adjacent/normal, primary tumor, and tissue from a metastatic site were analyzed. For mouse experiments, tissue was fixed in 4% PFA and cryoprotected in 30% sucrose before embedding in Tissue-Tek O.C.T. compound (4583). 4 μm human tissue sections were used and 10 μm murine sections were used for all applications. Anti-Rabbit HRP-DAB Tissue Staining Kit (R&D Systems, CTS005) was used. Primary antibodies used were: HSL (SAB4501762, Sigma), αSMA (ab5694, Abcam), and Ki67 (12202, Cell Signaling). Semi-quantitative scoring of human tissues was performed by a pathologist (Dr. Lizhi Zhang) in the Mayo Clinic Division of Anatomical Pathology. To label lipid droplets, sections were washed in 60% isopropanol for 30 seconds, 60% Oil Red O solution (5mg/ml in isopropanol) for 15 minutes, 60% isopropanol for 30 seconds, then washed in deionized water prior to hematoxylin counterstain. Further semi-quantitative analysis of tissue sections was completed using color deconvolution in ImageJ to obtain an optical density of the signal of interest.
Migration, invasion, and proliferation analysis
For transwell invasion analysis, 3 x 105 cells were seeded in 6-well transwell chambers with 8-μm pores (MCEP06H48, Millipore) and coated with 1 mg/mL Matrigel (356231, Corning). Cells were seeded in the upper chamber containing 0.1% serum and invaded towards the lower chamber containing 10% serum. Percent invasion was determined by counting nuclei on the bottom and top of the filter for each field and dividing the invaded nuclei (bottom) over the total. Nuclei were visualized using Hoechst 33342 (H3570, Invitrogen). For wound healing assays, cells were grown to confluency on gridded glass coverslips before a plastic pipette tip was used to scratch the monolayer. Brightfield images were taken at the T=0 and a timepoint determined for each cell line. Beginning and ending images were overlaid to determine migration distance using Adobe Photoshop. For matrix degradation assays cells were plated on fluorescently-labeled gelatin-coated coverslips and the percentage of cells that degraded the fluorescent gelatin was analyzed as described previously (25). Invadopodia were counted manually based on overlapping puncta of cortactin, actin, and gelatin degradation. For proliferation and viability assays, the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega, G5421) or propidium iodide (P4864, Sigma) were used.
Fluorescence and live-cell microscopy
Cells were washed in PBS and fixed in 3% formaldehyde. Phalloidin-TRITC (P1951, Sigma) was used to visualize actin Anti-cortactin 4F11 (05-180, Upstate Biotechnology) and Cy5 secondary antibody (115-175-166, Jackson ImmunoResearch) were used to visualize cortactin. To label lipid droplets, Oil Red O (O0625, Sigma) and C12-BODIPY (D3822, Thermo Fisher) were used and quantitated as described previously (26). Briefly, fixed samples were washed in 60% isopropanol for 30 seconds, 60% Oil Red O solution (5 mg/ml in isopropanol) for 1.75 minutes, and 60% isopropanol for an additional 30 seconds. Images were acquired on a Zeiss LSM 780 confocal microscope with a 40x oil objective lens and Zeiss Zen software. Lipid droplet quantitation was done using the auto local threshold and analyze particles tool within ImageJ as described previously (27).
qPCR mRNA analysis
RNA isolated using the RNeasy Plus Mini Kit (74134, Qiagen) was reverse-transcribed using oligo-(dT) primers and the Super Script III First Strand Kit (18080-51, Invitrogen). Quantitation of gene expression was performed using SYBR green fluorescence on a LightCycler 480 (04707516001, Roche). Primers used were: LIPE (HSL gene) forward: 5’–GATTTACGCACGATGACACAGT–3’, reverse: 5’–ACCTGCAAAGACATTAGACAGC–3’. 18S ribosomal RNA: forward: 5’–CGCTTCCTTACCTGGTTGAT–3’, reverse: 5’–GAGCGACCAAAGGAACCATA–3’. Expression was determined by normalizing LIPE to 18S by the ΔΔCt method.
In vivo experiments
Animal experiments were performed under Institutional Animal Care and Use Committee approval and in accordance with the approved protocol. For pancreatic orthotopic injections, six week old female C57BL/6J mice were purchased from The Jackson Laboratory. Mice were anesthetized by intraperitoneal injection of ketamine and xylazine, and 1 x 104 mKPC cells stably expressing control vector or HSL suspended in 100 μl PBS were injected into the tail of the pancreas. Mice were weighed every other day. At 6 weeks, or upon a weight loss of >10%, mice were sacrificed and both primary tumors and macrometastases were isolated and analyzed. Genetic mouse models, Ptf1a-Cre (023329) and KRAS LSL-G12D (008179, both from The Jackson Laboratory) were crossed to achieve a pancreas-specific KRASG12D/+ mouse that was compared to littermate Ptf1a-Cre mice. Both male and female mice were analyzed at 12 weeks, with no sex-specific differences in the results.
Metabolic analyses
For Seahorse metabolic analysis, a Seahorse XF24 Analyzer was used with Seahorse XF Cell Mito Stress Test Kit (103015-100, Agilent) and Seahorse XF24 FluxPak (100867-100, Agilent) reagents to measure oxygen consumption rate. Quantitation was normalized by cell count, determined by Hoechst 33342 positive cells imaged on a Celigo (Nexcelom Bioscience). Glycolytic activity was determined using a Glycolysis Cell-Based Assay Kit (600450, Cayman Chemical) and ATP levels were determined using an ATP Detection Assay Kit (700410, Cayman Chemical).
Optical Redox Ratio Analysis
To obtain optical redox ratios, endogenous autofluorescence of NADH and FAD were imaged on a Zeiss LSM 510 confocal microscope with a 40x oil objective lens with excitations of 364 and 458 nm and emissions of 460 ± 20 and >505 nm, respectively. Pixel-wise redox ratio maps were created in ImageJ using fluorescence intensities as FAD/(FAD + NADH). The mean redox ratio was acquired by averaging the redox ratio values within the cell cytoplasm, excluding the background and nucleus, when possible.
Statistical Analysis
Microscopy images were processed using ImageJ and Adobe Photoshop. Any adjustments were made uniformly across the whole image. Data were analyzed using GraphPad Prism. Graphed data represent mean +/− SEM, and statistical significance was determined using a Student’s t test, a Mann-Whitney U test, or a Wilcoxon Signed Rank test. For analysis of the TCGA-Pancreatic Cancer (PAAD) dataset, the UCSC Xenabrowser visualization tool (xenabrowser.net) was used to compare somatic mutation of KRAS and the gene expression of LIPE. The log2 expression values were graphed in GraphPad Prism as violin plots.
Results
Catabolism of stored lipids promotes PDAC invasion and migration
We first measured the basal LD content of pancreatic cancer cells, including Panc1, MiaPaCa2, BxPC3, and cells isolated from a mouse model of pancreatic cancer (mKPC) (28). Under basal conditions, PDAC cells are capable of storing FAs in intracellular LDs (Fig. 1A and Supplementary Fig. 1A). Exogenous FAs in the form of oleic acid (OA) stimulated lipid accumulation in the PDAC cells and dramatically increased the LD content of the cells (Fig. 1A–B and Supplementary Fig. 1A). Thus, PDAC cells are capable of storing intracellular lipids in LDs, which is exacerbated when exposed to elevated levels of FAs.
Figure 1.
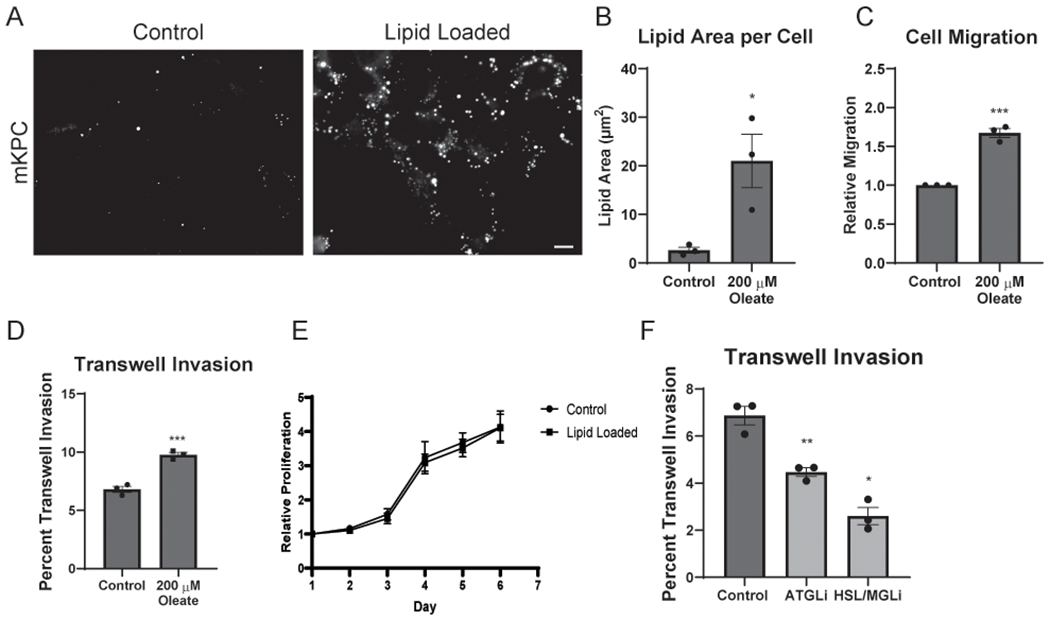
Excess lipid promotes PDAC invasion and migration through lipolysis. A, Control and oleic acid (OA) loaded (200 μM) mKPC cells were visualized by staining for LDs using Oil Red O (ORO). Scale bar, 10 μm. B, Quantitation of total LD area per cell from the conditions in (A). >125 cells scored in at least five fields for 3 independent biological replicates. C, Quantitation of distance migrated of control and OA-loaded mKPC cells in a wound healing assay over 12 hours. 10 measurements of three fields for 3 independent biological replicates represented. D, Quantitation of control and OA-loaded mKPC cells in a transwell invasion assay. Cells invaded for 16 hours through a Matrigel-coated filter toward high-serum media. >300 cells scored in each of 3 independent biological replicates. E, Quantitation of control and OA-loaded mKPC cell proliferation over the indicated number of days. 8 independent biological replicates, normalized to Day 1 for each experiment. F, Quantitation of control or drug-treated mKPC cells in a transwell invasion assay. Cells attached for 2 hours prior to drug treatment, and invaded for 14 hours. 10 μM Atglistatin (ATGLi) and 10 μM CAY 10499 (HSL/MGLi). >300 cells scored in each of 3 independent biological replicates. Graphs indicate mean ± SEM, analyzed by Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001.
As high fat diets correlate with increased metastasis and tumor progression (11,29), we tested how excess stored lipids impacted cellular invasion. Treatment of mKPC, BxPC3, and Panc04.03 cells with exogenous FAs stimulated cell migration in a wound healing assay (Fig. 1C and Supplementary Fig. 1B) and invasive ability in a transwell invasion assay (Fig. 1D and Supplementary Fig. 1C). A similar increase was observed following addition of linoleic acid, indicating that multiple FA species augment PDAC cell migration (Supplementary Fig. 1D). Metastasis requires tumor cells to degrade extracellular matrix for invasion into surrounding tissues. Following lipid-loading with exogenous FAs, we observed increased degradation of extracellular matrix as well as the formation of matrix-degrading invadopodia (Supplementary Fig. 1E–H). To test if lipid loading increased the metastatic capacity of PDAC cells by inducing an epithelial-to-mesenchymal transition (EMT), we analyzed the expression of a panel of EMT markers following OA treatment (Supplementary Fig. 1I). While the expression of EMT markers was largely unchanged by treatment with OA, there was a decrease in the tight junction marker ZO-1. Thus, while not likely driven by an EMT program, lipids promote multiple processes that support tumor cell invasion. There was not a significant change in mKPC cell proliferation or cell death following treatment with exogenous FAs (Fig. 1E and Supplementary Fig. 1J and K), indicating that stored lipids are more important for invasive migration than proliferation of tumor cells. Together, these results indicate that PDAC cells can utilize exogenous FAs and stored lipids to enhance their metastatic capacity.
Importantly, increased invasion only occurred after pre-loading of lipids, resulting in lipid storage, and not while exogenous FAs were available to the cells only during invasion (Supplementary Fig. 2A). We hypothesized that it was lipids stored in LDs that were required for tumor cell invasion. We therefore tested the requirement for diglyceride acyltransferases 1 and 2 (DGAT1/2) in tumor cell invasion, which are required for the packaging and storage of lipids in LDs. Inhibition of DGAT1/2 reduced the capacity to store lipids in lipid droplets, and consequently minimized the pro-invasive effects of lipid loading on PDAC cells (Supplementary Fig 2B and C). Thus, proper packaging of triglycerides into LDs is a crucial step in FA-mediated mediating augmented PDAC cell invasion.
As lipases liberate free FAs from LDs, we tested their role in lipid-regulated metastatic processes in tumor cells. Strikingly, treatment of mKPC cells with Atglistatin to inhibit ATGL, or CAY 10499 to inhibit HSL (with some inhibition of MGL), reduced transwell invasion (Fig. 1F), indicating that lipase activity is required for tumor cell invasion. Treatment with lipase inhibitors had no significant impact on proliferation (Supplementary Fig. 2D), suggesting that stored lipids are largely contributing to invasive potential, rather than proliferative capacity. Together, these data suggest that lipase dependent liberation of stored lipids from LDs is required during tumor cell invasion.
We hypothesized that LDs were undergoing lipolysis during the process of migration. Using live cell microscopy, we observed LDs in the highly motile leading-edge cells were lost as cells migrated (Fig. 2A and Movie S1), consistent with LD catabolism (30). In contrast, cells that were less motile and farther than two cell widths from the migratory edge appeared to retain their stored LDs (Supplementary Fig 2E and Movie S2). Loss of lipid droplets also occurred during invasion. Tumor cells pre-loaded with lipids were seeded in a transwell assay, and the filters were analyzed by confocal microscopy to image the LD content on the top (Non-Invasive) and bottom (Post-Invasion). Invasive cells had fewer lipid droplets remaining compared to noninvasive cells (Fig. 2B and C), consistent with LD loss and lipolysis during invasion.
Figure 2.
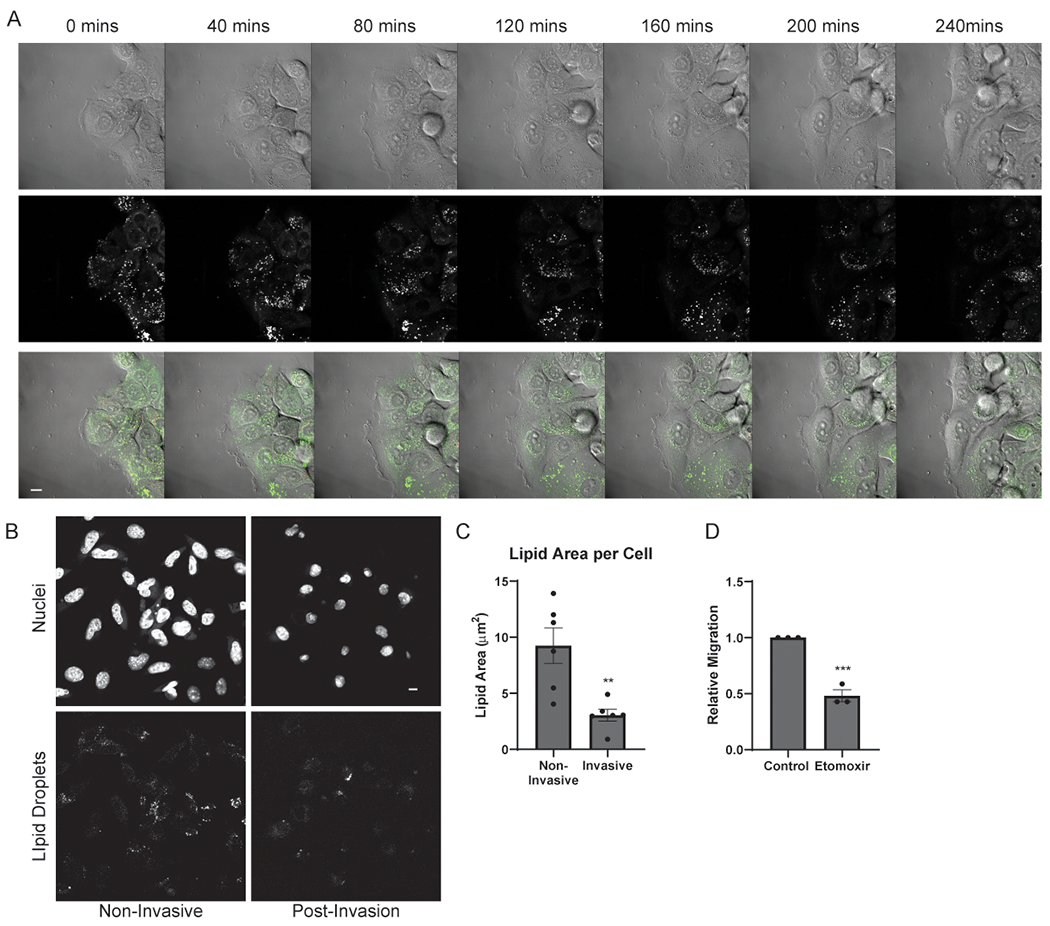
Lipid droplets are catabolized during invasive migration. A, Still images taken from Supplemental Video 1 depicting Panc04.03 cells at indicated time points migrating in a wound healing assay, with LDs visualized by BODIPY. Brightfield, lipid droplets (white), and composite images are shown. Scale bar, 10 μm. B, Representative images of LDs in mKPC cells in an invasion assay and pre-loaded with OA (200 μM). Non-Invasive cells (left) did not invade, whereas Invasive cells (right) did invade and were imaged on the bottom of the transwell. Cells were stained for nuclei (Hoechst, top) and LDs (ORO, bottom). Scale bar, 10 μm. C, Quantitation of lipid area per cell from (B). >50 cells scored in at least five fields from 6 independent biological replicates. D, Migration quantitation of control or etomoxir-treated (10 μM) mKPC cells. >10 measurements of 3 fields for 3 independent biological replicates shown. Graphs indicate mean ± SEM data, analyzed by Student’s t-test. **p < 0.01, ***p < 0.001.
We predicted that FAs liberated from LDs during lipolysis were acting as a fuel source for invasion. Etomoxir, an inhibitor of carnitine palmitoyltransferase 1 (CPT1), perturbs beta-oxidation of FAs by preventing FA mitochondrial import. Importantly, Etomoxir reduced the migration of mKPC cells, suggesting that lipid import and oxidation are required for migration (Fig. 2D). Similar results were observed following a knockdown of CPT1 (Supplementary Fig. 2F and G). As treatment with either lipase inhibitors (Fig. 1F) or a CPT1 inhibitor reduced tumor cell invasion and migration, the data suggest that tumor cells utilize lipolysis of LDs and subsequent FA uptake into mitochondria to promote invasive migration.
The lipase HSL is suppressed by the oncogenic GTPase KRAS
These data suggest that lipase activity is required for tumor cell invasion. Because lipolysis has been primarily studied in adipocytes, we first determined the levels of lipase expression in a panel of pancreatic cancer cell lines. Strikingly, we observed a correlation between HSL expression and the KRAS status of cultured pancreatic cancer cells (Fig. 3A). Both non-neoplastic HPDE cells and BxPC3 PDAC cells harbor wild-type KRAS (31,32) and showed significantly higher levels of HSL expression when compared to PANC1, CFPAC, and Panc04.03 cells, which have an activating mutation of KRAS (Fig. 3A). This suggested that HSL may be specifically downregulated in cells with oncogenic KRAS. Interestingly, the correlation between KRAS and expression level does not extend to ATGL or MGL (Fig. 3A and B).
Figure 3.
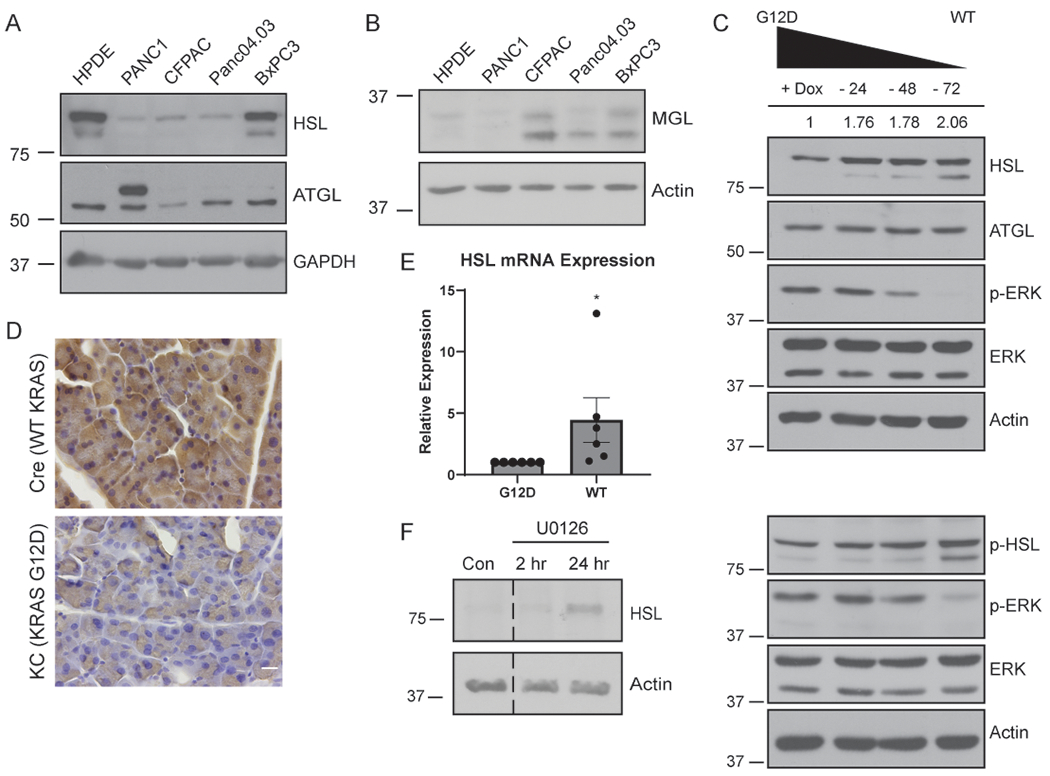
HSL is regulated by oncogenic KRAS. A, Western blot of the indicated cells for HSL, ATGL, and GAPDH from whole-cell lysates. B, Western blot of the indicated cells for MGL and Actin. C, Western blot of HSL, phospho-HSL, ATGL, phospho-ERK, ERK, and Actin expression from whole-cell lysates. iKRAS cells were cultured in doxycycline, or with doxycycline withdrawn for 24, 48, or 72 hours prior to lysis. Quantitative densitometry values for HSL expression from blot shown (both bands) are indicated above. D, Immunohistochemistry for HSL expression in pancreatic tissue sections isolated from Ptf1a-Cre (Cre) or littermate Ptf1a-Cre driven KRASG12D (KC) mice. n=3 mice per condition. Scale bar, 10 μm. E, Relative HSL mRNA levels by quantitative RT-PCR from iKRAS cells cultured with doxycycline (G12D) or following 72 hour doxycycline withdrawal (WT). 3 technical replicates for 6 independent biological replicates shown. Data analyzed by Wilcoxon Signed Rank test. F, Western blot of mKPC cells treated with vehicle control (Con) or the MEK inhibitor U0126 for the indicated time. Blots are representative of three independent experiments. Graphs indicate mean ± SEM. *p < 0.05.
Activating mutations of KRAS play a crucial role in the metabolism of pancreatic cancer cells (18,21,24,33). Therefore, we investigated the correlation between HSL and oncogenic KRAS using a doxycycline-inducible cell culture system (iKRAS) (34). Culture of these mouse pancreatic cancer cells in the presence of doxycycline drives expression of KRASG12D. Following removal of doxycycline, the cells revert to wild-type KRAS, as indicated by decreased phosphorylated ERK (Fig. 3C). Strikingly, reversion to wild-type KRAS also resulted in a significant increase in total and phosphorylated HSL (Fig. 3C), but not ATGL, consistent with the concept that oncogenic KRAS specifically suppresses HSL expression. To determine if expression of oncogenic KRAS was sufficient to downregulate HSL in vivo, we examined pancreatic tissue from mice expressing KRASG12D specifically in the pancreas using Ptf1a-Cre (“KC” mice). Indeed, immunohistochemistry for HSL revealed a striking downregulation of HSL in KC mice compared to mice expressing Ptf1a-Cre alone (Fig. 3D and Supplementary Fig 3A). To further examine the mechanism of KRAS-mediated HSL downregulation, we determined if KRASG12D decreased HSL transcription. Quantitative RT-PCR revealed that HSL mRNA was decreased in cells with oncogenic KRAS (+ Dox) compared to cells with wild-type KRAS (- Dox), suggesting that oncogenic KRAS suppresses HSL transcription (Fig. 3E).
KRAS signaling occurs through several downstream pathways (18). To determine which signaling pathway mediated the suppression of HSL by KRAS, we utilized the pharmacological MEK (mitogen-activated protein kinase kinase) inhibitor U0126. Interestingly, MEK inhibition reversed the KRAS-mediated suppression of HSL and increased HSL expression after 24 hours (Fig. 3F), indicating that KRAS-mediated HSL suppression occurs through the MEK/ERK signaling pathway. Together, these data reveal that expression of the lipase HSL is suppressed by oncogenic KRAS, and may represent a novel mechanism by which KRAS controls tumor cell metabolism.
HSL regulates the metastatic impact of oncogenic KRAS
We hypothesized that KRAS-mediated HSL regulation is a crucial mechanism for modulating lipid storage and metastatic ability. Decreased levels of HSL would be predicted to lead to LD accumulation due to decreased lipolysis. Indeed, cells with oncogenic KRAS had significantly higher levels of stored LDs than cells with wild-type KRAS (Fig. 4A and B), consistent with lower levels of HSL and increased lipid storage. Similarly, oncogenic KRAS led to increased levels of the LD protein PLIN-2, which is also consistent with lipid storage (Supplementary Fig. 3B). This was specific to PLIN-2, as we observed no change in PLIN-3 or PLIN-5 by oncogenic KRAS (Supplementary Fig. 3B).
Figure 4.
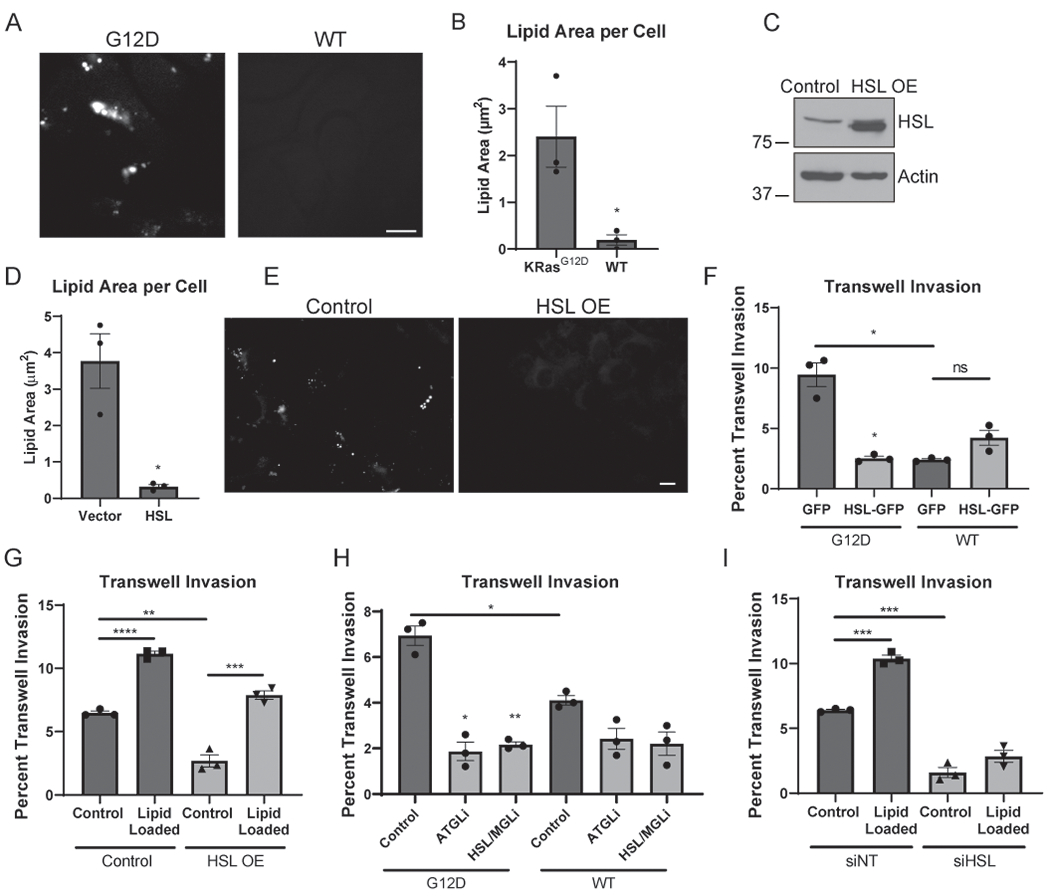
HSL regulates the invasive impact of oncogenic KRAS. A, Representative images of iKRAS cells cultured in doxycycline (KRASG12D) or following 72 hour doxycycline withdrawal (WT) and stained for LDs (ORO). Scale bar, 10 μm. B, Quantitation of total lipid droplet area per cell described in (A). >50 cells scored in each of five fields for 3 independent biological replicates. C, Western blot of HSL and Actin in mKPC whole-cell lysates constitutively overexpressing HSL or vector control. D, Quantitation of total lipid droplet area per cell of mKPC cells overexpressing HSL or a vector control. >75 cells scored in each of five fields for 3 independent biological replicates. E, Representative immunofluorescence images from cells described in (D) stained for LDs (ORO). Scale bar, 10 μm. F, Quantitation of iKRAS cells overexpressing GFP or HSL-GFP in the presence of doxycycline (G12D) or following 72 hour doxycycline withdrawal (WT) prior to seeding on a Matrigel-coated filter and allowed to invade for 16 hours. >40 cells scored in each of 3 independent biological replicates. G, Quantitation of mKPC overexpressing HSL or vector control and loaded with 200 μM BSA-conjugated OA or BSA control prior to invasion for 16 hours. >245 cells scored in each of 3 independent biological replicates. H, Quantitation of iKRAS cells cultured in the presence of doxycycline (G12D) or following 72 hour doxycycline withdrawal (WT) prior to seeding on a Matrigel-coated filter and allowed to invade. Cells attached for 2 hours prior to drug treatment, and invaded for 14 hours. 10 μM Atglistatin (ATGLi) and 10 μM CAY 10499 (HSL/MGLi) were used. >220 cells scored in each of 3 independent biological replicates. I, mKPC cells transfected with siRNA targeting HSL and loaded with 200μM BSA-conjugated OA or BSA control prior to seeding on a Matrigel-coated filter and invading for 16 hours. >260 cells scored in each of 3 independent biological replicates. Graphs indicate mean ± SEM and analyzed by Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To determine the functional consequence of HSL downregulation in PDAC cells, we disrupted the KRAS-mediated suppression by overexpressing HSL in both mKPC and iKRAS cells (Fig. 4C and Supplementary Fig. 3C). Because HSL catabolizes intracellular LDs, we first examined the LD content of the cells following HSL overexpression. As predicted, overexpression of the lipase HSL dramatically depleted the stored lipids (Fig. 4D–E and Supplementary Fig. 3D and E). Thus, bypassing the downregulation of HSL in cells with oncogenic KRAS reduces the capacity to store lipids in LDs.
Because stored lipids promote invasive potential (Fig. 1D), and HSL overexpression depletes stored lipids, we hypothesized that HSL may affect invasion. Indeed, HSL overexpression dramatically reduced the invasive potential of both the mKPC cells and the iKRAS cells (Fig. 4F and G). While KRASG12D significantly increased invasion in vitro, this effect was completely blocked by re-expression of HSL. Thus, even in the presence of oncogenic KRAS, overexpression of HSL depleted stored lipids and reduced invasion to the level of cells expressing wild-type KRAS. If the reduction in invasive ability caused by HSL overexpression is due to depletion of stored lipids available for the invasive cell to utilize, then providing additional lipids to the cell should overcome this inhibitory effect. Indeed, when HSL overexpressing cells were pre-loaded with excess OA, the defect in invasive ability was rescued (Fig. 4 G). These data support the model that lipolysis of stored LDs promotes tumor cell invasion.
Though HSL is downregulated by oncogenic KRAS, its expression is not completely lost (Fig. 3A and C). Therefore, we tested if KRASG12D-expressing cells were still sensitive to lipase inhibition. Indeed, pharmacological inhibition of the lipases ATGL or HSL/MGL blocked invasion in iKRAS cells, and reduced invasion to the level of WT KRAS cells (Fig. 4H). Similar to pharmacological inhibition, knockdown of HSL also reduced tumor cell invasion (Fig. 4I and Supplementary Fig. 3F). Therefore, even though cells with oncogenic KRAS promote lipid storage via decreased HSL expression, residual lipase activity is still required to utilize stored lipids during invasion. In contrast to HSL overexpression, the invasive defect in the HSL knockdown cells could not be rescued by pre-loading with excess OA as there was not sufficient HSL to catabolize this excess lipid (Fig. 4I). It initially seemed paradoxical that both HSL overexpression and pharmacological inhibition similarly inhibited tumor cell invasion (Fig. 1F, 4F–I). However, the net result of both treatments is the same in the reduced ability to use stored LDs: pharmacological inhibition or knockdown of HSL blocks mobilization of stored lipids from LDs, while HSL overexpression depletes LD content, resulting in a lack of stored lipids for subsequent utilization during invasion, but can be rescued by introduction of additional lipids. This important finding further emphasizes that the lipolysis-based utilization of the stored lipids is necessary for tumor cell invasion.
Previous investigations have established that HSL is phosphorylated and activated by Protein Kinase A (PKA) to promote lipolysis in adipocytes and hepatocytes (35). In support of this concept, inhibition of PKA signaling with H89 led to an accumulation of LDs consistent with defective lipolysis, and decreased PDAC cell invasion (Supplementary Fig. 3G and H). While PKA is known to affect cell migration by multiple mechanisms, these findings are consistent with the concept that HSL expression and activity are important for tumor cell invasion.
Interestingly, although the KRAS activation state profoundly impacts the proliferation of the iKRAS cells (Supplementary Fig. 3I), HSL overexpression did not significantly change the proliferative capacity of either cell model (Supplementary Fig. 3J and K). These observations indicate that lipid storage and utilization by the lipase HSL is critical for regulating the invasive ability of PDAC cells.
These data demonstrate the importance of HSL expression and activity for metastatic potential in vitro. We next utilized HSL overexpression to disrupt KRAS-mediated downregulation of HSL in vivo. mKPC cells constitutively overexpressing HSL (Supplementary Fig. 4A) were orthotopically injected into the pancreas of syngeneic C57BL/6 mice, and tumor progression and metastasis were compared to mice injected with vector control cells. Similar to our in vitro observations (Fig. 4F and G), overexpression of HSL blunted tumor progression and metastasis in vivo (Fig. 5A and B). HSL overexpression dramatically reduced metastasis, as the total number of metastases was reduced when compared to mice injected with control cells (Fig. 5A and Supplementary Fig. 4B). Interestingly, although HSL overexpression did not significantly alter cellular proliferation in vitro (Supplementary Fig. 3K), the primary tumor weight was reduced in mice injected with HSL overexpressing cells compared to that of control cells (Fig. 5B), suggesting additional systemic factors related to tumor lipid metabolism can contribute to tumor growth. These results indicated that KRAS-mediated HSL regulation is crucial to maintain metastatic ability in vivo.
Figure 5.
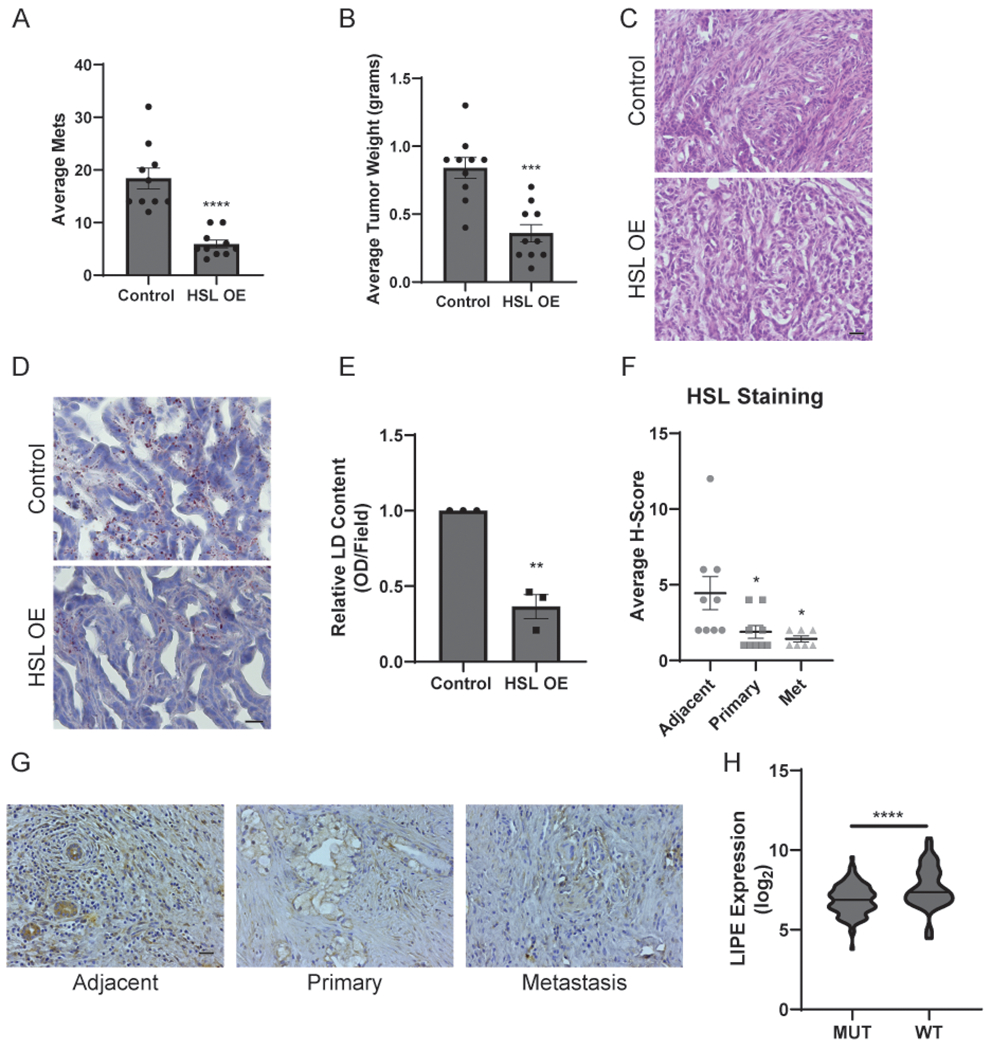
HSL regulates metastasis in vivo. A, The average total number of metastases from C57BL/6 mice following pancreatic orthotopic injections with mKPC cells constitutively overexpressing HSL or a vector control. n=10 mice per group. B, Average weights of the primary tumors isolated from the experiment described in (A). C, H&E staining of primary tumor sections isolated from the experiment described in (A). Scale bar, 20 μm. D, LD staining of primary tumor sections isolated from the experiment described in (A) using ORO and Hematoxylin as counterstain. Scale bar, 10 μm. E, Quantitation of histological sections stained for lipid droplets and depicted in (D). Data represent the relative change in the optical density (OD) of lipid content visualized by ORO staining in three distinct fields from 3 independent biological replicates. F, Immunohistochemistry analysis of patient-matched human pancreatic tissue sections stained for HSL. Data represent adjacent normal, primary tumor, and metastatic tissues isolated from each patient. G, Representative immunohistochemistry images of the tissues described in (F). Scale bar, 20 μm. Graphs indicate mean ± SEM and analyzed by Student’s t-test. H, Analysis of the TCGA-Pancreatic Cancer dataset comparing LIPE (HSL) gene expression in tumors with wild-type or mutant KRAS. Mean log2 expression values are indicated by a dashed line and significance was determined by a Mann-Whitney U test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
H&E staining revealed that both control and HSL-overexpressing tumors showed a similarly heterogeneous ductal and desmoplastic phenotype (Fig. 5C). Staining for intracellular LDs revealed reduced LD content in primary tumor tissue with HSL overexpression (Fig. 5D and E), similar to our in vitro observations and consistent with depletion of stored lipids (Fig. 4D–E). Staining for HSL confirmed increased expression in the primary and metastatic tissues of the mice injected with HSL overexpressing cells (Supplementary Fig. 4C). Ki67 staining indicated similar levels of proliferation in the primary and metastatic tumors of both experimental groups (Supplementary Fig. 4D). Finally, staining for α-smooth muscle actin revealed similar levels of activated fibroblasts in both experimental groups (Supplementary Fig. 4E). Thus, the primary difference resulting from HSL overexpression in the tumor cells is the depletion of stored lipids, and a reduction in tumor progression and metastasis.
In order to better understand the regulation of HSL in vivo, we assessed HSL protein levels in matched primary tumor, normal adjacent, and metastatic pancreatic tumor tissue from human pancreatic cancer patients. Quantitation and imaging of these tissues revealed downregulated HSL expression in both the primary and metastatic tissue when compared to benign (tumor-adjacent) tissue (Fig. 5F and G). Additionally, analysis of the TCGA-Pancreatic Cancer dataset revealed a significant inverse correlation between expression of the HSL gene (LIPE) and KRAS mutation (Fig. 5H). This is consistent with our previous observations that HSL expression is downregulated in tumor cells with oncogenic KRAS (Fig. 3A, C, E). Together, these results indicate that KRAS-dependent HSL expression is a crucial regulator of lipid storage, tumor progression, and metastasis in vivo.
HSL expression shifts PDAC cells to oxidative metabolism
Oncogenic KRAS controls metabolic reprogramming in cancer cells (19), including preferential utilization of glycolysis rather than oxidative respiration (21). Because of the profound impact of HSL in vitro and in vivo, we hypothesized that KRAS-mediated HSL regulation may play an uncharacterized role in PDAC metabolism. Since KRAS can shift cells towards glycolysis, we tested if the downregulation of HSL was a component of this metabolic shift by measuring the effects of HSL overexpression on both glycolysis and oxidative respiration. First, HSL overexpression in mKPC cells decreased glycolytic activity compared to control cells, as measured by lactate production (Fig. 6A). This suggests that HSL downregulation may contribute to the increased reliance on glycolysis. We next tested if HSL expression also impacts oxidative metabolism using Seahorse metabolic analysis. Interestingly, HSL overexpression resulted in increased basal and maximal respiration in both mKPC and doxycycline inducible iKRAS cell models (Fig. 6B and Supplementary Fig. 5A), suggesting that high HSL induces a metabolic shift towards oxidative respiration.
Figure 6.
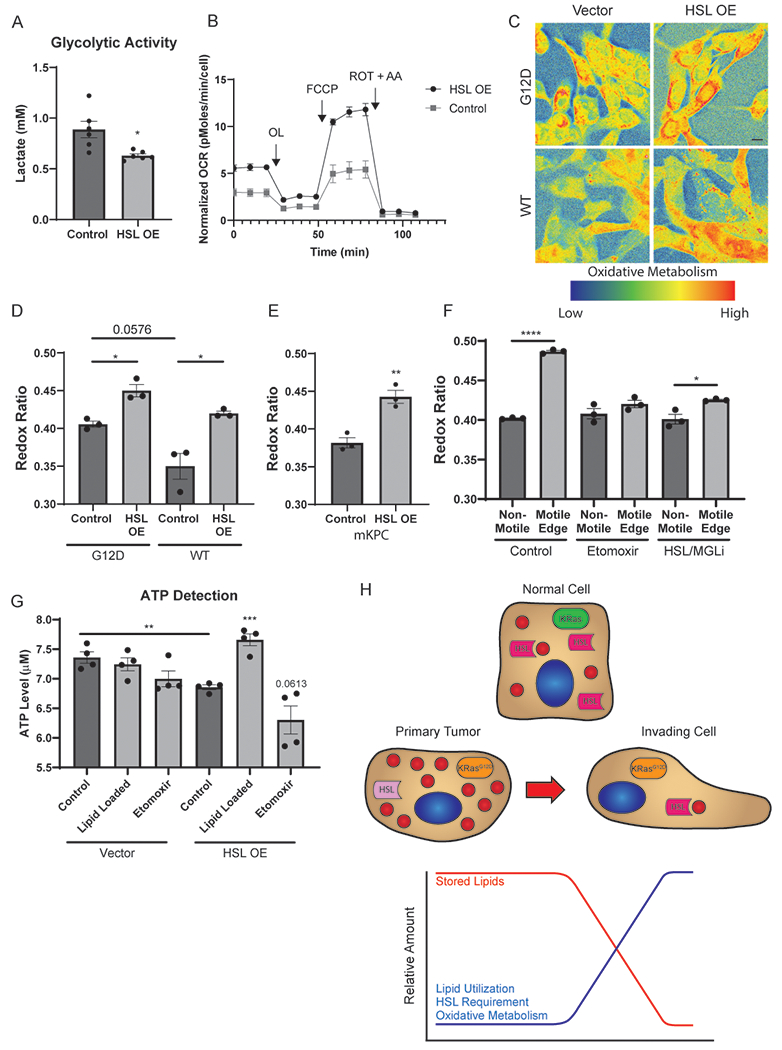
HSL shifts PDAC cells to oxidative metabolism. A, Analysis of glycolytic activity determined by lactate production of mKPC cells overexpressing HSL or a vector control. Data represent 6 independent biological replicates. B, Quantitation of the normalized oxygen consumption rate (OCR) of mKPC cells overexpressing HSL or a vector control via Seahorse metabolic analysis. Data represent 3 independent biological replicates. C, Representative composite images of the optical redox ratio of iKRAS cells overexpressing HSL or vector control. Cells were cultured in the presence of doxycycline (G12D) or following a 72 hour withdrawal of doxycycline (WT) prior to imaging. Scale bar, 10 μm. D, Optical redox ratio quantitation of the conditions depicted in (B). >40 cells scored in each of five fields for 3 independent biological replicates. E, Optical redox ratio quantitation of mKPC cells overexpressing HSL or vector control. >40 cells scored in each of five fields for 3 independent biological replicates. F, Optical redox ratio quantitation of mKPC cells in a wound healing assay. Cells were treated with DMSO (control), 10 μM Etomoxir, or CAY 10499 (HSL/MGLi) for 4 hours prior to imaging. Cells on the edge of the wound were classified as “Motile Edge” whereas cells at least 4 frames away from the edge were classified as “Non-Motile.” >35 cells scored in each of five fields for 3 independent biological replicates shown. G, mKPC cells treated with 200 μM OA, 10 μM Etomoxir, or grown under control conditions prior to lysis and use in an ATP Detection assay. H, Representation of lipid storage and catabolism during metastatic invasion. Graphs indicate mean ± SEM and analyzed by Student’s t-test. *p < 0.05, **p < 0.01, ***p<0.001, ****p < 0.0001.
To further analyze this phenomenon, we utilized the autofluorescence of flavin adenine dinucleotide (FAD) and nicotinamide adenine dinucleotide (NADH) to gain insights into the metabolic pathways being utilized by the cells (36). From the endogenous fluorescence of these two cofactors an optical redox ratio can be calculated as FAD/(NADH + FAD), with a higher optical redox ratio consistent with increased oxidative phosphorylation (37). As a proof of principle for this approach, we determined the impact of hypoxia on PDAC cells, which selectively blocks oxidative metabolism, resulting in enhanced utilization of glycolysis (38). This block in mitochondrial oxidative capacity would result in an increase of mitochondrial NADH due to lack of consumption by the electron transport chain, decreasing the optical redox ratio. Indeed, we observed a reduced optical redox ratio in hypoxic cells when compared to control cells (Supplementary Fig. 5B and C). Therefore, the optical redox ratio is a suitable method for the examination of PDAC cell metabolism.
Optical redox ratio analysis reveals that HSL overexpression in mKPC and iKRAS cells leads to an elevated redox ratio compared to control cells (Fig. 6C–E), consistent with the Seahorse based analysis and suggesting increased utilization of oxidative metabolism within PDAC cells. This is consistent with the concept that elevated HSL activity results in increased FA oxidation following lipolysis from stored LDs. Subsequently, increased oxidative phosphorylation by the electron transport chain results in production of FAD and consumption of NADH, leading to the observed elevation in the optical redox ratio. Thus, HSL overexpression shifts cells away from glycolysis and towards oxidative metabolism.
While metabolic analysis in populations of tumor cells highlights global metabolic changes, our data suggest that metabolism of stored lipids is crucially important during the acute process of tumor cell invasion. The mechanisms by which cancer cells transiently utilize metabolic resources during invasion and metastasis remain nearly completely undefined. To address this, we used the optical redox ratio to determine the metabolic state of tumor cells during migration using live cell imaging. Interestingly, leading-edge migratory cells had a significantly higher optical redox ratio than non-motile cells more than 5 cell widths within the cell mass (Fig. 6F and Supplementary Fig. 5D). Consistent with increased LD turnover in the leading-edge cells (Fig. 2A), these data indicate that tumor cells can catabolize and utilize stored lipids during the process of migration to transiently shift their metabolic program towards oxidative phosphorylation to fuel metastasis. We hypothesized that lipid utilization is critical for the shift towards oxidative metabolism in leading-edge migratory cells. To that end, we treated cells with pharmacological inhibitors to mitochondrial lipid uptake (Etomoxir) and HSL/MGL lipolytic activity, and imaged their redox ratio during migration. Both inhibition of mitochondrial lipid uptake and HSL/MGL ablated the utilization of oxidative metabolism in leading-edge cells (Fig. 6F and Supplementary Fig. 5D), indicating that lipolysis and lipid utilization is a critical component of the metabolic shift in highly dynamic, migratory cells. Thus, while KRAS-driven cancers may downregulate FA metabolism to rely on glycolysis for steady-state proliferation, a transient switch to oxidative metabolism occurs to support the use of stored lipids to enable invasion and metastasis.
A metabolic advantage of increased FA oxidation and oxidative phosphorylation could be increased generation of ATP, which is in high demand during tumor cell migration (8,39–41). We therefore measured ATP levels following HSL overexpression and in the presence of excess lipids. HSL overexpression, which depletes stored lipids and impairs invasive migration, also reduced ATP generation. Importantly, ATP production in the HSL overexpressing cells was significantly increased when cells were treated with excess lipid (Fig. 6G). These findings are consistent with Figure 4G, which showed that providing excess lipids replenished lipid stores and restored tumor cell invasion. Inhibition of mitochondrial lipid uptake with Etomoxir decreased ATP generation (Fig. 6G). Interestingly, ATP levels were not significantly altered by lipid loading in control conditions. Taken together, these results indicate that both lipids and HSL are required for augmented ATP generation.
Discussion
Altered cellular metabolism is a feature of cancer cells that enables unrestrained growth and motility. To date, the primary focus of metabolic rewiring has been on glycolysis (42), though recent studies have also implicated the importance of amino acids and lipids (14,43,44). Although metabolic reprogramming in cancer has been extensively studied, the majority of these investigations analyzed proliferating cells rather than metastatic cells (45,46). The focus of this study was to elucidate the resources metastatic cells utilize to enable invasive migration, and the mechanisms controlling the consumption of those resources. In PDAC, we observe that stored lipids can promote the pro-metastatic processes of migration, invasion, and matrix degradation. Lipid droplets are catabolized during invasive migration through the action of lipases, which allows mitochondrial import of FAs and upregulation of oxidative metabolism to promote metastatic migration (Fig. 6H). While oxidative metabolism is upregulated in actively migrating cells, it is important to note that overexpression of HSL did not increase tumor cell migration, even though it led to increased steady-state oxidative metabolism. Thus, elevated oxidative phosphorylation is not sufficient to drive invasive migration. Rather, we propose that tumor cell migration requires both the presence of stored LDs and a dynamic metabolic shift, allowing selective activation of oxidative phosphorylation coupled with LD breakdown in actively migrating cells.
Activating mutations of KRAS drive and maintain greater than 90% of pancreatic cancers (18,34,47). KRAS has been thought to promote a shift to aerobic glycolysis and anabolic glucose metabolism (48). However, our understanding of KRAS-driven metabolic reprogramming has evolved to include alterations in scavenging pathways, amino acid metabolism, and lipid metabolism (20,33,49). Here, we identified a novel mechanism by which KRAS modulates lipid metabolism through downregulation of HSL in both cell culture and in mice, consistent with decreased expression of HSL in human pancreatic cancers. We also report that oncogenic KRAS leads to increased protein levels of the lipid droplet protein PLIN-2, suggesting it may drive a larger program promoting lipid storage. Oncogenic KRAS led to decreased transcription of HSL through the MAPK/ERK signaling pathway, though the mechanism of this downregulation is unknown. It has been reported that ERK regulates lipolysis through phosphorylation of HSL (50); however, determining how this regulatory pathway controls HSL gene expression through transcriptional or epigenetic regulatory mechanisms requires further investigation. Importantly, this KRAS-HSL regulatory axis acts to promote lipid storage in PDAC cells for subsequent utilization during invasive migration. Re-expression of HSL depletes stored lipids, shifts cells to upregulated oxidative metabolism and decreased glycolysis, and reverses the invasive effects of oncogenic KRAS. It is likely that this shift is particularly important during the process of cell migration, when pancreatic tumor cells utilize lipid droplet turnover and increased oxidative metabolism. Understanding the metabolic requirements of metastatic cells remains extremely challenging due to the transient nature of invasive migration. Therefore, the optical redox ratio emerged as a powerful tool to analyze the relative metabolic pathways utilized by migratory and static cells. HSL, then, is a novel component of KRAS-mediated metabolic rewiring in pancreatic cancer.
Interestingly, genetic knockout of HSL was recently reported to increase pancreatic cancer incidence in mice (51). As KC;HSL−/− mice exhibited enhanced inflammation in the adipose tissue and pancreas, the prior study focused on the importance of HSL in the context of adipose tissue, whereas we investigated the tumor-cell specific effects of HSL. Interestingly, the authors reported enhanced survival in PDAC patients with higher expression of HSL based on transcript data from pancreatic tumor tissue. While this approach does not distinguish tumor cells from stromal adipocytes or islets, they are consistent with our findings that overexpression of HSL in pancreatic tumor cells decreased tumor burden and metastasis. Thus, both studies implicate HSL as an important regulator of pancreatic cancer progression, perhaps through multiple mechanisms.
Pancreatic cancer cells could access lipids from multiple sources. Obesity elevates the levels of available FAs, is commonly associated with pancreatic cancer, and worsens prognosis of patients and elevates metastatic burden (9,13,29). Further, mouse models of PDAC had earlier onset and more invasive pancreatic cancer when given an obesity-inducing diet (11,12,52,53). Cachexia is an additional comorbidity often present in advanced pancreatic cancer patients. This wasting condition promotes adipose breakdown, leading to elevated levels of FAs available for utilization by the tumor cells (15,54). The tumor microenvironment may also provide lipids for use by pancreatic cancer cells. Other cell types, such as adipocytes, cancer-associated fibroblasts, and activated stellate cells, may be in close proximity to tumor cells and facilitate tumor progression through elevated lipid availability (55,56). In these contexts, lipid uptake into PDAC cells would be required for excess lipids to impact metastasis, though the specific lipid transporters and mechanisms of uptake are still under investigation. Previous investigations in breast cancer have demonstrated that uptake of exogenous lipids promoted aggressive cellular phenotypes similar to our observations in pancreatic cancer cells (57). Therefore, import of FAs into PDAC cells represents a critical aspect upstream of our observations. Finally, PDAC cells have elevated levels of lipid synthesis (58), and can accumulate stored lipids in response to reactive oxygen species or hypoxia (59). Each of these factors may impact the availability of lipids to PDAC cells. Regardless of the source or uptake of the excess lipids, the data presented here support the hypothesis that lipid storage and utilization is a key mechanism that facilitates invasive migration of pancreatic cancer cells.
Our data indicate that coordinated breakdown of LDs is important for tumor cell migration and invasion. In other tissues, stimulation of lipolysis occurs following activation of PKA (35). Indeed, we observed changes in stored LDs and invasive migration following inhibition of PKA signaling, implicating the importance of PKA as an upstream signaling mechanism for the stimulation of lipid utilization and invasion. Further, the changes we observed in PLIN-2 levels following KRAS activation may also impact lipid utilization. While the specific stimulus to activate lipase-mediated breakdown during migration is unknown, our data support that stored lipids may be used for energy production to promote invasive migration, as inhibition of lipid transport into the mitochondria hinders migration. This is consistent with prior studies demonstrating increased oxidative phosphorylation and ATP demand in migratory cells (6–8) and that augmented ATP generation is critical to several aspects of invasive migration (8,40,41), though the source of this ATP is still not well defined. Interestingly, our data indicate that excess HSL levels and lipid are both required for augmented ATP production. This does not exclude the possibility that lipids may also be used for membrane dynamics and repair, or as signaling molecules (60). Additionally, KRAS activation, lipid metabolism, and mitochondrial respiration can all impact the levels of reactive oxygen species (61–63), which may contribute to tumor progression metastasis (64). While we observed lipolysis and HSL regulation to be critical to metastatic potential, other lipases and mechanisms of lipid utilization may also prove critical to metastasis. ATGL is thought to be the rate-limiting lipolytic enzyme (17), and MGL has been previously implicated in tumor aggressiveness (65). Indeed, pharmacological inhibition of all lipolytic enzymes perturbed tumor cell invasion, however the regulation of HSL by the oncogene KRAS may indicate its specific importance in the context of pancreatic cancer metastasis.
Taken together, the integrity of KRAS-mediated HSL regulation and the subsequent modulation of lipid storage, utilization, and metabolism reveal novel targets for suppression of metastasis and increased patient survival.
Supplementary Material
Significance.
KRAS-dependent regulation of HSL biases cells towards lipid storage for subsequent utilization during invasion of pancreatic cancer cells, representing a potential target for therapeutic intervention.
Acknowledgements:
This research was supported by the Mayo Clinic SPORE in Pancreatic Cancer (P50 CA102701), the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30 DK084567), the Mayo Clinic Cancer Center (P30 CA015083), the National Cancer Institute (R01 CA104125), the Hirshberg Foundation for Pancreatic Cancer Research, the Mayo Clinic Center for Biomedical Discovery, and the Mayo Clinic Graduate School of Biomedical Sciences (C. Rozeveld). We acknowledge Dr. Marina Pasca di Magliano for the inducible KRAS cell line (iKRAS), Dr. David Tuveson for the mouse KPC cell line (mKPC), and Dr. Kun Ling, Dr. Gregory Gores, Dr. Eugenia Trushina, Dr. Jason Doles, and Camden Daby for reagents, equipment, and technical support. We thank Mark McNiven, all members of the McNiven lab, and O. K. for technical support, helpful insight, and fortitude.
Footnotes
Competing Interests Statement: The authors declare no potential conflicts of interest.
References
- 1.Sleeman J, Steeg PS. Cancer metastasis as a therapeutic target. Eur J Cancer 2010;46:1177–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 2011;331:1559–64 [DOI] [PubMed] [Google Scholar]
- 3.Sahai E Illuminating the metastatic process. Nat Rev Cancer 2007;7:737–49 [DOI] [PubMed] [Google Scholar]
- 4.Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 2013;12:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elia I, Doglioni G, Fendt SM. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol 2018;28:673–84 [DOI] [PubMed] [Google Scholar]
- 6.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol 2014;16:992–1003, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin S, Huang C, Gunda V, Sun J, Chellappan SP, Li Z, et al. Fascin Controls Metastatic Colonization and Mitochondrial Oxidative Phosphorylation by Remodeling Mitochondrial Actin Filaments. Cell Rep 2019;28:2824–36 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunniff B, McKenzie AJ, Heintz NH, Howe AK. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell 2016;27:2662–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu M, Jung X, Hines OJ, Eibl G, Chen Y. Obesity and Pancreatic Cancer: Overview of Epidemiology and Potential Prevention by Weight Loss. Pancreas 2018;47:158–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eibl G, Cruz-Monserrate Z, Korc M, Petrov MS, Goodarzi MO, Fisher WE, et al. Diabetes Mellitus and Obesity as Risk Factors for Pancreatic Cancer. J Acad Nutr Diet 2018;118:555–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang HH, Moro A, Takakura K, Su HY, Mo A, Nakanishi M, et al. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS One 2017;12:e0184455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okumura T, Ohuchida K, Sada M, Abe T, Endo S, Koikawa K, et al. Extra-pancreatic invasion induces lipolytic and fibrotic changes in the adipose microenvironment, with released fatty acids enhancing the invasiveness of pancreatic cancer cells. Oncotarget 2017;8:18280–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Obesity Boden G. and free fatty acids. Endocrinol Metab Clin North Am 2008;37:635–46, viii–ix [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 2013;6:1353–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Das SK, Hoefler G. The role of triglyceride lipases in cancer associated cachexia. Trends Mol Med 2013;19:292–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol 2019;20:137–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lass A, Zimmermann R, Oberer M, Zechner R. Lipolysis - a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res 2011;50:14–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111:817–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016;531:110–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerr EM, Martins CP. Metabolic rewiring in mutant Kras lung cancer. FEBS J 2018;285:28–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012;149:656–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013;496:101–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM, et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun 2018;9:4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci 2014;39:91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Y, McNiven MA. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J Cell Biol 2012;196:375–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol 2019;218:3320–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schott MB, Rasineni K, Weller SG, Schulze RJ, Sletten AC, Casey CA, et al. beta-Adrenergic induction of lipolysis in hepatocytes is inhibited by ethanol exposure. J Biol Chem 2017;292:11815–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov 2016;6:852–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer 2017;16:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, et al. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010;39:425–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouyang H, Mou L-j, Luk C, Liu N, Karaskova J, Squire J, et al. Immortal Human Pancreatic Duct Epithelial Cell Lines with Near Normal Genotype and Phenotype. The American Journal of Pathology 2000;157:1623–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pupo E, Avanzato D, Middonti E, Bussolino F, Lanzetti L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Frontiers in Oncology 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012;122:639–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr 2007;27:79–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skala M, Ramanujam N. Multiphoton redox ratio imaging for metabolic monitoring in vivo. Methods Mol Biol 2010;594:155–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Z, Pouli D, Alonzo CA, Varone A, Karaliota S, Quinn KP, et al. Mapping metabolic changes by noninvasive, multiparametric, high-resolution imaging using endogenous contrast. Sci Adv 2018;4:eaap9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eales KL, Hollinshead KE, Tennant DA. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016;5:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber GF. Metabolism in cancer metastasis. Int J Cancer 2016;138:2061–6 [DOI] [PubMed] [Google Scholar]
- 40.Kelley LC, Chi Q, Caceres R, Hastie E, Schindler AJ, Jiang Y, et al. Adaptive F-Actin Polymerization and Localized ATP Production Drive Basement Membrane Invasion in the Absence of MMPs. Dev Cell 2019;48:313–28 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papalazarou V, Zhang T, Paul NR, Juin A, Cantini M, Maddocks ODK, et al. The creatine-phosphagen system is mechanoresponsive in pancreatic adenocarcinoma and fuels invasion and metastasis. Nat Metab 2020;2:62-+ [DOI] [PubMed] [Google Scholar]
- 42.Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol 2006;18:598–608 [DOI] [PubMed] [Google Scholar]
- 43.Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol 2018;20:782–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sullivan MR, Mattaini KR, Dennstedt EA, Nguyen AA, Sivanand S, Reilly MF, et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab 2019;29:1410–21 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012;491:364–73 [DOI] [PubMed] [Google Scholar]
- 46.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324:1029–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, et al. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One 2012;7:e49707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Racker E, Resnick RJ, Feldman R. Glycolysis and methylaminoisobutyrate uptake in rat-1 cells transfected with ras or myc oncogenes. Proc Natl Acad Sci U S A 1985;82:3535–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gouw AM, Eberlin LS, Margulis K, Sullivan DK, Toal GG, Tong L, et al. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci U S A 2017;114:4300–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenberg AS, Shen WJ, Muliro K, Patel S, Souza SC, Roth RA, et al. Stimulation of lipolysis and hormone-sensitive lipase via the extracellular signal-regulated kinase pathway. J Biol Chem 2001;276:45456–61 [DOI] [PubMed] [Google Scholar]
- 51.Xu M, Chang HH, Jung X, Moro A, Chou CEN, King J, et al. Deficiency in hormone-sensitive lipase accelerates the development of pancreatic cancer in conditional KrasG12D mice. BMC Cancer 2018;18:797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dawson DW, Hertzer K, Moro A, Donald G, Chang HH, Go VL, et al. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev Res (Phila) 2013;6:1064–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu M, Liu H, Duan Y, Zhang D, Li S, Wang F. Four types of fatty acids exert differential impact on pancreatic cancer growth. Cancer Lett 2015;360:187–94 [DOI] [PubMed] [Google Scholar]
- 54.Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016;5:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov 2019;9:617–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanford-Crane H, Abrego J, Sherman MH. Fibroblasts as Modulators of Local and Systemic Cancer Metabolism. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Antalis CJ, Uchida A, Buhman KK, Siddiqui RA. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin Exp Metastasis 2011;28:733–41 [DOI] [PubMed] [Google Scholar]
- 58.Sunami Y, Rebelo A, Kleeff J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers (Basel) 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, Saarinen AM, Hitosugi T, Wang Z, Wang L, Ho TH, et al. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. Elife 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 2008;9:112–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li LO, Klett EL, Coleman RA. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2010;1801:246–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Storz P KRas, ROS and the initiation of pancreatic cancer. Small GTPases 2017;8:38–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences 2010;107:8788–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nishikawa M Reactive oxygen species in tumor metastasis. Cancer Lett 2008;266:53–9 [DOI] [PubMed] [Google Scholar]
- 65.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010;140:49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.