

Abstract
We developed a novel treatment strategy for metastatic cancer by synergizing photothermal therapy (PTT), chemotherapy, and immunotherapy through a nanosystem to trigger host antitumor immunity. The nanosystem was constructed by loading mitoxantrone (MTX), a chemotherapeutic agent, and SB-431542 (SB), a transforming growth factor beta (TGF-β) inhibitor, onto reduced graphene oxide (rGO). Intratumoral administration of rGO/MTX/SB, followed by non-invasive irradiation of a near-infrared laser, destroyed local primary tumors and inhibited distant metastases in 4T1 mouse mammary tumor model, which is poorly immunogenic and highly metastatic. After treatment, 70% of the tumor-bearing mice became long-term survivors and developed a tumor type-specific immunity to resist rechallenged tumor cells. We found that rGO-based PTT provided an immunogenic antigen source, forming in situ vaccination with rGO as an immune-adjuvant. The use of SB changed the tumor microenvironment and improved the therapeutic effect of MTX-generated chemotherapy and rGO-based PTT. The immunological functions of MTX, SB, and rGO acted synergistically to induce an effective tumor vaccination, as evidenced by the increased infiltration of tumor-specific cytotoxic CD8+ T lymphocytes and decreased infiltration of regulatory T cells (Tregs) in distal tumors. Collectively, we demonstrated that rGO/MTX/SB combined with laser irradiation provided a synergistic chemo-immuno-photothermal effect against tumors by in situ vaccination and inhibition of immunosuppressive microenvironment. This unique combination embodies a promising approach to treat metastatic cancers by inducing a systemic antitumor response through a local intervention.
Keywords: metastatic cancers, chemotherapy, photothermal therapy, immunotherapy, graphene
1. Introduction
Triple-negative breast cancer (TNBC) accounts for 10-20% of diagnosed breast cancers, with limited options and poor prognosis [1–3]. More importantly, compared to hormone-positive breast cancer, TNBC is common in women younger than 40 years [4]. The cure rate of cancer has gradually increased in the past decades with the development of new cancer therapies, including target molecules, immune checkpoint inhibitors, vaccines, and CAR-T. However, the cure for TNBC is still elusive [5–8]. For the unsatisfactory outcomes in treating TNBC, the major reason is the poor tumor immunogenicity and the strong immunosuppressive tumor microenvironment [9, 10].
Chemotherapy has been one of the primary clinical modalities. However, the immunosuppressive effects of chemotherapeutic agents have been well known. As a few exceptions, doxorubicin (Dox) and mitoxantrone (MTX) induce topoisomerase II associated DNA damage in the cell nucleus, causing immunogenic cell death (ICD) that could trigger a tumor-specific immunity by releasing damage-associated molecular patterns (DAMPs) and tumor antigens [11–14]. However, drug resistance is still the big challenge in chemotherapy, resulting in recurrence and drug-resistant metastases in the treated TNBC patients [15].
TGF-β is a pleiotropic cytokine that maintains an immunosuppressive environment to help established tumor escape from the host immune system [16]. Many tumors overexpression of TGF-β correlate with metastasis and poor prognosis. Therefore, TGF-β was considered as an ideal target and TGF-β inhibitors were developed for the treatment of cancer [17–18]. One of the TGF-β inhibitors, SB-431542, was found to decrease lung metastasis, however, there was no effect on the growth of the primary tumor in 4T1 xenograft tumor model [17]. LY2157299 prevented reestablishment of tumors after paclitaxel treatment of 4T1 xenograft [18]. Either chemotherapy agents or TGF-β inhibitors face severe limitations in treating TNBC due to delivery challenges and toxicity profiles.
Nano-graphene oxide (nGO) has been widely used as a drug carrier and photo-agent for cancer theranostics, due to its principal characteristics of 2D film structure and near-infrared absorption spectrum. Especially, PEG-nGO was found to activate macrophages with a potential to enhance the antitumor immunity [19]. We hypothesized that a combination of an in situ tumor vaccine with improved tumor microenvironment, should lead to an effective antitumor immunity. In addition, local application of chemotherapeutic and immunotherapeutic agents could be effective against tumors while reducing the systemic side effect.
Herein, we developed a graphene-based nano-system by conjugating rGO with MTX and SB for photo-activated, synergistic chemo-immunotherapy to treat metastatic TNBC using 4T1 mouse breast tumor model. The therapeutic efficacy of photo-activated chemo-immunotherapy was investigated. The host immunity, including induced ICD, antitumor vaccination, tumor-specific T cells, and the abscopal effect, were also investigated.
2. Experimental
2.1. Synthesis and characterization of rGO/MTX/SB
The base of the nanoplatform, rGO, was conjugated with mPEG2000-NH2 following a well-established EDC/NHS protocol and purified by a 100 kDa filter through centrifugation. MTX and SB were then loaded onto rGO-PEG via π-π stacking interaction. The detailed protocol and analysis were shown in Supplemental Methods.
2.2. Cell culture and cellular treatment
4T1 murine breast cancer cell line and CT26 colon carcinoma cell line were both purchased from ATCC (Manassas, VA, EISA) , bone marrow macrophages and DCs were harvested from BALB/c mice, all the cells were cultured following standard protocol. After the co-culturation with different nano-materials, tumor cells were exposed to light (0.75 W/cm2 @5 min) that delivered using an optical fiber with a diffusion micro-lens, from an 805-nm semiconductor laser (AngioDynamics, Inc, NY, USA). Treated cells were harvested for cell death assay, ELISA assay, or flow cytometry assay.
2.3. Animal models and in vivo treatment
Female BALB/c mice in age of 6-8 weeks were purchased from Harlan Sprague Dawley Co. (Indianapolis, IN, USA). The feeding and treatment of the mice were conducted in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH) and approved by the University of Oklahoma Health Sciences Center (OUHSC) Institutional Animal Care and Use Committee (IACUC).
For the mouse orthotopic tumor model, 4T1 cells (1×105 cells) suspended in 50 μl PBS were injected into the breast pad of mice. For study of abscopal effect, the first tumor was established by subcutaneously injecting 1×105cells into the right hind thigh of the mouse, and the second tumor was established by subcutaneously injecting 5x104 cells into the left hind thigh of the same mouse 3 days later. For the study of anti-tumor immunity, cured mice were rechallenged by injecting 4T1 (2×105) or CT26 (2x105) cells into the flank of the mice. Tumor volume was estimated by length (mm)×width (mm)2/2, and ended till 2000 mm3.
For the treatment, mice were randomly divided into different groups: PBS control, rGO, rGO/MTX, and rGO/MTX/SB, with or without laser treatment. Tumor-bearing mice under anesthetization (4% isoflurane / 96% oxygen mixture) were injected by intratumorally with the solutions of nanosystem, followed by PTT (0.75 W/cm2 @5 min) treatment 2h later. The temperature increase during treatment was detected by an infrared thermal camera (T540sc, FLIR, Boston, MA, USA). Tumor tissue, spleens, and blood were harvested for further analysis. The detailed protocol and analysis were shown in Supplemental Methods.
2.4. Statistical analysis
Values are expressed as mean ± SEM. All of the data were analyzed with a 1-way ANOVA followed by Bonferroni posttest. A p value of <0.05 was considered statistically significant.
3. Result
3.1. Characterization of rGO/MTX/SB
The biocompatibility of rGO was improved by conjugating with mPEG2000-NH2, followed by MTX and SB loading via π-π stacking (Figs. S1–5). A UV-vis absorption spectrum of rGO/MTX/SB showed the characteristic absorption peaks of MTX and SB, indicating successful loading of MTX and SB, with a final size of 100~200 nm (Figs. 2A–B). The temperature increase of rGO/MTX/SB solutions detected under laser irradiation with an 805-nm laser indicated that rGO/MTX/SB kept the photothermal transfer effects and the stability of rGO under laser irradiation (Figs. S6).
Fig. 2.
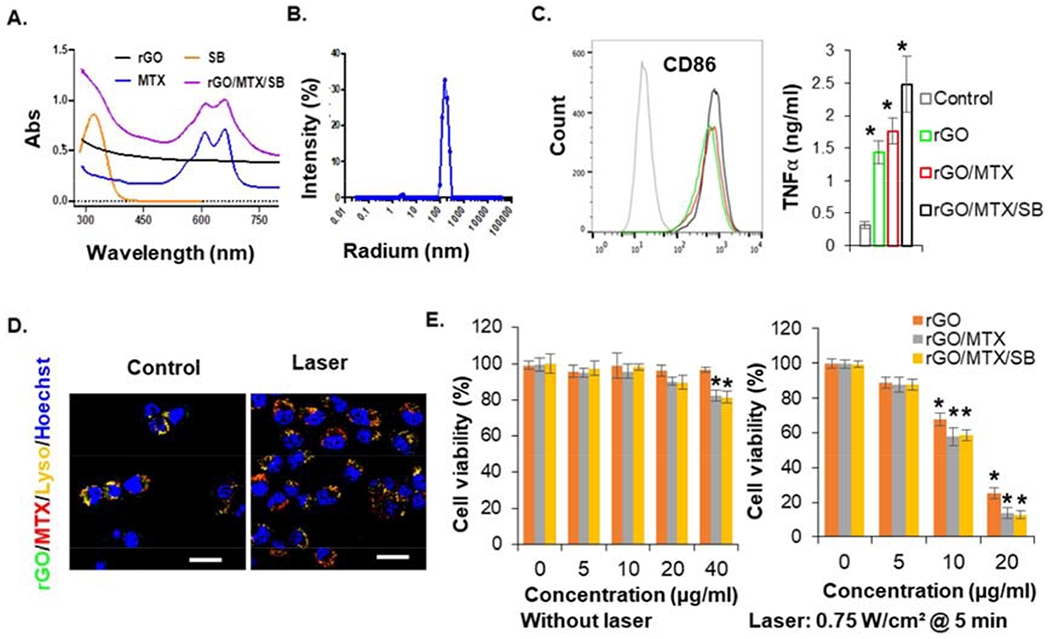
Characteristics of functionalized nanographenes. A. UV-vis-NIR spectra of different components, indicating the successful loading of MTX and SB onto rGO. B. Particle size analysis of rGO/MTX/SB. C. The expression of CD86 on BMDCs and secretion of TNFα from BMDCs, after the stimulation by different rGO derivatives, indicating the immune stimulation ability of rGO (n = 4, *p < 0.05). D. Cellular localization of FITC-rGO/MTX before and after laser irradiation. Bar=20 μm. E. Cytotoxic effects of different rGO derivatives on 4T1 cells at different doses, without or with irradiation of an 805-nm laser (0.75 W/cm2 for 5 min). Viability of tumor cells were analyzed by MTS assay 48 h after the treatments of rGO derivatives only, or 12 h after laser treatment with rGO derivatives, (n = 6, *P < 0.05).
It was found that rGO could stimulate the activation of RAW264.7 macrophages by producing high level TNFα [19]. We further confirmed the immunological effects of rGO by detecting high levels of IL6 and TNFα secretion from mouse primary macrophages treated with rGO (Fig. S7). As the important antigen-presenting cells (APCs), dendritic cells (DCs) initiate and regulate the innate and adaptive immunities. Therefore, we further determined the immunological effects of rGO on bone marrow-derived dendritic cells (BMDCs) harvested from BALB/c mice by analyzing the upregulation of CD86 with flow cytometer and detecting the secretion of TNFα by ELISA. As shown in Fig. 2C, rGO successfully stimulated TNFα secretion and CD86 up-expression of BMDCs.
To investigate whether rGO derivatives can enter into tumor cells, rGO was functioned with PEG-FITC (Fig. S8). 4T1 cells were incubated with FITC-tagged rGO derivatives and analyzed by flow cytometry. All the rGO derivatives were detected in 4T1 cells (Fig. S9). In addition, the cellular localization of MTX, rGO, rGO/MTX were analyzed by staining lysosome and nucleus. As shown in Fig. S10, MTX localized in lysosome and nucleus, rGO localized in lysosome. When conjugated with rGO, MTX mainly localized in lysosome with rGO, but released from lysosome after laser irradiation (Fig. S10 and Fig. 2D).
Viability of tumor cells treated by rGO derivatives for 48 h was assessed with MTT assay. There were no obvious cytotoxicity in all treated groups, except for a weak toxicity on 4T1 cells in the groups of rGO/MTX and rGO/MTX/SB with a high concentration (40 μg/ml), due to the cytotoxicity of MTX (the chemotherapeutic drug (Fig. 2E). To determine the photothermal effects, cells were incubated with rGO derivatives for 12 h and irradiated by an 805-nm laser (0.75 W/cm2 @ 5 min). Cell viability was detected 12 h after laser treatment and showed a significant reduction with concentrations of rGO above 10 μg/ml, to 20% at 20 μg/ml. However, at both 10 and 20 μg/ml, cell viability in the groups of rGO/MTX and rGO/MTX/SB were significant lower than that in the group of rGO. These results showed that rGO could act as both a photothermal agent and an immunostimulatory agent. On the other hand, as a drug carrier, rGO could increase tumor cell uptake of MTX by lysosome localization, to further enhance cell death by photo-activated release from lysosome.
3.2. Therapeutic efficacy of rGO/MTX/SB-based PTT on metastatic breast tumor
The highly tumorigenic and poorly immunogenic 4T1 orthotopic breast tumor model in mice was used to evaluate the efficacy of rGO/MTX/SB-based PTT. When the tumor size reached approximately 100 mm3, tumors were intratumorally injected with different rGO derivatives and locally irradiated by the laser (0.75 W/cm2 @5 min). In comparison, rGO or rGO/SB-treated mice had comparable tumor sizes, while rGO/MTX or rGO/MTX/SB-treated mice had reduced tumor sizes due to the cytotoxic effects of MTX (Figs. 3A–B, S11–12). When combined with laser treatment, all the primary tumors obviously shrank within 10 days in all of the groups of rGO derivatives (Fig. 3C). However, Laser + rGO led to a fast tumor recurrence within 20 days after treatment. Compared with that of the Laser + rGO group, the recurrence was delayed in the Laser + rGO/SB group. Notably, the tumor recurrences were significantly inhibited in both the Laser + rGO/MTX and Laser + rGO/MTX/SB groups (Figs. S11 and S13). All of the mice in the groups treated with PBS, rGO alone, rGO/MTX alone, or rGO/MTX/SB alone died within 21, 22, 25 and 29 days, respectively (Fig. 3D). When combined with laser treatment, administration of rGO and rGO/MTX significantly prolonged the mice survival time (Fig. 3E). Remarkably, 7 of 10 mice in the Laser+rGO/MTX/SB group survived and remained tumor free (Fig. 3E).
Fig. 3.
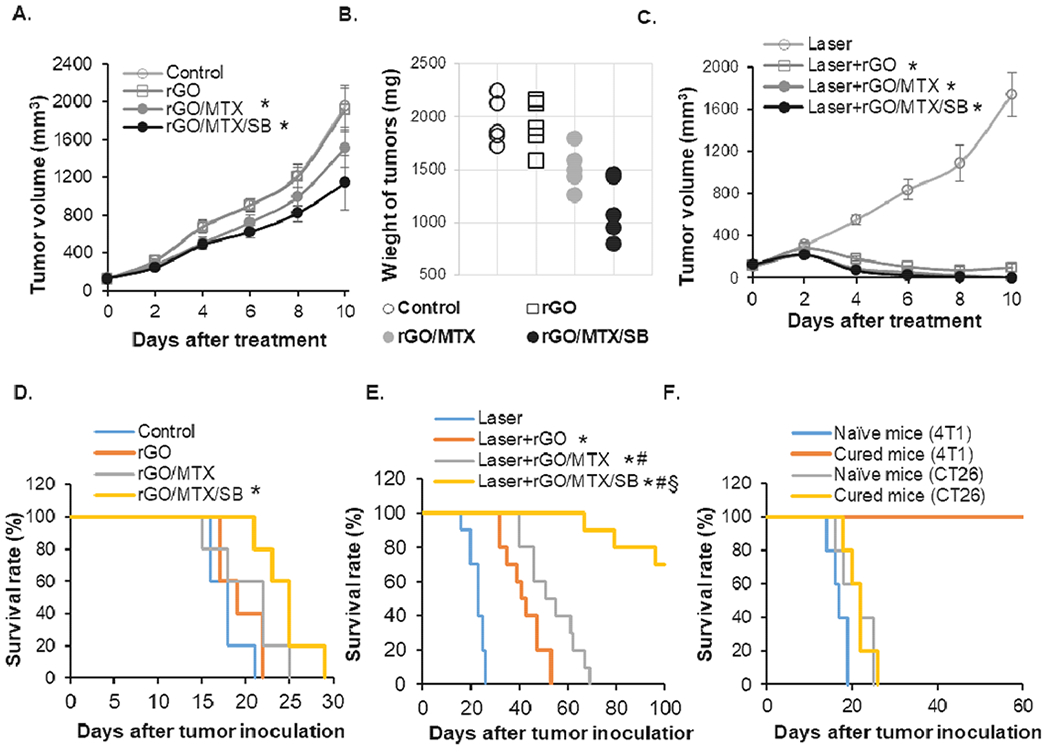
Therapeutic efficacy of different derivatives on 4T1 orthotopic tumors. 4T1 cells (5 × 104) were injected into the breast pad of female BLAB/c mice. Tumors, when reached a size of 100 mm3, were treated by an 805-nm laser (0.75 W/cm2 for 5 min), following the intratumoral injection of different rGO derivatives (50 mg/kg). A. Tumor growth curves of different groups following the treatments (n = 5, *P < 0.05). B. Weight of tumors harvested 10 days after treatments (n = 5, *P < 0.05). C. Tumor growth curves of different groups following the treatments (n = 5, *P < 0.05). D. Survival rates of tumor-bearing mice in the indicated treatment groups (without laser irradiation) up to 30 days after the initial tumor inoculation, (n = 5, *P < 0.05). E. Survival rates of tumor-bearing mice in the indicated treatment groups (with laser irradiation) up to 100 days after the initial tumor inoculation, (n = 10, *p < 0.001 vs Laser; #p < 0.05 vs Laser + rGO; §p<0.005 vs Laser + rGO/MTX). F. Survival rates of rechallenged mice, successfully treated by rGO/MTX/SB based PTT. (n = 5). 4T1 tumor-bearing mice ‘cured’ by rGO/MTX/SB based PTT were rechallenged with 2 × 10 · viable 4T1 or CT26 tumor cells 100 days after the initial challenge. Naive mice of the same age were used as controls.
To evaluate the long-term specific antitumor effects induced by Laser+rGO/MTX/SB, the cured tumor-bearing mice were rechallenged with 4T1 or CT26 tumor cells. As shown in Fig. 3F, all cured mice were resistant to 4T1 tumor rechallenge but not CT26 tumor rechallenge. These results suggested that rGO/MTX/SB based PTT not only prolonged the survival time, but also triggered a tumor-specific immune memory.
3.3. Immunogenic cell death (ICD) induced by rGO/MTX/SB-based PTT
To determine the synergistic reactions of rGO/MTX/SB-based PTT, cellular response were detected. Combined with laser irradiation, rGO treatment induced 86% cell death with 78% late apoptosis; however, rGO/MTX treatment increased the cell death rate to 95% with 88% late apoptosis (Fig. 4A), which showed the enhancement of MTX on the tumor killing effect.
Fig. 4.
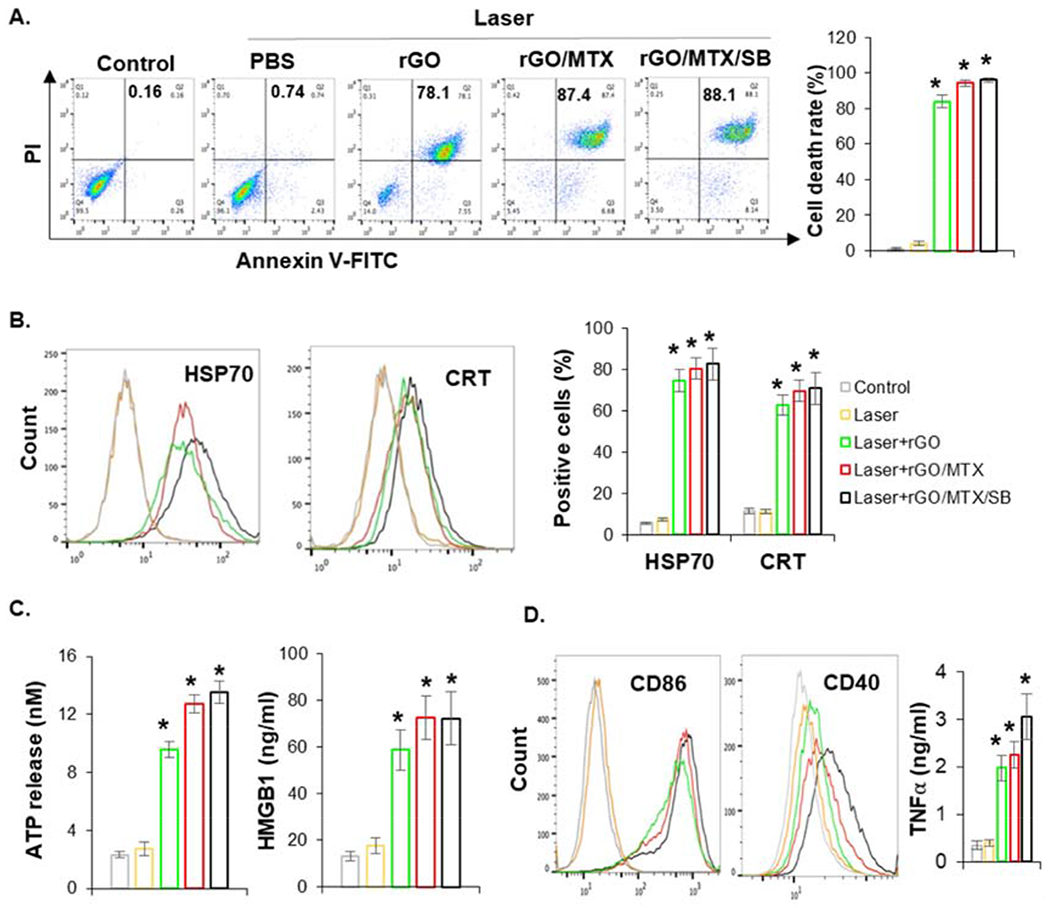
Immunogenic tumor cell death induced by rGO/MTX/SB based PTT. A. Cell death analysis by flow cytometry 12 h after the treatments, with Annexin V/PI staining. Bar graph depicts % of cell death, (n = 4, *P < 0.05). B. Surface expression of HSP70 and CRT on tumor cells determined by flow cytometer 3 h after treatments. Bar graph depicts % of cell with HSP70+ and CRT+ staining, (n = 4, *p < 0.05). C. Extracellular ATP and HMGB1 released from tumor cells detected 12 h after the treatments, (n = 4, *P < 0.05). D. The expression of CD86 and CD40 on the surface of BMDCs and secretion of TNFα from BMDCs, stimulated by treated tumor cells. BMDCs were co-cultured with treated tumor cells for 24 h, supernatants were collected and measured by ELISA, cells were collected and stained with PE-CD86/PacBlue-CD40 for analysis with flow cytometer.
Heat shock proteins (HSPs), calreticulin (CRT), high-mobility group box 1 (HMGB1) protein, and adenosine triphosphate (ATP) are DAMPs that reveal ICD. The expression and release of DAMPs induced by rGO and its derivatives were assessed by flow cytometry or ELISA. Combined with laser treatment, all the rGO derivatives remarkably increased the expression of HSP70 and CRT on the surface of treated tumor cells (Fig. 4B), increased the extracellular release of HMGB1 and ATP from treated tumor cells (Fig. 4C). To further confirm the immunological potential of treated tumor cells, the activation of DCs were evaluated by CD86/40 expression and TNFα production. As shown in Fig. 4D, rGO derivatives-PTT treated tumor cells stimulated TNFα secretion of DCs and up-expression of CD86/40 on the surface of DCs. To further confirm whether rGO can be used as an adjuvant to promote cancer vaccination, mice were vaccinated with heat shock treated cells. The specific host antitumor immunity was analyzed 7 days later by detecting the response of splenocytes. As shown in Fig. S14, rGO can enhance the cytotoxic effect of splenocytes and increased the IFN γ production of splenocytes, which indicated the specific host antitumor effects. Overall, rGO/MTX/SB-based PTT not only damaged the tumor cells, but also enhanced the expression and release of DAMPs, finally triggering immunogenic cell death that can stimulate the maturation of DCs. Moreover, rGO acted as an adjuvant to promote immunological response.
3.4. Effects of rGO/MTX/SB-based PTT on treated primary tumors
As monitored by an infrared thermal camera under laser irradiation, the surface temperature of the tumor injected with rGO derivatives increased to 58°C (Fig. 5A), which can cause tumor cell death and a local immune response.
Fig. 5.
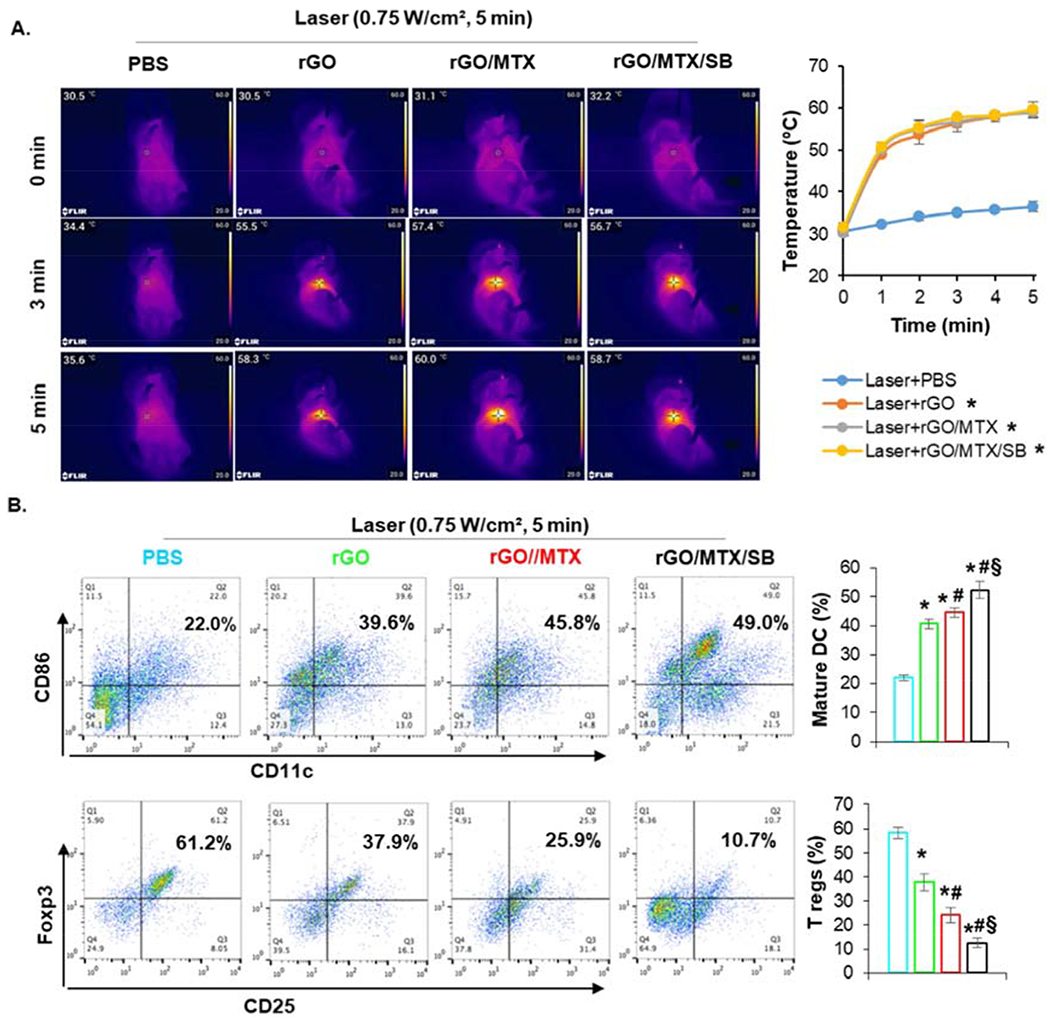
Effects of rGO/MTX/SB based PTT on treated primary tumors. A. Temperature increase in tumor tissue under different treatments. Left: IR thermal images of 4T1 tumor bearing mice treated with different rGO derivatives under an 805-nm laser irradiation (0.75 W/cm2 for 5 min). Right: Temperature increase on the surface of tumor tissue during laser irradiation. B. Infiltration of matured DCs and Tregs in treated tumor tissue. Dissociated single cells from tumors 24 h after treatments were stained with anti-CD11c, -CD86, -CD25, and -Foxp3 mAbs, analyzed by flow cytometry. Dot plots show CD86+/CD11c+ (marker of matured DCs) and Foxp3+CD25+ (marker of Tregs) cells, while bar graph shows % CD86+ CD11c+ and Foxp3+CD25+ cells for each treatment group, (n = 4, *p<0.001 vs Laser; #p < 0.05 vs Laser + rGO; §p < 0.005 vs Laser + rGO/MTX).
To analyze the infiltration and activation of DCs into treated primary tumors, single cells isolated from tumor tissue were stained with CD11c/CD86 for assessment by flow cytometry one day after treatments. Compared to laser-only treatment, all the rGO derivatives increased the infiltration of DCs (marked by CD11c) into treated tumors and enhanced the maturation of infiltrated DCs (marked by CD86) (Fig. 5B). Moreover, DC maturation in the group of rGO/MTX (~45.8%) and rGO/MTX/SB-based PTT (~49%) were much higher than that in the group of rGO based PTT (~39.6%) (Fig. 5B). To further analyze the impact of different treatments on the tumor microenvironment, cells from treated tumor were stained with CD3/CD4/CD25/Foxp3 for assessment by flow cytometry. About 61.2% of CD4 T cells were regulatory T cells (Tregs) in the laser-treated tumor; however, the percentage of Tregs was decreased in the tumors treated by rGO-based PTT (~37.9%) and further decreased by the treatment of rGO/MTX-based PTT (~25.9%) and rGO/MTX/SB-based PTT(~10.7%). These results demonstrated that rGO and its derivatives, particularly rGO/MTX/SB, enhanced the PTT treatment through infiltration and maturation of DCs in the treated primary tumors. In addition, the treatments also improved the tumor microenvironment by suppressing Tregs to further enhance the antitumor immune response.
3.5. Systemic immune response induced by rGO/MTX/SB-based PTT
To evaluate the antitumor immunity induced by rGO/MTX/SB-based PTT, cytokine production in the serum from treated mice was analyzed by ELISA. IFNγ and IL-12 levels in mice treated by all rGO derivatives-based PTT were higher than that treated by laser only (Fig. 6A). To determine the systemic antitumor immunity, the effector T cells (CD8+IFNγ+) in treated mice were analyzed by flow cytometry. Cells harvested from spleen (SPL) and draining lymph node (DLN) were co-stained with CD3/CD8/IFNγ. The number of IFNγ-producing CD8+T cells was increased in both SPL and DLN of the mice treated by rGO- and rGO/MTX-based PTT, and greatly increased by the treatment of rGO/MTX/SB-based PTT (Fig. 6B–C). Ten days after the treatment, lung tissues were harvested from treated mice and the metastases were assessed by H&E staining. As shown in Fig. 6D, there were numerous lung metastatic nodules in the mice treated by laser only, while in the mice treated by rGO- and rGO/MTX-based PTT, the lung tumor nodules were significantly reduced. In addition, there was almost no lung metastasis in the mice treated by rGO/MTX/SB-based PTT (Fig. 6D). These results suggested that rGO-based PTT could induce effective tumor-specific CTL that can be enhanced by MTX/SB.
Fig. 6.
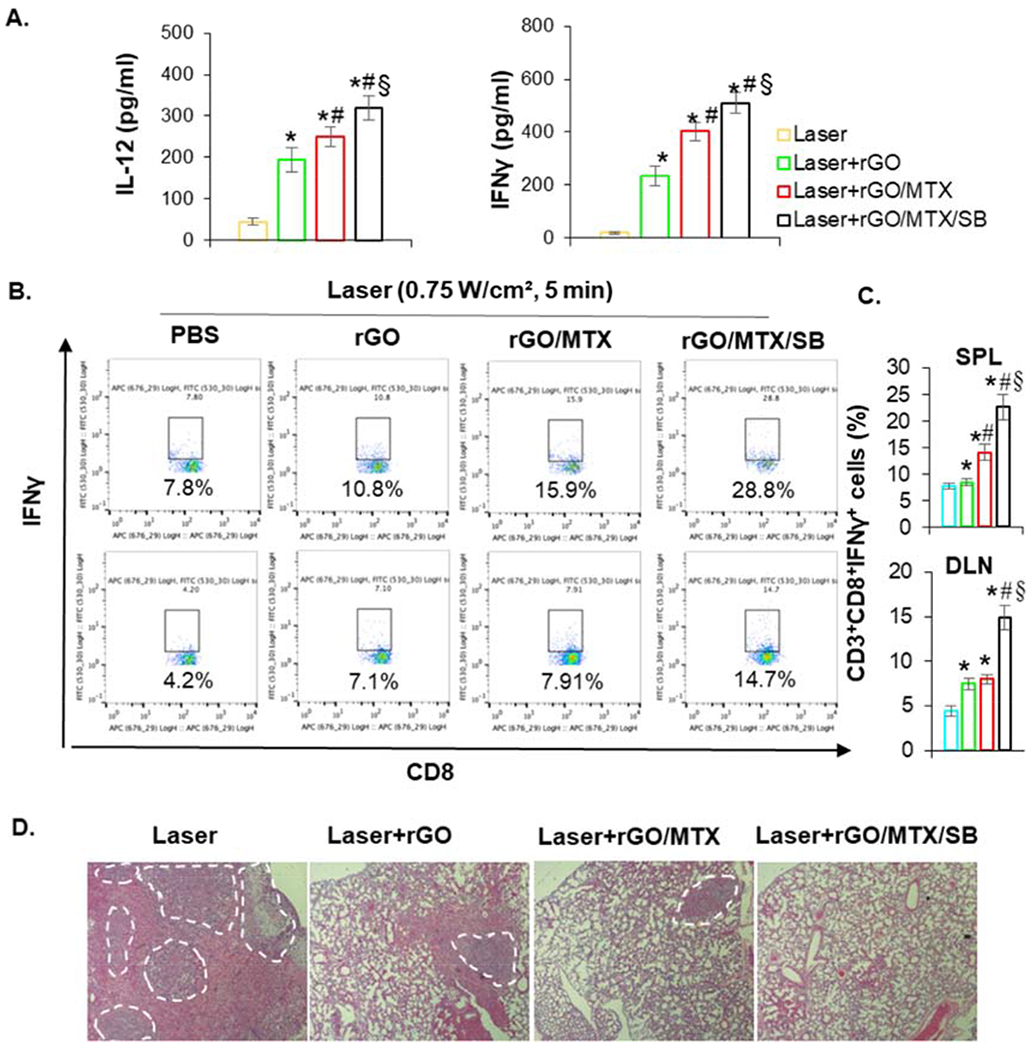
Systemic immune response induced by rGO/MTX/SB based PTT. A. IL-12 and IFN-γ levels in serum from mice isolated at 72 h post different treatments, (n = 4, *p < 0.001 vs Laser; #p < 0.05 vs Laser + rGO; §p < 0.01 vs Laser + rGO/MLX). B. Representative flow cytometry plots showing different groups of CD8+ IFN-γ+ cells in spleen (SPL) and draining lymph nodes (DLN) 7 days after the treatments. C. The percentage of effector L cells (CD8+IFN-γ+, gated on CD8+ cells) isolated from spleen (SPL) and draining lymph node (DLN) of treated mice 7 days after the treatments (n = 4). D. Histology of excised lungs 10 days after different treatments. Marked areas indicate the metastases.
3.6. Abscopal effect of rGO/MTX/SB-based PTT
We investigated whether the antitumor effects triggered by rGO/MTX/SB-based PTT can affect existing metastases with a bilateral tumor model (Fig. S15). The right tumors were designated as primary tumors for local treatment. The left secondary tumors, inoculated 3 days after that of the right tumors, served as surrogates of metastases without treatment. Compared to laser alone, both rGO- and rGO/MTX-based PTT on the primary tumors slowed the growth of the secondary tumors. However, after the treatment of rGO/MTX/SB-based PTT on the primary tumors, the growth of the secondary tumors was significantly inhibited (Figs. 7A–B).
Fig. 7.
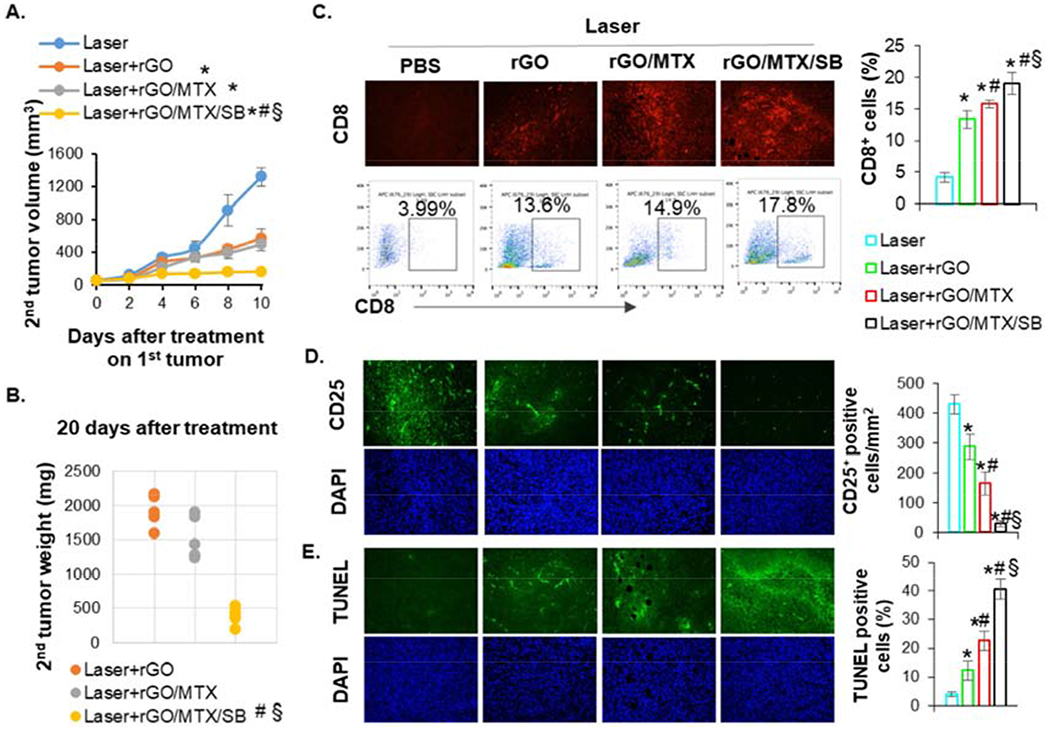
Abscopal effect induced by rGO/MTX/SB based PTT. A. Volume of secondary untreated tumors after indicated treatments on the primary tumors in mice that inoculated with two tumors, (n = 5, *p < 0.001 vs Laser; #p <0.001 vs Laser + rGO; §p < 0.001 vs Laser + rGO/MLX). B. Weight of secondary untreated tumors harvested 20 days after treatments of primary tumors, (n = 5, #p < 0.001 vs Laser + rGO; §p < 0.001 vs Laser + rGO/MLX). C. Representative IHC staining and flow cytometry plots showing CD8+ T cells in secondary tumors. Bar graph depicts the proportions of tumor-infiltrating CD8+ T cells (gated by CD3+) in secondary tumor, (n = 4, *p < 0.001 vs Laser; #p < 0.05 vs Laser+rGO; §p < 0.05 vs Laser + rGO/MLX). D. Representative IHC staining showing CD25+ cells in secondary tumors. Bar graph depicts the numbers of tumor-infiltrating CD25+ cells in secondary tumor. (n = 4, *p < 0.01 vs Laser; #p < 0.01 vs Laser+rGO; §p<0.001 vs Laser + rGO/MLX). E. Representative LUNEL staining showing cytotoxic effects in secondary tumors, (n = 4, *p < 0.005 vs Laser; #p < 0.005 vs Laser + rGO; §p <0.001 vs Laser + rGO/MLX).
To confirm the systemic antitumor effects triggered by rGO/MTX/SB-based PTT, the infiltration of immune cells in the secondary tumors were analyzed by immunofluorescence assay and flow cytometry. As shown in Figs. 7C–D, compared to laser alone, both rGO- and rGO/MTX-based PTT increased the infiltration of CTL (CD8+) and reduced the percentage of Tregs (CD25+) in the secondary tumors, showing the enhanced CTL effects and decreased immunosuppression. For mice treated with rGO/MTX/SB-based PTT, the percentage of CD8+ was significantly increased, and the percentage of Tregs was significantly deceased in the secondary tumors, compared with that in the mice treated with rGO- and rGO/MTX-based PTT (Figs. 7C–D). The increased CTL could directly kill target tumor cells and the decreased Tregs formed the foundation for the rGO/MTX/SB-PTT-induced abscopal effects. Higher cytotoxic effects were found in the secondary tumors of mice treated with rGO/MTX/SB-based PTT (Fig. 7E). These results demonstrated that the treatment of rGO/MTX/SB-based PTT could effectively suppress distal tumors by increasing the infiltration of CTL and decreasing the percentage of Tregs in distal tumors.
4. Discussion
The potent therapeutic strategies for metastatic cancer require efficient antitumor responses by eradicating primary tumors and controlling distal metastases. To this end, chemo-immunotherapy was developed as a combination therapeutic strategy by synergizing the tumor killing effect and antitumor immunomodulatory effect to control primary tumors and metastases with reduced drug doses. However, drug resistance and toxicity are still the big challenges due to the systemic delivery of chemotherapeutic agents. Photoimmunotherapy was proposed as a novel synergized strategy for metastatic cancer, by combining local laser irradiation and immunotherapy, with minor side effects. However, the limited light penetration cannot ensure removal of tumor tissue, which leads to tumor recurrence. To overcome these shortages, we developed a synergistic nanographene system combining three treatment modalities—photothermal therapy with rGO, chemotherapy by MTX, and immunotherapy with SB—to generate a tumor-specific, long-term immunity by local implement, in a highly aggressive 4T1 murine breast tumor model. To be specific, the primary tumor killing effect was achieved by rGO-PTT and MTX. MTX-induced ICD accompanied by rGO-driven vaccination effectively activate the systemic antitumor immune response. SB helped modification of the tumor microenvironment to enhance chemotherapy and immunotherapy. Overall, the present findings confirmed that rGO/MTX/SB-PTT, a synergized photoimmunotherapeutic strategy, could successfully eradicated primary tumors as well as distal metastases through the combination of in situ tumor killing and long-term immunity. Our approach has the advantages of both chemo-immunotherapy and photo-immunotherapy, while avoiding the major shortcomings of these two combination strategies.
The effective control of cancer depends on the immune surveillance and defense system. To improve host immune system, immunotherapy has been rapidly developed for cancer control, including immune-activating antibodies, check-point inhibitors, chimeric antigen receptor T cell therapy, and cancer vaccines [5, 7, 20, 21]. Although antibodies and check-point inhibitors have been approved by the FDA for the treatment of advanced solid tumors, the clinical response rate was just 10-15% due to the lack of a specific immunological response. Cancer vaccines have the unique advantage in inducing antigen-specific immunities, but their efficacy is limited due to weak immunogenicity of tumor antigens [22]. A novel strategy of an in situ autologous cancer vaccine was developed to resolve these issues, which is produced by targeted therapies, such as PTT, PDT, or RF [23–25]. These treatment modalities killed the tumor cells and triggered a local whole tumor cell vaccination using patients’ own tumor antigens, while improving the tumor microenvironment and immunogenicity. This type of in situ vaccination reduces the burden of ex vivo selection and processing.
Based on its intrinsic photoabsorption properties, rGO has been used as a photothermal agent to enhance the local thermal effects of PTT [26, 27]. As a local targeting therapy, PTT induces significant temperature increase in tumor tissue, resulting in ICD which not only significantly suppresses the growth of primary tumors but also modulates immune responses [28]. The immunogenicity of ICD relies on the exposure/release of DAMPs, such as ATP, HMGB1, HSPs and CRT [29]. The surface exposure of CRT and HSP serves as an ‘eat me’ signal to DCs, promoting uptake of dying cells and stimulating a T helper biased immune response [30, 31]. The release of ATP and HMGB1 can be recognized by receptors on DCs to enhancement DC maturation [32].
MTX is a FDA-approved chemotherapeutic drug for the treatment of many types of cancer and is known to induce an autophagy-dependent anticancer immune response by triggering ICD, as evidenced by ATP and HMGB1 release [12]. In our study, rGO-based PTT caused ICD, as evidenced by the high level of cell death, the surface exposed CRT and HSP on treated tumor cells, and the released ATP and HMGB1 from treated tumor cells. In addition, loading MTX onto rGO significantly enhanced the tumor killing effect and the sensitivity of 4T1 cells to rGO-based PTT. MTX augmented the tumor destruction by rGO-based PTT and, at the same time, complemented the ICD production, formulating a potent immunogenic cancer vaccine. More importantly, rGO was found to serve as an adjuvant to enhance the vaccination process by directly activating DCs during the treatment and indirectly enhancing the DCs activation through treated tumor cells. Integrating three functions of rGO, as a photothermal agent, a drug carrier, and an adjuvant, our findings highlight that rGO/MTX-based PTT can effectively drive a systemic antitumor immunity by producing a potent in situ immunogenic cancer vaccine.
Studies have shown that TGF-β antagonists have the effects on prevention or suppression metastases in a number of preclinical advanced tumor models, especially for 4T1 lung metastasis; however, the suppression effect primary tumor growth was limited. When TGF-receptor-I inhibitor (LY364947) was combined with the cytotoxic chemotherapeutic agent, doxorubicin, the effective against pancreatic and gastric carcinomas was improved [33]. In addition, it has been demonstrated that TGF-β neutralizing enhances RT induced T-cell responses in 4T1 metastatic breast cancer [34]. We hypothesized that the efficacy of local vaccination induced by rGO/MTX-based PTT can be further improved with TGF-β antagonists, leading to durable clinical benefits. Our hypothesis has been proven by strong evidence of rGO/MTX/SB-based PTT induced immune responses, including immunogenicity enhancement, microenvironment improvement, and development of a tumor-specific, long-term immunity. Specifically, compared to rGO/MTX-based PTT, rGO/MTX/SB-based PTT significantly increased the number of Tregs (an immunosuppressive marker) in tumor tissue and the antigen-specific IFNγ-producing CD8+ T cells in SPL and DLN. The CD8+ T cells infiltration into distal tumors and the adoptive immunity were enhanced by the induced systemic antitumor immunity. Finally, the treatment of rGO/MTX/SB-based PTT resulted in a 70% long-term survival rate in 4T1 tumor-bearing mice and induced an immune memory to resist tumor rechallenge.
In summary, we have developed an effective combination therapy for metastatic cancer, with the multi-functional rGO as a photothermal agent, an adjuvant, and a drug carrier, incorporating PTT, MTX-generated chemotherapy, and TGF-β inhibition induced immunotherapy. The combination therapy induced a superior antitumor immunity, synergizing the tumoricidal and immunological processes, to trigger an effective CTL for metastasis control. This designed synergistic nanographene offered a new strategy for treating many metastatic cancers with primary tumors accessible by PTT.
Supplementary Material
Fig. 1.
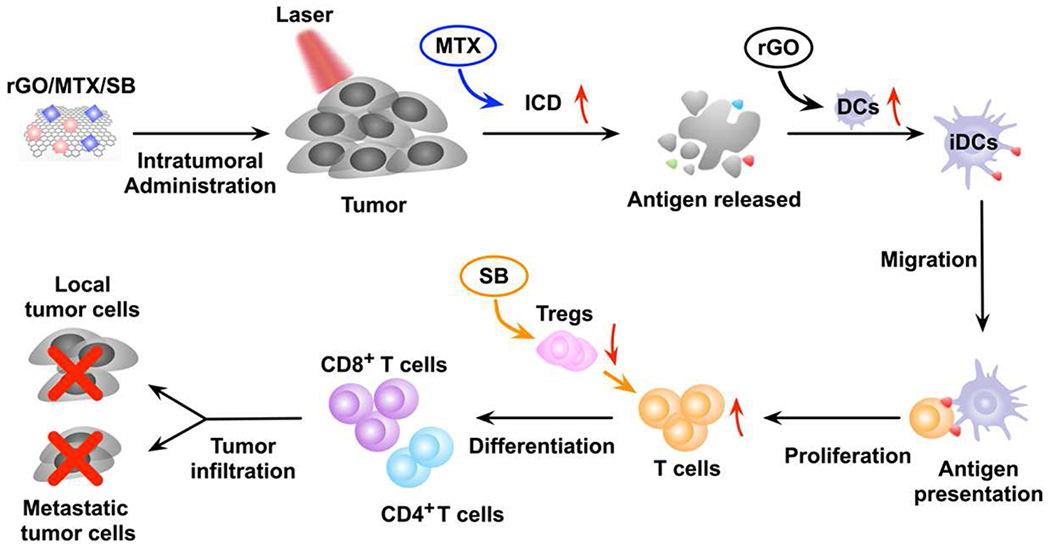
The mechanism of antitumor immune response induced by rGO/MTX/SB based PTT.
Acknowledgments
This study was supported in part by grants from the US National Institutes of Health R01 CA205348 (W.R.C.), the Oklahoma Center for Advancement of Science and Technology HR16-085 (W.R.C.), the National Natural Science Foundation of China (61722508/61525503/61620106016/61835009/81727804) (J.Q.), Guangdong Province Key Area R&D Program 2019B110233004 and Hainan University R&D Program (KYQD(ZR)20074) (F.Z.), Shenzhen Fundamental Research Program JCYJ20190808114609361 (M.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing financial interests
The authors declare no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References:
- [1].Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L, Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease, Nat. Rev. Clin. Oncol 13 (2016) 674–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, Ruddy K, Tsang J, Cardoso F, Breast cancer, Nat. Rev. Dis. Primers 5(1) (2019) 66. [DOI] [PubMed] [Google Scholar]
- [3].Waks AG, Winer EP, Breast cancer treatment: a review, Jama 321(3) (2019) 288–300. [DOI] [PubMed] [Google Scholar]
- [4].Ademuyiwa FO, Cyr A, Ivanovich J, Thomas MA, Managing breast cancer in younger women: challenges and solutions, Breast cancer 8 (2016) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Banchereau J, Palucka K, Immunotherapy: Cancer vaccines on the move, Nat. Rev. Clin. Oncol 15(1) (2018) 9–10. [DOI] [PubMed] [Google Scholar]
- [6].Lebert JM, Lester R, Powell E, Seal M, McCarthy J, Advances in the systemic treatment of triple-negative breast cancer, Curr. Oncol 25(Suppl 1) (2018) S142–sl50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sharma P, Allison JP, The future of immune checkpoint therapy, Science 348(6230) (2015)56–61. [DOI] [PubMed] [Google Scholar]
- [8].Zhou R, Yazdanifar M, Roy LD, Whilding LM, Gavriil A, Maher J, Mukheqee P, CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth, Front. Immunol 10 (2019) 1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bates JP, Derakhshandeh R, Jones L, Webb TJ, Mechanisms of immune evasion in breast cancer, BMC cancer 18(1) (2018) 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu Z, Li M, Jiang Z, Wang X, A Comprehensive Immunologic Portrait of Triple-Negative Breast Cancer, Trans. Oncol 11(2) (2018) 311–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Metivier D, Pichard E, Aucouturier P, Pierron G, Garrido C, Zitvogel L, Kroemer G, Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death, J. Exp. Med 202(12) (2005) 1691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, di Virgilio F, Zitvogel L, Kroemer G, Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice, Science 334(6062)(2011) 1573–7. [DOI] [PubMed] [Google Scholar]
- [13].Blankenstein T, Coulie PG, Gilboa E, Jaffee EM, The determinants of tumour immunogenicity, Nat. Rev. Cancer 12(4) (2012) 307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kroemer G, Galluzzi L, Kepp O, Zitvogel L, Immunogenic cell death in cancer therapy, Annu. Rev. Immunol 31 (2013) 51–72. [DOI] [PubMed] [Google Scholar]
- [15].O’Reilly EA, Gubbins L, Sharma S, Tully R, Guang MH, Weiner-Gorzel K, McCaffrey J, Harrison M, Furlong F, Kell M, McCann A, The fate of chemoresistance in triple negative breast cancer (TNBC), BBA Clin. 3 (2015) 257–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Massagué J, TGFβ in Cancer, Cell 134(2) (2008) 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sato M, Matsubara T, Adachi J, Hashimoto Y, Fukamizu K, Kishida M, Yang YA, Wakefield LM, Tomonaga T, Differential Proteome Analysis Identifies TGF-β-Related Pro-Metastatic Proteins in a 4T1 Murine Breast Cancer Model, PloS One 10(5) (2015) e0126483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, Stanford J, Cook RS, Arteaga CL, TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer, J. Clin. Invest 123(3) (2013) 1348–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luo N, Weber JK, Wang S, Luan B, Yue H, Xi X, Du J, Yang Z, Wei W, Zhou R, Ma G, PEGylated graphene oxide elicits strong immunological responses despite surface passivation, Nat. Commun 8 (2017) 14537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Curran KJ, Brentjens RJ, Chimeric antigen receptor T cells for cancer immunotherapy, J. Clin. Oncol 33(15) (2015) 1703–6. [DOI] [PubMed] [Google Scholar]
- [21].Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L, Immunostimulatory monoclonal antibodies for cancer therapy, Nat. Rev. Cancer 7(2) (2007) 95–106. [DOI] [PubMed] [Google Scholar]
- [22].van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ, Vaccines for established cancer: overcoming the challenges posed by immune evasion, Nat. Rev. Cancer 16(4) (2016)219–33. [DOI] [PubMed] [Google Scholar]
- [23].Formenti SC, Demaria S, Radiation therapy to convert the tumor into an in situ vaccine, Int. J. Radi. Oncol 84(4) (2012) 879–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, Agostinis P, Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma, Sci. Transl. Med 8(328) (2016) 328ra27. [DOI] [PubMed] [Google Scholar]
- [25].Zhou F, Yang J, Zhang Y, Liu M, Lang ML, Li M, Chen WR, Local Phototherapy Synergizes with Immunoadjuvant for Treatment of Pancreatic Cancer through Induced Immunogenic Tumor Vaccine, Clin. Cancer Res 24(21) (2018) 5335–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hu D, Zhang J, Gao G, Sheng Z, Cui H, Cai L, Indocyanine Green-Loaded Polydopamine-Reduced Graphene Oxide Nanocomposites with Amplifying Photoacoustic and Photothermal Effects for Cancer Theranostics, Theranostics 6(7) (2016) 1043–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Robinson JT, Tabakman SM, Liang Y, Wang H, Sanchez Casalongue H, Vinh D, Dai H, Ultrasmall Reduced Graphene Oxide with High Near-Infrared Absorbance for Photothermal Therapy, J. Am. Chem. Soc 133(17) (2011) 6825–6831. [DOI] [PubMed] [Google Scholar]
- [28].den Brok MH, Sutmuller RP, van der Voort R, Bennink EJ, Figdor CG, Ruers TJ, Adema GJ, In situ tumor ablation creates an antigen source for the generation of antitumor immunity, Cancer Res. 64(11) (2004) 4024–9. [DOI] [PubMed] [Google Scholar]
- [29].Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P, Immunogenic cell death and DAMPs in cancer therapy, Nat. Rev. Cancer 12 (2012) 860–875. [DOI] [PubMed] [Google Scholar]
- [30].Binder RJ, Functions of heat shock proteins in pathways of the innate and adaptive immune system, J. Immunol 193(12) (2014) 5765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh F, Perfettini JF, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Farochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel F, Kroemer G, Calreticulin exposure dictates the immunogenicity of cancer cell death, Nat. Med 13(1) (2007) 54–61. [DOI] [PubMed] [Google Scholar]
- [32].Apetoh F, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Fidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, Andre F, Tursz T, Kroemer G, Zitvogel F, The interaction between HMGB1 and TFR4 dictates the outcome of anticancer chemotherapy and radiotherapy, Immunol. Rev 220 (2007) 47–59. [DOI] [PubMed] [Google Scholar]
- [33].Kano MR, Bae Y, Iwata C, Morishita Y, Yashiro M, Oka M, Fujii T, Komuro A, Kiyono K, Kaminishi M, Hirakawa K, Ouchi Y, Nishiyama N, Kataoka K, Miyazono K, Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling, P. Natl. Acad. Sci. USA 104(9) (2007) 3460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, Barcellos-Hoff MH, Demaria S, TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity, Cancer Res. 75(11) (2015) 2232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.