

Abstract
The IL-2 receptor (IL-2R) is an attractive cancer immunotherapy target that controls immunosuppressive T regulatory cells (Tregs) and anti-tumor T cells. Here we used IL-2Rβ-selective IL-2/anti-IL-2 complexes (IL-2c) to stimulate effector T cells preferentially in the orthotopic mouse ID8agg ovarian cancer (OC) model. Despite strong tumor rejection, IL-2c unexpectedly lowered the tumor microenvironmental CD8+/Treg ratio. IL-2c reduced tumor microenvironmental Treg suppression and induced a fragile Treg phenotype, helping explain improved efficacy despite numerically increased Tregs without affecting Treg in draining lymph nodes. IL-2c also reduced Treg-mediated, high-affinity IL-2R signaling needed for optimal Treg functions, a likely mechanism for reduced Treg suppression. Effector T cell IL-2R signaling was simultaneously improved, suggesting that IL-2c inhibits Treg functions without hindering effector T cells, a limitation of most Treg depletion agents. Anti-PD-L1 antibody did not treat ID8agg, but adding IL-2c generated complete tumor regressions and protective immune memory not achieved by either monotherapy. Similar anti-PD-L1 augmentation of IL-2c and degradation of Treg functions were seen in subcutaneous B16 melanoma. Thus, IL-2c is a multifunctional immunotherapy agent that stimulates immunity, reduces immunosuppression in a site-specific manner, and combines with other immunotherapies to treat distinct tumors in distinct anatomic compartments.
Keywords: ovarian cancer, melanoma, immunotherapy, CD25, IL-2 complex, regulatory T cell, IL-2 receptor, PD-L1
Introduction
Despite recent successes of individual cancer immunotherapy agents, multiple, distinct immune pathways must be corrected for optimal treatment efficacy (1). Immunosuppressive Tregs are a fundamental obstacle to effective anticancer immunity in many cancer types (2,3), including ovarian cancer (OC) (4) and melanoma (5). However, the development of drugs that deplete or inhibit Tregs has been difficult, partly due to a scarcity of known targetable, Treg-specific molecules. The IL-2 receptor (IL-2R) is an attractive immunotherapy target due to its control over Treg development, homeostasis and functions (6–9), but also anti-tumor T cell effector functions (10,11). An ideal IL-2R-targeted agent would reduce Treg suppression, but simultaneously stimulate anti-tumor T cells and to serve as multifunctional cancer immunotherapy.
The IL-2R is comprised of three subunits: high-affinity IL-2Rα (CD25), intermediate-affinity IL-2Rβ (CD122), and a common gamma chain (CD132, γc). CD25 does not participate in signal transduction but markedly enhances IL-2 potency by augmenting IL-2 binding to the trimeric αβγ IL-2R (11,12). Mice lacking IL-2 (13), CD25 (14), or CD122 (15) die from lethal T cell-driven autoimmunity due to reduced functional Tregs, indicating IL-2 signaling dominantly supports Tregs and is dispensable for excessive T cell activation. Tregs constitutively express high CD25 (16) that is critical for their suppressive function (7) but also enables their preferential depletion by αCD25 antibodies (17). Unlike Tregs, memory-phenotype CD8+CD44+ T cells and natural killer (NK) cells mediating tumor cytotoxicity highly express CD122, not CD25 (11). Selective stimulation of medium-affinity βγ IL-2R can be achieved by complexing IL-2 with anti-IL-2 antibody clones that occlude the CD25-binding epitope of IL-2 (CD122-selective IL-2 complexes; IL-2c) (18–20); and reviewed in (21) to augment CD122+ effector T cell functions while largely avoiding Treg expansion (18,22). IL-2c induces CD8+ T cell proliferation associated with tumor rejection in several cancer models (20,21). However, the effects of CD122-selective IL-2c on Treg phenotype and functions are unreported.
Here, we show that IL-2c improves anti-tumor T cell functions as expected and expand the list of IL-2c-responsive tumors, but we unexpectedly found that IL-2c also significantly reduce Treg function specifically in the tumor microenvironment and induce a fragile Treg phenotype. We combined αCD25 with reportedly CD25-independent IL-2c after hypothesizing IL-2c would insulate effector T cells from negative off-target effects of αCD25. However, combination treatment with IL-2c plus αCD25 was less efficacious than IL-2c monotherapy in the OC model, indicating an unappreciated role for CD25 in IL-2c actions. Inhibiting programmed cell death-ligand 1 (PD-L1) signals with antagonistic monoclonal antibodies (αPD-L1) has generated high-profile clinical successes in several difficult-to-treat cancers (23–25). Despite these encouraging results, most patients do not benefit from PD-L1 blockade, due in part to the absence of preexisting tumor inflammation (26). Therefore, a strategy to sensitize the large proportion of patients unresponsive to PD-L1 blockade could include pretreatment with an immunotherapy that induces intratumoral T cell infiltration. Although αPD-L1 had no effect as monotherapy in ID8agg OC, combination treatment with αPD-L1 plus IL-2c produced complete tumor eradication and elicited durable, protective tumor-specific immunity. IL-2c increased intratumoral immune cell infiltration and improved αPD-L1 efficacy in ID8agg OC, and similarly improved the αPD-L1 response of subcutaneous B16 melanoma. We conclude that CD122-selective IL-2c reduce Treg immunosuppression, promote durable anti-tumor immunity and improve αPD-L1 treatment efficacy in distinct cancers and anatomic compartments. Thus, IL-2c is a strong candidate for clinical trials in OC, melanoma and other cancers alone and in rational combinations, including with αPD-L1.
Materials and Methods
Study design
We performed controlled preclinical experiments to test clinical efficacy and immune effects of selective stimulation of IL-2R using IL-2c in two tumor models: murine OC and melanoma in two anatomic compartments: peritoneum and skin, respectively. We tested αCD25 as an alternative therapy, and the efficacy of combining IL-2c with either αCD25 or αPD-L1. Outcomes included anti-tumor immunity, Treg effects, IL-2 signaling, tumor growth control and establishment and durability of effective anti-tumor immunity. All animals were randomized to control/treatment groups and tumor measurements were collected by experimenters blind to treatment conditions.
Proteins and antibodies
Carrier-free recombinant mouse IL-2 (IL-2) was purchased from Biolegend (San Diego, CA) and stored at −80°C in aliquots. αIL-2 (clone JES6–5H4), αCD25 (clone PC61.5.3), αPD-L1 (10F.9G2) and isotype control antibodies (clone LTF-2 rat IgG2b and clone HRPN rat IgG1) were purchased from BioXCell (Lebanon, NH) and stored at 4° C.
Mice and cell lines
Wild-type (WT) C57BL/6 mice (all female unless otherwise noted) were purchased from Jackson Labs (Bar Harbor, ME), bred in our animal facility and used at 7–20 weeks old. Female C57BL/6 FoxP3DTR mice were from Alexander Rudensky (then at University of Washington). WT BL6 mice that received diphtheria toxin were used as comparators for FoxP3DTR mice. FoxP3-IRES-RFP (FIR) mice from which viable RFP+ Tregs can be isolated$ were from Richard Flavell (Yale). Animals were maintained in specific pathogen-free conditions and provided a normal diet and water ad libitum. All tumor cells were cultured in RPMI-1640 medium containing 5% FBS, 10 U/mL penicillin, 10 µg/mL streptomycin, 2 mM L-glutamine, and 1 mM HEPES buffer (R5 medium) in a humidified 37° C incubator (5% CO2). The mouse OC cell line ID8 was from George Coukos (then at University of Pennsylvania), from which we developed the highly aggressive ID8agg subline (27). To track ID8agg tumor burden in vivo, we transfected ID8agg cells with the pGL4.51 vector encoding luciferase (Promega; Madison, WI) with Attractene transfection reagent (Qiagen; Valencia, CA) according to manufacturer instructions. Single-cell derived clones of luciferase-expressing ID8agg (ID8agg-Luc) were isolated by limiting dilution after 1 mg/mL selection with G418 antibiotic. One clone with optimal luciferase activity was cryopreserved and used in all experiments. Upon re-culture, ID8agg-Luc was continuously maintained in 0.2 mg/mL G418 to prevent the outgrowth of luciferase-negative clones. B16-F10 was purchased from ATCC (Manassas, VA). All cell lines were periodically tested and confirmed free of Mycoplasma by Mycoalert PLUS detection kit (Lonza Bioscience; Walkersville, MD) before experimental uses but were not genetically authenticated.
In vivo tumor challenge and treatments
Mice were challenged i.p. with 4 × 106 ID8agg or ID8agg-Luc cells, or s.c. on one flank with 5 × 105 B16-F10 cells. All cells were in log growth phase at challenge. Upon first tumor measurement and before randomizing into treatment groups, mice were occasionally excluded due to abnormal tumor growth that was verified as an outlier by Grubb’s test. 200 µg/mouse αCD25 was given i.p. 7 days after ID8agg challenge. 100ug/mouse αPD-L1 was given i.p. 11, 14, 17 and 20 days after ID8agg and B16 challenge. 1.5 µg/mouse IL-2 was complexed with 7.5 µg/mouse αIL-2 (clone JES6–5H4) at the optimal 1:2 molar ratio in PBS at 37°C for 15–30 min (18) before i.p. administration on days 7, 9, 11 and 13 after ID8agg or B16 challenge. Occasionally, bioluminescent measurements were excluded in the final measurement (week 5) due to obvious interference of ascites with light emission (28). Thus, week 5 bioluminescent mean values for control-treated mice are generally underestimated. For B16-F10 tumors, treatment was initiated when mean tumor volume was ~100 mm3 (7 days post challenge, as indicated) and measured every 2–4 days by calipers in a blinded fashion, with volume calculated as 0.5 × (width × length2). Survival was defined as spontaneous death, moribundity, tumor volume >1500 mm3 for B16, or weight >130% of baseline (ascites) in ID8agg. In vivo bioluminescence of ID8agg-Luc was assessed with an IVIS Lumina Imaging System (Perkin Elmer; Waltham, MA) 15 minutes after i.p. injection of 3 mg of PBS-dissolved d-luciferin K+ (Gold Biotechnology; St. Louis, MO) with 30 second exposures, medium binning, and F/stop = 1. Identical regions of interest were drawn over each subject’s abdomen and average radiance (photons/sec/cm2/sr) was quantified with Living Image software version 4.2. For FoxP3 DTR and WT mice, 5 µg/kg recombinant diphtheria toxin (Santa Cruz Biotechnologies; Santa Cruz, CA) was given i.p. every three days, as indicated.
Flow cytometry
Mice were sacrificed by cervical dislocation after induction of deep isoflurane anesthesia. Ascites and peritoneal exudate cells were isolated by recovering 5 mL of i.p. injected ice-cold PBS supplemented with 2% FBS and 1 mM EDTA. For intratumoral analysis, the large omental tumor was dissected, minced with a razor blade, incubated in serum-free RPMI-160 with 0.33 mg/mL DNAse I and 3 mg/mL collagenase IV (Worthington Biochemical, Lakewood, New Jersey) for 1 hour, and passed through a 70 µm filter. Ascites and dissected tumor-draining mesenteric lymph node cells were incubated with indicated antibodies and data acquired on a BD LSR II flow cytometer with FACSDiva software version 6.1.2. Dead cells were excluded by staining with Ghost Dye UV 450 (Tonbo; San Diego, CA). Non-specific labeling was pre-blocked by addition of anti-CD16/32 at 1:100 dilution (clone 2.4G2; Tonbo). Cells were stained for surface antigens by incubating at 4° C for 30–45 min with αCD3 (clone: 17A2; vendor: Tonbo), αCD4 (RM4–5; Tonbo), αCD8α (5H10; Invitrogen), αCD25 (eBio7D4; eBioscience), αCD107a (1D4B; Biolegend), αCTLA-4 (UC10–4F10–11; Tonbo), αPD-1 (RMP1–30; eBioscience), αCD103 (2E7; Biolegend), at 1:100 dilution. For intracellular staining, cells were fixed and permeablized with a FoxP3/transcription factor buffer kit (eBioscience; San Diego, CA) according to the manufacturer instructions, and incubated at 4° C for 45 min with αFoxP3 (FJK16s; eBioscience), αGranzyme B (NGZB; eBioscience), αIFN-γ (XMG1.2; Tonbo), TNF-α (MP6-XT22; Biolegend), αT-bet (4B10, Biolegend), αRorγt (Q31–378, BD Biosciences), αGata3 (L50–823, BD Biosciences), αIL-17 (TC11–18H10.1, Biolegend), αTCF-1 (S33–966, BD Biosciences) at 1:75 dilution in 1x FoxP3 permeabilization buffer. For detection of CD107a, granzyme B, TNF-α, and IFN-γ, cells were stimulated immediately after isolation with leukocyte activation cocktail + BD GolgiPlug (BD Biosciences; San Jose, CA) containing PMA, ionomycin, and brefeldin A at 2 µL cocktail/mL medium CR10 medium (RPMI-1640 with 10% FBS, L-glutamine, sodium pyruvate, non-essential amino acids, penicillin/streptomycin, and HEPES buffer) for 5 h in a 37° C incubator. Absolute numbers of cells were determined by multiplying the ratio of the cell of interest per live, singlet cell in each flow sample by the total number of viable cells from the sample specimen and then, as applicable, normalized to ascites volume (in cases where mice had no measurable ascites, a volume of 50 µL was used for calculations) or tumor weight. Fluorescence minus one (FMO) controls were used to set gates for all antibodies that did not produce clear, bimodal populations. All figures show geometric mean fluorescence intensity (MFI). Due to inter-experiment variability, pooled data show normalized MFI (where individual samples were normalized to the control MFI of their respective experiment).
In vitro Treg suppression assay
FIR mice were challenged with ID8agg, treated with IL-2c or isotype and sacrificed 5 weeks post tumor challenge. CD45+CD3+CD4+CD25hiRFP+ Tregs were sorted by flow cytometry (95–99% purity) from TDLN and ascites and incubated with 30,000 CFSE-stained, splenic CD45+CD3+CD4+CD25− responder T cells (95–99% purity) obtained by electronic cell sorting from naïve, aged matched syngeneic females. T cells from naïve WT mice, at graded concentrations plus αCD3/αCD28 Dynabeads (Invitrogen; 1 bead for every 5 effector cells) in a round-bottom 96-well plate for ~80 hours. Proliferation was analyzed by flow cytometry based on the dilution of CFSE.
Ex vivo Treg incubation
CD45+CD3+CD4+CD25hiRFP+ Treg from FIR mice spleens were sorted, incubated with mIL-2 (20 ng/mL) or IL-2c (20 ng/mL mIL-2 + 100 ng/mL αIL-2) in 100% CR10 medium or 50% CR10+50% ascites collected from ID8agg challenged WT mice in a 48 well plates coated with αCD3 and αCD28 antibodies. Five days later, Tregs from the culture were harvested and subjected to in vitro Treg suppression assays as described above
ELISA
Cell culture supernatants from Treg suppression assay were appropriately diluted and analyzed with an IL-2 ELISA Max Deluxe Kit (Biolegend), according to the manufacturer’s instructions.
In vitro STAT5 phosphorylation assay
CD3+ T cells were sorted from ascites and TDLN of ID8agg-bearing mice, plated at 100,000 cells/well in CR10 medium in a 96-well plate, and rested for 1 h. IL-2 was pulsed for 30 min and cells were immediately fixed and permeablized using BD Phosflow Lyse/Fix and Perm Buffer III, then stained with CD8, CD4, FoxP3 and pSTAT5 (BD Biosciences; clone: 47/Stat5(pY694)). The pSTAT5 gate was set with reference to a fluorescence minus one (FMO) control.
Statistics and data analysis
Data were analyzed and graphed with Graphpad Prism 6.01. All points with error bars represent the mean ± SEM and points without error bars represent individual mice, unless indicated otherwise. For comparison of two means, we used an unpaired Student’s t test. Three or more means were compared with one-way ANOVA and post hoc Sidak’s test. Tumor growth curves were compared by two-way ANOVA and first analyzed for an overall effect due to treatment (p value labeled on figure legend) followed by post-hoc Sidak’s test (p value labeled on individual graph points) of discrete time points. Log-rank test was used to compare Kaplan-Maier curves. Occasionally, data sets with suspected outliers were identified by Grubb’s test (used only once for a given data set). For all analyses, significance was based on a multiplicity-corrected, two-sided α of 0.05. In each figure, we provide p values to two significant digits and number of independent experiments performed.
Study approval
All animal work was done under UTHSA Institutional Animal Care and Use Committee approved studies in compliance with the Guide for the Care and Use of Laboratory Animal Resources.
Results
IL-2c promotes durable anti-tumor immunity in poorly-immunogenic ovarian cancer
We tested a CD122-selective IL-2c (18) that induces preferential expansion and activation of effector T cells to assess effects in mouse ID8agg OC, a poorly immunogenic tumor refractory to many forms of immunotherapy (29). IL-2c extended survival, durably reduced tumor burden, and reduced malignant ascites (Fig. 1A–C). Although IL-2c serum half-life is only ~24 hours (19), tumor control in mice treated with IL-2c alone persisted for several weeks after IL-2c administration, suggesting durably augmented anti-tumor immune function. Accordingly, ascites effector T cells isolated over 3 weeks after the final IL-2c dose exhibited increased expression of anti-tumor cytokines (Fig. 1D,E).
Figure 1. IL-2c durably reduces ID8agg OC burden and increases polyfunctional T cells.
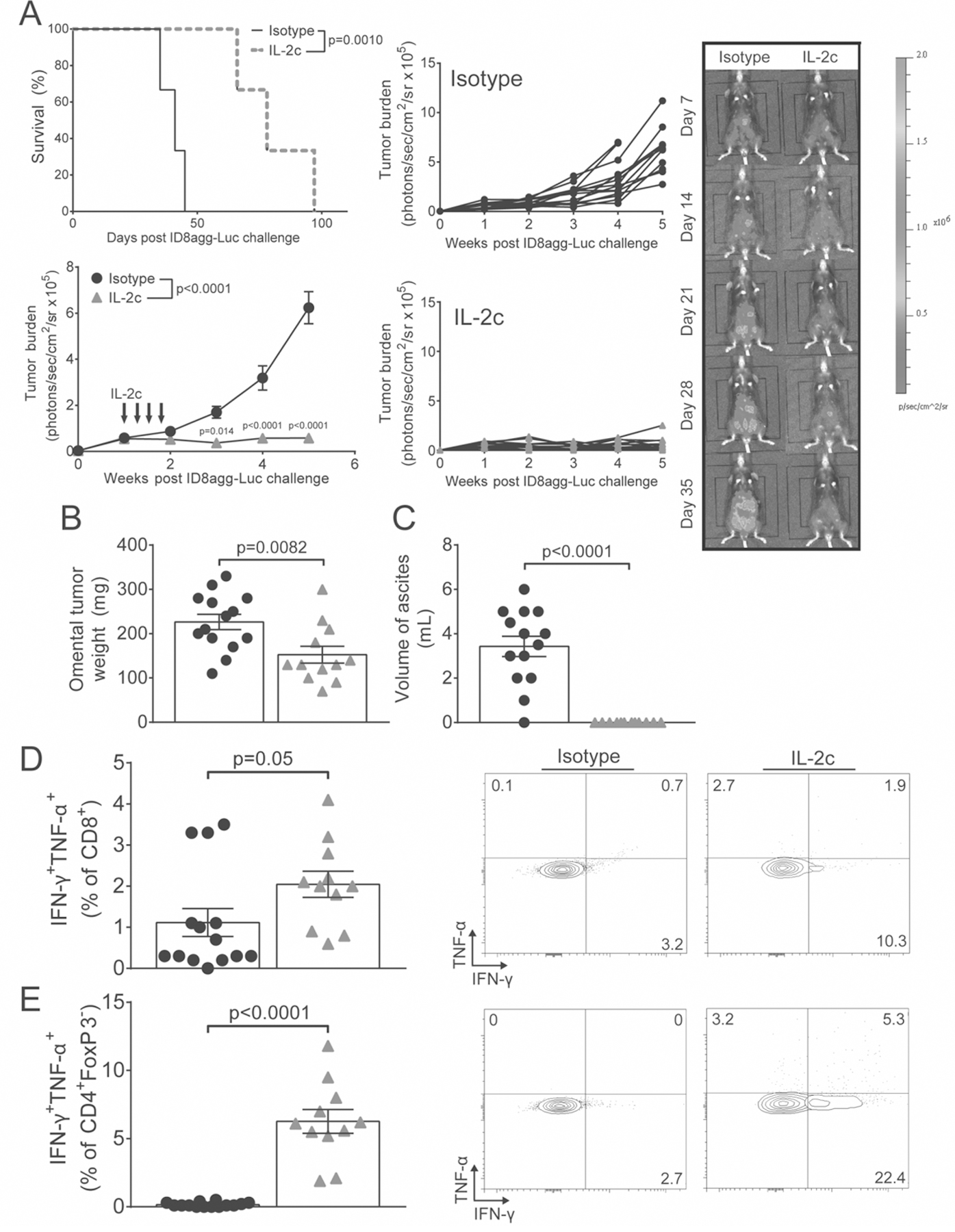
A. Kaplan-Maier plot of survival (top left; P value from log-rank test) and tumor burden by bioluminescence after challenge with ID8agg-Luc and IL-2c treatment as indicated (black arrows) (bottom left), with representative images of ID8agg-Luc-bearing mice ± IL-2c (right) and individual tumor growth curves (middle). Accumulating ascites starts to interfere with bioluminescence data acquisition 5 weeks after tumor challenge and thus data beyond 5 weeks is not acquired. B. Omental tumor weight 3 weeks after last IL-2c. C. Ascites volume 3 weeks after last IL-2c. D. Summary graph of ascites-infiltrating CD8+ IFN-γ+TNF-α+ cell frequency 3 weeks after last IL-2c (left) with representative flow plots (right). E. Summary graph of ascites CD4+FoxP3− IFN-γ+TNF-α+ T cell frequency 3 weeks after last IL-2c (left) with representative flow plots (right). P values from Student’s unpaired t test, except A (two-way repeated measures ANOVA (figure legend) with post hoc Sidak’s test (graph points)). N=14 for isotype, or 12 for IL-2c for all panels, except the Kaplan-Maier curve (N=3/group). All panels are pooled from two independent experiments, except the Kaplan-Maier curve (representative of at least 4 independent experiments). For all panels, black circles are isotype and grey triangles are IL-2c.
IL-2c surprisingly reduces CD8+/Treg ratio in the ascites and affects Treg and effector T cell numbers distinctly in tumor draining lymph nodes versus ascites
To assess how IL-2c altered effector and regulatory T cell numbers and thus their relative ratios in various compartments, we sacrificed ID8agg tumor-bearing mice ~3 weeks after the final IL-2c dose and measured prevalence and numbers of T cell subsets in ascites and tumor draining lymph nodes (TDLN). IL-2c greatly reduced overall ascites cellularity by preventing the formation of malignant ascites, greatly reducing the absolute number of both CD8+ T cells and Tregs in ascites (Fig. 2A, top). However, CD8+ T cell concentration was unchanged while Treg concentration was elevated (Fig. 2A, bottom). Although an increased CD8+/Treg cell ratio in the tumor microenvironment predicts enhanced survival for OC patients (30,31), we surprisingly found that IL-2c decreased the ascites CD8+/Treg ratio 3 weeks after the final IL-2c administration (Fig. 2B). Thus, a surprising IL-2c-mediated Treg increase accounted for the reduced CD8+/Treg ratio. IL-2c also increased prevalence of non-regulatory CD4+FoxP3− T cells in ascites (Fig. S1A). Such changes in T cell subsets were not observed one week after the final IL-2c dose (Fig. S1B, C). Therefore, we focused further immune studies three weeks after final IL-2c dose. Strong tumor rejection exhibited by IL-2c-treated mice suggested that the low CD8+/Treg ratio could be from improved CD8+ function, reduced Treg function, or both. In TDLN, we saw elevated numbers of CD8+ and non-regulatory CD4+FoxP3− T cells at 3 weeks after the last IL-2c dose (Fig. 2C, Fig S1A). However, Treg cell numbers in TDLN were not, or only slightly, elevated (Fig. 2C). Thus, IL-2c increased absolute numbers of non-Treg T cells reduced Treg prevalence in TDLN, but not ascites (Fig. 2C, 2D). We also observed an increase in both CD8+CD44hiCD62L+ central memory and CD8+CD44hiCD62L− effector memory T cell in ascites (Fig. S2, top) and an increase in CD8+CD44hiCD62L+ central memory T cells in TDLN (Fig. S2, bottom), suggesting IL-2c promotes long-term memory.
Figure 2. IL-2c reduces ascites CD8+/Treg ratio and affects Treg and effector T cell numbers distinctly in TDLN and ascites.
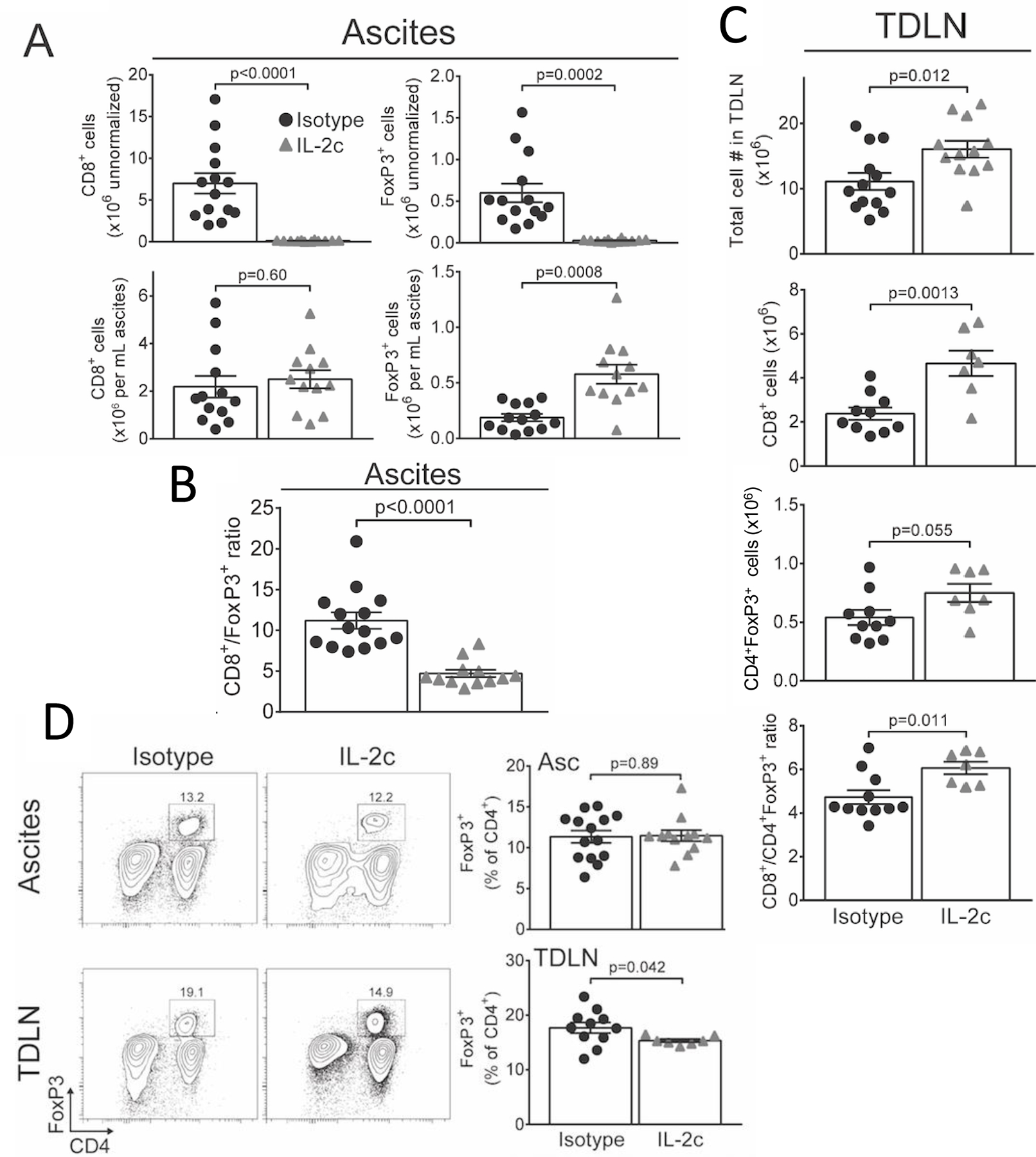
A. Absolute numbers of CD8+ T cells and CD45+CD3+CD4+FoxP3+CD25hi Tregs either raw (top) or normalized to ascites volume (bottom) 3 weeks after last IL-2c. N= 13 isotype, 12 IL-2c. B. Ratio of ascites CD8+/CD4+FoxP3+ T cells 3 weeks after last IL-2c. C. Absolute numbers of TDLN CD8+ and CD4+FoxP3+ Tregs isolated 3 weeks after last IL-2c. N=7–13/group. D. Representative flow plots (left; number indicates FoxP3+ prevalence in CD4+ gate) and summary graphs (right) showing FoxP3+ cell prevalence among CD4+ T cells 3 weeks after last IL-2c. P values from Student’s unpaired t test. All data are from two pooled independent experiments and are distinct from the cohort in Fig. 1.
IL-2c reduces ascites Treg suppressive function and induces a fragile Treg phenotype
Because IL-2c reduced both tumor burden and ascites CD8+/Treg ratio (Fig. 1A, Fig. 2B) due to increased Treg concentration in ascites (Fig. 2A), we hypothesized that IL-2c reduced Treg function in the tumor microenvironment. Thus, we examined IL-2c effects on expression of proteins that mediate Treg function. IL-2c increased FoxP3 mean fluorescence intensity (MFI [per cell expression]) among ascites, but not TDLN Tregs (Fig. 3A). However, despite increased FoxP3 MFI, IL-2c greatly reduced ascites Treg CD25 MFI, and slightly reduced CD25+ ascites Treg prevalence (Fig. 3B, Fig. S3A), suggesting reduced Treg function (7). Despite reducing Treg CD25, IL-2c increased CD25 expression in CD8+ and CD4+FoxP3− T cells (Fig. 3B) indicating differential effects on effector versus regulatory T cells, and suggesting favored IL-2 availability to anti-tumor T cells over Tregs as expected. IL-2c elicited a similar, opposing expression pattern for the cytotoxic effector protein granzyme B, important for Treg suppressive function (32,33) and anti-tumor T cell cytotoxicity, in effector versus regulatory T cells from ascites (Fig. 3C). IL-2c also reduced Treg CTLA-4 expression (Fig. 3D top), which Tregs use to prevent effector T cell co-stimulation (34), reduced CD103 (Fig. 3D bottom), a marker of highly suppressive Tregs (35) and mildly reduced CD39 (Fig. S3B), another marker of highly suppressive Tregs (36). We did not see a significant difference in expression of TIGIT, LAG3, the transcription factor HELIOS or production of IL-10 (Fig. S3C) in IL-2c treated ascites Tregs. These data indicate that IL-2c therapy durably reduces expression of proteins that mediate Treg function, while increasing some of those same proteins on effector T cells. Thus, CD122-selective IL-2c likely degrades Treg functions, but improves effector T cell functions.
Figure 3. IL-2c reduces ascites Treg effector proteins and induces a fragile Treg phenotype.
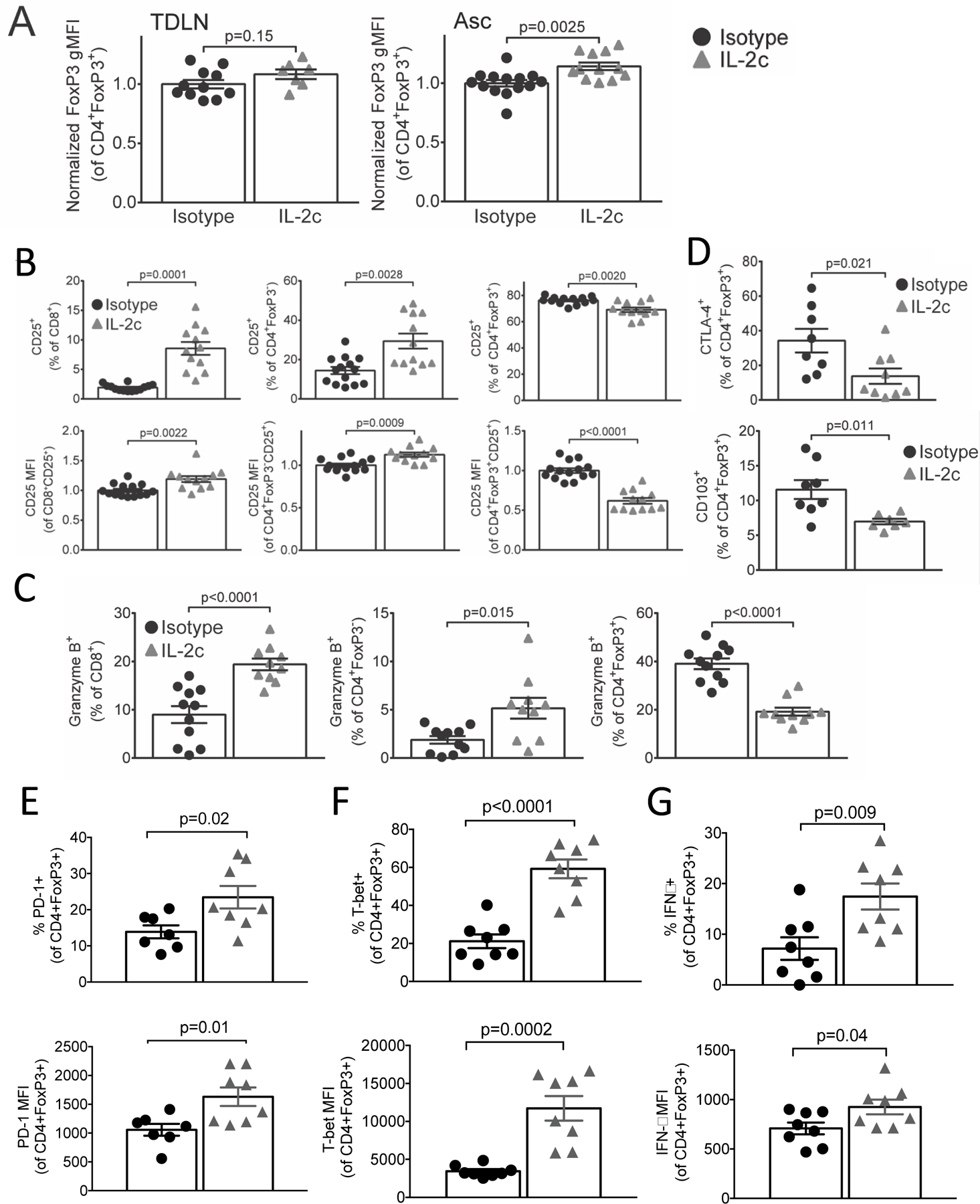
Mice were challenged with ID8agg-Luc, treated with IL-2c and sacrificed 3 weeks after the last IL-2c, but are a distinct cohort from Fig. 2. A. Normalized FoxP3 MFI of CD45+CD3+CD4+FoxP3+CD25hi Tregs from TDLN or ascites. N=10 for isotype, 7 for IL-2c in TDLN; N=13 for isotype, 12 for IL-2c in ascites. B. Summary graphs of CD25+ prevalence (top) or CD25 MFI (bottom) among CD8+ (left), CD4+FoxP3− (middle) and CD4+FoxP3+ (right) cells. N=14 for isotype, 12 for IL-2c. C. Summary graphs of granzyme B+ prevalence among CD8+ (left), CD4+FoxP3− (middle) and CD4+FoxP3+ cells (right). N= 11 isotype, 10 for IL-2c. D. Summary graphs of CTLA-4+ (top) and CD103+ (bottom) prevalence among CD4+FoxP3+ cells in the ascites. N= 8 for isotype, 7 for IL-2c. E-G Summary graphs of PD-1+ (E), T-bet+ (F) and IFNγ+ (G) prevalence (top) and MFI (bottom) among CD4+FoxP3+ cells in ascites. N=7 for isotype, 8 for IL-2c. All p values from Student’s unpaired t test.
Recent reports have described a fragile Treg phenotype in the tumor microenvironment which is defined as Tregs maintaining FoxP3 expression but with loss of suppressive function. These fragile Tregs produce IFNγ, upregulate the transcription factor Tbet, can have elevated PD-1 expression and are associated with responsiveness to αPD-1 cancer immunotherapy (37). We observed that IL-2c increased PD-1 (Fig. 3E), T-bet (Fig. 3F) and IFNγ production (Fig. 3G) in Tregs in ascites, but not TDLN (Fig. S3D), demonstrating that IL-2c induces a fragile Treg phenotype in ascites.
To assess Treg functions directly, we challenged FIR mice (38) with ID8agg, treated with IL-2c as before, recovered CD3+CD4+CD25hiRFP+ Tregs by electronic cell sorting and tested their ability to suppress cellular proliferation and cytokine secretion. Although IL-2c did not change TDLN Treg functions, IL-2c compromised ascites Treg suppression of naïve T cell proliferation and IL-2 secretion (Fig. 4A–C). Thus, IL-2c is a novel Treg inhibitor that does not compromise effector T cell functions, and specifically reduces tumor microenvironmental Treg functions in the ID8agg OC model. To determine whether IL-2c directly affects Treg suppressive function, we sorted live Tregs from naïve FIR mouse spleens and incubated them with IL-2 or IL-2c ex vivo. There was no significant difference in Treg numbers or suppressive function after ex vivo IL-2c incubation in complete culture medium (Fig. S4A), but we observed a minor reduction of Treg suppression after culturing in medium containing 50% ascites collected from ID8agg challenged mice treated with IL-2c (Fig. S4B). These data suggest an indirect mechanism for IL-2c-mediated reduction of Treg suppression, although direct mechanisms are not excluded.
Figure 4. IL-2c reduces ascites Treg suppressive function and high-affinity IL-2 signaling.
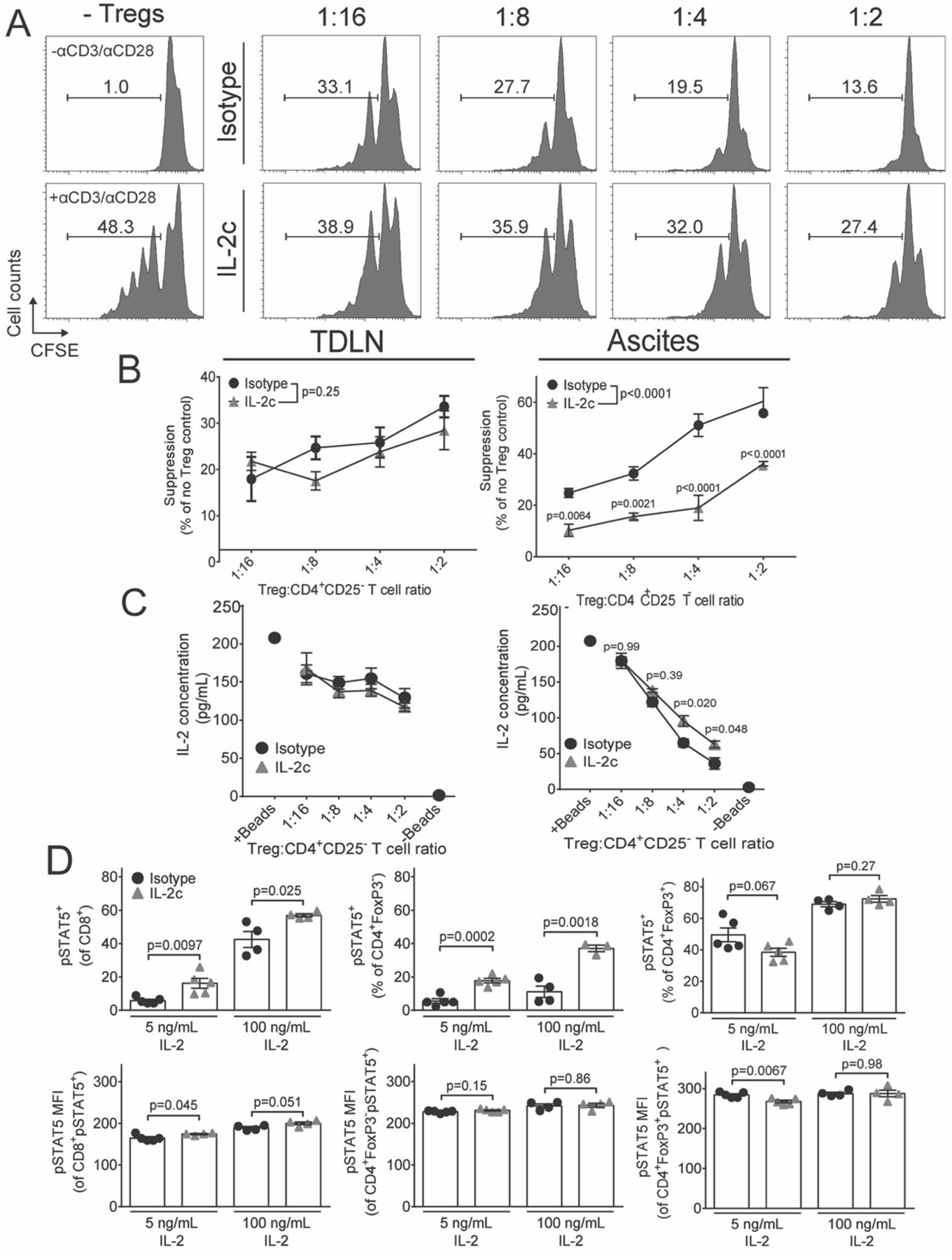
Mice were challenged with ID8agg-Luc, treated with IL-2c only, as in Fig 1A, and sacrificed CD45+CD3+CD4+FoxP3+CD25hi for studies dedicated to this figure only. A. Representative histograms of CFSE dye dilution by naïve T cells incubated with graded concentrations of isotype or IL-2c-treated CD45+CD3+CD4+CD25hiRFP+ Tregs isolated from ascites. B. Summary graphs showing percent suppression of proliferation by TDLN or ascites Tregs. αP values, two-way ANOVA Sidak’s post hoc test compared to isotype. C. Concentration of IL-2 in culture supernatants taken from B. P values, two-way ANOVA Sidak’s post hoc test compared to isotype. B is pooled from two independent experiments where CD45+CD3+CD4+CD25hiRFP+ Tregs were pooled from a total of 8 FIR mice/condition and N=4–5 total technical replicates/data point. This experiment is one of 3 such pooled experiments with similar results, all of which included αCD3/αCD28 (1 bead for every 5 effector cells). C is representative of two independent experiments. D. Mice were challenged with ID8agg-Luc, treated with IL-2c and sacrificed in cohorts dedicated specifically to such studies. pSTAT5+ prevalence or MFI were measured after incubating control or IL-2c-treated ascites CD45+CD3+ T cells with 100 ng/mL or 5 ng/mL or 30 min. N=4 mice/group. Data from one representative experiment of two.
IL-2c differentially regulates Treg and effector T cell IL-2-dependent STAT5 phosphorylation
Because both CD25 expression and Treg suppression are primarily STAT5-dependent (7), we assessed intracellular IL-2 signal transduction, as differential CD25 expression (Fig. 3B) suggested potentiation of signaling cascades in effector T cells but reduced IL-2 signaling in Tregs. We challenged mice with ID8agg and treated with IL-2c as in Fig. 1, sorted total CD3+ T cells from ascites and TDLN, and pulsed with IL-2 ex vivo. As expected, ascites-derived CD8+ and non-Treg CD4+FoxP3− T cells from IL-2c treated mice versus control treated mice exhibited increased STAT5 phosphorylation (Fig. 4D), indicating that IL-2c enhanced sensitivity to IL-2-dependent intracellular signaling. By contrast, tumor microenvironmental Tregs showed no change in pSTAT5 following incubation with up to 100 ng/mL IL-2 (Fig. 4D). However, Tregs exhibited downregulation of CD25 that mediates high-affinity IL-2 signaling, possibly only compromising their response to lower IL-2 concentrations. In support, after pulsing with low IL-2 ex vivo, we observed reduced pSTAT5 in Tregs in contrast to potentiated responses among CD8+ and CD4+FoxP3− effector T cells (Fig. 4D) while no difference was observed in pSTAT5 of Tregs from TDLN (Fig. S5). These data indicate that CD122-selective IL-2c compromises high-affinity IL-2 signal transduction in Tregs, which helps explain their reduced function, and supports a role for improved IL-2 signaling as a basis for IL-2c-mediated improved effector T cell functions as suggested (18,20–22,39), but not previously demonstrated.
IL-2c increases exhaustion receptor expression on anti-tumor T cells
IL-2c treatment promoted tumor rejection that greatly extended survival, but was not curative. As multiple immune pathways must be corrected for optimal anti-cancer efficacy, we hypothesized that IL-2c promoted chronic antigenic stimulation critical for the expression of exhaustion receptors that could impede cure. PD-1 expression was strongly upregulated on both CD8+ and CD4+FoxP3− effector T cells after IL-2c treatment (Fig. 5A,B). Co-expression of multiple inhibitory receptors could indicate greater T cell activation state or exhaustion (40). We observed increased prevalence of both double-positive lymphocyte activation gene (LAG)-3+PD-1+ CD8+ and CD4+FoxP3− T cells and triple-positive T cell immunoreceptor with Ig and ITIM domains (TIGIT)+LAG-3+PD-1+ CD8+ T cells after IL-2c treatment of ID8agg ascites (Fig. 5C,D). Beneficial IFN-γ expression was enriched among PD-1+ T cells in IL-2c-treated animals (Fig. 5E), consistent with enhanced effector function, proliferation and expression of costimulatory proteins on PD-1+ CD8+ T cells in the peripheral blood of lung cancer patients following αPD-1 treatment (41) and similar to enhanced T cell functions after treating tumor-bearing mice with a human IL-2c construct, although PD-1+ T cells had poor interferon-γ expression in that study (21). Thus, distinct IL-2c constructs appear to mediate distinct immune effects in different tumors.
Figure 5. IL-2c promotes PD-1 expression on tumor microenvironmental cytokine-producing effector T cells.
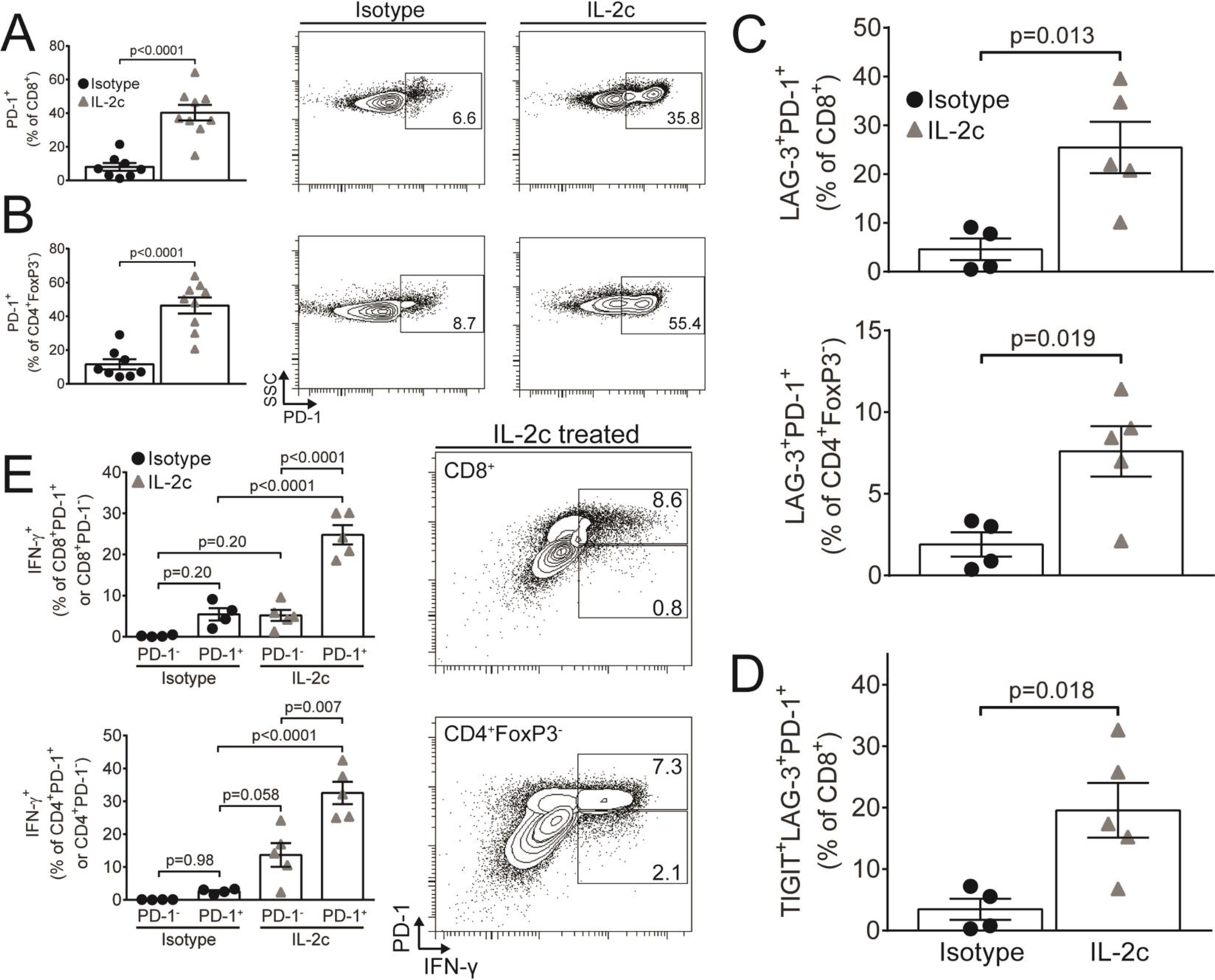
Mice were challenged with ID8agg-Luc, treated with IL-2c only, as in 2A, and 3 weeks after the last IL-2c for studies of ascites T cells. Summary graphs (left) and representative flow plots of PD-1+ prevalence among CD8+ (A) and CD4+FoxP3− (B) T cells. N= 8–9/group. C. LAG-3+PD-1+ prevalence among CD8+ and CD4+FoxP3− T cells after IL-2c treatment. N= 4–5/group. D. TIGIT+LAG-3+PD-1+ prevalence among CD8+ T cells. N= 4–5/group. E. IFN-γ expression among PD-1+ or PD-1− CD8+ (top) or CD4+FoxP3− (bottom) T cells. Numbers in flow plots represent %IFN-γ+ in the CD8+ gate. N= 4–5/group All p values from Student’s unpaired t test. All data are from one dedicated experiment, except A (pooled from two independent experiments).
IL-2c plus αPD-L1 promotes complete OC tumor response with durable, protective immune memory
We found that mice experienced complete tumor eradication after ID8agg challenge using FoxP3DTR mice for specific and near total Treg depletion (Fig. S6A–C), demonstrating the importance of Tregs to immunopathology in this model, as we showed in human OC (4). As IL-2c alone was effective but not curative against ID8agg, we considered adding αCD25 to improve Treg depletion by further preventing IL-2 access to CD25. However, αCD25 unexpectedly reduced IL-2c efficacy on ID8agg based on tumor growth, ascites accumulation and anti-tumor T cell functions (Fig. S7A–E) and thus was not considered further.
αPD-L1 immunotherapy is thought to be less effective in patients without preexisting T cell infiltration (26) and generally more effective in PD-L1+ tumors (42). We showed that both B16 mouse melanoma and ID8agg express PD-L1 (42). In ID8agg, while control mice had essentially no tumor infiltrating lymphocytes (TIL), IL-2c greatly increased intratumoral infiltration of effector T cells as reported in renal cell carcinoma (43) but also increased intratumoral Tregs, although the former to a greater degree (Fig. S8A). Because IL-2c promoted TILs and the expression of exhaustion receptors, particularly PD-1, on anti-tumor T cells we hypothesized that IL-2c-treated mice would derive additional benefit from PD-L1 blockade. Thus, we treated ID8agg-bearing mice with IL-2c as before plus αPD-L1 starting with the third IL-2c dose, to allow time for TIL to accumulate. While αPD-L1 had no effect on ID8agg tumor growth alone, we observed complete reduction of tumor bioluminescence with αPD-L1 addition in nearly all subjects and overall survival significantly exceeding that for IL-2c alone (Fig. 6A–C). For those IL-2c + αPD-L1 treated ID8agg-bearing mice with complete responses >60 days past the original tumor challenge, we re-challenged with ID8agg at the initial challenge inoculum. Tumors grew progressively in untreated, naïve wild type mice as expected. By contrast, tumors were fully rejected by complete responder mice in the absence of additional treatment (Fig. 6D), consistenjt with IL_2c-mediate induction of memory T cells (Fig. S2). Similarly, αPD-L1 improved IL-2c treatment response and survival in B16 melanoma (Fig. 6E–G). Similar to ID8agg, IL-2c increased B16-infiltrating Treg prevalence and numbers (Fig. S8B), but increased CD8+ T cells to a greater extent, thereby increasing the CD8/Treg ratio (Fig.S8C), contrasting with the reduced CD8/Treg ratio after IL-2c in ID8agg (Fig. 2A), but consistent with reported effects of a human IL-2c construct in B16 melanoma (21). IL-2c also reduced Treg functional markers (Fig. S9A,B) without affecting their FoxP3 expression (Fig. S9C) in B16 tumors, similar to effects in ID8agg tumors (Fig. 3B–E). We also observed increased TIGIT+LAG-3+PD-1+ CD8+ T cells in B16 tumors 1 day after IL-2c treatment (Fig. S10), differing from lower T cell exhaustion markers after treating B16 tumor-bearing mice with a human IL-2c construct (21). Comparable times after IL-2c treatment could not be assessed between B16 and ID8agg tumors owing to different kinetics of tumor progression. In both ID8agg and B16, αPD-L1 had little effect on tumor growth when used alone, suggesting pretreatment with IL-2c altered the immune milieu, including promoting TIL as shown, to promote αPD-L1 response. Thus, IL-2c sensitizes tumors to αPD-L1 in two distinct tumors in distinct anatomic compartments and is curative even in poorly immunogenic tumors with accompanying durable, protective immune memory.
Figure 6. IL-2c improves αPD-L1 to effect cures in ID8agg.
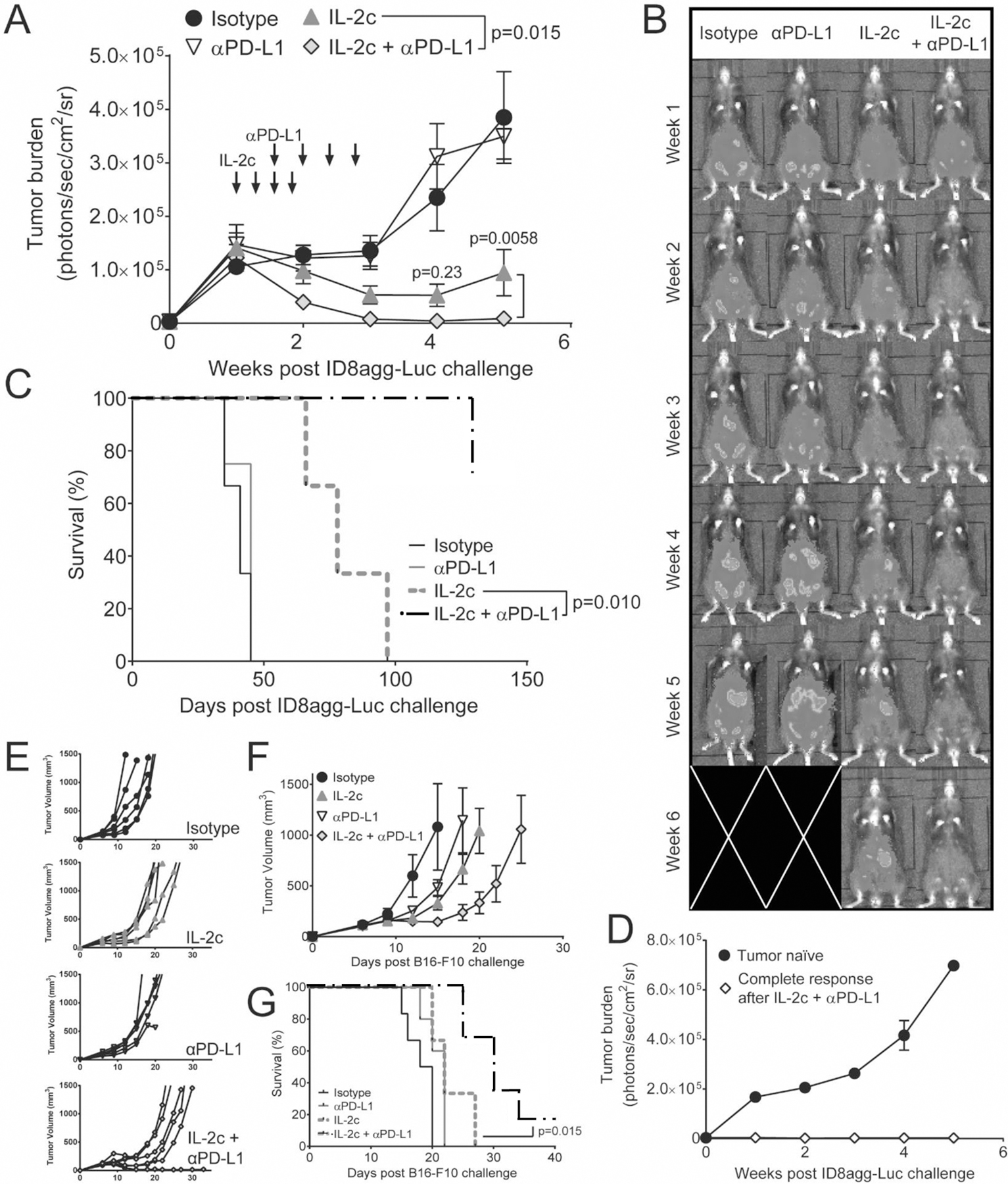
A. ID8agg tumor bioluminescence after challenge with ID8agg-Luc and treatment with IL-2c and/or αPD-L1 as indicated (arrows) (top left), with representative images (B) and survival (C). N=8–10 mice in A, N=3–4 mice in D. P values from two-way repeated measures ANOVA (figure legend) with post hoc Sidak’s test (graph points) (A) and log-rank test (C). D. ID8agg bioluminescence in WT tumor naïve mice or mice with a complete response lasting >60 days after the last IL-2c plus αPD-L1. N=4 tumor naïve, 1 CR mice. Individual tumor growth curves (E), group tumor growth (F), and survival (G) following challenge with B16-F10 and treatment with IL-2c and/or αPD-L1 as before. N=5–6 per group. P value from log-rank test. Panel A is pooled from two independent experiments, while C-G are representative of two independent experiments. Data in panel C for control or IL-2c was also used in Fig 1.
Discussion
Results from over three decades of OC immunotherapy clinical trials have largely been disappointing (44). OC has a relatively lower mutation load compared to other carcinomas that have exhibited immunotherapy trial successes (e.g., melanoma, lung, renal cell, and bladder cancer) (45), possibly making OC intrinsically more difficult to treat with immunotherapy. Nonetheless, intratumoral T cell infiltration predicts improved OC patient survival (46) that is reduced by intratumoral Tregs (4), providing a strong rationale for pursuit of effective anti-OC immunotherapy.
Despite its substantial side effects, treatment of cancer patients with IL-2 is among the first immunotherapies to generate cures in patients with treatment-resistant cancers (47). Nonetheless, aside from limited efficacy, IL-2 also suffers from significant toxicities and its ability to promote Tregs (47,48). The IL-2R is an excellent immunotherapy target for further studies of agents mediating multiple, beneficial immune outcomes, owing to its control of both anti-tumor and regulatory T cells. In preclinical studies, genetic ablation of the IL-2/IL-2R/STAT5 axis leads to life-threatening T cell-driven autoimmunity due to lack of functional Tregs (8,13–15), indicating dominant IL-2 control over Treg function and IL-2 dispensability to control excessive T cell activation. Thus, targeting the IL-2R for Treg ablation has a clear rationale to boost anti-tumor immunity, although an effective strategy in that regard remains to be defined.
Here we tested the effect of IL-2 complex comprised of IL-2 and αIL-2 (clone JES6–5H4), a S4B6-like clone, which selectively activated CD122 to improve anti-tumor effector functions while avoiding stimulating Tregs (20). IL-2c are known to promote anti-tumor immunity by direct stimulation of effector T cells (18,20–22,39), but we provide much insight into additional mechanisms that can improve clinical translation and optimal use of IL-2c and related agents. In contrast to studies in B16 melanoma, where tumor growth accelerates quickly after withdrawal of IL-2c treatment (22) (Fig. 6), we show that ID8agg tumor burden remains stable for weeks following IL-2c clearance, suggesting durably augmented anti-tumor immunity particularly for OC, the peritoneal microenvironment or both, or could reflect insufficient time for effective memory development in rapidly growing B16 tumors. Such effects could be directly on anti-tumor T cells, or through reducing microenvironmental immunosuppression (such as reducing Treg suppression) and could involve special features of the microenvironment of the cells circulating there, an area requiring additional study. Our data suggest that both mechanisms are operative, and that IL-2c promote long-lasting immunity to OC, a tumor notoriously refractory to immunotherapy (44). IL-2c reduces OC microenvironmental Treg CD25 expression and suppressive functions, and simultaneously increases CD25 expression and IL-2 responsiveness of effector T cells. Thus, IL-2c could promote anti-tumor immunity by decreasing IL-2 sequestration from anti-tumor T cells by Treg CD25 (7), inhibiting IL-2R-mediated Treg functions (7), promoting IL-2-driven anti-tumor effector cell functions (10) or some combination of these effects.
Multiple reports confirm that CD122-selective stimulation of the IL-2R promotes expansion of CD8+ T cells and NK cells much greater than proliferation of Tregs (18,22). However, none examined Treg functions in detail, despite the potential for IL-2c effects on all IL-2-dependent cells. We showed that IL-2c reduced Treg effector molecules for a prolonged period after the final dose in the peritoneum. CD122-selective αIL-2 antibodies block the CD25 binding epitope on IL-2 (39) which appears to deprive Tregs of IL-2 critical to differentiation and effector functions (6,7). We further found that IL-2c induced Treg fragility, which is associated with improved immune checkpoint blockade efficacy (37). As Treg fragility is induced by local IFN-γ (37), IL-2c-generated IFN-γ in local T cells or NK cells could contribute to this effect. Alternatively, improved effector T cell functions could promote sufficient inflammation to destabilize Treg differentiation secondarily in conjunction with IL-2c and ablate their suppressive functions as seen in toxoplasmosis (49). Finally, we cannot exclude the possibility that IL-2c selectively reduced a population of suppressive Tregs aside from inducing fragility. These issues and the basis for the site-specific effect of IL-2c on Tregs merit additional investigations.
IL-2c are thought to activate anti-tumor immunity in a CD25-independent manner, suggesting combination treatment with αCD25 could improve clinical responses. Unexpectedly, we found that αCD25 reduced IL-2c efficacy. Thus, either high-affinity IL-2 signaling via CD25 or the actions of CD25+ cells (which are depleted by αCD25) are crucial for optimal anti-tumor activities of IL-2c. Recent work has shown that CD25 is responsible for intracellular recycling of IL-2 and prolonging STAT5 activation when anti-tumor T cells leave IL-2-rich TDLN and enter relatively IL-2-poor peripheral tissues such as the tumor microenvironment (10). αCD25 depletion of newly formed CD25+ effector cells induced by IL-2c treatment likely helps explain our finding of αCD25-mediated reduction in IL-2c efficacy
We also found other differences here versus prior studies of IL-2c. For example, we found that IL-2c promoted T cell exhaustion markers, whereas others report little increase in these with a human IL-2c construct (22), which could be from differences in details of the constructs, including actions of human versus mouse IL-2 on mouse immune cells, the fact that we studied tumors later in their evolution and different timing of treatment initiation and interval until immune studies were performed. Nonetheless, our studies here accord with prior work showing improved CD8+ T cell functions using similar IL-2c constructs.
The use of combinations of multiple chemotherapy agents, radiation, surgery, and/or other specific targeted therapies is used to achieve optimal responses in cancer patients. αPD-L1 (and other) immunotherapy works best in inflamed tumors with T cell enriched TIL (50). We identified improving TIL numbers as another novel IL-2c mechanism and thus tested the ability of IL-2c to improve αPD-L1. We showed that in two distinct tumors (ID8agg OC and B16 melanoma) in two distinct anatomic compartments (peritoneum and skin, respectively), αPD-L1, which alone at these doses and schedule had little effect on tumor growth, effectively improved tumor responses in each model. Further, αPD-L1 was curative in the aggressive, poorly antigenic ID8agg model for OC when combined with IL-2c. Increased PD-1 expression on anti-tumor T cells could improve response to αPD-L1 (51). IL-2c also augmented local T cell expression of exhaustion markers including PD-1, although we and others (50,52) have shown that some PD-1+ cells have significant effector functions after immunotherapy for OC, including data presented here. IL-2c differentially affected the intratumoral CD8/Treg ratio in the OC versus melanoma models studied here suggesting tumor or compartment effects of IL-2c that merit additional investigations. IL-2c induction of fragile Tregs which fragile Tregs promotes αPD-1 efficacy in other tumor models (37,53) and could contribute to IL-2c-mediated potentiation of αPD-L1 efficacy in these studies.
Our study has some limitations. We only tested IL-2c effects in vivo in mice. However, there is significant homology between rodent and human IL-2 and IL-2R subunits confirmed in vivo by effectiveness of human IL-2 in mouse models of melanoma (21), consistent with relevance of these data to humans. A potential obstacle in the translation of IL-2c could be discordance in the distribution of high- or medium-affinity IL-2R among effector and regulatory T cells between cancer patients and rodents (e.g., if human Tregs have a greater ability to bind CD122-selective IL-2c) that could undermine the selectivity of IL-2c to promote the strongest stimulation in effector T cells and thus mediate anti-tumor immunity. Targeting IL-2 to medium-affinity IL-2R with human IL-2c was recently reported that replicated major mouse IL-2c effects in murine cancer models (21). Nonetheless, no Treg effects were reported. There is additional preclinical evidence for the use of CD122-selective IL-2 and related strategies to reduce IL-2/CD25 interactions in clinical trials, including pegylated IL-2, IL-2 with the CD25 binding site genetically deleted and IL-2 bound to CD25 (21,54–56). Pegylated IL-2 (NKTR214, bempegaldesleukin) shows promise in metastatic melanoma when combined with nivolumab (αPD-1) (57), and just received FDA Breakthrough designation on that basis. Based on our data, these specific constructs could have differential effects on Tregs or other immune cells, a subject meriting additional studies. Our data support a trial of IL-2c or similar agent in the treatment of OC, especially in combination with αPD-L1.
Supplementary Material
Significance:
Findings present CD122-targeted IL-2 complexes as an advancement in cancer immunotherapy, as they reduce Treg immunosuppression, improve anti-cancer immunity, and boost PD-L1 immune checkpoint blockade efficacy in distinct tumors and anatomic locations.
Acknowledgements
This study was supported by grants from NCI (CA164122, CA054174, CA205965), the Ovarian Cancer Research Fund Alliance (290498), the Skinner endowment, The Holly Beach Public Library, The Owens Foundation, and The Barker Foundation to TJC; the NCATS (TL1 TR001119) to JMD, the NCI (CA205568) to CAC and CPRIT (RP170345) and Ovarian Cancer Research Alliance to YD. We thank the UTHSA MD/PhD program for financial and administrative support to JMD and CAC. We thank Shunhua Lao and June Deng for animal colony management.
Funding: This study was supported by grants from NCI (CA164122, CA054174, CA205965), the Ovarian Cancer Research Fund Alliance (290498), the Skinner endowment, The Holly Beach Public Library, The Owens Foundation, and The Barker Foundation to TJC; the NCATS (TL1 TR001119) to JMD, the NCI (CA205568) to CAC, the CPRIT Research Training Award (RP170345) and Ovarian Cancer Research Alliance to YD, JC-G and TJC.
Footnotes
Conflict of interest statement: All authors have declared no conflict of interest exists.
References
- 1.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med 2016 [DOI] [PMC free article] [PubMed]
- 2.Zou W Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 2006;6:295–307 [DOI] [PubMed] [Google Scholar]
- 3.Curiel TJ. Tregs and rethinking cancer immunotherapy. J Clin Invest 2007;117:1167–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004;10:942–9 [DOI] [PubMed] [Google Scholar]
- 5.Jacobs JF, Nierkens S, Figdor CG, de Vries IJ, Adema GJ. Regulatory T cells in melanoma: the final hurdle towards effective immunotherapy? The Lancet Oncology 2012;13:e32–42 [DOI] [PubMed] [Google Scholar]
- 6.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nature immunology 2005;6:1142–51 [DOI] [PubMed] [Google Scholar]
- 7.Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nature immunology 2016 [DOI] [PMC free article] [PubMed]
- 8.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol 2007;178:280–90 [DOI] [PubMed] [Google Scholar]
- 9.Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med 2014;211:121–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su EW, Moore CJ, Suriano S, Johnson CB, Songalia N, Patterson A, et al. IL-2Ralpha mediates temporal regulation of IL-2 signaling and enhances immunotherapy. Science translational medicine 2015;7:311ra170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malek TR. The biology of interleukin-2. Annual review of immunology 2008;26:453–79 [DOI] [PubMed] [Google Scholar]
- 12.Nakamura Y, Russell SM, Mess SA, Friedmann M, Erdos M, Francois C, et al. Heterodimerization of the IL-2 receptor beta- and gamma-chain cytoplasmic domains is required for signalling. Nature 1994;369:330–3 [DOI] [PubMed] [Google Scholar]
- 13.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 1993;75:253–61 [DOI] [PubMed] [Google Scholar]
- 14.Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 1995;3:521–30 [DOI] [PubMed] [Google Scholar]
- 15.Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science (New York, NY) 1995;268:1472–6 [DOI] [PubMed] [Google Scholar]
- 16.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995;155:1151–64. [PubMed] [Google Scholar]
- 17.Setiady YY, Coccia JA, Park PU. In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. European journal of immunology 2010;40:780–6 [DOI] [PubMed] [Google Scholar]
- 18.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science 2006;311:1924–7 [DOI] [PubMed] [Google Scholar]
- 19.Letourneau S, van Leeuwen EM, Krieg C, Martin C, Pantaleo G, Sprent J, et al. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor alpha subunit CD25. Proc Natl Acad Sci U S A 2010;107:2171–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: Biology, Design and Application. Trends Immunol 2015;36:763–77 [DOI] [PubMed] [Google Scholar]
- 21.Arenas-Ramirez N, Zou C, Popp S, Zingg D, Brannetti B, Wirth E, et al. Improved cancer immunotherapy by a CD25-mimobody conferring selectivity to human interleukin-2. Sci Transl Med 2016;8:367ra166. [DOI] [PubMed] [Google Scholar]
- 22.Krieg C, Letourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci U S A 2010;107:11906–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ribas A, Hu-Lieskovan S. What does PD-L1 positive or negative mean? The Journal of experimental medicine 2016;213:2835–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark CA, Gupta H, Sareddy GR, Pandeswara S, Lao S, Yuan B, et al. Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis and autophagy in ovarian cancer and melanoma. Cancer research 2016 [DOI] [PMC free article] [PubMed]
- 28.Baert T, Verschuere T, Van Hoylandt A, Gijsbers R, Vergote I, Coosemans A. The dark side of ID8-Luc2: pitfalls for luciferase tagged murine models for ovarian cancer. Journal for immunotherapy of cancer 2015;3:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Downs-Canner S, Berkey S, Delgoffe GM, Edwards RP, Curiel T, Odunsi K, et al. Suppressive IL-17A+Foxp3+ and ex-Th17 IL-17AnegFoxp3+ Treg cells are a source of tumour-associated Treg cells. Nat Commun 2017;8:14649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Preston CC, Maurer MJ, Oberg AL, Visscher DW, Kalli KR, Hartmann LC, et al. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PloS one 2013;8:e80063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A 2005;102:18538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007;27:635–46 [DOI] [PubMed] [Google Scholar]
- 33.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 2004;21:589–601 [DOI] [PubMed] [Google Scholar]
- 34.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008;322:271–5 [DOI] [PubMed] [Google Scholar]
- 35.Anz D, Mueller W, Golic M, Kunz WG, Rapp M, Koelzer VH, et al. CD103 is a hallmark of tumor-infiltrating regulatory T cells. International journal of cancer Journal international du cancer 2011;129:2417–26 [DOI] [PubMed] [Google Scholar]
- 36.Gu J, Ni X, Pan X, Lu H, Lu Y, Zhao J, et al. Human CD39(hi) regulatory T cells present stronger stability and function under inflammatory conditions. Cell Mol Immunol 2017;14:521–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Interferon-gamma Drives Treg Fragility to Promote Anti-tumor Immunity. Cell 2017;169:1130–41 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 2007;445:766–70 [DOI] [PubMed] [Google Scholar]
- 39.Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation through Distinct Conformational Mechanisms. Immunity 2015;42:815–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature immunology 2009;10:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A 2017;114:4993–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res 2016;76:6964–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han KH, Kim KW, Yan JJ, Lee JG, Lee EM, Han M, et al. Effects of stimulating interleukin −2/anti- interleukin −2 antibody complexes on renal cell carcinoma. BMC Urol 2016;16:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drerup JM, Liu Y, Padron AS, Murthy K, Hurez V, Zhang B, et al. Immunotherapy for ovarian cancer. Curr Treat Options Oncol 2015;16:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003;348:203–13 [DOI] [PubMed] [Google Scholar]
- 47.Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. Journal of immunology (Baltimore, Md : 1950) 2014;192:5451–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei S, Kryczek I, Edwards RP, Zou L, Szeliga W, Banerjee M, et al. Interleukin-2 administration alters the CD4+FOXP3+ T-cell pool and tumor trafficking in patients with ovarian carcinoma. Cancer Res 2007;67:7487–94 [DOI] [PubMed] [Google Scholar]
- 49.Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity 2009;31:772–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16:275–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Massard C, Gordon MS, Sharma S, Rafii S, Wainberg ZA, Luke J, et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. J Clin Oncol 2016;34:3119–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hurez V, Dao V, Liu A, Pandeswara S, Gelfond J, Sun L, et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell 2015;14:945–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Overacre AE, Vignali DA. T(reg) stability: to be or not to be. Curr Opin Immunol 2016;39:39–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22:680–90 [DOI] [PubMed] [Google Scholar]
- 55.Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, Ju W, et al. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity 2015;42:826–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature 2012;484:529–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hurwitz M, Cho D, Balar A, Curti B, Siefker-Radtke A, Sznol M, et al. Baseline Tumor Immune Signatures Associated with Response to Bempegaldesleukin (NKTR-214) and Nivolumab. American Society of Clinical Oncology 2019:Abstract 2623
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.