

Abstract
Ectopic hamartomatous thymoma is a rare neck lesion originally thought to represent a non-neoplastic hamartoma, even though thymic origin has been questioned, and there is uncertainty about whether the lesion is a neoplasm. We investigated the genetics by performing targeted next generation sequencing (NGS). Three cases were identified from the authors’ consultation files. A custom, targeted NGS panel including 1385 pan-cancer‐related genes was performed on all cases. Three patients included 2 males and 1 female, aged 50, 58 and 70 years, respectively (mean 59.3 years), with tumors arising in the low anterior neck. All cases showed classical histologic features of EHT, with one case showing intraductal carcinoma in association with the EHT. By targeted NGS, one case harbored a hotspot HRAS mutation (p.Gln61Lys), while the other two cases only showed non oncogenic variants. Dual mesoderm and endoderm derivation/differentiation (biphenotypic) has been previously recognized, with epithelial and myoepithelial components, and arising from the apparatus contributing to neck development (branchial apparatus). Thus, EHT has been shown to have genetic alterations in HRAS. These findings, without evidence of thymic derivation or an ectopic tissue location, strongly support that EHT is a true neoplasm. The name biphenotyic branchioma more correctly reflects the true nature of this dual mesoderm and endoderm derived tumor occurring in the lower neck.
Keywords: Ectopic hamartomatous thymoma, Biphenotypic branchioma, Carcinoma, Branchial pouch, NGS, Neoplasm
Introduction
Ectopic hamartomatous thymoma (EHT) is a rare benign neoplasm that almost exclusively occurs in the lower neck of middle-aged patients with a remarkable male predominance as reported in a recent review the literature [1]. These lesions show an admixture of spindle cells, epithelial islands, and adipocytes. Since the original unifying descriptions [2–4], there has been continued discussion about nomenclature. The term biphenotypic branchioma was recently introduced as a better name for this lesion [1]. There has been no definitive genetic evaluation of this lesion and thus we sought to further understand the genetic features of this tumor by performing next generation sequencing on a series of cases.
Methods
Case Selection
Cases fulfilling the criteria for EHT were selected from the authors’ consultation files (one previously reported [1]). These criteria include low anterior neck presentation of a well circumscribed mass lesion, composed of an admixture of spindle cells, epithelial islands, and adipocytes, with plump spindled cells. Immunophenotypic analysis was performed in cases with sufficient suitable material by a standardized Envision™ method employing 4 µm-thick, formalin fixed, paraffin embedded sections. The commercially available immunohistochemical antibodies included: pancytokeratin (AE/AE3, Dako and Becton-Dickson, 1:40 and 1:8), p63 (Leica Microsystems, 1:40), p40 (Biocare, 1:200), CK5/6 (Dako, 1:25), SOX10 (Epitomics, 1:250), S100 protein (polyclonal, Dako, 1:2000) and androgen receptor (Biogenex, 1:2000). The analysis was performed on a single representative block for each primary tumor. Epitope retrieval was performed, as required by the manufacturer guidelines. Standard positive controls were used throughout, with serum used as the negative control. One of the cases showed intraductal carcinoma within the neoplasm, but confined to the capsule of the tumor, as previously described [1].
Next-Generation Sequencing
DNA and RNA were isolated from formalin-fixed, paraffin-embedded tissue. Sequencing libraries were generated using Kapa Biosystems and Illumina chemistry. A custom panel of DNA probes were used to produce an enriched library containing all exons from over 1385 cancer-related genes, which were sequenced on Illumina HiSeq 4000, NextSeq 550 or MiSeq instruments. DNA and RNA sequence analyses were done using custom germline, somatic and mRNA bioinformatics pipelines run on the UTSW Bio-High Performance Computer cluster and optimized for detection of single nucleotide variants, indels and known gene fusions. Reports were generated in the Philips IntelliSpace Genomics system (Philips Healthcare, 2 Canal Park, Cambridge, MA). Median target exon coverage for the assay is 900X with 94% of exons at > 100X. The minor allele frequency limit of detection is 5% for single nucleotide variants and 10% for indels and known gene fusions. The assay is not informative for mutations outside the 1385 cancer-related genes or for those regions for which the assay achieves limited coverage. Full details of the genes tested, exon coverage and the bioinformatics pipeline are available at https://www.utsouthwestern.edu/sites/genomics-molecular-pathology/. Somatic variants were identified on the basis of their variant allele frequencies (VAF), as well as their presence in databases of germline variants including dbSNP and gnomAD. All variants were reviewed in the Integrated Genomics Viewer (IGV) software prior to reporting.
Results
Three cases of EHT were retrieved from the consultation files of the authors. They were identified in the low anterior neck (Fig. 1) of 2 men and one woman, aged 50, 58 and 70 years, respectively (mean 59.3 years). The tumors were 4.4, 3.7 and 3.5 cm in greatest dimension, respectively (mean 3.9 cm). Patients presented with a slowly-growing, painless mass, between 1 and 10 years in duration. No patients had syndrome association and none had documented malignancies elsewhere. Follow-up is short, but there has been no recurrence or metastasis (in the malignant case), with a mean follow-up of 2.3 years.
Fig. 1.
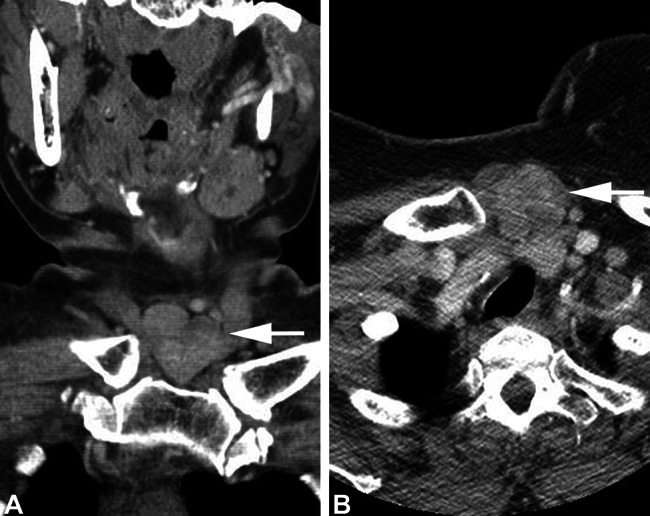
Computed tomography imaging of the low anterior neck tumor (a, white arrow), identified between the heads of the clavicle, showing (b) a mixed heterogeneous soft tissue density mass (white arrow)
All cases revealed the characteristic diagnostic findings of EHT, showing a well circumscribed or encapsulated haphazard proliferation of spindled cells, epithelial islands and adipocytes (Fig. 2). The epithelial cells formed non-keratinizing squamoid islands or glandular structures (Fig. 3). The spindled cells were fine and delicate or plump and coarse, the latter arranged in a fascicular pattern (Fig. 4). The adipocytic tissue was identified haphazardly within the proliferation. The epithelial islands were immunoreactive with pancytokeratin, CK5/6, p40 and p63 in all of the cases. SOX10 and S100 protein highlighted spindled myoepithelial cells. One of the cases showed an intraductal carcinoma, with features that ranged from an atypical ductal hyperplasia, to atypical hyperplasia to more pleomorphism as would be seen in an intraductal carcinoma (Fig. 5). No desmoplasia was identified, but the glandular cells were strongly positive with androgen receptor, while S100 protein negative. The remaining tumors did not show androgen receptor reactivity.
Fig. 2.
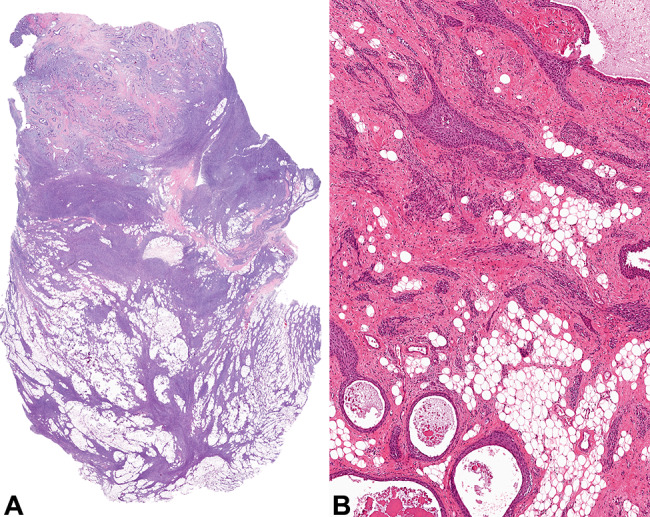
a A well circumscribed tumor demonstrating epithelial and spindled cells associated with adipocytic tissue. b Squamous epithelial lined cysts merge with elongated, anastomosing epithelial islands associated with fat
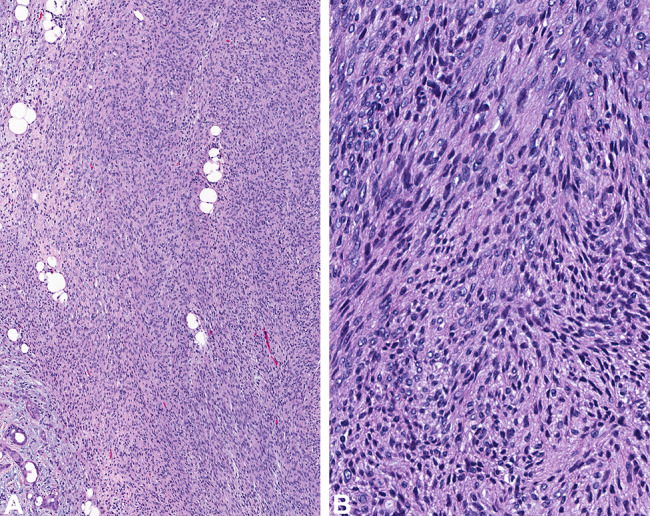
Fig. 3.
The epithelial component demonstrates a squamoid epithelial lined spaces, while b a more glandular appearance is seen in other areas. Note the background fibrous connective tissue
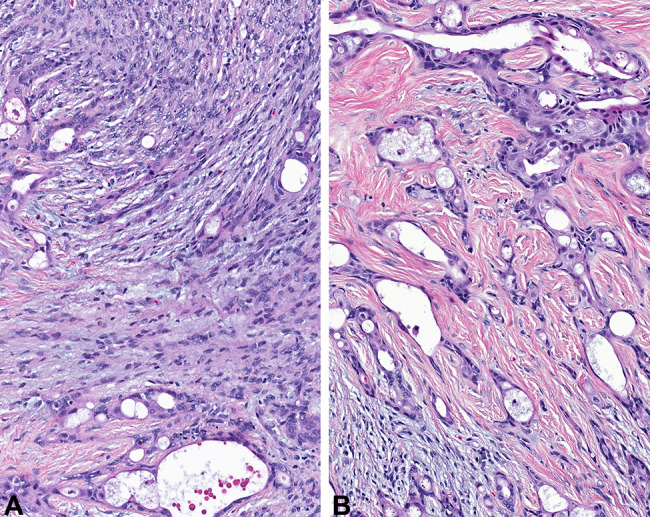
Fig. 4.
The stromal component was composed of a spindled cell proliferation, arranged in a haphazard (a) to fascicular (b) architecture
Fig. 5.
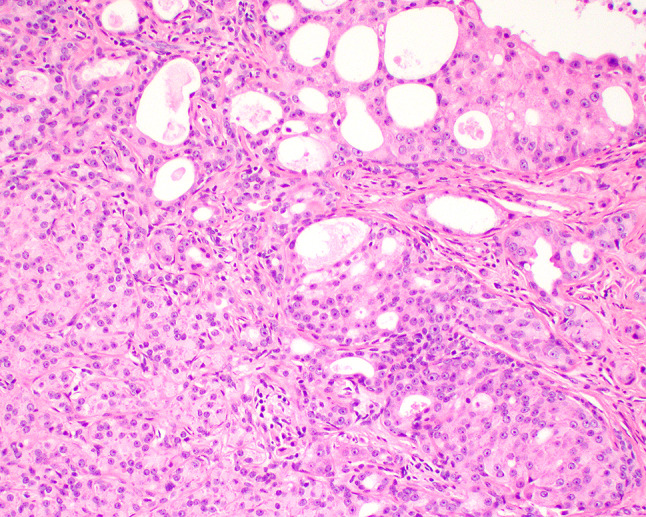
Within the epithelial compartment, a transformation to intraductal carcinoma is noted with a Roman-ridge formation, showing ample eosinophilic cytoplasm
Targeted NGS results are summarized in Table 1. One case featured a hotspot mutation in HRAS (pGln61Lys). No definitive oncogenic drivers were detected in the other two cases. There were also no copy number alterations found. Fusions were not found by RNA sequencing in any of the cases for which RNA was successfully isolated.
Table 1.
Next-generation sequencing results for the biphenotypic branchioma cases
| Case # | Clinical information | Variant | Coordinate | Tumor allele frequency (%) | Favor Germline versus Somatic |
|---|---|---|---|---|---|
| 1 |
50 year old male; 4 year history of left low anterior neck mass, 4.4 cm; A, NED 4 years |
RET c.166C > A (p.Leu56Met) |
Chr10:43595999 | 46.73 | Germline |
| NF1 (p.Thr2335Ala) | Chr17:29667604 | 54.17 | Germline | ||
| RPA3 | Chr7:7677493 | 47.20 | Germline | ||
| 2 |
58 year old male; 10 year history of left supraclavicular mass, 3.7 cm; A, NED < 1 year |
ARID1A (p.Ala1757Ser) | Chr1:26779167 | 48.23 | Germline |
| MIB1 (p.Asp455Gly) | Chr18:21799967 | 52.19 | Germline | ||
| HSP90AB1 (p.Arg449His) | Chr6:44251768 | 46.89 | Germline | ||
| SERPINF1 (p.Glu324Lys) | Chr17:1776715 | 44.11 | Germline | ||
| RFC2 (p.Leu261fs) | Chr7:74237422 | 41.74 | Germline | ||
| 3 |
70 year old female; 1 year history of midline suprasternal mass, 3.5 cm; A, NED, 2 years |
HRAS c.181C > A (p.Gln61Lys) | Chr11:533875 | 29.13 | Somatic |
| PI4KA (p.Arg1941Ter) | Chr22:21065731 | 38.89 | Germline | ||
| SFRP4 (p.Met1?) | Chr7:37956138 | 49.77 | Germline |
A, NED alive, no evidence of disease
Discussion
When aggregating all of the cases from the literature, as previously reported (1), middle aged male patients (mean 46 years; M:F = 3.4:1) present with a low anterior supraclavicular/suprasternal neck mass (mean size, 4.9 cm). The present series of 3 cases fits well within these clinical findings. The branchial apparatus embryologically gives rise to a very complex and coordinated development of the neck structures, with significant anatomic variation and anomalies. There are many neck structures and organs derived from the branchial pouches, clefts and arches, respectively. Thus, during embryologic development, it is easy to extrapolate a derivative of these “branchial” apparatus as responsible for the origin of these tumors, as was postulated in the original papers, with the 3rd branchial arch responsible for thymus origin. These apparatus give rise to fat, myoepithelial and epithelial cells, including squamous and glandular epithelium, and thus have “branchial” origin. The etymology of “branchioma” encompass the likely origin of the neoplasm (branchi), as well as using the Greek derived suffix “əʊmə” (“oma”), to indicate “morbid growth, tumor.”
Previous molecular evaluation has been limited to showing the absence of PLAG1 rearrangements, helping to exclude pleomorphic adenoma as a potential candidate diagnosis [5]. There are no consistent molecular findings in these three cases tested. Thus, with only three cases tested, perhaps additional cases should be evaluated to aid in further clarifying the molecular findings of this neoplasm. The lesion is not ectopic, as the components of the tumor are normal for the location embryologically (eutopic). PAX8 is absent in any of the neoplastic cells of cases tested, while it is normally present in thymic epithelial cells and neoplasms [6]. Myoepithelial differentiation is seldom seen in thymic tumors, which is seen in the spindled cells of this tumor. These myoepithelial cells demonstrate a plump appearance, highlighted by p63, p40, CK5/6, SOX10 and S100 protein, although to a variable degree and intensity within each category. This finding confirms that of previous authors, although we did not repeat the CD34, which is known to be positive in these specialized myofibroblasts spindle cells and not the squamous epithelial cells [6]. The entity is not seen in the mediastinum, thymus, or is thymic tissue seen within or adjacent to the lesion. Thus, “thymoma” or “thymic anlage tumor” cannot be applied to this tumor when thymic histology or phenotype cannot be supported [6].
One of the last hurdles has been around hamartoma versus neoplasm (it is obviously a tumor in the “mass” sense of the word). The presence of the HRAS variant in one case may represent the carcinoma component rather than the benign components, but as the atypical component was blended and mingled throughout, this distinction cannot be reliable reported. Still, is there enough data to infer neoplasm or malignancy from molecular data? Oncogenic drivers being detected in otherwise benign conditions is a well-described phenomenon [7]. For example, PIK3CA mutations can be found in seborrheic keratosis [8] and low level mosaic mutations have been reported in lipomatosis of nerve-associated macrodactyly, which is considered a lipofibromatous hamartoma by some [9]. The RET p.L56M is possibly a rare germline variant, and thus may not be an oncogenic driver (https://gnomad.broadinstitute.org/variant/10-43595999-C-A?dataset=gnomad_r2_1. Accessed 28 November, 2019), with similar findings for the NF1 p.T2335A as a possible single nucleotide polymorphism (SNP). Similarly, ARID1A may be a variant of uncertain significance and may be a private SNP, not necessarily adding to the known disease associations with this genetic finding as it SMARCB1 and its related genes encoding the SWI/SNF subunits (specifically ARID1A) [10]. As such, somatic mutations are clearly documented in the proliferation evaluated, but we do not intend to imply a new disease association by documenting the genetic findings in these neoplasms. The oncogenic nature is too unclear in this tumor at this time.
Data suggests that intraductal carcinoma is actually comprised of at least two variants: (1) the intercalated duct-like type which is S100 protein-positive, negative for androgen receptor and usually harbors RET rearrangements, and (2) the apocrine type which is androgen receptor positive, S100 protein-negative, and has salivary duct carcinoma-like genetic changes [11, 12]. When non-invasive (i.e., intraductal carcinoma) in salivary glands, the tumor has a very indolent behavior. The tumor in patient #3 showed histologic features of intraductal carcinoma, revealed a positive androgen receptor in the epithelial cells, myoepithelial cells negative with S100 protein, and a RET mutation, thus providing support for an intraductal carcinoma diagnosis in this case.
“Branchial anlage mixed tumor” [3, 13] was proposed to encompass the epithelial and myoepithelial features, but the term “mixed tumor” is too encompassing of many other lesions, with salivary gland mixed tumor excluded by the known molecular findings. With the anatomic site of the supraclavicular neck in the vast majority of cases, it seems that a branchial apparatus origin is supported. The term biphenotypic branchioma more closely encompasses the origins and tissue types seen in this tumor (which include epithelium, spindled cells and fat, but from only two primordial layers). Taxonomy is always a source of frustration and controversy, with more time devoted to the rare and uncommon entities than is probably worthwhile given the overall clinical incidence of these lesions. However, when you are the patient with a diagnosis, not having a uniform classification of the tumor is disturbing at least, and may at worst result in delayed or incomplete management. Towards a more anatomically, biologically and histologically accurate term, biphenotypic branchioma should replace EHT, branchial anlage mixed tumor and thymic anlage tumor. Clearly, the authors encourage others to perform additional evaluation of these neoplasms, with next generation sequencing, which may provide additional support for our hypothesis or add other molecular findings. Biphenotypic is based on the presence of mesoderm and endodermal germ layer derivatives even though represent by three components histologically (epithelium, spindled myoepithelial cells and fat). “Branchi” implies branchial apparatus origin while “oma” supports the concept of a neoplasm. When carcinoma develops, the type of carcinoma can be added to the term, such as “intraductal carcinoma arising within biphenotypic branchioma”.
Acknowledgements
The views expressed are those of the authors solely and do not represent endorsement from Southern California Permanente Medical Group or the University of Texas Southwestern Medical Center.
Funding
This study was funded in part by the Jane B. and Edwin P. Jenevein M.D. Endowment for Pathology at UT Southwestern Medical Center.
Compliance with Ethical Standards
Conflict of interest
All authors declare that he/she has no conflict of interest as it relates to this research project.
Ethical Approval
All procedures performed in this retrospective data analysis involving human participants were in accordance with the ethical standards of the institutional review board (IRB #5968), which did not require informed consent.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sato K, Thompson LDR, Miyai K, Kono T, Tsuda H. Ectopic hamartomatous thymoma: a review of the literature with report of new cases and proposal of a new name: biphenotypic branchioma. Head Neck Pathol. 2017;12(2):202–209. doi: 10.1007/s12105-017-0854-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosai J, Limas C, Husband EM. Ectopic hamartomatous thymoma. A distinctive benign lesion of lower neck. Am J Surg Pathol. 1984;8(7):501–513. doi: 10.1097/00000478-198407000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Fetsch JF, Weiss SW. Ectopic hamartomatous thymoma: clinicopathologic, immunohistochemical, and histogenetic considerations in four new cases. Hum Pathol. 1990;21(6):662–668. doi: 10.1016/S0046-8177(96)90014-3. [DOI] [PubMed] [Google Scholar]
- 4.Chan JK, Rosai J. Tumors of the neck showing thymic or related branchial pouch differentiation: a unifying concept. Hum Pathol. 1991;22(4):349–367. doi: 10.1016/0046-8177(91)90083-2. [DOI] [PubMed] [Google Scholar]
- 5.Liang PI, Li CF, Sato Y, Lee VK, Bahrami A, Chuang SS. Ectopic hamartomatous thymoma is distinct from lipomatous pleomorphic adenoma in lacking PLAG1 aberration. Histopathology. 2013;62(3):518–522. doi: 10.1111/his.12022. [DOI] [PubMed] [Google Scholar]
- 6.Weissferdt A, Kalhor N, Petersson F, Moran CA. Ectopic Hamartomatous thymoma-new insights into a challenging entity: a clinicopathologic and immunohistochemical study of 9 cases. Am J Surg Pathol. 2016;40(11):1571–1576. doi: 10.1097/PAS.0000000000000699. [DOI] [PubMed] [Google Scholar]
- 7.Kato S, Lippman SM, Flaherty KT, Kurzrock R. The conundrum of genetic "drivers" in benign conditions. J Natl Cancer Inst. 2016;108(8):djw036. doi: 10.1093/jnci/djw036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hafner C, Lopez-Knowles E, Luis NM, Toll A, Baselga E, Fernandez-Casado A, et al. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc Natl Acad Sci USA. 2007;104(33):13450–13454. doi: 10.1073/pnas.0705218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu J, Tian W, Tian G, Sumner K, Hutchinson DT, Ji Y. An investigation of PIK3CA mutations in isolated macrodactyly. J Hand Surg Eur. 2018;43(7):756–760. doi: 10.1177/1753193418770366. [DOI] [PubMed] [Google Scholar]
- 10.Le Loarer F, Zhang L, Fletcher CD, Ribeiro A, Singer S, Italiano A, et al. Consistent SMARCB1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by FISH in archival material. Genes Chromosom Cancer. 2014;53(6):475–486. doi: 10.1002/gcc.22159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skalova A, Vanecek T, Uro-Coste E, Bishop JA, Weinreb I, Thompson LDR, et al. Molecular profiling of salivary gland intraductal carcinoma revealed a subset of tumors harboring NCOA4-RET and novel TRIM27-RET fusions: a report of 17 cases. Am J Surg Pathol. 2018;42(11):1445–1455. doi: 10.1097/PAS.0000000000001133. [DOI] [PubMed] [Google Scholar]
- 12.Skalova A, Ptakova N, Santana T, Agaimy A, Ihrler S, Uro-Coste E, et al. NCOA4-RET and TRIM27-RET are characteristic gene fusions in salivary intraductal carcinoma, including invasive and metastatic tumors: is "intraductal" correct? Am J Surg Pathol. 2019;43(10):1303–1313. doi: 10.1097/PAS.0000000000001301. [DOI] [PubMed] [Google Scholar]
- 13.Fetsch JF, Laskin WB, Michal M, Remotti F, Heffner D, Ellis G, et al. Ectopic hamartomatous thymoma: a clinicopathologic and immunohistochemical analysis of 21 cases with data supporting reclassification as a branchial anlage mixed tumor. Am J Surg Pathol. 2004;28(10):1360–1370. doi: 10.1097/01.pas.0000135518.27224.3f. [DOI] [PubMed] [Google Scholar]