

Abstract
Background
CXCL13 is a B and T lymphocyte chemokine that mediates neuroinflammation through its receptor CXCR5. This chemokine is highly expressed by motoneurons (MNs) in Amyotrophic Lateral Sclerosis (ALS) SOD1G93A (mSOD1) mice during the disease, particularly in fast-progressing mice. Accordingly, in this study, we investigated the role of this chemokine in ALS.
Methods
We used in vitro and in vivo experimental paradigms derived from ALS mice and patients to investigate the expression level and distribution of CXCL13/CXCR5 axis and its role in MN death and disease progression. Moreover, we compared the levels of CXCL13 in the CSF and serum of ALS patients and controls.
Findings
CXCL13 and CXCR5 are overexpressed in the spinal MNs and peripheral axons in mSOD1 mice. CXCL13 inhibition in the CNS of ALS mice resulted in the exacerbation of motor impairment (n = 4/group;Mean_Diff.=27.81) and decrease survival (n = 14_Treated:19.2 ± 1.05wks, n = 17_Controls:20.2 ± 0.6wks; 95% CI: 0.4687–1.929). This was corroborated by evidence from primary spinal cultures where the inhibition or activation of CXCL13 exacerbated or prevented the MN loss. Besides, we found that CXCL13/CXCR5 axis is overexpressed in the spinal cord MNs of ALS patients, and CXCL13 levels in the CSF discriminate ALS (n = 30) from Multiple Sclerosis (n = 16) patients with a sensitivity of 97.56%.
Interpretation
We hypothesise that MNs activate CXCL13 signalling to attenuate CNS inflammation and prevent the neuromuscular denervation. The low levels of CXCL13 in the CSF of ALS patients might reflect the MN dysfunction, suggesting this chemokine as a potential clinical adjunct to discriminate ALS from other neurological diseases.
Funding
Vaccinex, Inc.; Regione Lombardia (TRANS-ALS)
Keywords: Amyotrophic lateral sclerosis, SOD1G93A mice, Motor neurons, Immune response, Chemokines
Research in context.
Evidence before this study
Amyotrophic lateral sclerosis (ALS) is the most common neuromuscular disorder, affecting individuals from all ethnic backgrounds, with an incidence of 2–3 cases per 100,000 population per year.
CXCL13 is a chemokine physiologically involved in the organisation of follicles within lymphoid organs. Besides its physiological role, the ectopic expression of CXCL13 in the CNS is strictly associated with neuroinflammation, a pathological feature of several neurodegenerative diseases including ALS.
Our preliminary evidence showed a significant upregulation of CXCL13 in the CNS of the ALS mice, indicating a direct correlation between the activation of the chemokine and a faster disease progression.
Added value of this study
Unexpectedly, the neutralisation of the chemokine in fast progressing ALS mice worsened the disease exacerbating the motor neuron (MN) impairment and the atrophy of skeletal muscles suggesting a beneficial effect of CXCL13 in ALS. Indeed, we established that the CXCL13 silencing enhanced MNs loss and inflammation in vitro whilst its administration preserved MNs from degeneration.
Noteworthy, CXCL13 levels are also altered in ALS patients, who showed strong activation of the chemokine in the spinal cord but a significant downregulation in the CSF if compared to non-neurological and Multiple Sclerosis patients.
Implications of all the available evidence
Our evidence introduced an unexpected and peculiar role of CXCL13 in ALS, expressed by MNs to prevent inflammation and translocated along with motor axons to prevent their degeneration. Besides, the reduced chemokine levels in the CSF of ALS patients might be an index of the MNs dysfunction, putting CXCL13 forward as a clinical adjunct for the early discrimination of the disease compared to ALS-mimic disorders such as Multiple Sclerosis.
Alt-text: Unlabelled box
1. Introduction
Amyotrophic lateral sclerosis (ALS) is an incurable neurodegenerative disorder of unknown cause arising from progressive degeneration of motor neurons (MNs) and resulting in paralysis and death, usually within 2–4 years from diagnosis [1]. Its incidence is between 1.5 and 2.5 per 100.000 per year: approximately 90% of cases are sporadic, and the remaining 10% are familial [1]. The diagnosis is based on a clinical assessment on the history of progression of symptoms, with a delay of more than a year from symptom onset, quite beyond the therapeutic window of a disease-modifying drug [2]. Moreover, the clinical course varies widely with phenotypic variability involving several clinical aspects such as the site of onset, age of onset, rate of progression, and response to therapy [3]. No ALS biomarkers are currently in clinical use, but they would be valuable in supporting early diagnosis, monitoring disease progression, and assessing the efficacy of any new treatment [4].
The multisystemic nature of ALS pathology, which distinctively encompasses central and peripheral biological systems, makes it even more challenging to identify a proper therapeutic target [1]. Besides, ALS is a multifactorial disease where different cell types, i.e., astrocytes, neurons, microglia, and oligodendrocytes, contribute to the pathological mechanism [5,6]. Along with this, growing evidence indicates a prominent role of the immune system in the pathogenesis and progression of ALS [7]. This influence is multifaceted with different immune cells affecting the disease progression and having a positive or negative effect depending on the disease stage [7]. The selective ablation of mSOD1 (mSOD1) in microglial cells and neonatal bone marrow (BM) transplantation significantly increased MN survival and lifespan of mSOD1 transgenic mice [5]. Conversely, T cell depletion accelerated the disease progression in these mouse models [8,9]. The analysis of the immune system becomes even more complicated if we consider that different molecules classically associated with an immune activity also have a pleiotropic role within CNS and peripheral nervous system (PNS) that influences their development in normal and pathological conditions [10], [11], [12].
Chemokines are a family of small-secreted proteins that are well-known regulators of peripheral immune cell trafficking [13]. It has become evident that chemokines are also expressed at the central level, where they regulate CNS function in both physiological and pathological conditions, including neuronal development, synaptic transmission, and disease-associated neuroinflammation [14,15].
In ALS, increased levels of several chemokines, amongst which CCL2, CCL3, CCL4, and CXCL10, in the serum or cerebrospinal fluid (CSF) of patients, had a negative correlation with the disease progression rate [16,17]. Besides, heightened levels CCL2, CX3CL1 or CXCR4, have been described in the spinal cord of transgenic ALS mice [18,19].
C-X-C motif chemokine 13 (CXCL13), also known as B lymphocyte chemoattractant, was initially identified in stromal cells and follicular dendritic cells as regulating homing of CXCR5 expressing cells, such as B cells and subsets of T cells [20,21]. CXCL13 is not expressed in the healthy CNS but is upregulated in the brain and spinal cord under pathological conditions, such as Lyme neuroborreliosis (LMN), autoimmune demyelination, and primary CNS lymphoma [22], [23], [24], [25]. In mice with experimental autoimmune encephalomyelitis (EAE), CXCL13 was found in infiltrating dendritic cells in the inflamed brain meninges and spinal cord [22,23].
Several studies have shown that CXCL13 levels correlate directly with the number of B cells in brain tissue and CSF [26,27], while successful treatment of multiple sclerosis (MS) patients is associated with parallel declines of CXCL13 levels and CSF B cell counts [26], [27], [28]. However, it was also reported that CXCL13-deficient animals showed a mild, self-limited form of EAE [22,28], indicating that CXCL13/CXCR5 in the CNS may act via B cell-independent mechanisms. Jiang et al. [29] showed that CXCL13 induction plays an essential role in driving CXCR5-mediated astrocyte activation and promoting neuropathic pain following experimental spinal nerve ligation. Conversely, CXCL13 plays an indifferent role in the outcome of mice with lethal alphavirus encephalitis [30], while it has been involved in neuroprotection following stroke [31]. These results suggest that the information about CXCL13/CXCR5 axis is still incomplete and hitherto, we lack data on its implication in chronic neurodegenerative diseases other than MS.
In the present study, we started from our evidence that CXCL13 dramatically increased in laser captured MNs of 129Sv-mSOD1 mice, which had earlier motor function impairment and a reduced survival than C57-mSOD1 mice, despite carrying the same amount of human mSOD1 transgene [32]. Nevertheless, the neutralisation of CXCL13 expression in the CSF and the spinal cord of fast-progressing mice exacerbated MN impairment and motor ability, and reduced survival. Interestingly, CXCL13 and CXCR5 are persistently increased in MNs of mSOD1 mice, and the administration of CXCL13 to primary co-cultures prevented MN loss while reducing astrogliosis. Our results also demonstrated that spinal MNs express CXCL13 in ALS patients, and the reduced CXCL13 concentration in their CSF might reflect the MN dysfunction during the disease progression.
2. Methods
2.1. Human samples
Spinal cord: samples from five sporadic ALS patients, four familial ALS patients and four controls were used. Further details are provided in Supplementary Table 1. Spinal cords were fixed in formalin, embedded in paraffin, and tissue slices (6 μm) were collected on poly-l-lysine coated slides (VWR).
CSF: samples from 30 sporadic ALS patients (17 males and 13 females) with definite ALS according to revised El Escorial criteria [33], aged 26–80 years (mean ± SD: 58 ± 13.5), with a duration of the disease of 11–192 months (mean ± SD: 20 ± 10) at the time of the lumbar puncture Further details are provided in the Supplementary Table 2. Control CSF samples were from i) 10 normative subjects (5 males and No. 5 females) aged 53–67 years (mean ± SD: 60 ± 5 years) without neurological signatures at the time of withdrawal; ii) 16 patients (6 males and 10 females), aged 31–77 years (mean = SD: 53 ± 13) with multiple sclerosis. CSF samples were collected, centrifuged at 450 × g for 10 min, and stored at −80°C.
Serum: patients enroled in three Italian population-based registries (Lombardia, Piemonte, Emilia Romagna), were newly diagnosed definite, probable, or possible ALS, according to the El Escorial criteria [33]. To be eligible, patients had to be ≤ 55 years of age (early ALS) or ≥ 75 years of age (late ALS) at diagnosis. Controls were residency-, sex-, and age-matched (± five years), randomly chosen within the same hospital of the patient amongst subjects admitted for surgery for a non-spontaneously evolving disease. Subjects enroled for the discovery phase study were 20 early ALS, 20 late ALS, and 40 controls (early and late controls). All cases were sporadic. Samples of blood were collected in EDTA pre-coated tubes (BD Vacutainer K2EDTA), and serum was isolated from EDTA blood by centrifugation at 12,000 x g for 10 min at 18–20°C.
2.2. Animals
Female transgenic SOD1G93A mice on C57BL/6 J or 129SvHsd genetic background, hereafter indicated as C57-mSOD1 and 129Sv-mSOD1, respectively, and corresponding Ntg littermates were used. Mice were housed 4/5 per standard cages in specific pathogen-free conditions and in a controlled environmental condition (temperature: 22±2°C; relative humidity: 55±10% and 12 h of light). Food (standard pellets) and water were supplied ad libitum.
2.3. Intra-cerebral-ventricular administration of monoclonal antibodies
mAb 5261-muIg anti-CXCL13 antibody (VX5/5378; Patent number: US 8277,809 B2 licensed on Oct. 2, 2012) [34] or irrelevant Mouse Immunoglobulin G (IgG) was provided by Vaccinex, Inc. (New York, U.S.A.), and administered to 129Sv-mSOD1 though 1004 Alzet® Osmotic pump (in accordance to manufacturer's instruction. http://www.alzet.com) to ensure continuous delivery and constant mAb levels within CSF. CXCL13-neutralizing mAb was delivered at a concentration of 4.6 ng/mL, considering CXCL13 concentration in the CSF of 129Sv-mSOD1 mice (1500 pg/mL), the replacement speed of CSF in mice (18 μL/h) and the release speed of Alzet® Osmotic pump (0.11 μL/h).
2.4. Disease progression and survival
Disease progression and survival was monitored bi-weekly, starting from twelve weeks in anti-CXCL13 mAb- and IgG-treated 129 Sv mSOD1 mice. Bodyweight and paw grip strength were recorded for each session, as previously described [35]. The Paw Grip Endurance (PaGE) test involved placing the mouse on the wire-lid of a conventional housing cage. The mice are placed on a horizontal grid at 30 cm from the table, and the tail is gently pulled until they grasp the grid with their fore and hind paws. The lid is gently turned upside down, and the latency time of the mouse to fall on the table is recorded for a maximum of 90 s. Each mouse is given up to three attempts, and the most prolonged latency is recorded. The onset of hind limb force motor deficit is considered when the mice showed the first signs of impairment (latency less than 90 s) in the PaGE test. The mice are euthanized when they are unable to right themselves within ten seconds after being placed on each side according to the institutional ethical committee guidelines. The age at the euthanasia was considered as the time of survival
2.5. Laser-captured microdissection and microarray data analysis
We extrapolated data for CXCL13 messenger RNA (mRNA) levels in C57Ntg; 129SvNtg, 129Sv-mSOD1, and C57-mSOD1 laser-captured MNs from microarray analysis (GEO Series accession number GSE46298); the detailed procedure has been previously described in Nardo et al. [32].
2.6. Real-Time PCR
Spinal cords were freshly collected and immediately frozen on dry ice after mouse perfusion with 0.1 M PBS. The total RNA from the spinal cord was extracted using the Trizol method (Invitrogen) and purified with PureLink RNA columns (Life Technologies). For real-time PCR, we used the Taq Man Gene expression assay (Applied Biosystems) following the manufacturer's instructions on cDNA specimens in triplicate, using 1X Universal PCR master mix (Life Technologies) and 1X mix containing specific receptor probes. The following probes were used for the real-time PCR: CXCL13 (Cxcl13; Mm04214185_s1; Life Technologies); interleukin-1β (Il-1β; Mm01268569_m1; Life Technologies); interleukin-23 (Il-23; Mm00519943_m1; Life Technologies); interleukin-17 (Il17a; Mm00439618_m1; Life Technologies). Relative quantification was calculated from the ratio between the cycle number (Ct) at which the signal crossed a threshold set within the logarithmic phase of the given gene and that of the reference β-actin gene (4310881E; Life Technologies). The mean values of the triplicate results for each animal were mused as individual data for 2-ΔΔCt statistical analysis.
2.7. Western blot
Spinal cords were freshly collected and immediately frozen on dry ice after mouse perfusion with 0.1 M PBS. Tissues were processed as previously described [35]. Equal amounts of total protein homogenates were loaded on polyacrylamide gels and electroblotted onto PVDF membrane (Millipore). Membranes were immunoblotted with the following primary antibodies: rabbit anti-CXCR5 (1:1000; Abcam); mouse anti-β-actin (1:30,000; Chemicon); mouse anti-GFAP (1:10,000; Millipore); mouse anti-Chat (1:1000; Millipore); rabbit anti-Iba1 (1:1000; Wako); rabbit anti- pERK (1:1000; Cell signalling); mouse anti-ERK (1:1000; Cell signalling); rabbit anti-pAKT (1:1000; Cell signalling); rabbit anti-AKT (1:1000; Cell Signalling); mouse anti-GAPDH (1:10,000; Millipore) followed by HRP-conjugated secondary antibodies (Santa Cruz) and developed with Luminata Forte Western Chemiluminescent HRP Substrate (Millipore) on the Chemi-Doc XRS system (Bio-Rad). Densitometric analysis was performed with Progenesis PG240 v2006 software (Nonlinear Dynamics). Protein levels were normalised to the total amount of protein detected by red Ponceau (Sigma Aldrich) or to GAPDH, as previously published [35]. pERK, and pAKT levels were respectively normalized to total ERK, and total AKT.
2.8. Immunohistochemistry
Mouse: spinal cords and sciatic nerves were processed as previously described [35]. Briefly, the mice were perfused with 0.1 M PBS, followed by 4% PFA at 20°C. Tissues were quickly dissected out and left in the same fixative overnight at 4°C, rinsed, and stored 24 h in 10% sucrose with 0.1% sodium azide in 0.01 M PSB at 4°C for cryoprotection, before mounting in optimal cutting temperature compound (OCT). The spinal cords and sciatic nerves were cut in 30 μm and 20 μm sections, respectively. The following primary antibodies and staining were used: goat anti-CXCL13 (1:50; R&D), rabbit anti CXCR5 (1:100; Abcam), rabbit anti-CXCR5 (1:200; Bioss Antibodies); rabbit anti-NF200 (1:200; Sigma-Aldrich); anti CD68 (1:200; Bio-Rad); mouse anti GFAP (1:2500; Millipore); mouse anti S100β (1:400; Sigma); mouse anti Smi32 (1:200; Stenberg) and, Neuro- Trace conjugated with Alexa-594 (1:500; Invitrogen). Secondary antibodies were as follows: Alexa 488 or Alexa 594 goat anti-rat, Alexa 488 or Alexa 594 goat anti-rabbit, and Alexa 594 goat anti-mouse (Invitrogen). The Alexa 647-Tyramide amplification (Perkin Elmer) protocol was used for detecting CXCL13 in the spinal cord. Mab 5378-biotin, Mouse IgG2a (Vaccinex, Inc. New York, U.S.A.) and Alexa 647-Tyramide amplification (Perkin Elmer) were used to identify αCXCL13 mAb within the parenchyma of mSOD1mice at 16 weeks of age, 24 h after Alzet® Osmotic pump infusion at a concentration of 7.2 mg/mL.
Human: lumbar spinal cord sections on polysinated slides were deparaffinised for 15 min at 60°C and then rehydrated in xylene and ethanol. Antigen retrieval was done in citrate buffer (Dako), and Alexa 647-Tyramide amplification (Perkin Elmer) or peroxidase-diaminobenzidine (DAB) reaction were used for detecting CXCL13 using anti-human rabbit CXCL13 (1:100; Bioss Antibodies); anti-phospho TD43(1:5000; Cosmo Bio Co., LTD); anti-CXCR5 (1:100; Abcam) and Neuro-Trace conjugated with Alexa-594 (1:500; Invitrogen) were used in immunofluorescence for detecting pTD43 inclusions and neuronal cells, respectively. Images were acquired under Olympus virtual slide system VS110 (Olympus, centre Valley, PA, USA) at 20X-magnification
2.9. Muscle denervation
Tibialis anterior (TA) was dissected out, and snap-frozen in isopentane cooled in liquid nitrogen. 20-μm serial longitudinal cryosections were collected on poly-lysine objective slides (VWR International). Five serial sections (average ~ 70 NMJs) per animal were analysed. Muscle sections were stained with anti-synaptic vesicle protein (SV2; 1:100; Developmental Studies Hybridoma Bank), mouse anti-neurofilament 165 kDa (2H3; 1:50; Developmental Studies Hybridoma Bank), followed by 647 anti-mouse secondary antibody (1:500; Invitrogen). α-Bungarotoxin coupled to Alexa Fluor 488 (1:500) (Invitrogen) was then added and left for 2 h at room temperature. Innervation analysis was performed directly. Images of all genotypes for the innervation analysis were obtained with an Olympus virtual slide system VS110 (Olympus, centre Valley, PA, USA) at 40X magnification. The percentage of neuromuscular innervation was quantified in OlyVIA (Olympus) based on the overlay between neurofilament (SV2/2H3) staining and α-BTX labelled endplates. Endplates were quantified as occupied when any neurofilaments staining were overlying the endplate and as vacant when there was no overlay. The endplate area was determined using Fiji software (ImageJ, National Institutes of Health).
2.10. Lentiviral vectors production
Two shRNAs targeting the sequence of mice Cxcl13 (Gene Bank Accession: NM_018866) were designed according to Jang et al. [29]. An additional scrambled (SCR) sequence was also designed as a negative control (NC). The recombinant lentivirus containing Cxcl13 shRNA (LV-Cxcl13 shRNA), or NC shRNA (LV-NC) was packaged using pGreenPuro (System Biosciences, Palo Alto, CA). The shRNA-1 and shRNA-2 sequences were reported in Jang et al. [29]. The knockdown effect of the above lentivirus was examined by ELISA on primary Ntg co-cultures of MNs, astrocytes, and microglia.
2.11. In vitro analysis of motor neuron loss and microglia activation in primary co-cultures
Primary cultures were obtained from the spinal cord of 12-day-old (E12) C57 Ntg, 129 Sv Ntg, and 129 Sv mSOD1 mouse embryos, as previously described [35,36]. The glial fraction was plated at a density of 25,000 cells/cm2 into flasks already pre-coated with poly-l-lysine (Sigma-Aldrich) and maintained in incubators for 3–4 weeks. To prepare a “physiological” feeder glial layer for spinal cord co-cultures, astrocytes were collected from confluent mixed glial cultures (from which the microglia had previously been removed) and seeded at a density of 40,000 cells/cm2 into 24-well plates, while microglia (obtained by shaking overnight the mixed glial cultures at 275 rpm) was seeded at a density of 10,000 cells/cm2 onto a mature astrocyte layer. Finally, to establish neuron/glia co-cultures, Ntg or mSOD1 motor neurons (obtained from an iodixanol-based separation) were seeded at a density of 15,000 cells/cm2 onto the mature mixed glial layers (Ntg astrocytes/microglia; mSOD1 astrocytes/microglia).
Co-culture treatments: LV: after 6 days in vitro (DIV), primary C57 Ntg co-cultures were treated with 1.6 × 106 ifu/cells of LV-Cxcl13 shRNA or NC for 48 h. After 72 h, cells were fixed and, conditional media were collected. Cultures maintained with standard medium served as the controls (untreated; UT).
Acute/Chronic: after 6 DIV, primary 129 Sv Ntg and mSOD1 co-cultures were exposed to: i) 1 μg/mL LPS (from Escherichia coli 0111:B4); ii) 1 μg/mL LPS+100 ng/mL rCXLC13 (Peprotech, Rocky Hills, NJ); 1 μg/mL LPS+ 100 ng/mL rCXLC13 + 100 ng/mL αCXCL13 mAb (Vaccinex, Inc.; NJ) for 24 h (acute). Alternatively, primary co-cultures were exposed to rCXLC13 or 100 ng/mL rCXLC13 + 100 ng/mL of αCXCL13 mAb, for six days (chronic treatment, the media were renewed twice a day). At the end of the treatments cell were fixed. Cultures maintained with normal medium served as the controls (UT).
Immunocytochemical and immunofluorescent assays: cells were fixed with 4% paraformaldehyde and permeabilised by 0.2% Triton X-100 (Sigma-Aldrich). Primary antibodies were: goat anti-CXCL13 (1:200, R&D); mouse anti-nonphosphorylated neurofilament H (Smi32, 1:1000; Covance); mouse anti-GFAP (1:1000; Millipore) mouse anti-Iba1 (1:200; Wako). Appropriate fluorescent secondary antibodies conjugated to different fluorochromes were used at 1:1000 dilution. Pictures of stained cells were obtained with an Olympus virtual slide system VS110 (Olympus) at 10X magnification, and images were analysed with Fiji (Image J, US National Institutes of Health).
CXCL13 levels: six non-overlapping stereological 0.5 × 0.5 - mm fields for each well were analysed to determine the CXCL13 immunofluorescence signal in Ntg and, mSOD1 co-cultures treated or untreated. The Mean grey Value was measured for each field with Fiji (Image J, US National Institutes of Health) following background subtraction.
MN viability: the viability of MNs was assayed by counting SMI32-positive cells with typical morphology (triangular shape, single well-defined axon) and intact axons and dendrites, considering five non-overlapping 2 × 12-mm fields (total area analysed: about 45% of each well). This number was normalised to the mean of SMI32+ cells counted in the appropriate control wells. Astrocytosis: astrocyte activation was evaluated on the same area field of MNs, analysing the percentage of the covered area or the Mean grey Value of GFAP+cells.
Microglia activation: to determine the activation status of immunocompetent cells, the area (μm2) of Iba1-positive cells was measured with Fiji (Image J, US National Institutes of Health) considering from six to nine non-overlapping stereological 0.5 × 0.5 - mm fields for each well.
2.12. ELISA
CSF was collected from the cisterna magna of mice, centrifuged at 13.500 X g for 5 min at 4°C, and the supernatant was stored at −80°C until analysis. Blood was collected in EDTA pre-coated tubes (BD Vacutainer K2EDTA), and serum was isolated from EDTA blood by centrifugation at 12,000 x g for 10 min at 18–20°C.
CXCL13 was measured in serum and CSF of mice and human respectively by mouse CXCL13 ELISA kit (Thermo Fisher) and human CXCL13 ELISA kit (Quantikine; R&D Systems), following manufacturer's instructions.
2.13. Statistical analysis
GraphPad v7.03 (GraphPad Software) was used for was used for the statistical analysis. For each analysis, the dependant and group variable are respectively named on the y- and x-axis of the graph.
The sample size for behavioural analysis was defined according to the “Guidelines for preclinical animal research in ALS/MND: A consensus meeting” [37]. The Mantel-Cox log-rank test and Kaplan-Meier survival plots were used for comparing disease onset and survival between groups. The number of animals at risk at each stage of follow-up were reported in the Supplementary Table 1, and Supplementary Table 2.
The survival analysis was done with the following criteria: i) the follow up started at the age of treatment (12 weeks) and ended at the time of the euthanasia (see the Disease Duration and survival section). The dependant variable was the age (in weeks) at the time of the euthanasia and the independent variable was the treatment; ii) no censored subjects; iii) no confounding adjustment; the mice were collected through a block-randomization in which the blocks are defined on the body weight, sex and sibling separation.
Paw Grip Strength and body weight were analysed by repeated-measures ANOVA with Sidak's post-analysis checking for normality in the residual and homoscedasticity trough the Geisser-Greenhouse's epsilon to evaluate potential violations.
Parametric unpaired t-test and the One-way ANOVA with Tukey's post-analysis were used to compare the differences between two or more mouse groups, respectively. D'Agostino & Pearson omnibus normality test and relative QQ plots were used to assess the assumption of normality. Alternatively, the non-parametric Kruskall-Wallis ANOVA with uncorrected Dunn's post analysis was used. The non-parametric Mann and Whitney test was used to compare the human samples. Receiver operating characteristics (ROC) curve and analysis of the area under the curve (AUC) with a 95% confidence interval were used to find the discriminatory power for CXCL13 levels in the CSF.
All experiments were done a minimum of three times unless otherwise indicated. P < 0.05 was considered statistically significant. Further details, including P values and number of samples, are documented in the Results, Figures, and relative captions.
2.14. Ethics statement
Humans: the Ethics Committees approved the study of the clinical centres involved: the Second University of Naples, Naples, Italy; Fondazione Salvatore Maugeri, Brescia, Italy; Comitato Etico Interaziendale A.O.U. San Giovanni Battista di Torino – AO." CTO. Maria Adelaide di Torino”, Torino, Italy; Comitato etico degli Spedali Civili di Brescia, Brescia, Italy; Comitato Etico Provinciale di Modena, Modena, Italy. All human biofluids (CSF; serum) had been previously obtained for diagnostic purposes, after informed consent of the Second University of Naples, Naples, Italy, and stored at the Mario Negri biorepositories. All experimental protocols and procedures were carried out following the ethical standards of the Mario Negri Institute at which the studies were conducted.
Formalin-fixed, paraffin-embedded spinal cord tissues was collected at post-mortem in the pathology department of the Academic Medical Centre (AMC), the Netherlands, with the approval of the AMC Medical Ethical Committee and according to local legal and ethical regulations. Patients or relatives gave informed consent for autopsy and use of spinal cord tissue for research purposes.
Animals: Procedures involving animals and their care were conducted according to the Mario Negri institutional guidelines. The IRFMN adheres to the principles set out in the following laws, regulations, and policies governing the care and use of laboratory animals: Italian Governing Law (D.lgs 26/2014; Authorisation n.19/2008-A issued March 6, 2008, by Ministry of Health); Mario Negri Institutional Regulations and Policies providing internal authorisation for persons conducting animal experiments (Quality Management System Certificate – UNI EN ISO 9001:2015 – Reg. N° 6121); the NIH Guide for the Care and Use of Laboratory Animals (2011 edition) and EU directives and guidelines (EEC Council Directive 2010/63/UE). All sections of this report adhere to the ARRIVE Guidelines for reporting animal research [38]. An ARRIVE guidelines checklist is included in the Supplementary Checklist 1.
3. Role of funding sources
Vaccinex Inc. granted part of the study and gave its contribution to the production and delivery of materials (anti-CXCL13 neutralising antibody), interpretation of results, and protocol design. No contribution was given in the writing of the report. The "Translating molecular mechanisms into ALS risk and patient's well- being" (TRANS-ALS) - Regione Lombardia (no. 2015–0023) granted part of the study, and it was involved neither in the study design and data collection analysis nor in the interpretation of the results and writing of the report.
4. Results
4.1. Motor neurons strongly activate CXCL13-CXCR5 signalling in the spinal cord of fast mSOD1 mice, and the chemokine is released in the CSF during the disease progression
We previously found that CXCL13 is significantly upregulated in laser captured motor neurons of the mSOD1 mouse model with a more rapid disease progression 129Sv-mSOD1 compared to slow-progressing mice C57-mSOD1 starting from the presymptomatic phase of the disease [32]. Noteworthy, the basal levels of CXCL13 mRNA expression is two-fold higher in 129Sv-Ntg than C57-Ntg MNs (Supplementary Fig. 1) while in pathological conditions, it dramatically increased reaching a peak at the disease onset with a fold change of 125 ± 3.2 and 5 ± 3.5 in 129Sv-mSOD1 and C57-mSOD1, respectively (Fig. 1a). This trend was further validated on total lumbar spinal cord RNA extracts from both fast and slow mSOD1 progressing mice (Fig. 1b, c). Also, the immunohistochemistry showed a progressive and marked upregulation of CXCL13 (Fig. 1d-g; Supplementary Fig. 2a-d) in correspondence of MNs and surrounding axons (Fig. 1h) as well as in efferent motor axons in the ventral portion of the spinal cord (Fig. 1i). Besides, microglia (Fig. 1j), but not astrocytes (Fig. 1k), expressed high levels of CXCL13. Noteworthy, the CXCL13 receptor, CXCR5, increased its expression during the disease progression, mainly in MNs (Fig. 2a-f; Supplementary Figure 3a-c). Our results are discrepant with Jiang et al. [29], who showed spread labelling of CXCR5 in astrocytes and partially in neuronal cells of the dorsal horn of the spinal cord of mice after spinal nerve ligation. Accordingly, we used a supplemental anti-CXCR5 antibody (Bioss Antibodies), whose specificity was validated in mice deficient for the CXCL13 receptor [39]. Results mirrored what we found with the other antibody (Abcam) showing upregulation of CXCR5 by MNs, but not astrocytes, in the spinal cord of fast mSOD1 mice at the disease onset (Supplementary Fig. 4).
Fig. 1.

Motor neurons strongly activate Fig. 1 CXCL13 in the spinal cord of fast mSOD1 mice, and the chemokine is released in the CSF during the disease progression. (a) Cxcl13 mRNA fold change levels from laser captured MNs of C57-mSOD1 and 129Sv-mSOD1 mice. mRNA levels are expressed as mean fold change ratio (± SEM) between C57-mSOD1 versus C57Ntg (n= 4) mice and between 129Sv-mSOD1 (n= 4) versus 129SvNTg (n= 4) mice. (b, c) Real-time PCR for CXCL13 transcript in the lumbar spinal cord of (b) 129Sv-mSOD1 and (c) C57-mSOD1 mice. Data are normalised to β-actin and expressed as the mean ± SEM fold-change ratio between C57-mSOD1 and 129Sv-mSOD1 mice compared to respective age-matched Ntg littermates. ⁎⁎P < 0.01; ⁎⁎⁎⁎P < 0.0001 (Vs. Ntg) by unpaired t-test. (d-g) Fast progressing mice showed a striking activation of CXCL13 in the spinal cord at the disease onset, although the chemokine is upregulated in both (e) 129Sv-mSOD1 and (g) C57-mSOD1 mice compared to respective (d, f) Ntg littermates; scale bar, 50 μm. (h) Representative co-localization of CXCL13 (green) with the neuronal marker (Neurotrace; NT; red) in the ventral horn of the spinal cord of fast-progressing mice; scale bar: 20 μm. (i) Confocal representative image showing CXCL13 staining in correspondence of efferent motor axons (white arrowheads); scale bar: 20 μm. (j) Representative co-localization of CXCL13 (green) with the microglia marker (CD68) in the ventral horn of the spinal cord of fast-progressing mice. (k) Confocal image of the ventral portion of the spinal cord showing the lack of co-localisation of CXCL13 (green) with the astrocyte marker GFAP (red). (l-o) CXCL13 levels (pg/mL) in the CSF and serum of (l, n) 129Sv-mSOD1 and (m, o) C57-mSOD1 mice during the disease progression. Data are expressed as the mean ± SEM. *P < 0.05; **P < 0.001; ****P < 0.0001 (Vs. Ntg); °P < 0.05; °°P < 0.01; (between groups) by one-way ANOVA with Tukey's post-analysis.
Fig. 2.

CXCR5 is upregulated in the motor neurons of mSOD1 mice during the disease progression. (a) Real-time PCR for CXCL13 transcript in the lumbar spinal cord of 129Sv-mSOD1 mice. Data are normalised to β-actin and expressed as the mean ± SEM fold-change ratio between 129Sv-mSOD1 mice compared to respective age-matched Ntg littermates. *P < 0.05; ⁎⁎P < 0.01 (Vs. Ntg) by one-way ANOVA with Tukey's post-analysis. (b) Representative co-localization of CXCR5 (green) with NT (purple) but not with GFAP (red) in the ventral horn of the spinal cord of fast-progressing mice; scale bar: 20 μm. (c) Confocal representative images showing the progressive increase in CXCR5 staining in the lumbar spinal cord of 129Sv-mSOD1 mice.
The increase of CXCL13 mRNA within the spinal cord was associated with a massive progressive increase of CXCL13 in the CSF (Fig. 1l), but not in the serum (Fig. 1n), until the chemokine reached a concentration of 2.98 ± 1.6 ng/μL at the symptomatic stage of the disease.
Slow progressing mSOD1 mice showed a consistent and progressive upregulation of CXCL13 in the spinal cord (Fig. 1c, f, g), albeit not comparable to the levels detected in fast-progressing mice. Noteworthy, this did not translate in the release of the chemokine in biofluids since CXCL13 concentration was lower in the CSF of C57-mSOD1 mice than Ntg mice from the presymptomatic disease stage (Fig. 1m). A lower level of CXCL13 was also registered in the serum of C57-mSOD1 (Fig. 1o). Given the pivotal role played by CXCL13 in the biogenesis of secondary lymphoid organs [20,21], our result is in line with peripheral lymphoid deficits previously reported in C57-mSOD1 mice [40].
4.2. The intra-cerebroventricular neutralisation of CXCL13 accelerates the symptom onset and reduced the survival of mSOD1 mice
Given the detrimental role exerts by the unconstrained expression of CXCL13 in several neuroinflammatory and autoimmune pathologies [25], we next examined the effect of a chronic intra-cerebroventricular (i.c.v.) administration of the CXCL13 neutralising monoclonal antibody (α-CXCL13 mAb) to neutralise the activity of the chemokine during the disease progression in 129Sv-mSOD1 mice. Based on the infusion capacity of 4.5 weeks of the Alzet® osmotic pumps, we implanted animals from the presymptomatic stage (12 weeks), when the CXCL13 levels start to increase, until the symptomatic stage 16.5 weeks to examine the effect on the onset of motor impairment, the disease progression, and survival. Irrelevant IgG- i.c.v.-treated 129Sv-mSOD1 mice were used as controls.
No difference in terms of weight loss between the two experimental groups was observed during the treatment (Fig. 3a). This indicates that the neutralisation of CXCL13 chemokine within CNS did not induce significant side effects in ALS mice. Surprisingly, we found an earlier impairment in grip strength (Fig. 3b) and the extension reflex of hind limbs (Fig. 3c) in anti-αCXCL13 mAb_mSOD1 mice than controls, indicating that the progressive inhibition of the chemokine resulted in a worsening of the motor activity of mSOD1 mice. This translated in an earlier onset of motor impairment (Fig. 3d) and reduced survival (Fig. 3e) of αCXCL13 mAb_mSOD1-treated mice compared to controls. Indeed, the computation of the days of survival after the end of the treatment (from 17 weeks to the sacrifice) highlighted that αCXCL13 mAb_mSOD1-treated mice had a higher percentage of death between 21 and 28 days than controls (Fig. 3f).
Fig. 3.

The intra-cerebroventricular neutralisation of CXCL13 accelerates the symptom onset and reduced the survival of mSOD1 mice. (a) Body weight loss and (b) Paw Grip Endurance (PaGE) test for mAb-αCXCL13- (n= 14) and IgG- treated (n= 17) 129Sv-mSOD1 mice. Data are reported as mean ± SEM for each time point. *P < 0.05; ⁎⁎⁎⁎P < 0.0001 by repeated-measures ANOVA with Sidak's post-analysis. (c) mAb-αCXCL13-treated mice show an earlier impairment in the extension reflex of hindlimbs *P < 0.05; ⁎⁎P < 0.01; ⁎⁎⁎P < 0.001 ⁎⁎⁎⁎P < 0.0001 by repeated-measures ANOVA with Sidak's post-analysis. (d) mAb-αCXCL13-treated mice have an earlier onset of motor impairment than IgG- treated mice. P < 0.0055 by Mantel-Cox log-rank test. (e) mAb-αCXCL13-treated 129Sv-mSOD1 mice display a reduced survival in respect IgG- treated 129Sv-mSOD1 mice. P < 0.0303 by Mantel-Cox log-rank test. (f) Percentage of surviving mAb α-CXCL13 treated mice and controls at different time points (days) after the end of the treatment (16 weeks).
4.3. Intra-cerebroventricular neutralisation of CXCL13 exacerbated motor neuron impairment, and astrocytosis in the lumbar spinal cord of mSOD1 mice
We next treated a supplemental group of 129Sv-mSOD1 mice treated with the neutralising mAb, or irrelevant IgG, at 12 weeks of age and sacrificed at 16 weeks of age. This approach allowed us to evaluate the effect of the treatment at the end of the therapeutic window or 3–4 weeks after the infusion (Endstage), comparing CXCL13 signalling and pathological ALS signatures.
First, we evaluated the expression level of CXCL13 and CXCR5 in the lumbar spinal cord. At 16 weeks of age, we found a reduction in both chemokine and its receptor counterpart in αCXCL13 mAb_mSOD1 mice compared to IgG_mSOD1-treated mice (Fig. 4a, b; Supplementary Fig. 4a, b). Nevertheless, this effect was lost 3–4 weeks after the infusion (Supplementary Fig. 5a, b). Noteworthy, after 24 h from ICV administration, αCXCL13 mAb was present in the spinal cord parenchyma (Supplementary Figure 5a-d) of 129Sv-mSOD1 mice and in microvessels surrounding MNs (Supplementary Figure 5e-g) suggesting that αCXCL13 mAb crossed the blood-brain barrier or is cleared via bulk flow and transferred to blood circulation due to the CSF turnover [41].
Fig. 4.

Intra-cerebroventricular neutralisation of CXCL13 exacerbated motor neuron impairment, and astrocytosis in the lumbar spinal cord of mSOD1 mice. (a, b) Real-time PCR for (a) Cxcl13 and (b) Cxcr5 transcripts in the lumbar spinal cord of mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. Data are normalised to β-actin and expressed as the mean ± SEM fold-change ratio between mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice and controls. ⁎⁎⁎⁎P < 0.0001 (Vs Ntg); °P <0.05 by one-way ANOVA with Tukey's post-analysis. (c-f) Representative Immunoblot images and densitometric analysis of (c, d) ChAT (c, e) GFAP (c, f) P-AKT expression in spinal cord extracts from mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. Data are reported as percentages of the relative Ntg (mean ± SEM). *P < 0.05; ⁎⁎P < 0.01: ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 (Vs. Ntg); °P < 0.05 (between groups) by one-way ANOVA with Tukey's post-analysis.
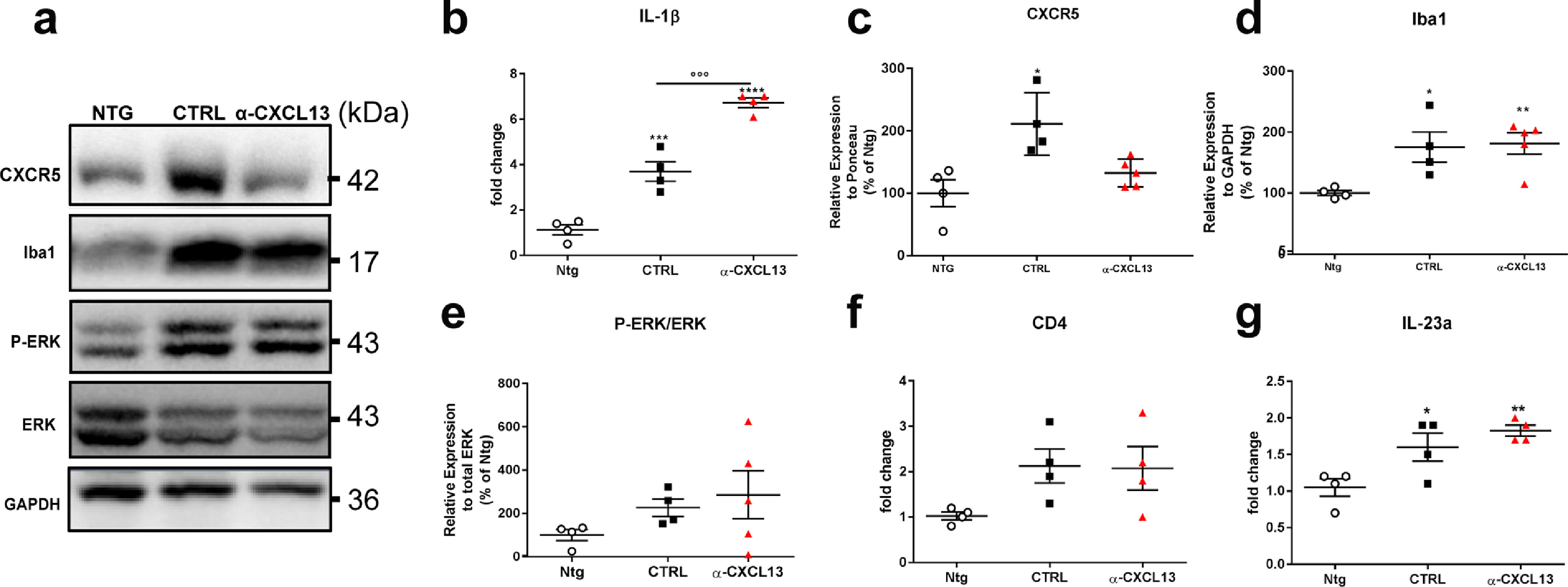
Next, we evaluated the effect of the inhibition of CXCL13 on MN impairment and inflammation at 16 weeks of age. Immunoblot and qRT-PCR analysis on the lumbar spinal cord lysates showed a downregulation of the Choline acetyltransferase (ChAT) in the αCXCL13 mAb-mSOD1-treated mice compared to controls (Fig. 4c, d), suggesting an increased MN dysfunction. On the contrary, the GFAP protein expression (Fig. 4c, e) and the levels of IL-1β mRNA (Supplementary Fig. 6b), were increased, suggesting an enhanced inflammatory response. No difference between the two experimental groups was found in terms of microglia expansion as assessed by immunoblot for Iba1 (Supplementary Fig. 6a, d).
Given that CXCL13-CXCR5 axis is directly involved in the activation of the pro-survival AKT signalling in cancer [42], we evaluated the level of phosphorylation of AKT in the spinal cord of αCXCL13 mAb-treated mSOD1 mice compared to controls. We found that αCXCL13 mAb treatment inhibited AKT phosphorylation in mSOD1 mice (Fig. 4c, f). Conversely, although CXCL13 was reported to induce astrocytic activation trough CXCR5/ERK signalling [29], we did not find any significant variation in ERK phosphorylation in the spinal cord αCXCL13 mAb_mSOD1- and IgG_mSOD1-treated mice, (Supplementary Fig. 6a, e).
We evaluated whether the CXCL13 inhibition may have affected miR-186–5p, which has been reported to tightly control CXCL13 levels within the spinal cord in a neuropathic pain condition induced by spinal nerve ligation (SNL) [29]. Nevertheless, miR-186–5p showed no variation in pathological conditions in mSOD1 mice compared to Ntg mice, and the use of αCXCL13 mAb did not influence miR-186–5p expression (Supplementary Figure 7).
One of the main pathological hallmarks of CXCL13 is the direct recruitment of CD4+Th17 cells [34], which actively produces a robust inflammatory factor, IL17 [43]. We found that the levels of CD4 mRNA were only partially, but not significantly, increased in the spinal cord of mSOD1 mice and this effect was unchanged by the anti-αCXCL13 mAb treatment (Supplementary Fig. 6f). Differently, the level of IL23 was slightly higher in mSOD1 than Ntg mice, but this effect was not significantly modified by the anti-αCXCL13 mAb_treatment (Supplementary Fig. 6 g). Levels of IL17 were undetectable in all groups (not shown).
As expected, we found intense gliosis in mSOD1 mice at the endstage compared to non-transgenic (Ntg) littermates. However, the analysis did not show any difference in astrocytosis (Supplementary Fig. 8c) and microgliosis (Supplementary Fig. 8d) between the two experimental groups. These results reflected in the downregulation of ChAT in both αCXCL13 mAb_mSOD1- and IgG_mSOD1-treated mice compared to Ntg mice (Supplementary Fig. 8f).
4.4. Intra-cerebroventricular neutralisation of CXCL13 exacerbated the denervation atrophy of hind limb skeletal muscles of mSOD1 mice
Given the presence of CXCL13 in correspondence of efferent motor axons in the spinal cord of mSOD1 mice, we investigated the level of activation of CXCL13 and CXCR5 in the sciatic nerves of 129 Sv mSOD1 mice and respective Ntg littermates, at the disease onset. Our results suggested an involvement of the CXCL13/CXCR5 axis during axonal regeneration in the PNS. Indeed, in pathological conditions, we found high levels of CXCL13 (Fig. 5a, b) and CXCR5 (Fig. 5c-e) in the sciatic nerves, co-localising in correspondence of peripheral axons (Fig. 5f, g; Supplementary Fig. 9a-c). We then examined whether the more severe motor function impairment observed in αCXCL13 mAb_mSOD1- treated mice was related to earlier denervation atrophy of hind limbs muscles.
Fig. 5.

Intra-cerebroventricular neutralisation of CXCL13 exacerbated the denervation atrophy of hindlimb skeletal muscles of mSOD1 mice. (a, b) Confocal micrographs showing higher levels of CXCL13 (red) in the sciatic nerve of 129Sv-mSDO1 mice at disease onset and co-localisation with the axonal marker NF200 (green); scale bars 200 μm. (c-e) Confocal micrographs showing higher levels of CXCR5 (green) in the sciatic nerve of 129Sv-mSDO1 mice than Ntg mice at disease onset; scale bar: 100 μm. CXCR5 is present in the bundles of Schwann cells (S100β, red), as evidenced by (e) the volume view of Fig. 5e. (f, g) Confocal micrographs showing the colocalisation of CXCL13 (red), and CXCR5 (green) in correspondence of peripheral axons (Smi32; purple) in the sciatic nerves of 129Sv-mSDO1 mice at the disease onset Scale bar: (f) 50 μm; (g) 10 μm. (k) Percent muscle atrophy (muscle wasting) calculated by measuring of the tibialis anterior muscle weight of mAb-αCXCL13- and IgG-treated 129Sv-mice at 16 weeks and endstage compared to relative Ntg littermates. Data are presented as mean ± SEM. °P < 0.05 by unpaired t-test. (h-j) Analysis of muscle denervation on tibialis anterior (TA) muscle of both mAb-αCXCL13-, IgG-treated 129Sv-mice and Ntg littermates at 16 weeks of age. α-Bungarotoxin (BTX, green) was used to identify the postsynaptic domain, synaptic vesicle glycoprotein 2A (SV2, red) + neurofilaments (2H3, red) were used to identify presynaptic terminals. Scale bar 20 μm. (l) For each mouse group, the percentage of occupied endplates was calculated. Data are reported as mean ± SEM. ⁎⁎⁎⁎P < 0.0001 (Vs. Ntg) °P < 0.05 (between groups) by one-way ANOVA with Tukey's post-analysis.
Measurements of tibialis anterior (TA) muscle weight at 16 weeks of age showed that αCXCL13 mAb_mSOD1 mice had more significant hind limb muscle wasting than IgG-treated mSOD1 mice (Fig. 5k). The TA muscle of treated mice lost 21.9 ± 7.4% of its mass compared with the Ntg littermates, while in the control group we recorded a reduction of 12.5 ± 6.4%. At the endstage, the weight loss of TA fallen further in both the experimental groups but without any significant difference between them.
To evaluate a correlation between muscle atrophy and denervation, we analysed the percentage of innervated neuromuscular junctions (NMJs) in the TA of both the experimental groups at 16 weeks of age (Fig. 5h, i, j, l). NMJs in the TA showed more marked denervation in αCXCL13 mAb_mSOD- treated mice with 67.3 ± 6.4% remaining innervated compared to 80.0 ± 1.7% in IgG_mSOD1- treated mice (Fig. 5l).
4.5. Silencing of CXCL13 expression enhanced MN loss and astrocytosis in non-transgenic and mSOD1 primary co-cultures
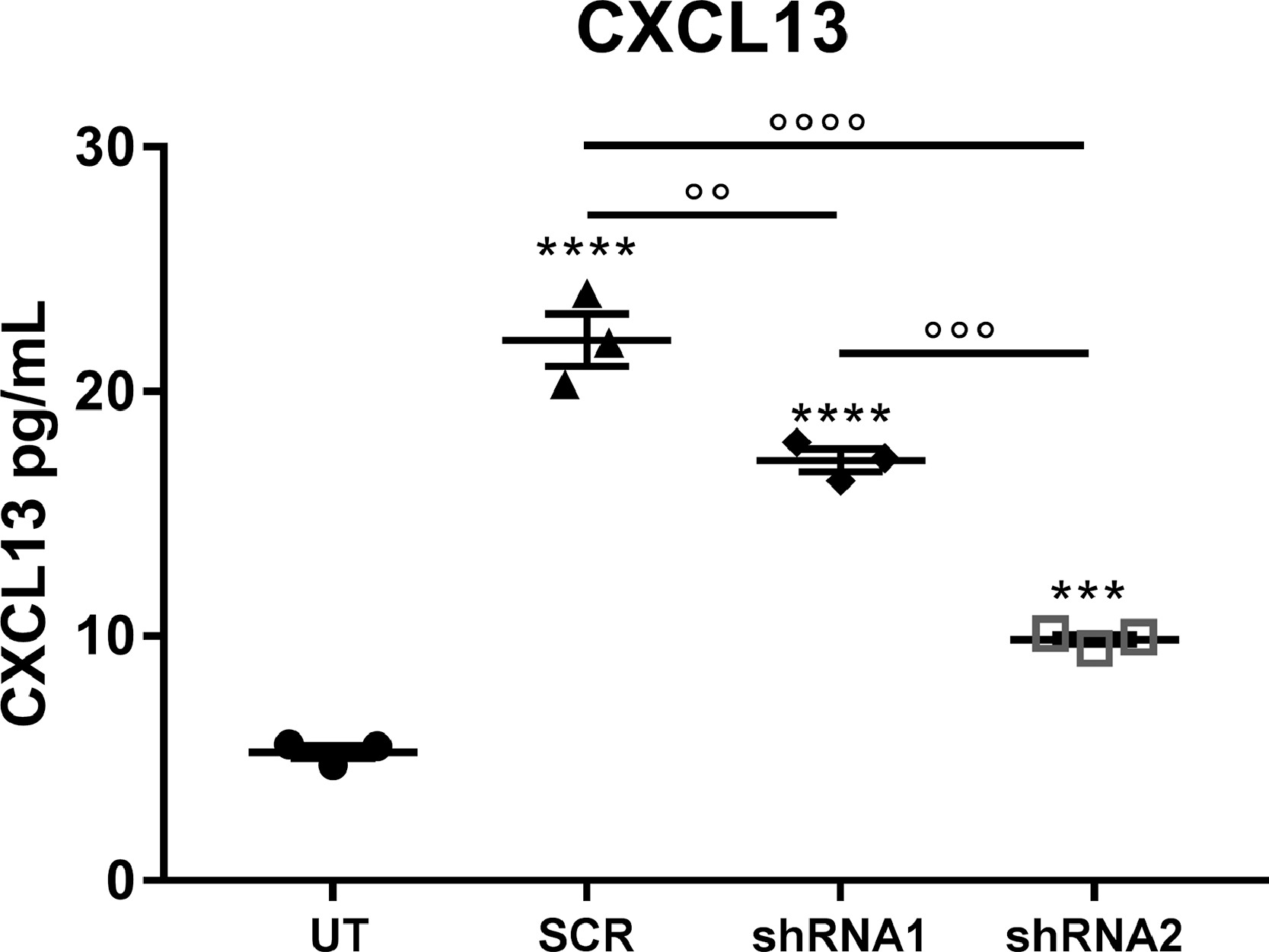
In keeping with in vivo data, CXCL13 was expressed by SMI-32 positive MNs (Fig. 6a, b) in both mSOD1 and NTg primary co-cultures and partially by microglial cells (Fig. 6b, c) but only in mSOD1 primary co-cultures. We then examined the role of CXCL13 in vitro by testing CXCL13 shRNA lentiviral constructs for their effects in persistently knocking down the chemokine expression. For the target sequences of shRNA we referred to Jiang et al. [29], producing two constructs LV-CXCL13 shRNA-1 and LV-CXCL13 shRNA-2. We chose LV-CCX13 shRNA-2 since this sequence showed stronger knockdown effects in reducing CXCL13 protein levels in the medium of primary co-cultures of MNs, astrocytes, and microglia from E12 Ntg mouse embryos (Supplementary Fig. 10).
Fig. 6.

Silencing of CXCL13 expression heightened MN loss and astrocytosis in non-transgenic and mSOD1 primary co-cultures. (a-c) Confocal micrographs showing the co-localisation of CXCL13 (red) with (a, b) the neuronal marker Smi32 (green) and with (c) the microglia marker Iba1; Scale bar, (a, b): 100 μm; (c): 200 μm. (d) CXCL13 concentration (pg/mL) in the conditional media of shRNA-CXCl13- and shRNA- scramble (SCR)-treated or untreated (UT) mSOD1 and Ntg co-cultured with mSOD1 and Ntg microglia/astrocytes, respectively. Data are expressed as the mean ± SEM from three independent experiments for each experimental group. *P < 0.05; ⁎⁎P < 0.001; ⁎⁎⁎P < 0.001 Kruskall-Wallis ANOVA with uncorrected Dunn's post analysis. (e) Representative SMI-32 immunostaining images (white) of shRNA-CXCl13- and shRNA-SCR-treated mSOD1 and Ntg motor neurons co-cultured with mSOD1 and Ntg microglia/astrocytes, respectively; scale bar 200 μm. (f) Cell count analysis showing greater MN loss in both shRNA-CXCl13 treated mSOD1 and Ntg co-cultures than or shRNA-SCR- treated or untreated (UT) co-cultures. Data are reported as mean ± SEM of the No. fields analysed from three independent experiments for each experimental group. *P < 0.05; ⁎⁎P < 0.001; ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 by Kruskall-Wallis ANOVA with uncorrected Dunn's post analysis. (g) Representative GFAP immunostaining images (green) of shRNA-CXCl13- and shRNA-SCR-treated mSOD1 and Ntg astrocytes co-cultured with mSOD1and Ntg motor neurons/microglia, respectively; scale bar 100 μm. (h) Cell Area Fraction (percentage) analysis showing greater astrocytosis in shRNA-CXCl13 treated mSOD1 co-cultures than shRNA-SCR-treated or UT mSOD1 co-cultures. Data are reported as mean ± SEM of the No. fields analysed from three independent experiments for each experimental group. *P < 0.05; ⁎⁎P < 0.01; ⁎⁎⁎P < 0.001 ⁎⁎⁎⁎P < 0.0001 by Kruskall-Wallis ANOVA with uncorrected Dunn's post analysis. (i) Representative Iba1 immunostaining images (red) of shRNA-CXCl13- and shRNA-SCR-treated mSOD1 and Ntg microglia co-cultured respectively with mSOD1and Ntg motor neurons/astrocytes; scale bar 100 μm. (j) Morphometric parameter of Iba1-positive microglia showing higher activation (mean microglial cell area) of shRNA-CXCl13-treated mSOD1 microglia than shRNA-SCR- treated or UT mSOD1 co-cultures. Data are reported as mean ± SEM of the No. fields analysed from three independent experiments for each experimental group. *P < 0.05; ⁎⁎P < 0.001; ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 by Kruskall-Wallis ANOVA with uncorrected Dunn's post analysis.
The shRNA-2 or its negative control lentivirus (LV-shRNA_scrumble; SCR) was used at a concentration of 1.4 × 106 ifu/cells to treat spinal co-cultures from E12 Ntg or mSOD1 129 Sv mouse embryos for 48 h. After 72 h, cells were fixed, and the conditioned medium (CM) collected. We found a significant reduction of CXCL13 at the cellular level (Supplementary Fig. 11) and in the CM (Fig. 6d) of both LV-CCX13-shRNA-treated Ntg and mSOD1 co-cultures. This effect correlated with a decrease in SMI32+ MN number compared to LV-SCR treated or untreated co-cultures (Fig. 6e, f), suggesting a protective role of CXCL13 on MNs. We observed an increased astrocytosis (Fig. 6g, h) and microgliosis (Fig. 6i, j) in co-cultures from mSOD1, but not Ntg mice, after the treatment with LV-CXCL13-shRNA compared to LV-SCR. This result suggested that gliosis was not a consequence of MN loss, which occurred in both Ntg and mSOD1 co-cultures. Instead, CXCL13 was essential to supporting MN survival in normal or pathological conditions and modulating gliosis in the presence of mSOD1 overexpression.
4.6. CXCL13 preserved motor neuron survival under acute and chronic treatment
To clarify the direct effect influence of CXCL13 on MN survival and gliosis, we next examined the effect of murine recombinant CXCL13 (rCXCL13) added to primary co-cultures. At first, we evaluated the effect of rCXCL13 under the inflammatory condition triggered by the LPS load in co-cultures of microglia, astrocytes, and MNs derived from Ntg mice. This is a consolidated experimental paradigm inducing MN loss and inflammation as previously showed [35,36]. To better evaluate the exogenous contribution of rCXCL13, C57 Ntg co-cultures were exposed to i) LPS (1 μg/mL); ii) LPS (1 μg/mL) + rCXCL13 (100 ng/mL) or, iii) LPS (1 μg/mL) + rCXCL13 (100 ng/mL) + α-CXCL13 mAb (100 ng/mL) for 24 h. We observed a reduced LPS-dependant MN death in co-cultures treated with rCXCL13 compared to the LPS group, and this effect was abolished by the neutralising α-CXCL13 mAb (Fig. 7a, d). In keeping with previous findings, the neutralisation of rCXCL13 with α-CXCL13 mAb resulted in an exacerbated astrocytosis (Fig. 7b, e) but not microgliosis (Fig. 7c, f).
Fig. 7.

CXCL13 protects motor neurons under acute and chronic treatment. (a, g) Representative SMI-32 immunostaining images (white) of rCXCL13- and rCXCL13+ mAb-αCXCL13- treated co-cultures in the presence (a) or (g) absence of LPS stimulus; scale bar 200 μm. (d, j) Cell count analysis showing the protective effect of rCXCL13 in the presence (d) or (j) absence of LPS stimulus compared to CXCL13+ mAb-αCXCL13-treated co-cultures. Data are reported as the percentage of UT co-cultures (mean ± SEM of the No. fields analysed from three independent experiments for each experimental group). *P < 0.05; ⁎⁎P < 0.01 (Vs. UT); °°°P < 0.01 (between groups) by one-way ANOVA with Tukey's post-analysis. (b, h) Representative GFAP immunostaining images (blue) of rCXCL13- and rCXCL13+ mAb-αCXCL13- treated co-cultures in the presence (b) or (h) absence of LPS stimulus; scale bar 100 μm. (e, k) Fluorescence Integrated Density (Int_Den) analysis showing greater astrocytosis in rCXCL13 + mAb-αCXCL13-treated co-cultures in the presence (e) or (k) absence of LPS stimulus compared to rCXCL13-treated or UT co-cultures. Data are reported as mean ± SEM of the No. fields analysed from three independent experiments for each experimental group. *P < 0.05; ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 (Vs. UT); °°°°P < 0.01 (between groups) by one-way ANOVA with Tukey's post-analysis. (c, i) Representative Iba1 immunostaining images (green) of rCXCL13- and rCXCL13+ mAb-αCXCL13- treated co-cultures in the presence (c) or (i) absence of LPS stimulus; scale bar 100 μm. (f, l) Morphometric parameter of Iba1-positive microglia showing no difference in activation (mean microglial cell area) between the experimental groups. Data are reported as mean ± SEM of the No. fields analysed from three independent experiments for each experimental group. *P < 0.05; ⁎⁎P < 0.01; ⁎⁎⁎⁎P < 0.0001 (Vs. UT) by one-way ANOVA with Tukey's post-analysis.
Then we examined whether adding rCXCL13 in untreated co-cultures could promote MN survival after 6 DIV in the absence of toxic stimuli. This was done by chronically treating co-cultures twice a day with rCXCL13 (100 ng/mL) or rCXCL13 (100 ng/mL) + mAb α-CXCL13 (100 ng/mL) for six days (in the absence of LPS). Results showed that rCXCL13 increased MN survival compared to untreated co-cultures, and this effect was abolished by mAb α-CXCL13 (Fig. 7g, j). Once again, treatment with mAb markedly activated astrocytes (Fig. 7h, k) but not microglia (Fig. 7i, l).
4.7. CXCL13 and CXCR5 are upregulated in spinal cord motor neurons of ALS patients, whose chemokine levels are low in the CSF
To evaluate the expression and distribution of CXCL13 in humans, we immunostained with an anti-human CXCL13 antibody, post-mortem spinal cords of familial C9orf72 and sporadic ALS and ALS-TDP3 patients, characterised by phosphorylated TDP-43 inclusions within MNs, a typical hallmark of ALS-TDP pathology [44]. Tissues from individuals with other pathologies were used as controls. (Supplementary Table 3).
Results showed that of eight ALS patients analysed; seven showed a remarked increased CXCL13 levels within MNs of the ventral portion of the spinal cord compared to controls (Fig. 8a-l).
Fig. 8.

CXCL13 is upregulated in the motor neurons of the spinal cord of familial and sporadic ALS patients. (a-l) Representative confocal images of immunofluorescence staining for pTDP43, CXCL13, and Neurotrace (neuronal marker) in the post-mortem lumbar spinal cord of (e-l) n = 4 sporadic (sALS) and n = 4 familial (fALS) ALS patients showing the upregulation of the CXCL13 staining in motor neurons compared to (a-d) n = 4 controls; Scale bar: 100 μm; Inset: 50 μm.
Noteworthy, CXCL13, likewise transgenic mouse model, is also present in axons surrounding MNs (Supplementary Fig. 12a, b) and efferent motor axons (Supplementary Fig. 12c, d).
Consistently with mouse data, we also found that the CXCL13 counterpart, CXCR5, was overexpressed by MNs of both sporadic and familial ALS patients compared to controls (Supplementary Fig. 13a-i).
Next, we evaluated the concentration of CXCL13 in the CSF and serum of ALS patients by ELISA. The comparison of CSF of ALS patients compared to controls (CTRL; subjects without neurological alterations) showed a significant reduction of the chemokine (ALS, 2.04 ± 4.17 pg/mL; CTRL, 3.57 ± 3.4 pg/mL; p = 0.0128) (Fig. 9a). Surprisingly, of no. 30 CSF analysed (Supplementary Table 4), CXCL13 was undetectable in no. 15, no. 11 had a concentration under 4 pg/mL, no. 2 under 7 pg/mL and no. 2 between 15 and 17 pg/mL. The lower CXCL13 levels were predominant in patients with a spinal onset (2.03 ± 5.2 pg/mL) (Fig. 9b).
Fig. 9.

CXCL13 has a low concentration in the CSF of ALS patients (a, b) CXCL13 concentration (pg/mL) in the CSF of (a) total ALS patients and (b) sub-grouped by disease onset and normative controls. (c) CXCL13 concentration (pg/mL) in the CSF of ALS patients and MS patients. Data are reported as mean ± SEM. P < 0.05 by Mann-Whitney test. (d) ROC curve and analysis of the area under the curve (AUC) were used to find the discriminatory power of CXCL13 CSF levels between ALS and MS patients. A 95% Confidence Interval (CI) was used and results are reported as percentage.
CXCL13 reduction in the CSF of ALS patients was even more significant when compared with patients affected by multiple sclerosis, who showed a remarked increase of CXCL13 in the CSF (98.77 pg/mL ± 106.52; p < 0.0001) (Fig. 9c). The ROC analysis done on the CSF showed that CXCL13 concentration discriminated ALS from MS patients with a sensitivity of 97.56% (Fig. 9d)
We next examined the concentration of CXCL13 in the serum of a different cohort of ALS patients divided into sub-groups with early (E_ALS) and late (L_ALS) based on age at the disease onset, and compared with age and sex-matched controls. Results showed no difference either between the respective early (E_ALS: 67 ± 35.4 pg/mL; E_CTRL: 75, 96 ± 60.4 pg/mL; p= 0.758) and late (L_ALS: 124.23 ± 131.3 pg/mL; L_CTRL: 96.7 ± 118.03 pg/mL; p= 0.165) subgroups or between early and late ALS patients (p= 0.235) (Supplementary Fig. 14).
5. Discussion
In this study, we examined the possible involvement of the chemokine CXCL13 in ALS by virtue of in vitro and in vivo experimental paradigms on mSOD1 ALS mouse models and tissue samples from human subjects with ALS.
We identified an unexpected and peculiar role of CXCL13, expressed by ALS MNs to attenuate inflammation in the CNS and prevent the neuromuscular denervation in the periphery. Besides, we found that monitoring CXCL13 levels in the CSF of ALS patients might be a useful parameter for diagnosing the disease.
We recently reported that CXCL13 had a trend of expression that well correlated with the fast disease progression of mSOD1 mice as it dramatically increased in laser captured MNs of 129 Sv fast-progressing mice compared to C57 slow-progressing mice [32]. We here further confirm this evidence showing that CXCL13 is persistently upregulated in spinal MNs and microglia of fast progressing ALS mice, and it is released progressively and remarkably in the CSF.
CXCL13 was initially classified into a lymphoid chemokine, as it plays an essential role in the establishment and maintenance of lymphoid tissue microarchitecture [20]. Over the past years, it has become evident that, in addition to its role in peripheral lymphoid tissue, CXCL13 is also implicated in neuroinflammation in a plethora of diseases, including autoimmune demyelination, neuroborreliosis, MS and neuropathic pain [25]. These data supported our preliminary findings surmising a prominent role of CXCL13 in the early neuroinflammation of fast-progressing mice compared to slow-progressing mice [45,46].
Nevertheless, here we showed that CXCL13 inhibition resulted in the exacerbation of motor impairment and decrease survival of fast progressing ALS mice. This evidence was corroborated in primary cultures, where the inhibition of CXCL13 release in the conditioned medium resulted in a heightened MN loss and a remarked astrocytosis. On the contrary, CXCL13 administration fully preserved MNs under acute proinflammatory stimulus or chronic condition.
Our data deviates significantly from previous studies, which showed a nonneuronal induction of CXCL13 in microglia, macrophages, and endothelial cells in the CNS after infection [47,48] or in infiltrating dendritic cells in EAE mice [22,23]. Notably, Jiang et al. [29] recently found that spinal neurons produced CXCL13 in the dorsal horn after spinal nerve ligation. However, in this study, CXCR5 was mainly colocalised with the astrocytes and partially with neurons, and the CXCL13/CXCR5 axis drove neuropathic pain via neuronal-astrocytic interaction in the spinal cord. On the contrary, we found that spinal MNs mainly activated the CXCL13/CXCR5 axis in the ventral horn of mSOD1 mice driving neuroprotection through the AKT signalling, which we previously showed to be a key mechanism for the preservation of MN in ALS models [49,50].
CXCL13 is a crucial regulator of B cell recruitment to the CSF in acute Lyme neuroborreliosis [51,52] and MS [26]. However, previous studies have shown that B cells do not infiltrate the CNS [9] and do not have a significant role in the pathogenesis of ALS-like disease in transgenic mSOD1 mice [53].
Recent works have suggested that CXCR5+ CD4+ Th17 cells, rather than B cells or Th1 cells, may play a pivotal role in the pathogenesis of immune-related diseases, including MS [34] and ALS [54,55]. The harmful effect of CD4+ Th17 is mainly due to the expression of IL-17, a potent proinflammatory cytokine that amplifies ongoing inflammation by inducing expression of TNFα, IL-1β, and IL-6 [43]. We here showed that CXCL13 inhibition did not affect CD4+ T cells infiltration and IL-17 expression in the spinal cord mSOD1 mice.
Our data suggest a peculiar, neuroprotective, immune-unrelated role of CXCL13 in mSOD1 mice.
A large number of proteins that were first discovered in the immune system have since been detected in the nervous system, raising the possibility that these proteins have pleiotropic functions in neurons [10]. Indeed, accumulating evidence indicates that several immune proteins have a novel, nonimmune functions in the CNS. These include proinflammatory cytokines (e.g. TNFα, IL-6), proteins of the innate immune system (e.g. complement C1q and C3), proteins of the adaptive immune system (e.g. members of the major histocompatibility complex class I [MHCI] family) [10,56].
Based on our findings, CXCL13 would fall into the category of the immune molecules activated by MNs to manage gliosis [57]. For example, it was recently showed how increasing MHCI expression on MNs increased survival and motor performance in a mouse model of ALS and protected MNs against astrocyte toxicity [58].
We previously showed that immune response in peripheral axons delayed disease progression in mSOD1 mice, pointing out that immune molecules activated by MNs, such as MHCI and CCL2, have an intrinsic role in promoting axonal regeneration during the disease progression [59]. CXCL13 may be involved in axonal regeneration in the PNS given that the activation of the CXCL13/CXCR5 axis in the peripheral motor axons in the sciatic nerves of mSOD1 mice correlated with lower denervation atrophy of hind limb skeletal muscles. In this context, CXCL12, which, along with CXCL13, mediates the dark zone and light zone organisation of germinal centres in lymphatic tissue [60], also has a complex function in neurobiology. It is crucial for neuronal guidance in development [61,62] and expressed in the mature brain [63]. Besides, Negro et al. [64] recently found that recombinant CXCL12, via binding to the neuronal receptor, CXCR4, accelerated neurotransmission rescue upon damage in vivo, and very effectively stimulated the axon growth of spinal cord MNs in vitro. These findings indicate that the CXCL12/CXCR4 axis plays an essential role in the regeneration of the NMJs after motor axon injury. Although further investigations are needed, it is likely that CXCL13, likewise CXCL12, can promote axonal regeneration in the PNS of mSOD1 mice trough an immune-independent activity. This evidence might be easily translatable to ALS patients, where CXCL13 is present in correspondence of MN somata and efferent motor axons in the white matter of the ventral spinal cord.
CXCL13 dramatically increased in the CSF of 129Sv-mSOD1 mice reflecting the high activation of the CXCL13/CXCR5 axis in the spinal cord during the disease progression. Noteworthy, the basal levels of CXCL13 mRNA expression is two-fold higher in 129 Sv than C57 MNs, suggesting an intrinsic strain-dependant aptitude of 129 Sv MNs to strongly activate CXCL13/CXCR5 axis to counteract the harmful activity of mSOD1 protein, mostly during the early disease stages. Besides, given that MNs of 129Sv-mSOD1 mice expressed 25 fold greater levels of CXCL13 than the C57-mSOD1 mice, the release of the chemokine in consequence of MN death is higher in these mice. On the contrary, CXCL13 and its receptor counterpart increased in the spinal cord of ALS patients while the chemokine is lower in the CSF of ALS patients than controls, remarking the trend we found in slow-progressing mice.
The dysregulated expression of the chemokine in fALS and sALS patients with C9orf72 and TDP43 mutations suggested that CXCL13 activity is not peculiar to mSOD1 ALS cases. Moreover, the lower CXCL13 levels in the CSF might reflect the MN dysregulation in the spinal cord of both C57-mSOD1 mice and ALS patients.
We previously showed that 129Sv-mSOD1 mice had a fast disease progression which is more related to an exacerbated axonal dysregulation and muscle denervation than to MN loss [59].
Accordingly, it is possible to surmise that the remarked MN upregulation of CXCL13 and its translocation in the peripheral motor axons might be a compensatory mechanism aimed to preserve the PNS mainly. This may explain why the neutralization of the chemokine in 129Sv-mSOD1 mice anticipated the denervation atrophy of skeletal muscles. Therefore, we can envisage that treatments aimed at increasing CXCL13 may be useful strategies for slowing down the disease in mice and eventually in patients.
Reduced chemokine levels are unusual in ALS, considering that other proinflammatory chemokines, such as CCL2 and CXCL10, greatly heightened in the CSF of patients [16]. Most importantly, here, we showed net discrimination between ALS and MS patients.
MS and ALS share similar symptoms, especially in the early phase, which makes their diagnosis and differentiation a challenging and time-consuming task. Despite the progress made in imaging techniques and biomarker identification over the last few decades, misdiagnosis still occurs and sometimes involves 10% of the examined patients. [65].
Measuring CXCL13 concentration in the CSF could be a clinical adjunct to discriminate ALS from other neurological diseases. This because, unlike ALS, CXCL13 dramatically increased in the CSF of patients with other inflammatory diseases such as encephalitis [66], chronic inflammatory demyelinating polyneuropathy [67], MS [26,68], LMN [51] or rheumatoid meningitis [69].
In conclusion, we here introduced a chemokine with a peculiar role in ALS, whose activity might be relevant in the pathology and diagnostics of the disease.
First, we demonstrate that spinal MNs robustly express CXCL13, which possibly attenuates astrocytic activation in the CNS and promotes axonal regeneration in the PNS to counteract ALS-like disease progression in mSOD1 mice. Thus, strategies that stimulate CXCL13/CXCR5 signalling may lead to novel therapies for the treatment of ALS.
Second, we identified and verified CXCL13 as a clinical parameter of ALS that may be useful in clinical studies given its low concentration in CSF that might directly reflect the MN dysfunction in the spinal cord.
In the future, a proper multi-centric large-scale validation, including an appropriate stratification of patients based on the rate of disease progression, will be necessary to confirm CXCL13 as an attractive candidate for the introduction into clinical practice. In parallel, a more extensive analysis of the processes underlying motoneuronal activation of CXCL13 in ALS mice will allow comprehending: i) the (direct or indirect) mechanism hindering astrocyte activation in the spinal cord, once MNs are stimulated by CXCL13; ii) the active involvement of CXCL13 in the PNS during axonal regeneration.
Author contributions
MCT treated mSOD1 mice with the help of FS. Besides, she recruited mouse tissues, did the behavioural, immunohistochemical, biomolecular, and biochemical analysis of tissues and biofluids of transgenic mice and ALS patients with the help of GN, under the supervision of GN. AM produced and treated the primary cell cultures and acquired the images under the supervision of MdP. MT and MK produced the LV constructs. MCT did the in vitro morphometric analysis of microglia and MN loss, under the supervision of GN. PF did the immunostaining analysis of sciatic nerves and the imaging of CXCL13 levels of in vitro. SB analysed miR186–5p under the supervision of SM and PB. FT provided the CSF samples and information on ALS patients. EA provided tissue slices from ALS patients. GN and CB designed the experiments. GN wrote the manuscript. All authors have read and approved the final version of the manuscript.
Data sharing
Data, materials and software information supporting the conclusions of this article are included within the article and its additional file.
Declaration of Competing Interest
Dr. Bendotti reports grants from Vaccinex Inc.; Dr. Nardo has nothing to disclose. Dr. Trolese has nothing to disclose. Dr Mariani has nothing to disclose. Dr. Terao has nothing to disclose. Dr. de Paola has nothing to disclose. Dr. Fabbrizio has nothing to disclose. Dr. Sironi has nothing to disclose. Dr. Kurosaki has nothing to disclose. Dr. Bonanno has nothing to disclose. Dr. Marcuzzo has nothing to disclose. Dr. Bernasconi has nothing to disclose. Dr. Trojsi has nothing to disclose. Dr. Aronica has nothing to disclose.
Acknowledgements
We thank Dr. Luca Porcu of the Lab. “Methodology for Clinical Research” at the Mario Negri Institute for the statistical consultancy. We thank the ALS Stichting grant "The Dutch ALS Tissue Bank" (A.E.) and the team, who helped in the collection of ALS tissue samples. This work was mainly supported by Vaccinex Inc. (1895 Mount Hope Avenue, Rochester, NY 14620), together with the "Translating molecular mechanisms into ALS risk and patient's well- being" (TRANS-ALS) - Regione Lombardia (no. 2015–0023).
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ebiom.2020.103097.
Contributor Information
Caterina Bendotti, Email: caterina.bendotti@marionegri.it.
Giovanni Nardo, Email: giovanni.nardo@marionegri.it.
Appendix. Supplementary materials
Supplementary Figure 1. The basal levels of CXCL13 are higher in MN of 129 Sv than C57 mice.Cxcl13 mRNA levels from laser captured MNs of 129 Sv Ntg versus C57 Ntg mice. mRNA levels are expressed as mean fold change ratio (± SEM) between C57Ntg (n = 4) and 129SvNTg (n = 4) mice. ⁎⁎P-value < 0.01 by Mann and Whitney's test.
{kind=link}
Supplementary Figure 2 CXCL13 is progressively upregulated in the spinal cord of fast progressing mice. (a-d) Confocal images showing a progressive heightened of CXCL13 immunostaining in the spinal cord of 129Sv-G93A mice; NC: negative control; OS: Onset; SYMPT: Symptomatic; Scale bar: 20 μm.
{kind=link}
Supplementary Figure 3 CXCR5 is progressively upregulated in the spinal cord of fast progressing mice. (a-c) Confocal images showing the progressive heightened of CXCR5 immunostaining in the spinal cord of 129Sv-G93A mice; Scale bar 20 μm.
{kind=link}
Supplementary Figure 4 CXCR5 is upregulated by MNs of fast progressing mice at the disease onset. (a-d) Confocal micrographs at 10X and 20X magnification showing the upregulation of CXCR5 in the spinal cord of 129SvmSOD1 mice at the disease onset compared to NTg mice. (d, e) The CXCR5 immunostaining colocalised with the Neurotrace (neuronal marker; red) but not with astrocytes (GFAP; purple). For this analysis we used a rabbit anti-CXCR5 antibody (Bioss), previously tested on CXCR5 knockout mice [39]. Scale Bar: a-d: 100 μm; e: 10 μm.
{kind=link}
Supplementary Figure 5 Twenty four hours after intracerebroventricular infusion mAb-αCXCL13 is present in the parenchyma and microvasculature surrounding motor neurons. (a) Representative microscope image showing the longitudinal lumbar spinal cord stained with neurotrace (NT; neuronal marker). Red circles identified the area with motor neurons. Scale bar: 1 mm. (b-d) Magnifications showing the presence (orange arrows) of biotynilated mAb-αCXCL13 (Alexa 647-Strepatvidin; blu) in the parenchyma surrounding MNs (NT; green). Scale bar: (b) 200 μm; (c, d) 50 μm. (e-g) Representative microscope image showing (e) the distribution of the biotinylated mAb-αCXCL13 in the (longitudinal) spinal cord parenchyma of 129Sv-mSOD1 mice after i.c.v. infusion. (f, g) Magnifications showing the presence of the mAb in correspondence of microvessels. Scale bar: (a) 200 μm; (b, c) 50 μm.
{kind=link}
Supplementary Figure 6 The intra-cerebroventricular neutralisation of CXCL13 increased inflammation, which is not associated to microgliosis, T cell infiltration or to the activation of P-ERK signalling. (a) Representative Immunoblot images of CXCR5, Iba1, and P-ERK/ERK mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. (b) Real-time PCR for IL-1β transcript in the lumbar spinal cord of mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. Data are normalised to β-actin and expressed as the mean ± SEM fold-change ratio between mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice and controls. ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 (Vs Ntg); °P <0.05 (between groups) by one-way ANOVA with Tukey's post-analysis. (c-e) Densitometric immunoblot analysis of (c) CXCR5; (d) Iba1, and (e) P-ERK/ERK in the lumbar spinal cord of mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. Data are reported as percentages of the relative Ntg (mean ± SEM). *P < 0.05; ⁎⁎P < 0.01 (Vs Ntg) by one-way ANOVA with Tukey's post-analysis.
{kind=link}
Supplementary Figure 7 miR-186–5p expression is unchanged in both fast and slow-progressing mice, and it was not modified by the Intra-cerebroventricular neutralisation of CXCL13 Real-time PCR for miR-186–5p transcript in the lumbar spinal cord of fast and slow progressing mice and relative Ntg littermates during the disease progression, and of mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. Data are normalised to RNU6B (U6) and expressed as the mean ± SEM fold-change ratio with respect to controls. ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 (Vs Ntg); °P <0.05 (between groups) by one-way ANOVA with Tukey's post-analysis.
{kind=link}
Supplementary Figure 8 The intra-cerebroventricular neutralisation of CXCL13 did not modify the pathological signatures in the lumbar spinal cord of mSOD1 mice at the endstage of the disease. (a) Real-time PCR for Cxcl13 transcript in the lumbar spinal cord of mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at the endstage of the disease. Data are normalised to β-actin and expressed as the mean ± SEM fold-change ratio between mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice and controls. ⁎⁎⁎⁎P < 0.0001 (Vs. Ntg) by one-way ANOVA with Tukey's post-analysis. (b-f) Densitometric Immunoblot analysis of (b) CXCR5; (c) GFAP; (d) Iba1, and (f) ChAT in spinal cord extracts from mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at the endstage of the disease. Data are reported as percentages of the relative Ntg (mean ± SEM). *P < 0.05; ⁎⁎P < 0.01; ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 (Vs. Ntg); by one-way ANOVA with Tukey's post-analysis.
{kind=link}
Supplementary Figure 9 CXCL13 and CXCR5 colocalised in correspondence of peripheral neurofilaments of fast mSOD1 mice. (a) Confocal micrograph showing the expression of CXCL13 (red) and CXCR5 (green) in the sciatic nerves of 129Sv-mSOD1 mice, and (b, c) their colocalisation in correspondence of peripheral neurofilaments (purple). Scale bar: (a) 50 μm; (b, c) 10 μm.
{kind=link}
Supplementary Figure 10 shRNA-2-CXCL13 Lentiviral construct showed the best efficiency of inhibition in primary co-cultures. After six days in vitro (DIV), primary co-cultures were treated with 1.6 × 106 ifu/cells of LV-Cxcl13 shRNA-1 LV-Cxcl13 shRNA-1 or shRNA-scramble (SCR) for 24 h and after 72 h, conditional media were collected. Cultures maintained with normal medium served as the controls (UT). The graph illustrates the CXCL13 concentration (pg/mL) in the conditional media of shRNA-1-CXCl13- shRNA-2-CXCl13- and shRNA-SCR-treated or untreated (UT) Ntg MNs co-cultured with Ntg microglia/astrocytes. Data are expressed as the mean ± SEM from three independent experiments for each experimental group. ⁎⁎⁎P < 0.05; ⁎⁎⁎⁎P < 0.0001 (Vs. Ntg); °°P < 0.01; °°°P < 0.001; °°°°P < 0.0001 (between groups) by one-way ANOVA with Tukey's post-analysis.
{kind=link}
Supplementary Figure 11 shRNA-CXCL13-treated co-cultures had a reduced CXCL13-immunofluorescence. (a, b) Representative CXCL13 immunostaining images (white) of shRNA-CXCl13- and shRNA-SCR-treated mSOD1 and Ntg motor neurons co-cultured with mSOD1 and Ntg microglia/astrocytes, respectively; scale bar 200 μm. h) Mean grey Value analysis showing greater a lower CXCL13 fluorescence signal in shRNA-CXCL13 treated Ntg and mSOD1 co-cultures than shRNA-SCR-treated Ntg and mSOD1 co-cultures. Data are reported as mean ± SEM of the No. fields analysed from three independent experiments for each experimental group. *P < 0.05; ⁎⁎P < 0.01; ⁎⁎⁎P < 0.001 (Vs. UT); °P < 0.05; °°°°P < 0.0001 (between groups) by one-way ANOVA with Tukey's post-analysis.
{kind=link}
Supplementary Figure 12 CXCL13 is expressed in axons surrounding motor neurons, and efferent motor axons in the spinal cord of ALS patients. (a-b) Representative images of DAB immunostaining for CXCL13 in the post-mortem lumbar spinal cord of an ALS patient showing the distribution of the chemokine in motor neurons and axons (red arrows) surrounding motor neurons; scale bar, A, B: 100 μm; c, d: 20 μm. (c) Representative microscope image and (d) magnification of the ventral horn (VH) of the spinal cord of the ALS patient showing CXCl13 staining in correspondence of efferent motor axons of motor neurons (MNs); scale bar; e: 100 μm; F: 20 μm.
{kind=link}
Supplementary Figure 13 CXCR5 is upregulated in the motor neurons of the spinal cord of familial and sporadic ALS patients. (a-i) Representative confocal images of immunofluorescence staining for CXCR5, GFAP, and Neurotrace (neuronal marker) in the post-mortem lumbar spinal cord of (d-i) n = 4 sporadic (sALS) and n = 3 familial (fALS) ALS patients showing the upregulation of the CXCL13 staining in motor neurons compared to (a-c) n = 3 controls; Scale bar: 100 μm; Inset: 50 μm.
{kind=link}
Supplementary Figure 14. CXCL13 is unvaried in the serum of ALS patients. CXCL13 concentration (pg/mL) in the serum of early and late-onset ALS patients and age-matched controls. Data are reported as mean ± SEM. P < 0.05 Mann-Whitney test.
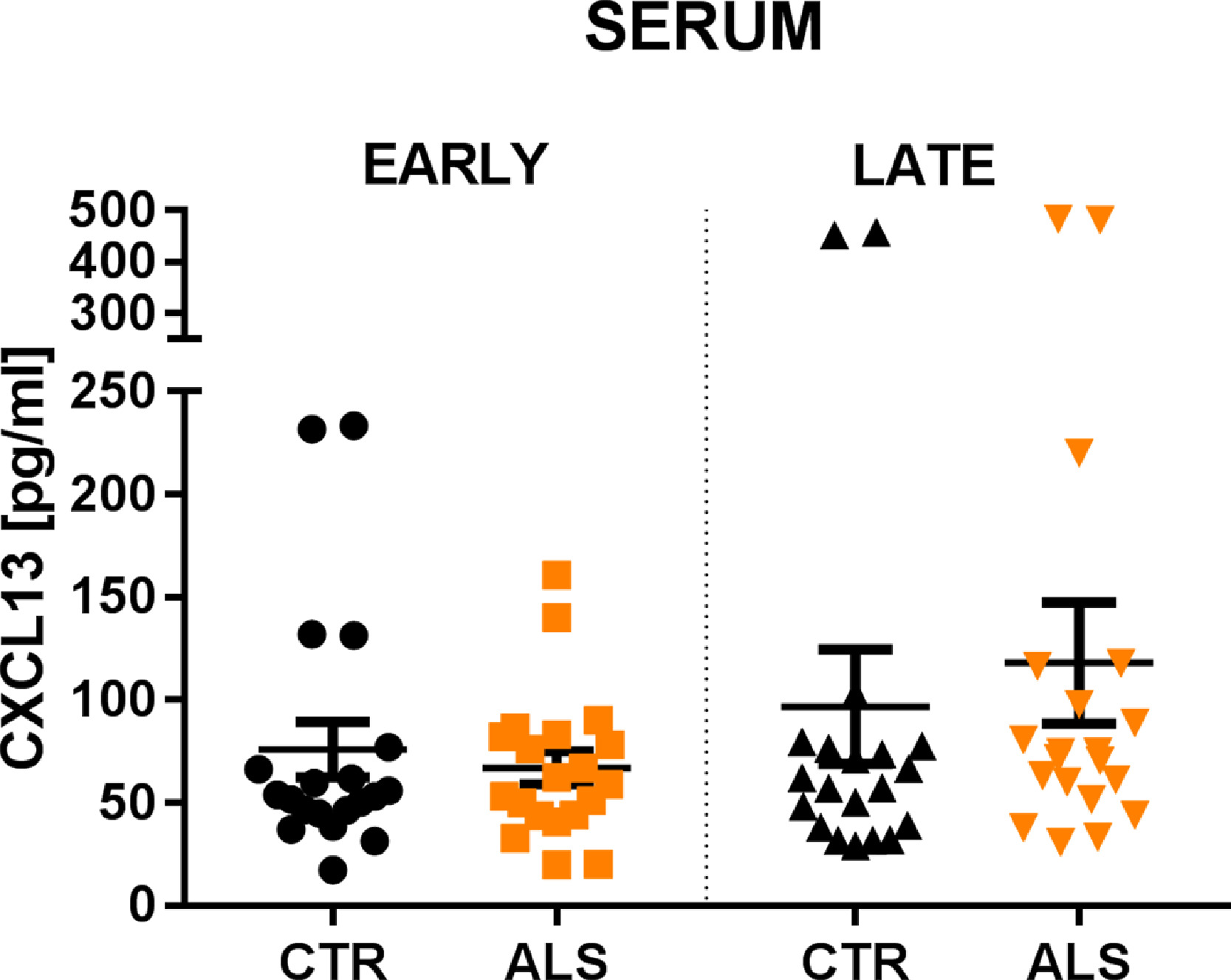{kind=link}
References
- 1.Hardiman O., Al-Chalabi A., Chio A., Corr E.M., Logroscino G., Robberecht W. Amyotrophic lateral sclerosis. Nat Rev Dis Prim [Internet] 2017 Dec 5;3(1):17071. doi: 10.1038/nrdp.2017.71. http://www.nature.com/articles/nrdp201771 Available from: [DOI] [PubMed] [Google Scholar]
- 2.Hardiman O., van den Berg L.H., Kiernan M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol [Internet] 2011 Nov 11;7(11):639–649. doi: 10.1038/nrneurol.2011.153. http://www.nature.com/articles/nrneurol.2011.153 Available from: [DOI] [PubMed] [Google Scholar]
- 3.Beghi E., Chiò A., Couratier P., Esteban J., Hardiman O., Logroscino G. The epidemiology and treatment of ALS: focus on the heterogeneity of the disease and critical appraisal of therapeutic trials. Amyotroph Later Scler [Internet] 2011 Jan 11;12(1):1–10. doi: 10.3109/17482968.2010.502940. http://www.tandfonline.com/doi/full/10.3109/17482968.2010.502940 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner M.R., Bowser R., Bruijn L., Dupuis L., Ludolph A., McGrath M. Mechanisms, models and biomarkers in amyotrophic lateral sclerosis. Amyotroph Later Scler Front Degener [Internet] 2013 May 16;14(sup1):19–32. doi: 10.3109/21678421.2013.778554. http://www.tandfonline.com/doi/full/10.3109/21678421.2013.778554 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boillée S., Vande Velde C., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron [Internet] 2006 Oct 5;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. http://www.ncbi.nlm.nih.gov/pubmed/17015226 [cited 2015 Sep 22]. Available from: [DOI] [PubMed] [Google Scholar]
- 6.Yamanaka K., Chun S.J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D.H. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci [Internet] 2008 Mar 3;11(3):251–253. doi: 10.1038/nn2047. http://www.nature.com/articles/nn2047 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thonhoff J.R., Simpson E.P., Appel S.H. Neuroinflammatory mechanisms in amyotrophic lateral sclerosis pathogenesis. Curr Opin Neurol [Internet] 2018 Oct;31(5):635–639. doi: 10.1097/WCO.0000000000000599. http://journals.lww.com/00019052-201810000-00018 Available from: [DOI] [PubMed] [Google Scholar]
- 8.Beers D.R., Henkel J.S., Zhao W., Wang J., Appel S.H. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A [Internet] 2008 Oct 7;105(40):15558–15563. doi: 10.1073/pnas.0807419105. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2547419&tool=pmcentrez&rendertype=abstract [cited 2015 Dec 12]. Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiu I.M., Chen A., Zheng Y., Kosaras B., Tsiftsoglou S a, Vartanian T.K. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A [Internet] 2008 Nov 18;105(46):17913–17918. doi: 10.1073/pnas.0804610105. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2581614&tool=pmcentrez&rendertype=abstract Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boulanger L.M. Immune proteins in brain development and synaptic plasticity. Neuron [Internet] 2009 Oct 15;64(1):93–109. doi: 10.1016/j.neuron.2009.09.001. http://www.ncbi.nlm.nih.gov/pubmed/19840552 [cited 2013 Mar 14]Available from: [DOI] [PubMed] [Google Scholar]
- 11.Nardo G., Trolese M.C., Bendotti C. Major histocompatibility complex I expression by motor neurons and its implication in amyotrophic lateral sclerosis. Front Neurol [Internet] 2016 Jun 13;7(JUN) doi: 10.3389/fneur.2016.00089. http://journal.frontiersin.org/Article/10.3389/fneur.2016.00089/abstract Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kwon M.J., Shin H.Y., Cui Y., Kim H., Thi A.H.L., Choi J.Y. CCL2 mediates neuron-macrophage interactions to drive proregenerative macrophage activation following preconditioning injury. J Neurosci [Internet] 2015 Dec 2;35(48):15934–15947. doi: 10.1523/JNEUROSCI.1924-15.2015. http://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.1924-15.2015 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charo I.F., Ransohoff R.M. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med [Internet] 2006 Feb 9;354(6):610–621. doi: 10.1056/NEJMra052723. http://www.nejm.org/doi/abs/10.1056/NEJMra052723 Available from: [DOI] [PubMed] [Google Scholar]
- 14.de Haas A.H., van Weering H.R.J., de Jong E.K., Boddeke H.W.G.M., Biber K.P.H. Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol [Internet] 2007 Oct 10;36(2):137–151. doi: 10.1007/s12035-007-0036-8. http://link.springer.com/10.1007/s12035-007-0036-8 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Savarin-Vuaillat C., Ransohoff R.M. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics [Internet] 2007 Oct;4(4):590–601. doi: 10.1016/j.nurt.2007.07.004. http://link.springer.com/10.1016/j.nurt.2007.07.004 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhle J., Lindberg R.L.P., Regeniter A., Mehling M., Steck A.J., Kappos L. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur J Neurol [Internet] 2009 Jun;16(6):771–774. doi: 10.1111/j.1468-1331.2009.02560.x. http://doi.wiley.com/10.1111/j.1468-1331.2009.02560.x Available from: [DOI] [PubMed] [Google Scholar]
- 17.Endo F., Komine O., Yamanaka K. Neuroinflammation in motor neuron disease. Clin Exp Neuroimmunol [Internet] 2016 May;7(2):126–138. http://doi.wiley.com/10.1111/cen3.12309 Available from: [Google Scholar]
- 18.de Oliveira G.P., Alves C.J., Chadi G. Early gene expression changes in spinal cord from SOD1G93A amyotrophic lateral sclerosis animal model. Front Cell Neurosci [Internet] 2013;7 doi: 10.3389/fncel.2013.00216. http://journal.frontiersin.org/article/10.3389/fncel.2013.00216/abstract Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J., Liu Y., Liu X., Li S., Cheng C., Chen S. Dynamic changes of CX3CL1/CX3CR1 axis during microglial activation and motor neuron loss in the spinal cord of ALS mouse model. Transl Neurodegener [Internet] 2018 Dec 21;7(1):35. doi: 10.1186/s40035-018-0138-4. https://translationalneurodegeneration.biomedcentral.com/articles/10.1186/s40035-018-0138-4 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ansel K.M., Ngo V.N., Hyman P.L., Luther S.A., Förster R., Sedgwick J.D. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature [Internet] 2000 Jul;406(6793):309–314. doi: 10.1038/35018581. http://www.nature.com/articles/35018581 Available from: [DOI] [PubMed] [Google Scholar]
- 21.Förster R., Mattis A.E., Kremmer E., Wolf E., Brem G., Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell [Internet] 1996 Dec;87(6):1037–1047. doi: 10.1016/s0092-8674(00)81798-5. https://linkinghub.elsevier.com/retrieve/pii/S0092867400817985 Available from: [DOI] [PubMed] [Google Scholar]
- 22.Bagaeva L.V, Rao P., Powers J.M., Segal B.M. CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol [Internet] 2006 Jun 15;176(12):7676–7685. doi: 10.4049/jimmunol.176.12.7676. http://www.jimmunol.org/lookup/doi/10.4049/jimmunol.176.12.7676 Available from: [DOI] [PubMed] [Google Scholar]
- 23.Magliozzi R., Columba-Cabezas S., Serafini B., Aloisi F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol [Internet] 2004 Mar;148(1–2):11–23. doi: 10.1016/j.jneuroim.2003.10.056. https://linkinghub.elsevier.com/retrieve/pii/S0165572803004983 Available from: [DOI] [PubMed] [Google Scholar]
- 24.Smith J.R., Braziel R.M., Paoletti S., Lipp M., Uguccioni M., Rosenbaum J.T. Expression of B-cell-attracting chemokine 1 (CXCL13) by malignant lymphocytes and vascular endothelium in primary central nervous system lymphoma. Blood [Internet] 2003 Feb 1;101(3):815–821. doi: 10.1182/blood-2002-05-1576. https://ashpublications.org/blood/article/101/3/815/88835/Expression-of-Bcellattracting-chemokine-1-CXCL13 Available from: [DOI] [PubMed] [Google Scholar]
- 25.Irani D.N. Regulated production of CXCL13 within the central nervous system. J Clin Cell Immunol [Internet] 2016;7(5) doi: 10.4172/2155-9899.1000460. https://www.omicsonline.org/open-access/regulated-production-of-cxcl13-within-the-central-nervous-system-2155-9899-1000460.php?aid=80680 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krumbholz M., Theil D., Cepok S., Hemmer B., Kivisäkk P., Ransohoff R.M. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain [Internet] 2006 Jan 1;129(1):200–211. doi: 10.1093/brain/awh680. http://academic.oup.com/brain/article/129/1/200/311844/Chemokines-in-multiple-sclerosis-CXCL12-and-CXCL13 Available from: [DOI] [PubMed] [Google Scholar]
- 27.Sellebjerg F., Bornsen L., Khademi M., Krakauer M., Olsson T., Frederiksen J.L. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology [Internet] 2009 Dec 8;73(23):2003–2010. doi: 10.1212/WNL.0b013e3181c5b457. http://www.neurology.org/cgi/doi/10.1212/WNL.0b013e3181c5b457 Available from: [DOI] [PubMed] [Google Scholar]
- 28.Fischer L., Korfel A., Pfeiffer S., Kiewe P., Volk H.-.D., Cakiroglu H. CXCL13 and CXCL12 in central nervous system lymphoma patients. Clin Cancer Res [Internet] 2009 Oct 1;15(19):5968–5973. doi: 10.1158/1078-0432.CCR-09-0108. http://clincancerres.aacrjournals.org/cgi/doi/10.1158/1078-0432.CCR-09-0108 Available from: [DOI] [PubMed] [Google Scholar]
- 29.Jiang B.-.C., Cao D.-.L, Zhang X., Zhang Z.-.J., He L.-.N, Li C.-.H. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest [Internet] 2016 Jan 11;126(2):745–761. doi: 10.1172/JCI81950. https://www.jci.org/articles/view/81950 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esen N., Rainey-Barger E.K., Huber A.K., Blakely P.K., Irani D.N. Type-I interferons suppress microglial production of the lymphoid chemokine, CXCL13. Glia [Internet] 2014 Sep;62(9):1452–1462. doi: 10.1002/glia.22692. http://doi.wiley.com/10.1002/glia.22692 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monson N.L., Ortega S.B., Ireland S.J., Meeuwissen A.J.M., Chen D., Plautz E.J. Repetitive hypoxic preconditioning induces an immunosuppressed B cell phenotype during endogenous protection from stroke. J Neuroinflamm [Internet] 2014;11(1):22. doi: 10.1186/1742-2094-11-22. http://jneuroinflammation.biomedcentral.com/articles/10.1186/1742-2094-11-22 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nardo G., Iennaco R., Fusi N., Heath P.R., Marino M., Trolese M.C. Transcriptomic indices of fast and slow disease progression in two mouse models of amyotrophic lateral sclerosis. Brain [Internet] 2013 Nov;136(11):3305–3332. doi: 10.1093/brain/awt250. https://academic.oup.com/brain/article-lookup/doi/10.1093/brain/awt250 Available from: [DOI] [PubMed] [Google Scholar]
- 33.Brooks B.R. El escorial world federation of neurology criteria for the diagnosis of amyotrophic lateral sclerosis. J Neurol Sci [Internet] 1994 Jul;124:96–107. doi: 10.1016/0022-510x(94)90191-0. https://linkinghub.elsevier.com/retrieve/pii/0022510X94901910 Available from: [DOI] [PubMed] [Google Scholar]
- 34.Klimatcheva E., Pandina T., Reilly C., Torno S., Bussler H., Scrivens M. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol [Internet] 2015;16(1):6. doi: 10.1186/s12865-015-0068-1. http://www.biomedcentral.com/1471-2172/16/6 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nardo G., Trolese M.C., Verderio M., Mariani A., de Paola M., Riva N. Counteracting roles of MHCI and CD8+ T cells in the peripheral and central nervous system of ALS SOD1G93A mice. Mol Neurodegener [Internet] 2018 Dec 9;13(1):42. doi: 10.1186/s13024-018-0271-7. https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-018-0271-7 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Paola M., Mariani A., Bigini P., Peviani M., Ferrara G., Molteni M. Neuroprotective effects of toll-like receptor 4 antagonism in spinal cord cultures and in a mouse model of motor neuron degeneration. Mol Med [Internet] 2012 Jun 1;18(6):971–981. doi: 10.2119/molmed.2012.00020. https://molmed.biomedcentral.com/articles/10.2119/molmed.2012.00020 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ludolph A.C., Bendotti C., Blaugrund E., Chio A., Greensmith L., Loeffler J.-.P. Guidelines for preclinical animal research in ALS/MND: a consensus meeting. Amyotroph Later Scler [Internet] 2010 Jan 26;11(1–2):38–45. doi: 10.3109/17482960903545334. http://www.tandfonline.com/doi/full/10.3109/17482960903545334 Available from: [DOI] [PubMed] [Google Scholar]
- 38.Kilkenny C., Browne W.J., Cuthill I.C., Emerson M., Altman D.G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol [Internet] 2010 Jun 29;8(6) doi: 10.1371/journal.pbio.1000412. https://dx.plos.org/10.1371/journal.pbio.1000412 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang H., Liu Y., Wang L., Li W. Age-related macular degeneration phenotypes are associated with increased tumor necrosis-alpha and subretinal immune cells in aged Cxcr5 knockout mice. Boulton ME, editor. PLoS One [Internet] 2017 Mar 10;12(3) doi: 10.1371/journal.pone.0173716. https://dx.plos.org/10.1371/journal.pone.0173716 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerjee R., Mosley R.L., Reynolds A.D., Dhar A., Jackson-Lewis V., Gordon P.H. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. McCabe BD, editor. PLoS One [Internet] 2008 Jul 23;3(7):e2740. doi: 10.1371/journal.pone.0002740. https://dx.plos.org/10.1371/journal.pone.0002740 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noguchi Y., Kato M., Ozeki K., Ishigai M. Pharmacokinetics of an intracerebroventricularly administered antibody in rats. MAbs [Internet] 2017 Oct 3;9(7):1210–1215. doi: 10.1080/19420862.2017.1345834. https://www.tandfonline.com/doi/full/10.1080/19420862.2017.1345834 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hussain M., Adah D., Tariq M., Lu Y., Zhang J., Liu J. CXCL13/CXCR5 signaling axis in cancer. Life Sci [Internet] 2019 Jun;227:175–186. doi: 10.1016/j.lfs.2019.04.053. https://linkinghub.elsevier.com/retrieve/pii/S0024320519303157 Available from: [DOI] [PubMed] [Google Scholar]
- 43.Waite J.C., Skokos D. Th17 response and inflammatory autoimmune diseases. Int J Inflam [Internet] 2012;2012:1–10. doi: 10.1155/2012/819467. http://www.hindawi.com/journals/iji/2012/819467/ Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brettschneider J., Del Tredici K., Toledo J.B., Robinson J.L., Irwin D.J., Grossman M. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol [Internet] 2013 Jul;74(1):20–38. doi: 10.1002/ana.23937. http://doi.wiley.com/10.1002/ana.23937 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marino M., Papa S., Crippa V., Nardo G., Peviani M., Cheroni C. Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol Aging [Internet] 2015 Jan;36(1):492–504. doi: 10.1016/j.neurobiolaging.2014.06.026. http://www.ncbi.nlm.nih.gov/pubmed/25085783 [cited 2015 Dec 12]Available from: [DOI] [PubMed] [Google Scholar]
- 46.Nardo G., Trolese M.C., Tortarolo M., Vallarola A., Freschi M., Pasetto L. New insights on the mechanisms of disease course variability in ALS from mutant SOD1 mouse models. Brain Pathol [Internet] 2016 Mar;26(2):237–247. doi: 10.1111/bpa.12351. http://doi.wiley.com/10.1111/bpa.12351 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramesh G., Borda J.T., Gill A., Ribka E.P., Morici L.A., Mottram P. Possible role of glial cells in the onset and progression of Lyme neuroborreliosis. J Neuroinflamm [Internet] 2009;6(1):23. doi: 10.1186/1742-2094-6-23. http://jneuroinflammation.biomedcentral.com/articles/10.1186/1742-2094-6-23 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Narayan K., Dail D., Li L., Cadavid D., Amrute S., Fitzgerald-Bocarsly P. The nervous system as ectopic germinal center: CXCL13 and IgG in lyme neuroborreliosis. Ann Neurol [Internet] 2005 Jun;57(6):813–823. doi: 10.1002/ana.20486. http://doi.wiley.com/10.1002/ana.20486 Available from: [DOI] [PubMed] [Google Scholar]
- 49.Peviani M., Tortarolo M., Battaglia E., Piva R., Bendotti C. Specific induction of Akt3 in spinal cord motor neurons is neuroprotective in a mouse model of familial amyotrophic lateral sclerosis. Mol Neurobiol [Internet] 2014 Feb 20;49(1):136–148. doi: 10.1007/s12035-013-8507-6. http://link.springer.com/10.1007/s12035-013-8507-6 Available from: [DOI] [PubMed] [Google Scholar]
- 50.Vallarola A., Sironi F., Tortarolo M., Gatto N., De Gioia R., Pasetto L. RNS60 exerts therapeutic effects in the SOD1 ALS mouse model through protective glia and peripheral nerve rescue. J Neuroinflamm [Internet] 2018 Dec 1;15(1):65. doi: 10.1186/s12974-018-1101-0. https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-018-1101-0 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kowarik M.C., Cepok S., Sellner J., Grummel V., Weber M.S., Korn T. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J Neuroinflamm [Internet] 2012 Dec 16;9(1):624. doi: 10.1186/1742-2094-9-93. http://jneuroinflammation.biomedcentral.com/articles/10.1186/1742-2094-9-93 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rupprecht T.A., Plate A., Adam M., Wick M., Kastenbauer S., Schmidt C. The chemokine CXCL13 is a key regulator of B cell recruitment to the cerebrospinal fluid in acute Lyme neuroborreliosis. J Neuroinflamm [Internet] 2009;6(1):42. doi: 10.1186/1742-2094-6-42. http://jneuroinflammation.biomedcentral.com/articles/10.1186/1742-2094-6-42 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Naor S., Keren Z., Bronshtein T., Goren E., Machluf M., Melamed D. Development of ALS-like disease in SOD-1 mice deficient of B lymphocytes. J Neurol [Internet] 2009 Aug 12;256(8):1228–1235. doi: 10.1007/s00415-009-5097-3. http://link.springer.com/10.1007/s00415-009-5097-3 Available from: [DOI] [PubMed] [Google Scholar]
- 54.Rentzos M., Rombos A., Nikolaou C., Zoga M., Zouvelou V., Dimitrakopoulos A. Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation? Acta Neurol Scand [Internet] 2010 Dec;122(6):425–429. doi: 10.1111/j.1600-0404.2010.01333.x. http://doi.wiley.com/10.1111/j.1600-0404.2010.01333.x Available from: [DOI] [PubMed] [Google Scholar]
- 55.Fiala M., Chattopadhay M., La Cava A., Tse E., Liu G., Lourenco E. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J Neuroinflammation [Internet] 2010;7(1):76. doi: 10.1186/1742-2094-7-76. http://jneuroinflammation.biomedcentral.com/articles/10.1186/1742-2094-7-76 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boulanger L.M., Shatz C.J. Immune signalling in neural development, synaptic plasticity and disease. Nat Rev Neurosci [Internet] 2004 Jul;5(7):521–531. doi: 10.1038/nrn1428. http://www.ncbi.nlm.nih.gov/pubmed/15208694 [cited 2013 Mar 12]. Available from: [DOI] [PubMed] [Google Scholar]
- 57.Pehar M., Vargas M.R., Cassina P., Barbeito A.G., Beckman J.S., Barbeito L. Complexity of astrocyte-motor neuron interactions in amyotrophic lateral sclerosis. Neurodegen Dis [Internet] 2005;2(3–4):139–146. doi: 10.1159/000089619. https://www.karger.com/Article/FullText/89619 Available from: [DOI] [PubMed] [Google Scholar]
- 58.Song S., Miranda C.J., Braun L., Meyer K., Frakes A.E., Ferraiuolo L. Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat Med [Internet] 2016 Apr 29;22(4):397–403. doi: 10.1038/nm.4052. http://www.nature.com/articles/nm.4052 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nardo G., Trolese M.C., de Vito G., Cecchi R., Riva N., Dina G. Immune response in peripheral axons delays disease progression in SOD1G93A mice. J Neuroinflamm [Internet] 2016 Dec 7;13(1):261. doi: 10.1186/s12974-016-0732-2. http://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-016-0732-2 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allen C.D.C., Ansel K.M., Low C., Lesley R., Tamamura H., Fujii N. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol [Internet] 2004 Sep 1;5(9):943–952. doi: 10.1038/ni1100. http://www.nature.com/articles/ni1100 Available from: [DOI] [PubMed] [Google Scholar]
- 61.Lazarini F., Tham T.N., Casanova P., Arenzana-Seisdedos F., Dubois-Dalcq M. Role of the ?-chemokine stromal cell-derived factor (SDF-1) in the developing and mature central nervous system. Glia [Internet] 2003 Apr 15;42(2):139–148. doi: 10.1002/glia.10139. http://doi.wiley.com/10.1002/glia.10139 Available from: [DOI] [PubMed] [Google Scholar]
- 62.Klein R.S., Rubin J.B. Immune and nervous system CXCL12 and CXCR4: parallel roles in patterning and plasticity. Trends Immunol [Internet] 2004 Jun;25(6):306–314. doi: 10.1016/j.it.2004.04.002. https://linkinghub.elsevier.com/retrieve/pii/S1471490604001188 Available from: [DOI] [PubMed] [Google Scholar]
- 63.Stumm R.K., Rummel J., Junker V., Culmsee C., Pfeiffer M., Krieglstein J. A dual role for the SDF-1/CXCR4 chemokine receptor system in adult brain: isoform-selective regulation of SDF-1 expression modulates CXCR4-dependent neuronal plasticity and cerebral leukocyte recruitment after focal ischemia. J Neurosci [Internet] 2002 Jul 15;22(14):5865–5878. doi: 10.1523/JNEUROSCI.22-14-05865.2002. http://www.jneurosci.org/lookup/doi/10.1523/JNEUROSCI.22-14-05865.2002 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Negro S., Lessi F., Duregotti E., Aretini P., La Ferla M., Franceschi S. <scp>CXCL</scp>12α/ <scp>SDF</scp>‐1 from perisynaptic Schwann cells promotes regeneration of injured motor axon terminals. EMBO Mol Med [Internet] 2017 Aug 30;9(8):1000–1010. doi: 10.15252/emmm.201607257. https://onlinelibrary.wiley.com/doi/abs/10.15252/emmm.201607257 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.El Khoury Y., Collongues N., De Sèze J., Gulsari V., Patte-Mensah C., Marcou G. Serum-based differentiation between multiple sclerosis and amyotrophic lateral sclerosis by Random Forest classification of FTIR spectra. Analyst [Internet] 2019;144(15):4647–4652. doi: 10.1039/c9an00754g. http://xlink.rsc.org/?DOI=C9AN00754G Available from: [DOI] [PubMed] [Google Scholar]
- 66.Leypoldt F., Höftberger R., Titulaer M.J., Armangue T., Gresa-Arribas N., Jahn H. Investigations on CXCL13 in Anti - N -methyl- D - aspartate receptor encephalitis. JAMA Neurol [Internet] 2015 Feb 1;72(2):180. doi: 10.1001/jamaneurol.2014.2956. http://archneur.jamanetwork.com/article.aspx?doi=10.1001/jamaneurol.2014.2956 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim Y.H., Jang S.Y., Shin Y.K., Jo Y.R., Yoon B.-.A., Nam S.H. Serum CXCL13 reflects local B-cell mediated inflammatory demyelinating peripheral neuropathy. Sci Rep [Internet] 2019 Dec 11;9(1):16535. doi: 10.1038/s41598-019-52643-2. http://www.nature.com/articles/s41598-019-52643-2 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alvarez E., Piccio L., Mikesell R.J., Klawiter E.C., Parks B.J., Naismith R.T. CXCL13 is a biomarker of inflammation in multiple sclerosis, neuromyelitis optica, and other neurological conditions. Mult Scler J [Internet] 2013 Aug 15;19(9):1204–1208. doi: 10.1177/1352458512473362. http://journals.sagepub.com/doi/10.1177/1352458512473362 Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nissen M.S., Nilsson A.C., Forsberg J., Milthers J., Wirenfeldt M., Bonde C. Use of cerebrospinal fluid biomarkers in diagnosis and monitoring of rheumatoid meningitis. Front Neurol [Internet] 2019 Jun 26;10 doi: 10.3389/fneur.2019.00666. https://www.frontiersin.org/article/10.3389/fneur.2019.00666/full Available from: [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. The basal levels of CXCL13 are higher in MN of 129 Sv than C57 mice.Cxcl13 mRNA levels from laser captured MNs of 129 Sv Ntg versus C57 Ntg mice. mRNA levels are expressed as mean fold change ratio (± SEM) between C57Ntg (n = 4) and 129SvNTg (n = 4) mice. ⁎⁎P-value < 0.01 by Mann and Whitney's test.
Supplementary Figure 2 CXCL13 is progressively upregulated in the spinal cord of fast progressing mice. (a-d) Confocal images showing a progressive heightened of CXCL13 immunostaining in the spinal cord of 129Sv-G93A mice; NC: negative control; OS: Onset; SYMPT: Symptomatic; Scale bar: 20 μm.
Supplementary Figure 3 CXCR5 is progressively upregulated in the spinal cord of fast progressing mice. (a-c) Confocal images showing the progressive heightened of CXCR5 immunostaining in the spinal cord of 129Sv-G93A mice; Scale bar 20 μm.
Supplementary Figure 4 CXCR5 is upregulated by MNs of fast progressing mice at the disease onset. (a-d) Confocal micrographs at 10X and 20X magnification showing the upregulation of CXCR5 in the spinal cord of 129SvmSOD1 mice at the disease onset compared to NTg mice. (d, e) The CXCR5 immunostaining colocalised with the Neurotrace (neuronal marker; red) but not with astrocytes (GFAP; purple). For this analysis we used a rabbit anti-CXCR5 antibody (Bioss), previously tested on CXCR5 knockout mice [39]. Scale Bar: a-d: 100 μm; e: 10 μm.
Supplementary Figure 5 Twenty four hours after intracerebroventricular infusion mAb-αCXCL13 is present in the parenchyma and microvasculature surrounding motor neurons. (a) Representative microscope image showing the longitudinal lumbar spinal cord stained with neurotrace (NT; neuronal marker). Red circles identified the area with motor neurons. Scale bar: 1 mm. (b-d) Magnifications showing the presence (orange arrows) of biotynilated mAb-αCXCL13 (Alexa 647-Strepatvidin; blu) in the parenchyma surrounding MNs (NT; green). Scale bar: (b) 200 μm; (c, d) 50 μm. (e-g) Representative microscope image showing (e) the distribution of the biotinylated mAb-αCXCL13 in the (longitudinal) spinal cord parenchyma of 129Sv-mSOD1 mice after i.c.v. infusion. (f, g) Magnifications showing the presence of the mAb in correspondence of microvessels. Scale bar: (a) 200 μm; (b, c) 50 μm.
Supplementary Figure 6 The intra-cerebroventricular neutralisation of CXCL13 increased inflammation, which is not associated to microgliosis, T cell infiltration or to the activation of P-ERK signalling. (a) Representative Immunoblot images of CXCR5, Iba1, and P-ERK/ERK mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. (b) Real-time PCR for IL-1β transcript in the lumbar spinal cord of mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. Data are normalised to β-actin and expressed as the mean ± SEM fold-change ratio between mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice and controls. ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 (Vs Ntg); °P <0.05 (between groups) by one-way ANOVA with Tukey's post-analysis. (c-e) Densitometric immunoblot analysis of (c) CXCR5; (d) Iba1, and (e) P-ERK/ERK in the lumbar spinal cord of mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. Data are reported as percentages of the relative Ntg (mean ± SEM). *P < 0.05; ⁎⁎P < 0.01 (Vs Ntg) by one-way ANOVA with Tukey's post-analysis.
Supplementary Figure 7 miR-186–5p expression is unchanged in both fast and slow-progressing mice, and it was not modified by the Intra-cerebroventricular neutralisation of CXCL13 Real-time PCR for miR-186–5p transcript in the lumbar spinal cord of fast and slow progressing mice and relative Ntg littermates during the disease progression, and of mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at 16 weeks of age. Data are normalised to RNU6B (U6) and expressed as the mean ± SEM fold-change ratio with respect to controls. ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 (Vs Ntg); °P <0.05 (between groups) by one-way ANOVA with Tukey's post-analysis.
Supplementary Figure 8 The intra-cerebroventricular neutralisation of CXCL13 did not modify the pathological signatures in the lumbar spinal cord of mSOD1 mice at the endstage of the disease. (a) Real-time PCR for Cxcl13 transcript in the lumbar spinal cord of mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at the endstage of the disease. Data are normalised to β-actin and expressed as the mean ± SEM fold-change ratio between mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice and controls. ⁎⁎⁎⁎P < 0.0001 (Vs. Ntg) by one-way ANOVA with Tukey's post-analysis. (b-f) Densitometric Immunoblot analysis of (b) CXCR5; (c) GFAP; (d) Iba1, and (f) ChAT in spinal cord extracts from mAb-αCXCL13- and IgG- treated 129Sv-mSOD1 mice compared to Ntg littermates at the endstage of the disease. Data are reported as percentages of the relative Ntg (mean ± SEM). *P < 0.05; ⁎⁎P < 0.01; ⁎⁎⁎P < 0.001; ⁎⁎⁎⁎P < 0.0001 (Vs. Ntg); by one-way ANOVA with Tukey's post-analysis.
Supplementary Figure 9 CXCL13 and CXCR5 colocalised in correspondence of peripheral neurofilaments of fast mSOD1 mice. (a) Confocal micrograph showing the expression of CXCL13 (red) and CXCR5 (green) in the sciatic nerves of 129Sv-mSOD1 mice, and (b, c) their colocalisation in correspondence of peripheral neurofilaments (purple). Scale bar: (a) 50 μm; (b, c) 10 μm.
Supplementary Figure 10 shRNA-2-CXCL13 Lentiviral construct showed the best efficiency of inhibition in primary co-cultures. After six days in vitro (DIV), primary co-cultures were treated with 1.6 × 106 ifu/cells of LV-Cxcl13 shRNA-1 LV-Cxcl13 shRNA-1 or shRNA-scramble (SCR) for 24 h and after 72 h, conditional media were collected. Cultures maintained with normal medium served as the controls (UT). The graph illustrates the CXCL13 concentration (pg/mL) in the conditional media of shRNA-1-CXCl13- shRNA-2-CXCl13- and shRNA-SCR-treated or untreated (UT) Ntg MNs co-cultured with Ntg microglia/astrocytes. Data are expressed as the mean ± SEM from three independent experiments for each experimental group. ⁎⁎⁎P < 0.05; ⁎⁎⁎⁎P < 0.0001 (Vs. Ntg); °°P < 0.01; °°°P < 0.001; °°°°P < 0.0001 (between groups) by one-way ANOVA with Tukey's post-analysis.
Supplementary Figure 11 shRNA-CXCL13-treated co-cultures had a reduced CXCL13-immunofluorescence. (a, b) Representative CXCL13 immunostaining images (white) of shRNA-CXCl13- and shRNA-SCR-treated mSOD1 and Ntg motor neurons co-cultured with mSOD1 and Ntg microglia/astrocytes, respectively; scale bar 200 μm. h) Mean grey Value analysis showing greater a lower CXCL13 fluorescence signal in shRNA-CXCL13 treated Ntg and mSOD1 co-cultures than shRNA-SCR-treated Ntg and mSOD1 co-cultures. Data are reported as mean ± SEM of the No. fields analysed from three independent experiments for each experimental group. *P < 0.05; ⁎⁎P < 0.01; ⁎⁎⁎P < 0.001 (Vs. UT); °P < 0.05; °°°°P < 0.0001 (between groups) by one-way ANOVA with Tukey's post-analysis.
Supplementary Figure 12 CXCL13 is expressed in axons surrounding motor neurons, and efferent motor axons in the spinal cord of ALS patients. (a-b) Representative images of DAB immunostaining for CXCL13 in the post-mortem lumbar spinal cord of an ALS patient showing the distribution of the chemokine in motor neurons and axons (red arrows) surrounding motor neurons; scale bar, A, B: 100 μm; c, d: 20 μm. (c) Representative microscope image and (d) magnification of the ventral horn (VH) of the spinal cord of the ALS patient showing CXCl13 staining in correspondence of efferent motor axons of motor neurons (MNs); scale bar; e: 100 μm; F: 20 μm.
Supplementary Figure 13 CXCR5 is upregulated in the motor neurons of the spinal cord of familial and sporadic ALS patients. (a-i) Representative confocal images of immunofluorescence staining for CXCR5, GFAP, and Neurotrace (neuronal marker) in the post-mortem lumbar spinal cord of (d-i) n = 4 sporadic (sALS) and n = 3 familial (fALS) ALS patients showing the upregulation of the CXCL13 staining in motor neurons compared to (a-c) n = 3 controls; Scale bar: 100 μm; Inset: 50 μm.
Supplementary Figure 14. CXCL13 is unvaried in the serum of ALS patients. CXCL13 concentration (pg/mL) in the serum of early and late-onset ALS patients and age-matched controls. Data are reported as mean ± SEM. P < 0.05 Mann-Whitney test.