

Abstract
In this study we investigated the effect of linsitinib on the reversal of multidrug resistance (MDR) mediated by the overexpression of the ATP-binding cassette (ABC) subfamily members ABCB1, ABCG2, ABCC1 and ABCC10. Our results indicate for the first time that linsitinib significantly potentiate the effect of anti-neoplastic drugs mitoxantrone (MX) and SN-38 in ABCG2-overexpressing cells; paclitaxel, docetaxel and vinblastine in ABCC10-overexpressing cells. Linsitinib moderately enhanced the cytotoxicity of vincristine in cell lines overexpressing ABCB1, whereas it did not alter the cytotoxicity of substrates of ABCC1. Furthermore, linsitinib significantly increased the intracellular accumulation and decreased the efflux of [3H]-MX in ABCG2-overexpressing cells and [3H]-paclitaxel in ABCC10-overexpressing cells. However, linsitinib, at a concentration that reversed MDR, did not significantly alter the expression levels of either the ABCG2 or ABCC10 transporter proteins. Furthermore, linsitinib did not significantly alter the intracellular localization of ABCG2 or ABCC10. Moreover, linsitinib stimulated the ATPase activity of ABCG2 in a concentration-dependent manner. Overall, our study suggests that linsitinib attenuates ABCG2- and ABCC10-mediated MDR by directly inhibiting their function as opposed to altering ABCG2 or ABCC10 protein expression.
Keywords: Linsitinib, Multi-drug resistance, ABCG2, ABCC10, Tyrosine kinase inhibitor
1. Introduction
Resistance to cancer chemotherapy can be mediated by an increased efflux of chemotherapeutic drugs, thereby leading to a reduction of intracellular drug concentration and subsequent drug insensitivity. The resistance to mechanistically and structurally unrelated drugs is called multi-drug resistance (MDR) (Fletcher et al., 2010). One of the most common causes of MDR is the increased expression of some members of the ATP binding cassette (ABC) transporter superfamily. The ABC transporters involved in mediating MDR in cancer cells include the ABCB1 (also known as MDR1 or P-glycoprotein), ABCG2 (also known as BCRP or MXR), ABCC1 (also known as MRP1) and ABCC10 transporters (also known as MRP7) (Kathawala et al., 2013; Kruh et al., 2007; Sodani et al., 2012a; Yang et al., 2013). The human ABCB1 transporter, a 170-kDa plasma membrane glycoprotein encoded by the human MDR1 gene, catalyzes the efflux of a wide range of structurally dissimilar compounds, including various anticancer drugs such as vinca alkaloids, anthracyclines, taxanes, epipodophyllotoxins, camptothecins and anthracenes (Sharom, 2008). The human ABCG2, a 72-kDa half transporter, transports anti-neoplastic drugs such as mitoxantrone [MX], epipodophyllotoxins, camptothecins and anthracyclines (Doyle and Ross, 2003). Furthermore, ABCG2 has been identified as a molecular determinant for bone marrow stem cells and has been hypothesized to be a marker for cancer stem cells (de Paiva et al., 2005). Mutations within ABCG2 causes varied substrate profiles within the mutant and wild type variants. Mutations at position 482 are critical for substrate specificity (Chen et al., 2003). The amino acid arginine (Arg or R), located on the carboxy terminal of the third transmembrane segment of the membrane spanning domain in ABCG2 is involved in drug binding through the salt bridge formation (Honjo et al., 2001). The replacement of Arg with threonine (Thr or T) or glycine (Gly or G) causes differences in substrate or drug binding affinity, ultimately resulting in changes in substrate profiles among the variants (Ejendal and Hrycyna, 2002; Mao and Unadkat, 2005). Rhodamine 123, daunorubicin, and lyso-tracker green are substrates for the Gly and Thr mutants, although these are not substrates of the wild type ABCG2 (Mao and Unadkat, 2005). In contrast, MX, BODIPY-prazosin and almost all nucleoside inhibitors are substrates of both the wild type and the mutant ABCG2 transporter (Ejendal and Hrycyna, 2002). The human ABCC1 transporter, a 190-kDa transmembrane protein, confers resistance to a broad spectrum of anticancer drugs such as vinca alkaloids, anthracyclines, epipodophyllotoxins, camptothecins and methotrexate, but not to taxanes (Morrow et al., 2006; Sodani et al., 2012a). Another important member of the ABCC family is ABCC10, a 171-kDa membrane protein that confers resistance to certain anticancer drugs, such as vinca alkaloids and taxanes (Kathawala et al., 2013; Kruh et al., 2007). Numerous studies have been conducted to isolate compounds that could either block or inactivate ABC transporters in cancer cells in order to increase the concentration of anti-cancer drugs within the cells, without producing significant adverse effect (Kathawala et al., 2014; Sodani et al., 2014).
Linsitinib is a novel, highly selective and orally bioavailable dual insulin-like growth factor 1 (IGF-1R)/insulin receptor (IR) kinase inhibitor with a favorable preclinical profile (Mulvihill et al., 2009). A phase III clinical study of linsitinib in patients with locally advanced or metastatic adrenocortical carcinoma is currently being conducted, along with several phase II clinical studies (Demeure et al., 2011; Scagliotti and Novello, 2012). Here, we investigate the effect of linsitinib on the cells that overexpress the ABC transporters mediated MDR. Furthermore, we determine the previously unknown effect of linsitinib in antagonizing ABCG2- and ABCC10-mediated MDR.
2. Materials and methods
2.1. Chemicals and reagents
[3H]-MX (4 Ci/mmol) and [3H]-paclitaxel (15 Ci/mmol) were purchased from Moravek Biochemicals, Inc. (Brea, CA). MX, SN-38, paclitaxel, vincristine, verapamil, cisplatin, docetaxel, vinblastine, penicillin/streptomycin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), Triton X-100, sodium dodecyl sulphate (SDS), Tris–HCl, NaCl, and Tween20 were purchased from Sigma–Aldrich (St. Louis, MO). Linsitinib was purchased from LC laboratories (Woburn, MA). Fumitremorgin C (FTC) was synthesized by Thomas McCloud, Developmental Therapeutics Program, Natural Products Extraction Laboratory, NCI, NIH (Bethesda, MD). Cepharanthine was generously provided by Kakenshoyaku Co. (Tokyo, Japan). PAK-104P was a gift of Prof. Shin-Ichi Akiyama (Kagoshima University, Kagoshima, Japan) from Nissan Chemical Ind. Co. Ltd (Chiba, Japan). The monoclonal antibody BXP-21 for ABCG2 was obtained from Signet Laboratories, Inc. (Dedham, MA). The polyclonal antibody PA5–23652 for ABCC10 was obtained from Thermo scientific (Rockford, IL). The anti-β-actin monoclonal antibody (sc-8432) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Dulbecco Modified Eagle Medium (DMEM) was purchased from Hyclone Thermo Scientific (Logan, UT). Ammonium molybdate, MES hydrate, antimony potassium tartrate, sodium azide and N-methyl-d-glucamine were purchased from Sigma–Aldrich (St. Louis, MO). Potassium phosphate, ethylene glycol tetraacetic acid (EGTA) and ATP were products of AMRESCO (Solon, OH). Sulfuric acid (37 N) was purchased from Fisher Scientific (Pittsburgh, PA). KCl was product of Avantor Performance Materials (Center Valley, PA). Ouabain was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY). Dithiothreitol was product of Promega Corporation (Madison, WI). MgCl2 was purchased from EMD Millipore (Billerica, MA). Ascorbic acid was product of VWR International (West Chester, PA). Sodium orthovanadate was purchased from Alfa Aesar (Ward Hill, MA).
2.2. Cell lines and cell culture
The human non-small cell lung carcinoma cell line (NSCLC) H460 was maintained in DMEM and its MX resistant cell line, H460/MX20, was maintained in DMEM in the presence of 20 nM MX (Guo et al., 2013; Henrich et al., 2006). The human colon carcinoma SW620 cell line was maintained in DMEM and its resistant cell line, SW620/AD300, was maintained in DMEM in the presence of 300 ng/ml of doxorubicin (Alvarez et al., 1995; Henrich et al., 2006). Wild-type ABCG2-482-R2, mutant ABCG2-482-G2 and mutant ABCG2-482-T7 cells were established as previously described (Robey et al., 2003). HEK293/ABCC1 and HEK293/ABCC10 cells were generated by transfecting the HEK293 cells with vectors expressing ABCC1 or ABCC10, respectively (Deng et al., 2013; Shukla et al., 2009; Sun et al., 2013). The transfected cell lines were used to obtain transporter specific effect and avoid involvement of other pathways due to long-term exposure to anticancer drugs as observed in drug-selected cancer cells. All cell lines were grown as adherent monolayers in DMEM culture medium supplemented with penicillin, streptomycin, and 10% fetal bovine serum at 37 °C in a humidified incubator containing 5% CO2.
2.3. Cytotoxicity assays
Drug sensitivity was analyzed using a modified MTT colorimetric assay (Bradford, 1976). Cells were collected and seeded into 96-well plates. Aliquots of each cell suspension were evenly distributed into 96-well plates. In the reversal experiments, concentrations of chemotherapeutic drugs (20 μl/well) were added into the designated wells after preincubation with specified concentrations of reversal agents (20 μl/well) for 1 h. After 68 h, 20 μl of the MTT solution (4 mg/ml) was added into each well and then further incubated for 4 h. Subsequently, the medium was removed, and 100 μl/well of DMSO was added to dissolve the formazan crystals. Finally, the absorbance of the samples was read at 570 nm using an Opsys microplate reader (Dynex Technologies, Chantilly, VA). The resistance-fold was calculated by dividing the IC50 for the MDR cells with or without inhibitors by that of the parental cells without the inhibitor.
2.4. [3H]-MX and [3H]-paclitaxel accumulation
The effect of linsitinib on intracellular drug accumulation was determined as previously described (Patel et al., 2013). Cells were collected and pre-incubated with or without the reversing agents for 1 h at 37 °C, then incubated with 0.2 μM [3H]-MX or 0.01 μM [3H]-paclitaxel for 2 h in the presence or absence of linsitinib at 37 °C. After washing three times with ice-cold PBS, the cells were lysed in lysis buffer (pH 7.4, containing 1% Triton X-100 and 0.2% SDS). Each sample was placed in scintillation fluid and radioactivity was measured using a Packard TRI-CARB 1900CA liquid scintillation analyzer from Packard Instrument Company, Inc. (Downers Grove, IL).
2.5. [3H]-MX and [3H]-paclitaxel efflux
Cells were pretreated in the same manner as in the drug accumulation experiment and further incubated for a period of 2 h. Subsequently, the cells were washed in ice-cold PBS and supplemented with fresh medium with or without the reversal compounds at 37 °C. After 0, 30, 60 or 120 min, aliquots of cells were removed and immediately washed twice with 10 ml of ice-cold PBS. The cells were collected and lysed for the detection of radioactivity. Each sample was placed in scintillation fluid and radioactivity was measured as described previously (Deng et al., 2013; Patel et al., 2013).
2.6. Immunoblot analysis
Cells in T-25 flask were incubated with linsitinib for different time periods (0, 24, 48 and 72 h) and were harvested and rinsed twice with cold PBS. The cell extracts were lysed by a lysis buffer (pH 7.4, containing 1% Triton X-100 and 0.2% SDS) for 20 min on ice, with occasional rocking, followed by centrifugation at 12,000 rpm at 4 °C for 15 min (Sodani et al., 2012b). The supernatant containing the total cell lysates was stored at −80 °C. The protein concentration was determined using the bicinchoninic acid (BCATM)-based protein assay (Thermo Scientific, Rockford, IL). Equal amounts of total cell lysate (60 μg protein) were resolved by SDS-PAGE and transferred onto PVDF membranes through electrophoresis. The membrane was then blocked using TBST buffer (10 mM Tris–HCl, 150 mM NaCl, and 0.1% Tween20 pH 8.0) with 5% non-fat milk for 2 h at room temperature. The membranes were incubated overnight with the primary monoclonal antibody against ABCG2 (at a 1:200 dilution), ABCC10 (at a 1:100 dilution) or β-actin (at a 1:1000 dilution) at 4 °C and were then incubated with horseradish peroxidase-conjugated secondary antibody (1: 1000 dilution) for 2 h at room temperature. After washing the membrane three times with TBST, the protein–antibody complex was detected using enhanced chemi-luminescence detection system (Amersham, NJ). The β-actin was used to confirm equal loading in each lane in the samples prepared from cell lysates. The protein expression level was quantified using Image J Software.
2.7. Immunofluorescence assay
Cells (2 × 103 per well) were seeded in 96-well plates and incubated with 2 μM linsitinib for 24, 48 and 72 h at 37 °C. Subsequently, cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.25% TritonX-100 in PBS for 10 min at room temperature. Cells were rinsed three times with PBS and incubated with BSA (2 mg/ml) for 1 h at 37 °C. Subsequently, cells were incubated with monoclonal antibody BXP-21 against ABCG2 (at a 1:200 dilution) or polyclonal antibody D-19 against ABCC10 (at a 1:200 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4 °C, followed by a goat anti-mouse IgG (1:100, Molecular Probes, Engene, OR) or rabbit anti-goat IgG (1:100, Molecular Probes, Engene, OR) for 1 h. Nuclei were visualized using 4, 6-diamidine-2-phenylindole (1:2000, Sigma–Aldrich, St. Louis, MO). Finally, images were taken by an IX70 microscope with IX-FLA fluorescence and CCD camera from Olympus America Inc. (Center Valley, PA) (Zhao et al., 2012).
2.8. ATPase assay of ABCG2
The vanadate (Vi)-sensitive ATPase activity of ABCG2 in the membrane vesicles of high five insect cells was measured as previously described (Ambudkar, 1998). The membrane vesicles (10–20 μg protein/reaction) were incubated in ATPase assay buffer (50 mM MES–Tris, pH 6.8, 50 mM KCl, 5 mM sodium azide, 1 mM EGTA, 1 mM ouabain, 2 mM dithiothreitol and 10 mM MgCl2) at 37 °C for 5 min with or without 0.3 mM vanadate. The membrane vesicles in ATPase assay buffer were incubated with different concentrations of linsitinib ranging from 0 to 80 μM at 37 °C for 3 min and then 5 mM ATP was added at 37 °C. After 20 min incubation the reaction was terminated by adding 0.1 ml of a 5% SDS solution. The amount of Pi released was quantified using a colorimetric method (Yang et al., 2013).
2.9. Molecular modeling of ABCG2
Linsitinib structure and human ABCG2 homology model along with various grids and docking simulations were carried out as per our previous protocols (Sun et al., 2012; Tiwari et al., 2013). All computations were carried out on a Dell Precision 490n dual processor with the Linux OS (Ubuntu 12.04 LTS).
2.10. Statistical analysis
All experiments were repeated at least thrice. Microsoft Office Excel 2010, and Image J were used in data processing and analyzing. The data were analyzed using Student’s two-tailed t-test. The a priori significance level was p < 0.05.
3. Results
3.1. Linsitinib sensitizes ABCG2- and ABCC10-overexpressing cells to antineoplastic drugs
Prior to investigating the reversal potential of linsitinib on ABC transporters, we first determined the cytotoxicity of linsitinib in ABCB1-, ABCG2-, ABCC1- and ABCC10-overexpressing cell lines and their parental sensitive cells using the MTT assay. Notably, the IC50 values of linsitinib in these cells were >50 μM and more than 90% of the cells survived at a concentration of 2 μM (Fig. 1A–F). Therefore, linsitinib at 2 μM was chosen as the maximum concentration for experiments with ABCB1, ABCG2, ABCC1 or ABCC10 substrate antineoplastic drugs.
Fig. 1.
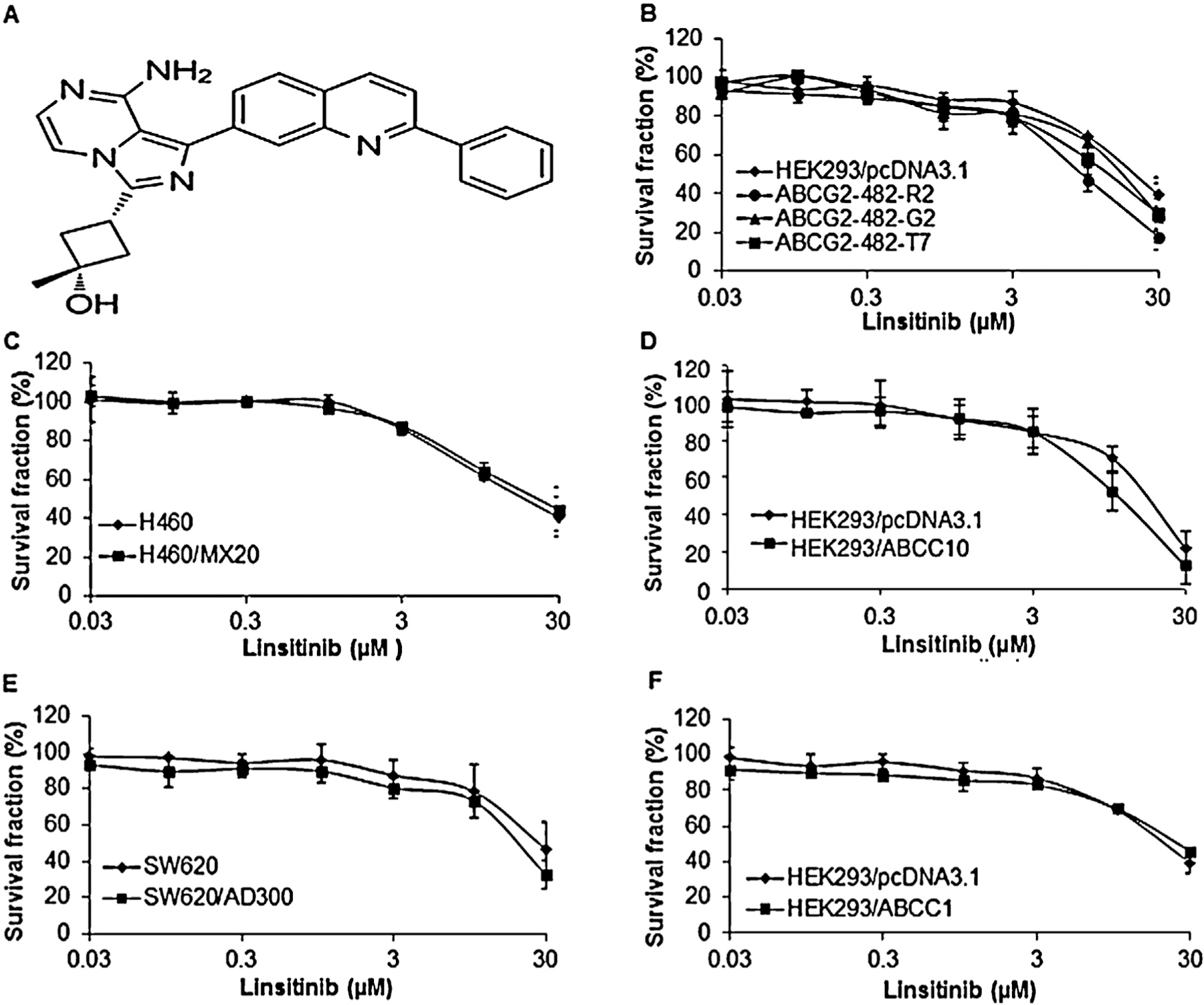
The chemical structure of linsitinib and the effect of linsitinib on the cell lines used in the study. (A) The chemical structure of linsitinib (cis-3-[8-amino-1-(2-phenyl-7-quinolinyl)imidazo[1,5-a]pyrazin-3-yl]-1-methylcyclobutanol). (B) Cytotoxicity of linsitinib in HEK293/pcDNA3.1, ABCG2-482-R2, ABCG2-482-G2 and ABCG2-482-T7 cell lines. (C) Cytotoxicity of linsitinib in H460 and H460/MX20 cell lines. (D) Cytotoxicity of linsitinib in HEK293/pcDNA3.1 and HEK293/ABCC10 cell lines. (E) Cytotoxicity of linsitinib in SW620 and SW620/AD300 cell lines. (F) Cytotoxicity of linsitinib in HEK293/pcDNA3.1 and HEK293/ABCC1 cell lines.
Subsequently, we determined whether linsitinib could sensitize the MDR cells to certain chemotherapeutic drugs. The ABCG2-overexpressing H460/MX20 cells had significantly higher IC50 values for ABCG2 substrates, such as MX and SN-38, than the parental H460 cells (Table 1). In contrast, linsitinib significantly decreased the IC50 values of these drugs in H460/MX20 cells (Table 1). Linsitinib produced a concentration-dependent increase in the sensitivity of H460/MX20 cells to MX and SN-38 (Table 1). In fact, the increase in cytotoxicity produced by 2 μM linsitinib was comparable to FTC, a well-known highly potent ABCG2 inhibitor (Garimella et al., 2004). It has been reported that mutations at amino acid 482 in ABCG2 alter the substrate and antagonist specificity of the ABCG2 transporter (Honjo et al., 2001; Robey et al., 2003). Thus, in order to determine whether linsitinib increases the drug sensitivity in both wild-type and mutant ABCG2-overexpressing cells, we used HEK293 transfected wild-type ABCG2-482-R2 (Arg482), mutant ABCG2-482-G2 (Arg482Gly) and mutant ABCG2-482-T7 (Arg482Thr) cell lines. Linsitinib, at 1 and 2 μM, significantly sensitized the ABCG2-overexpressing cells to the above-mentioned substrates (Table 2). Linsitinib did not significantly alter the sensitivity of parental HEK293/pcDNA3.1 cells. However, in the aforementioned cells, the IC50 values of cisplatin, which is not a substrate of ABCG2, were not affected by linsitinib (Table 2). These results indicated that linsitinib inhibits the activity of both wild-type and mutant Arg482Gly/Thr ABCG2. The ABCC10-gene transfected cells HEK293/ABCC10 were determined to be highly resistant to its substrates paclitaxel, docetaxel and vinblastine, as compared to the parental cells (Table 3). However, the resistance decreased significantly in presence of linsitinib (1 and 2 μM) in HEK293/ABCC10 cells (Table 3). The IC50 values of HEK293/pcDNA3.1 cells to these substrates were not altered significantly by linsitinib (Table 3). There was no significant change in IC50 values of cisplatin, which is not a substrate of ABCC10, in the presence or absence of linsitinib (Table 3).
Table 1.
The effect of linsitinib on reversal of ABCG2-mediated MDR in H460 and H460/MX20 cell lines.
| Drugs | IC50 ± SDa (nM) (resistance fold)b |
|
|---|---|---|
| H460 | H460/MX20 | |
| Mitoxantrone | 30.85 ± 4.43 (1.00) | 2106.97 ± 318.33 (68.30) |
| +Linsitinib 1 μM | 22.90 ± 3.37 (0.74) | 539.50 ± 59.20* (17.49) |
| +Linsitinib 2 μM | 15.05 ± 1.96** (0.49) | 87.31 ± 5.14** (2.83) |
| +FTC 2 μM | 14.69 ± 2.61** (0.48) | 83.26 ± 8.83** (2.70) |
| SN-38 | 47.75 ± 3.29 (1.00) | 8350.36 ± 615.79 (174.88) |
| +Linsitinib 1 μM | 24.62 ± 2.58* (0.52) | 864.40 ± 43.79** (18.10) |
| +Linsitinib 2 μM | 17.43 ± 0.95* (0.37) | 151.91 ± 11.37** (3.18) |
| +FTC 2 μM | 16.82 ± 1.64* (0.35) | 148.97 ± 15.40** (3.12) |
| Cisplatin | 3238.15 ± 325.66 (1.00) | 3977.54 ± 410.61 (1.23) |
| +Linsitinib 2 μM | 3179.94 ± 332.19 (0.98) | 4156.63 ± 379.47 (1.28) |
IC50: concentration that inhibited cell survival by 50% (means ± SD).
Resistance-fold was determined by dividing the IC50 values of substrate in H460/MX20 cells by the IC50 of substrate in H460 cells in the absence of linsitinib; or the IC50 of substrate in H460 cells in the presence of linsitinib divided by the IC50 of substrate in H460 cells in the absence of linsitinib. Values in table are means ± SD of three independent experiments performed in triplicate.
p < 0.05,
p < 0.01 versus that obtained in the absence of inhibitor
Table 2.
The effect of linsitinib on reversal of ABCG2-mediated MDR in gene-transfected cell lines.
| Drugs | IC50 ± SDa (nM) (resistance fold)b |
|||
|---|---|---|---|---|
| HEK293/pcDNA3.1 | ABCG2-482-R2 | ABCG2-482-G2 | ABCG2-482-T7 | |
| Mitoxantrone | 24.27 ± 2.72 (1.00) | 170.58 ± 17.35 (7.03) | 234.93 ± 20.53 (9.68) | 232.59 ± 17.95 (9.58) |
| +Linsitinib 1 µM | 21.24 ± 2.94 (0.88) | 33.79 ± 3.75** (1.39) | 89.27 ± 59.90* (3.68) | 27.94 ± 1.84** (1.15) |
| +Linsitinib 2 µM | 17.78 ± 1.61 (0.73) | 14.12 ± 1.61** (0.58) | 23.15 ± 3.29** (0.95) | 76.81 ± 10.21 (824) |
| +FTC 2 µM | 21.67 ± 2.04 (0.89) | 14.55 ± 1.28** (0.60) | 15.80 ± 1.55** (0.65) | 104.47 ± 25.37** (4.30) |
| SN-38 | 9.32 ± 1.32 (1.00) | 122.99 ± 20.60 (13.20) | 146.93 ± 16.07 (15.77) | 25.65 ± 2.63** (1.06) |
| +Linsitinib 1 μM | 7.89 ± 0.78 (0.85) | 33.85 ± 3.16** (3.63) | 19.79 ± 1.66** (2.12) | 23.99 ± 1.40** (2.57) |
| +Linsitinib 2 μM | 6.69 ± 0.97 (0.72) | 9.47 ± 1.22** (1.02) | 10.07 ± 1.28** (1.08) | 10.30 ± 2.32** (1.11) |
| +FTC 2 μM | 6.76 ± 1.08 (0.73) | 9.70 ± 0.94** (1.04) | 9.07 ± 0.82** (0.97) | 9.74 ± 1.20** (1.05) |
| Cisplatin | 2450.39 ± 223.91 (1.00) | 1323.84 ± 120.22 (0.54) | 1684.76 ± 177.60 (0.69) | 1516.75 ± 180.73 (0.62) |
| +Linsitinib 2 μM | 2225.60 ± 301.60 (0.91) | 1219.85 ± 156.30 (0.50) | 1609.99 ± 153.07 (0.66) | 1690.92 ± 166.04 (0.69) |
IC50: concentration that inhibited cell survival by 50% (means ± SD).
Resistance-fold was determined by dividing the IC50 values of substrate in ABCG2–482-R2, ABCG2–482-T7 and ABCG2–482-G2 cells by the IC50 of substrate in HEK293/pcDNA3.1 cells in the absence of linsitinib; or the IC50 of substrate in HEK293/pcDNA3.1 cells in the presence of linsitinib divided by the IC50 of substrate in HEK293/pcDNA3.1 cells in the absence of linsitinib. Values in table are means ± SD of three independent experiments performed in triplicate.
p < 0.05,
p < 0.01 versus that obtained in the absence of inhibitor.
Table 3.
The effect of linsitinib on reversal of ABCC10-mediated MDR.
| Drugs | IC50 ± SDa (nM) (resistance fold) b |
|
|---|---|---|
| HEK293/pcDNA3.1 | HE HEK293/ABCC10 | |
| Paclitaxel | 6.35 ± 1.11 (1.00) | 76.53 ± 8.51 (12.05) |
| +Linsitinib 1 μM | 7.66 ± 0.63 (1.21) | 34.36 ± 3.42** (5.41) |
| +Linsitinib 2 μM | 6.07 ± 1.02 (0.96) | 8.50 ± 1.16** (1.34) |
| +Cepharanthine 2 μM | 4.69 ± 1.20 (0.74) | 9.02 ± 1.30** (1.42) |
| Docetaxel | 2.52 ± 0.45 (1.00) | 33.30 ± 4.20 (13.21) |
| +Linsitinib 1 μM | 2.62 ± 0.32 (1.04) | 7.22 ± 0.89** (2.87) |
| +Linsitinib 2 μM | 2.58 ± 0.39 (1.02) | 5.25 ± 0.77** (2.08) |
| +Cepharanthine 2 μM | 3.01 ± 0.19 (1.19) | 6.01 ± 1.01** (2.38) |
| Vinblastine | 2.04 ± 0.53 (1.00) | 9.13 ± 0.89 (4.48) |
| +Linsitinib 1 μM | 2.13 ± 0.42 (1.04) | 3.20 ± 0.70** (1.57) |
| +Linsitinib 2 μM | 1.89 ± 0.33 (0.93) | 2.51 ± 0.62** (1.23) |
| +Cepharanthine 2 μM | 1.97 ± 0.22 (0.97) | 3.72 ± 0.71** (1.82) |
| Cisplatin | 1784.25 ± 89.24 (1.00) | 1045.20 ± 197.41 (0.59) |
| +Linsitinib 2 μM | 1952.52 ± 221.90 (1.09) | 963.76 ± 63.33 (0.54) |
IC50: concentration that inhibited cell survival by 50% (means ± SD).
Resistance-fold was determined by dividing the IC50 values of substrate in HEK293/ABCC10 cells by the IC50 of substrate in HEK293/pcDNA3.1 cells in the absence of linsitinib; or the IC50 of substrate in HEK293/pcDNA3.1 cells in the presence of linsitinib divided by the IC50 of substrate in HEK293/pcDNA3.1 cells in the absence of linsitinib. Values in table are means ± SD of three independent experiments performed in triplicate.
p < 0.01 versus that obtained in the absence of inhibitor.
Linsitinib, at 2 μM, moderately increased the cytotoxicity of vincristine in ABCB1-overexpressing cells SW620/AD300 as the IC50 of vincristine in SW620/AD300 cells decreased from 909.60 to 274.24 nM. However, linsitinib did not significantly increase the cytotoxicity of vincristine in SW620 cells (Table 4). In addition, linsitinib did not significantly reverse ABCC1-mediated MDR (Table 4). Based on the above results, we conclude that linsitinib significantly surmounts ABCG2- and ABCC10-mediated MDR, and it moderately sensitizes ABCB1-overexpressing cells to its substrate antineoplastic drugs, but has no effect on ABCC1-mediated MDR.
Table 4.
The effect of linsitinib on reversal of ABCB1- and ABCC1-mediated MDR.
| Drugs | IC50 ± SDa (nM) (resistance fold) b |
|
|---|---|---|
| SW620 | SW620/AD300 | |
| Vincristine | 7.80 ± 0.08 (1.00) | 909.60 ± 8.91 (116.62) |
| +Linsitinib 1 μM | 7.06 ± 0.07 (0.91) | 885.08 ± 7.92 (113.47) |
| +Linsitinib 2 μM | 6.42 ± 0.09 (0.82) | 274.24 ± 22.88** (35.16) |
| +Verapamil 2 μM | 7.23 ± 0.11 (0.93) | 70.23 ± 9.40** (9.00) |
| HEK293/pcDNA3.1 | HEK293/ABCC1 | |
| Vincristine | 1.45 ± 0.20 (1.00) | 19.29 ± 2.08 (13.30) |
| +Linsitinib 1 μM | 1.41 ± 0.22 (0.97) | 19.86 ± 1.55 (13.70) |
| +Linsitinib 2 μM | 1.39 ± 0.17 (0.96) | 21.02 ± 2.10 (14.49) |
| +PAK-104P 2 μM | 1.20 ± 0.08 (0.83) | 3.83 ± 0.42** (2.64) |
IC50: concentration that inhibited cell survival by 50% (means ± SD).
Resistance-fold was determined by dividing the IC50 values of substrate in SW620/AD300 cells by the IC50 of substrate in SW620 cells in the absence of linsitinib; or the IC50 of substrate in SW620 cells in the presence of linsitinib divided by the IC50 of substrate in SW620 cells in the absence of linsitinib. The resistance-fold for HEK293/pcDNA3.1 and HEK293/ABCC1 cells was obtained in the similar manner. Values in table are means ± SD of three independent experiments performed in triplicate.
p < 0.01 versus that obtained in the absence of inhibitor.
3.2. Linsitinib significantly increases the accumulation of [3H]-MX and [3H]-paclitaxel
To ascertain how linsitinib sensitizes ABCG2- and ABCC10-overexpressing cells to chemotherapeutic drugs, we examined the effect of linsitinib on the accumulation of chemotherapeutic drugs in ABCG2- or ABCC10-overexpressing cells. The intracellular level of [3H]-MX in ABCG2-overexpressing cells was significantly lower than that of HEK293/pcDNA3.1 cells. After 2 h of incubation, linsitinib at 1 or 2 μM, significantly increased the intracellular level of [3H]-MX in ABCG2-overexpressing cells and this effect was comparable to that of 2 μM of FTC. In contrast, linsitinib did not significantly alter [3H]-MX levels in the parental HEK293/pcDNA3.1 cells (Fig. 2A). Similarly, the intracellular levels of [3H]-paclitaxel were significantly increased in ABCC10-overexpressing cells in the presence or absence of linsitinib (Fig. 2B).
Fig. 2.
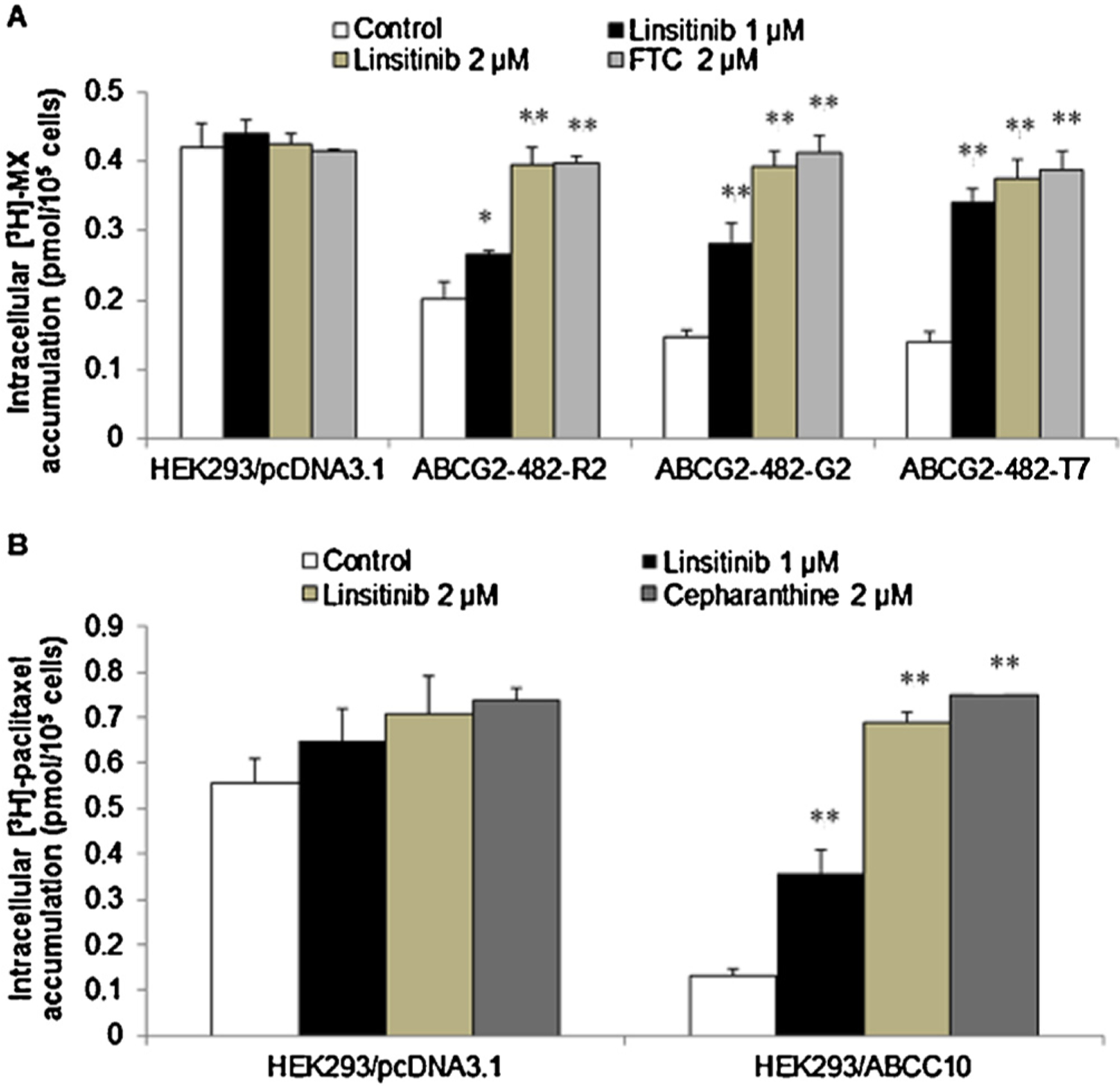
The effect of linsitinib on the accumulation of [3H]-MX and [3H]-paclitaxel, respectively. (A) The accumulation of [3H]-MX was significantly increased in ABCG2-482-R2, ABCG2-482-G2 and ABCG2-482-T7 cell lines in the presence of linsitinib. (B) The accumulation of [3H]-paclitaxel was increased in HEK293/ABCC10 cell line in the presence of linsitinib. *indicates p < 0.05 or **indicates p < 0.01 versus the control group. Error bars represent the SD. The experiments were performed at least three independent times.
3.3. Linsitinib significantly decreases the efflux of [3H]-MX and [3H]-paclitaxel
To ascertain whether the increase in the intracellular accumulation of [3H]-MX or [3H]-paclitaxel in the presence of linsitinib was due to the inhibition of efflux function, we performed a time course study to measure intracellular [3H]-MX or [3H]-paclitaxel levels in the presence or absence of linsitinib. A 2 μM concentration of linsitinib significantly blocked [3H]-MX efflux from the ABCG2-482-R2 cells at different time periods (0, 30, 60, 120 min) (Fig. 3A). However, linsitinib did not significantly alter [3H]-MX efflux in the parental cells (Fig. 3A). Similarly, linsitinib at 2 μM, significantly decreased [3H]-paclitaxel efflux at different time periods (0, 30, 60, 120 min) from the HEK293/ABCC10 cells (Fig. 3B). These results further validate the efficacy of linsitinib to inhibit the drug efflux function of ABCG2 and ABCC10.
Fig. 3.
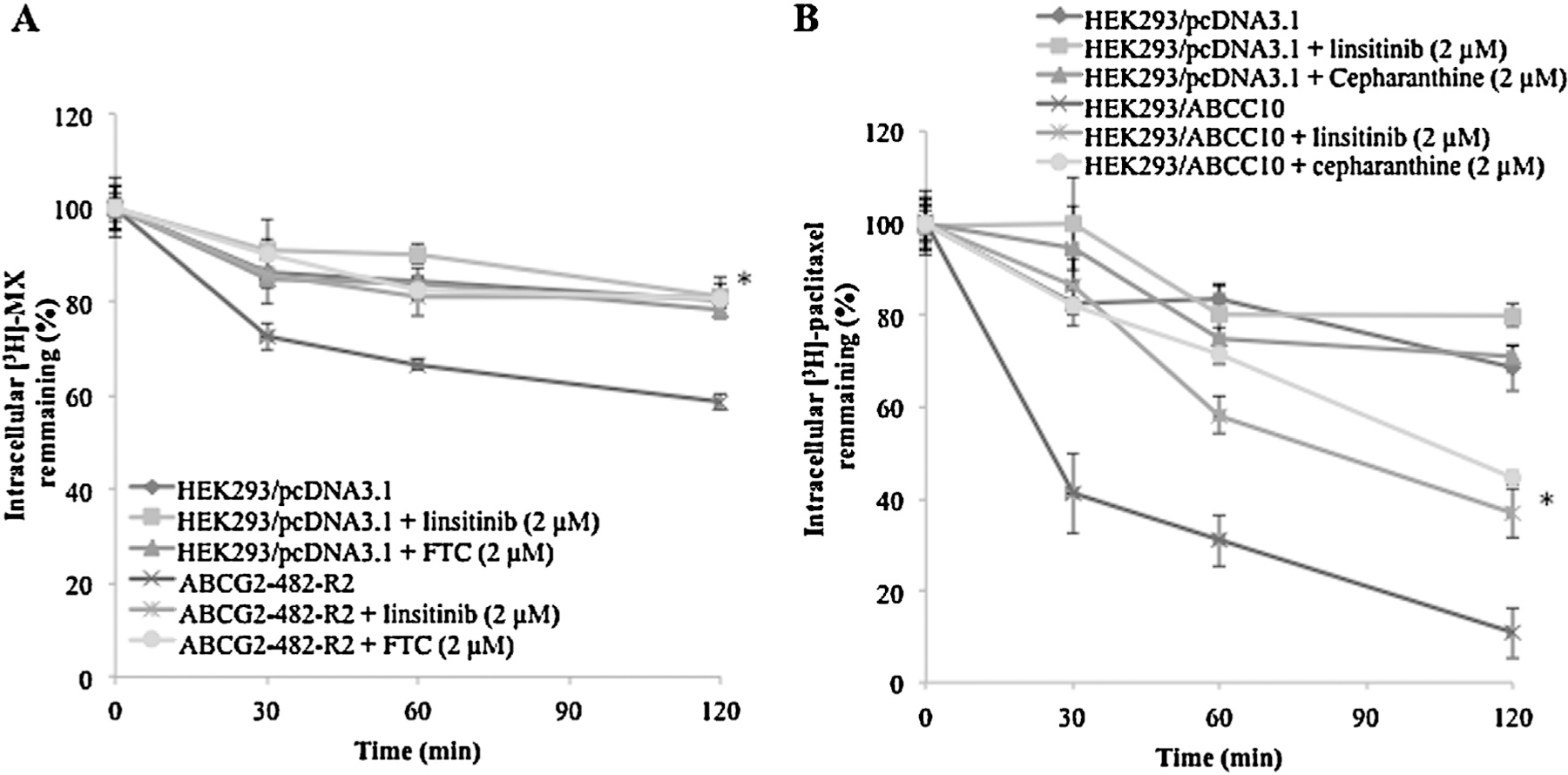
The effect of linsitinib on the efflux of [3H]-MX and [3H]-paclitaxel, respectively. (A) A time course (0, 30, 60, 120 min) versus percentage of intracellular [3H]-MX remaining (%) was plotted using HEK293/pcDNA3.1 and ABCG2-482-R2 cell lines in presence or absence of linsitinib or FTC; (B) A time course (0, 30, 60, 120 min) versus percentage of intracellular [3H]-paclitaxel remaining (%) was plotted using HEK293/pcDNA3.1 and HEK293/ABCC10 cell lines in presence or absence of linsitinib or cepharanthine. *indicates p < 0.05 versus the control group. Error bars represent the SD. The experiments were performed at least three independent times.
3.4. Linsitinib does not significantly alter the expression levels and cellular localization of the ABCG2 or ABCC10 transporter proteins
ABCG2- and ABCC10-mediated MDR could be reversed either by decreasing the protein expression of ABCG2 and ABCC10 or by inhibiting their transport function. The results indicated that linsitinib, at 2 μM, did not significantly alter the expression levels of the ABCG2 or ABCC10 proteins (Fig. 4A–D). Immunoblot analysis indicated bands in the ABCG2-482-R2, ABCG2-482-G2, ABCG2-482-T7, SW620/AD300, HEK293/ABCC1 cell lysates, suggesting the presence of the ABCG2, ABCB1 and ABCC1 protein, respectively. However, this band was not present in the parental HEK293/pcDNA3.1 and SW620 cells (Supplemental Fig. 1).
Fig. 4.
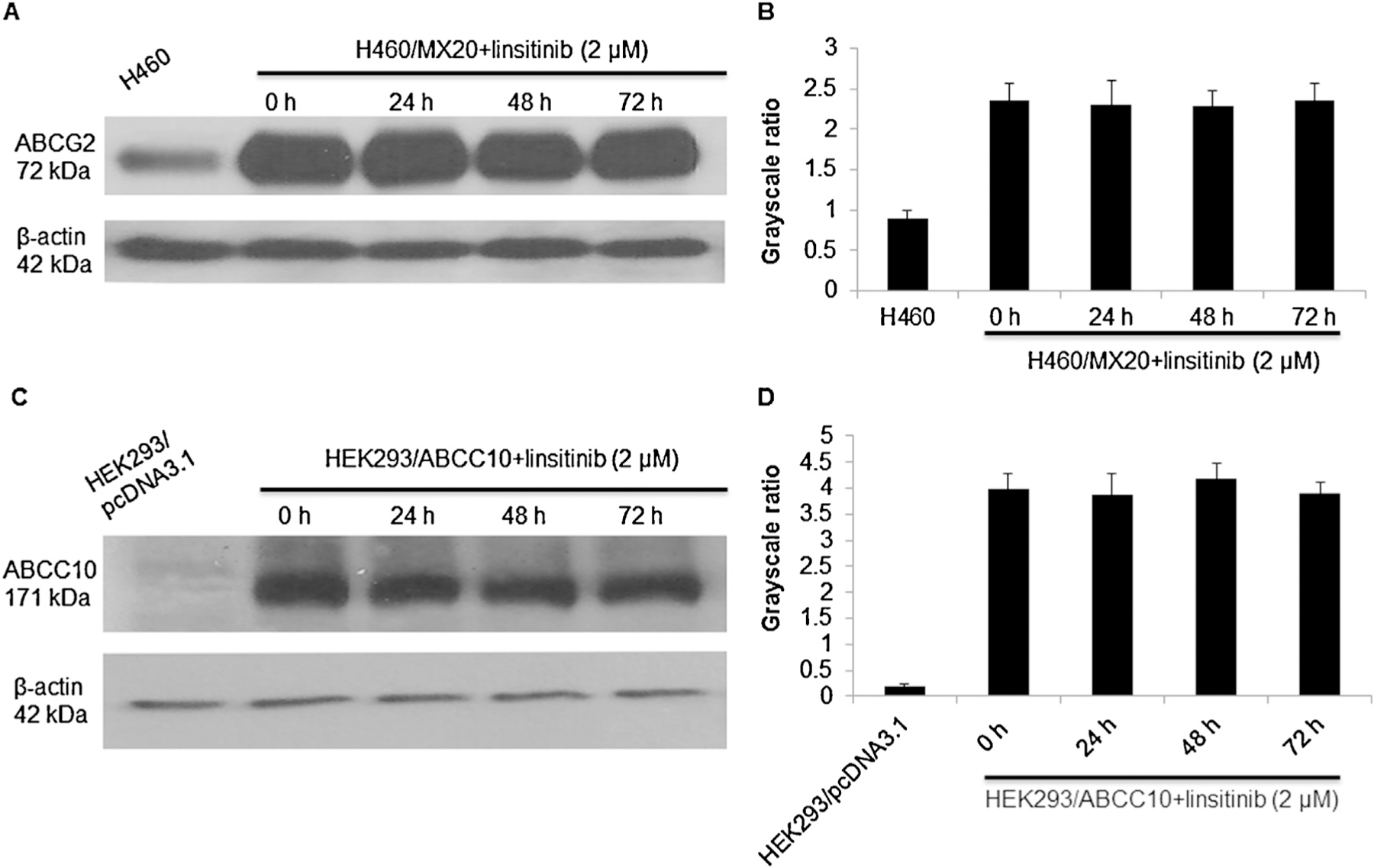
The effect of linsitinib on the expression levels of ABCG2 and ABCC10 transporters, respectively. (A and B) The expression levels of ABCG2 protein in H460 and H460/MX20 cell lysates were shown. (C and D) The expression levels of ABCC10 protein in HEK293/pcDNA3.1 and HEK293/ABCC10 cell lysates were shown. Image J was used to analyze the grayscale ratios. The grayscale ratios were proportional to the ABCG2 or ABCC10 protein levels. The differences were statistically non-significant (p > 0.05). A representative result was shown and similar results were obtained in two other experiments.
Furthermore, the results of immunofluorescence assay indicated that the incubation of H460/MX20 and HEK293/ABCC10 cells with 2 μM linsitinib for 72 h did not alter cellular localization of ABCG2 or ABCC10 (Fig. 5A and B) protein. Thus, these results suggest that the reversal effect of linsitinib is not related to its effect on the expression or the localization of the ABCG2 or ABCC10 protein.
Fig. 5.
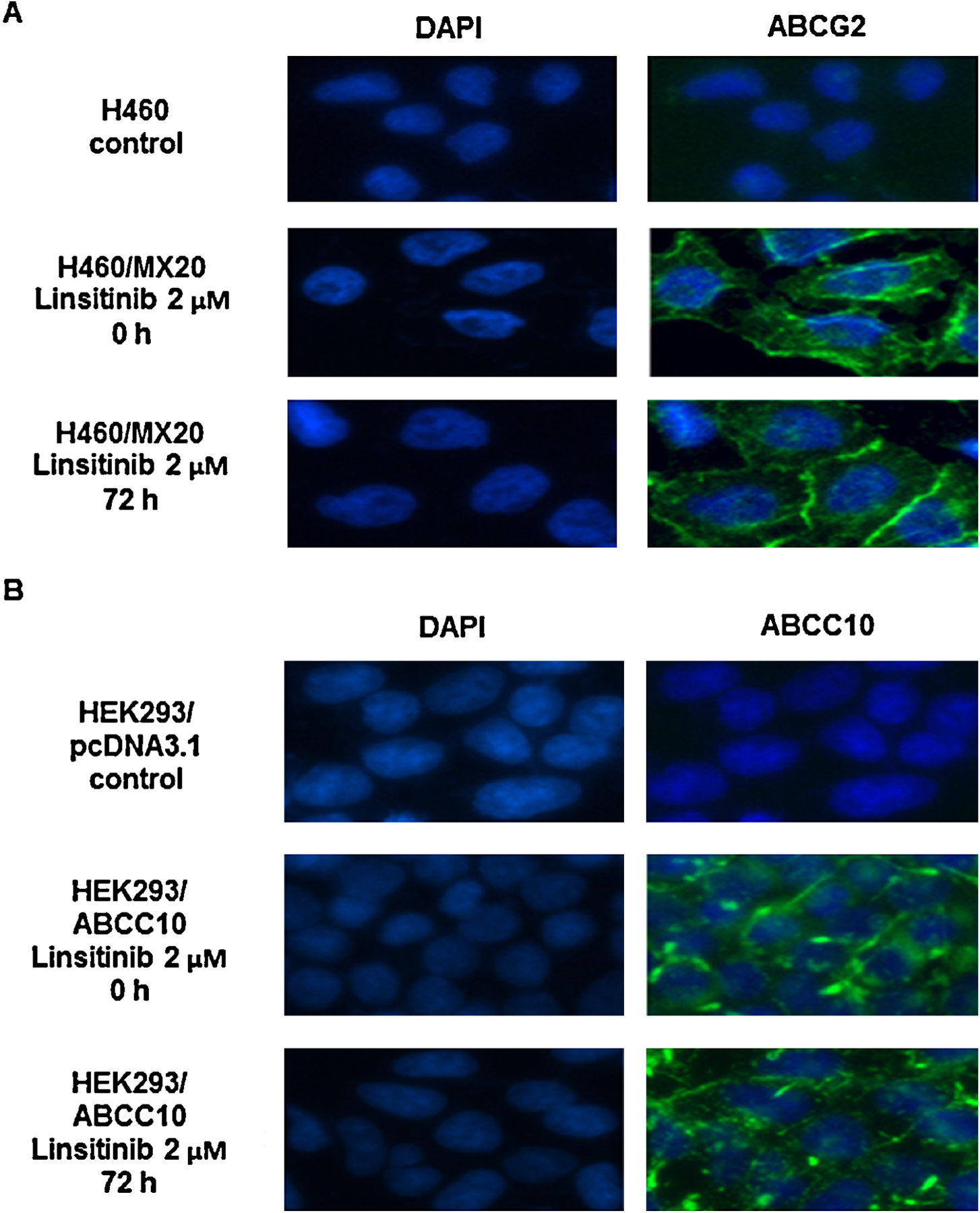
The effect of linsitinib treatment on the subcellular localization of ABCG2 and ABCC10, respectively. (A) H460/MX20 cells were treated with 2 μM linsitinib for 72 h. The subcellular localization of ABCG2 was analyzed by immunofluorescence. ABCG2 staining is shown in green. DAPI (blue) counterstains the nuclei. (B) HEK293/ABCC10 cells were treated with 2 μM linsitinib for 72 h. The subcellular localization of ABCC10 was analyzed by immunofluorescence. ABCC10 staining is shown in green. DAPI (blue) counterstains the nuclei. A representative result was shown and similar results were obtained in two other experiments.
3.5. Linsitinib stimulates ATPase activity of ABCG2
ABCG2 utilizes the energy of ATP hydrolysis for efflux of drug-substrates, and substrates/modulators either stimulate or inhibit ATPase activity. The stimulatory effect of test compounds on ATPase activity of ABCG2 indicates that the compound is interacting at the drug-substrate-binding site on the transporter. For this reason, we measured ABCG2-mediated ATP hydrolysis in the presence of different concentrations of linsitinib. Linsitinib increased the vanadate-sensitive ATPase activity in a concentration-dependent manner with a fold-stimulation of ~2-fold at higher concentrations. The fact that linsitinib stimulated the ATPase activity suggests that it interacts with the drug-substrate-binding site, possibly similar to other substrates. However, whether linsitinib is transported by ABCG2 needs to be validated by doing transport assays using a fluorescent or radiolabeled linsitinib (Fig. 6).
Fig. 6.
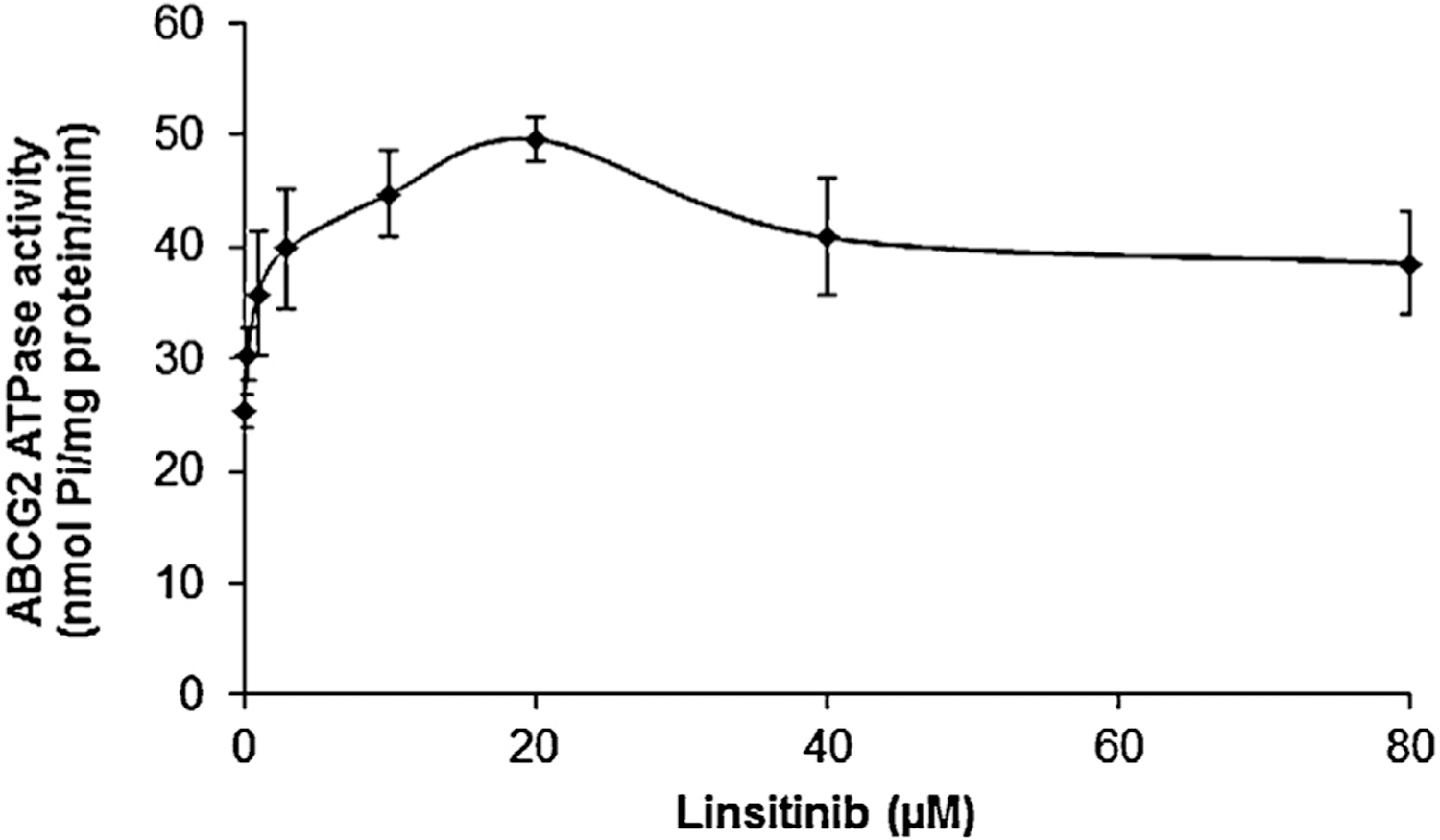
The effect of linsitinib on ATP hydrolysis by ABCG2. Crude membranes (10 μg protein/reaction) from High-five cells expressing ABCG2 were incubated with increasing concentrations of linsitinib (0–80 μM) in the presence and absence of 0.3 mM vanadate, in ATPase assay buffer as described in Section 2. The mean values are plotted and error bars represent the SD. The experiments were performed at least three independent times.
3.6. Model for binding of linsitinib to ABCG2
The highest scored docking pose for linsitinib was found at Asn629-centroid grid and this pose revealed the importance of hydrophobic interactions in linsitinib binding. The phenyl ring was stabilized by nearby residues Leu626, Trp627, His630 and Val631 through hydrophobic interactions. The quinoline ring was stabilized into a large hydrophobic cavity formed by residues Phe507, Phe511, Ala580, Leu581, Leu626 and the backbone of Asn629. The quinoline ring nitrogen atom formed electrostatic interaction with the side chain of Asn629 (N1⋯H2N-Asn629, 3.6 Å). The imidazo[1,5-a]pyrazine ring formed π–π interaction with phenyl ring of Phe511. The 1-methyl-cyclobutanol group was stabilized by backbone atoms of nearby residues Val508, Thr512 and Met515. The hydroxyl group formed a hydrogen bond with the hydroxyl group of Thr512 (OH⋯OH-Thr512, 2.1 Å) (Fig. 7).
Fig. 7.
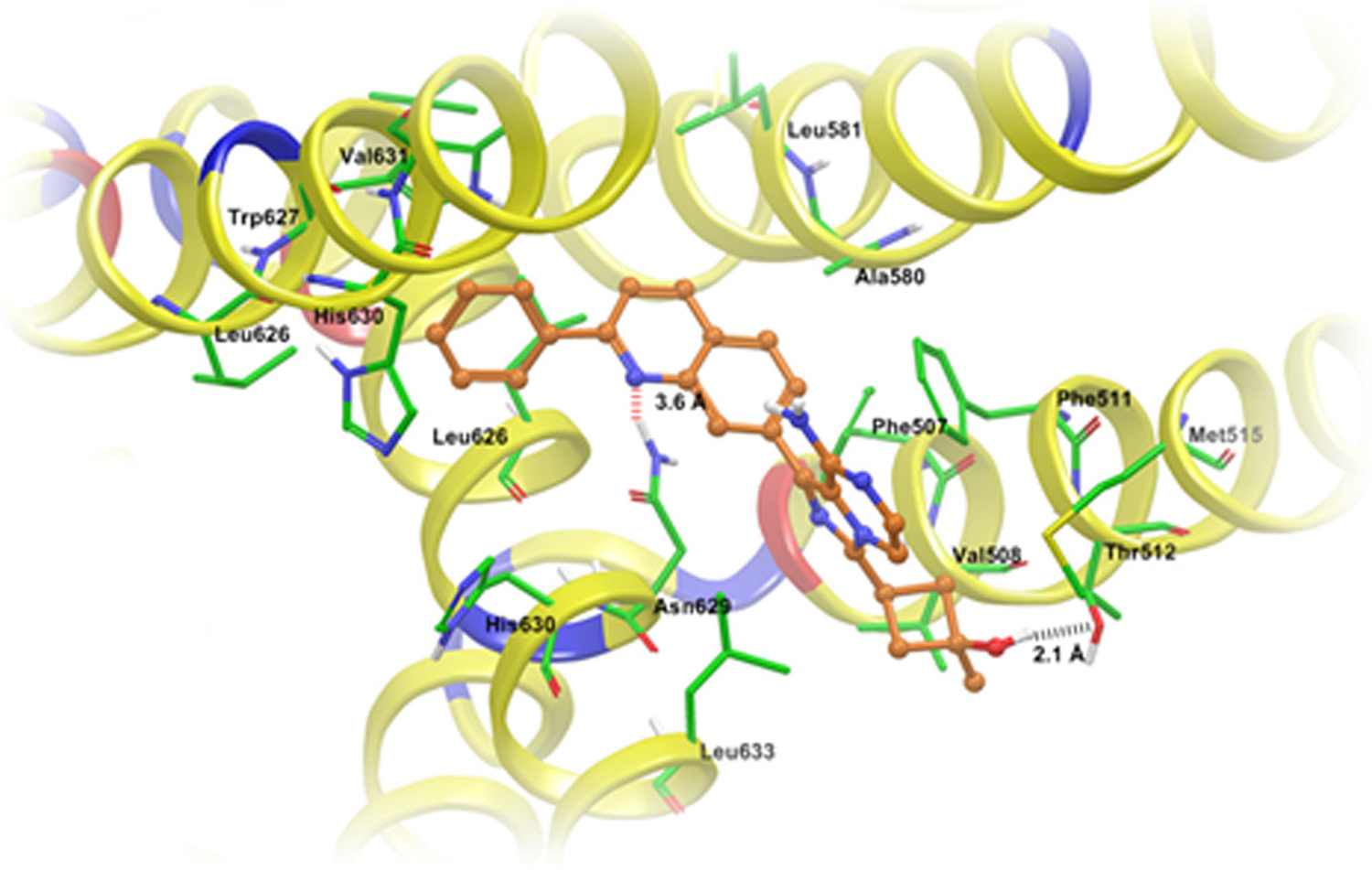
XP Glide predicted binding mode of linsitinib with homology modeled ABCG2. The docked conformation of linsitinib as ball and stick model is shown within the large cavity of ABCG2. Important amino acids are depicted as sticks with the atoms colored as carbon – green, hydrogen – white, nitrogen – blue, oxygen – red and sulfur – yellow, whereas linsitinib is shown with the same color scheme as above except carbon atoms are represented in orange. Dotted black line indicates hydrogen bonding interactions, whereas dotted red line indicates electrostatic interactions. ABCG2 is represented as ribbons based on residue charge (hydrophobic-yellow, basic-blue, acidic-red).
4. Discussion
One of the major findings of the study was that linsitinib could significantly reverse ABCG2-mediated MDR. The combination of linsitinib at nontoxic concentrations (1 and 2 μM) and ABCG2-substrate anticancer drugs could enhance the cytotoxic effect in ABCG2-overexpressing H460/MX20, ABCG2-482-R2, ABCG2-482-G2 and ABCG2-482-T7 cells (Tables 1 and 2). The inhibition of ABCG2 drug efflux function was observed in both wide-type and mutant ABCG2-overexpressing cells. In addition, linsitinib could slightly sensitize H460 cells to ABCG2-substrates (Table 1). This phenomenon was consistent with the low expression levels of ABCG2 protein in the H460 cells as indicated by the Western blot analysis (Fig. 4A and B). We know that the reversal effect of linsitinib could be due in part by inhibition of the efflux ability of ABC transporters or by reducing ABCG2 protein expression. Therefore, we examined the effect of linsitinib on the intracellular accumulation of [3H]-MX in ABCG2-overexpressing cells. Consistent with the cytotoxicity data, the results of drug accumulation assay showed that linsitinib significantly enhanced the intracellular accumulation of [3H]-MX (Fig. 2A). In addition, time course of efflux studies revealed that linsitinib could block the efflux function of ABCG2 (Fig. 3A). Western blot analysis results demonstrated that there was no evident alteration of ABCG2 protein expression levels in MDR cells incubated with linsitinib up to 72 h at 2 μM concentration. Furthemore, immunofluorescence assay indicated that the cellular localization of ABCG2 did not alter when treated with linsitinib (2 μM) for 72 h. Thus, we conclude that the reversal effect of linsitinib might result from inhibition of the efflux function of ABCG2 transporter as opposed to alteration in protein expression levels. As energy is utilized by ABC transporters from ATP hydrolysis, we investigated the effect of linsitinib on ATPase activity of ABCG2 transporter. Here, we found that linsitinib stimulated ATPase activity of ABCG2 transporter in a concentration-dependent manner, indicating that linsitinib might be a substrate of ABCG2.
Another major finding of this study was that linsitinib (2 μM) significantly enhanced the cytotoxicity of the ABCC10 substrates like paclitaxel, docetaxel and vinblastine in ABCC10-overexpressing HEK293/ABCC10 cells (Table 3). However, linsitinib (2 μM) did not significantly sensitize the parental HEK293/pcDNA3.1 to the anticancer drugs used in this study. Thus, we conducted experiments to ascertain if linsitinib affected the intracellular accumulation and efflux of [3H]-paclitaxel, a substrate for the ABCC10 transporter. Linsitinib (1 and 2 μM), as well as cepharanthine (2 μM), a known ABCC10 inhibitor, produced a significant concentration-dependent increase in the intracellular accumulation of [3H]-paclitaxel in HEK293/ABCC10 cells (Fig. 2B). In addition, linsitinib and cepharanthine significantly decreased the efflux of [3H]-paclitaxel from cells overexpressing the ABCC10 transporter (Fig. 3B). These findings tentatively suggest that linsitinib increases the sensitivity of ABCC10 overexpressing cells to paclitaxel (and potentially other substrate drugs) by inhibiting their efflux from the cells. It could be argued that linsitinib increases the sensitivity of ABCC10 transfected cells to docetaxel, paclitaxel and vinblastine via a decrease in the expression of the ABCC10 transporter protein. However, this is unlikely as the incubation of cells with linsitinib (2 μM for 0, 24, 48 and 72 h) did not significantly alter the expression of the ABCC10 protein (171-kDa protein on in the immunoblot assay) in HEK293 cells transfected with the ABCC10 transporter. This finding is consistent with our results indicating that there is no significant alteration in the sensitivity of parental HEK293/pcDNA3.1 cells to the ABCC10 substrate drugs in the presence of linsitinib.
Furthermore, linsitinib could moderately enhance the cytotoxicity of vincristine in SW620/AD300 cells but not its parental SW620 cells (Table 4). We also measured the reversal effect of linsitinib in another important ABC transporter ABCC1, but the results showed that linsitinib did not significantly alter the IC50 values of ABCC1-substrates in HEK293/ABCC1 cells (Table 4). These results indicated that linsitinib at 1 μM–2 μM could significantly sensitize ABCG2- and ABCC10-overexpressing cells to their substrate chemotherapeutic drugs.
In order to understand the binding interaction of linsitinib, we performed docking simulations at several grids based on previous mutation studies on ABCG2 (Robey et al., 2003). The highest-score docked model of linsitinib was found at Asn629-centroid grid, which may explain its non-selective reversal effect for ABCG2-482-G2, ABCG2-482-R2 and ABCG2-482-T7 cell lines. Molecular structure of lintisinib exhibited the pharmacophoric features such as hydrophobic groups (Clog P value = 3.42) and/or aromatic ring centers (quinoline, phenyl, imidazopyrazine rings), hydrogen bond donors and acceptors (quinoline ring nitrogen and hydroxy group) (Supplemental Table 1) that had been identified as essential for ABCG2 inhibition (Nicolle et al., 2009).
In conclusion, linsitinib significantly inhibits ABCG2 and ABCC10 efflux function, without altering protein expression levels and reverses the ABCG2- and ABCC10-mediated MDR, respectively. Furthermore, linsitinib could moderately reverse ABCB1 mediated MDR. Linsitinib stimulates ATPase activity of ABCG2 suggesting that it might be a substrate of ABCG2 transporter. These results tentatively suggest that linsitinib has previously unknown function of augmenting the clinical response to conventional chemotherapeutic agents that are substrates of ABCG2 or ABCC10, in patients with MDR.
Supplementary Material
Acknowledgements
This work was supported by funds from NIH (No. 1R15CA143701) and St. John’s University Research Seed Grant (No. 579-1110-7002) to Z.S. Chen, The Major Science and Technology Project of The National Natural Sciences Foundation of China Grant (No. 81072669 and No. 81061160507) to L.W. Fu. Drs. S. Shukla, R.W. Robey, S.V. Ambudkar and S.E. Bates were supported by the Intramural Research Program, National Institutes of Health, National Cancer Institute, Center for Cancer Research. We are grateful to late Dr. Gary Kruh (University of Illinois at Chicago) for providing ABCC10 plasmid. We thank Dr. Mark F. Rosenberg (University of Manchester, Manchester, UK) and Dr. Zsolt Bikádi (Virtua Drug Ltd., Budapest, Hungary) for providing coordinates of ABCG2 homology model.
Footnotes
Conflict of interest
No potential conflicts of interest were disclosed.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.biocel.2014.03.026.
References
- Alvarez M, Paull K, Monks A, Hose C, Lee JS, Weinstein J, et al. Generation of a drug resistance profile by quantitation of mdr-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. J Clin Invest 1995;95:2205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol 1998;292:504–14. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54. [DOI] [PubMed] [Google Scholar]
- Chen ZS, Robey RW, Belinsky MG, Shchaveleva I, Ren XQ, Sugimoto Y, et al. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res 2003;63:4048–54. [PubMed] [Google Scholar]
- de Paiva CS, Chen Z, Corrales RM, Pflugfelder SC, Li DQ. ABCG2 transporter identifies a population of clonogenic human limbal epithelial cells. Stem Cells 2005;23:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeure MJ, Bussey KJ, Kirschner LS. Targeted therapies for adrenocortical carcinoma: IGF and beyond. Horm Cancer 2011;2:385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Dai CL, Chen JJ, Kathawala RJ, Sun YL, Chen HF, et al. Tandutinib (MLN518) reverses multidrug resistance by inhibiting the efflux activity of the multidrug resistance protein 7 (ABCC10). Oncol Rep 2013;29:2479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle L, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003;22:7340–58. [DOI] [PubMed] [Google Scholar]
- Ejendal KF, Hrycyna CA. Multidrug resistance and cancer: the role of the human ABC transporter ABCG2. Curr Protein Pept Sci 2002;3:503–11. [DOI] [PubMed] [Google Scholar]
- Fletcher JI, Haber M, Henderson MJ, Norris MD. ABC transporters in cancer: more than just drug efflux pumps. Nat Rev Cancer 2010;10:147–56. [DOI] [PubMed] [Google Scholar]
- Garimella TS, Ross DD, Bauer KS. Liquid chromatography method for the quantitation of the breast cancer resistance protein ABCG2 inhibitor fumitremorgin C and its chemical analogues in mouse plasma and tissues. J Chromatogr B Analyt Technol Biomed Life Sci 2004;807:203–8. [DOI] [PubMed] [Google Scholar]
- Guo HQ, Zhang GN, Wang YJ, Zhang YK, Sodani K, Talele TT, et al. Beta-elemene, a compound derived from Rhizoma zedoariae, reverses multidrug resistance mediated by the ABCB1 transporter. Oncol Rep 2013, Epub ahead of print. [DOI] [PubMed]
- Henrich CJ, Bokesch HR, Dean M, Bates SE, Robey RW, Goncharova EI, et al. A high-throughput cell-based assay for inhibitors of ABCG2 activity. J Biomol Screen 2006;11:176–83. [DOI] [PubMed] [Google Scholar]
- Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res 2001;61:6635–9. [PubMed] [Google Scholar]
- Kathawala RJ, Sodani K, Chen K, Patel A, Abuznait AH, Anreddy N, et al. Masitinib Antagonizes ATP-binding cassette subfamily C Member 10-mediated paclitaxel resistance: a preclinical study. Mol Cancer Therapeut 2014, Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Kathawala RJ, Wang YJ, Ashby CR Jr., Chen FZS Recent advances regarding the role of ABC subfamily C member 10 (ABCC10) in the efflux of antitumor drugs. Chin J Cancer 2013, Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Kruh GD, Guo Y, Hopper-Borge E, Belinsky MG, Chen ZS. ABCC10, ABCC11, and ABCC12. Pflugers Arch 2007;453:675–84. [DOI] [PubMed] [Google Scholar]
- Mao Q, Unadkat JD. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J 2005;7:E118–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow CS, Peklak-Scott C, Bishwokarma B, Kute TE, Smitherman PK, Townsend AJ. Multidrug resistance protein 1 (MRP1, ABCC1) mediates resistance to mitoxantrone via glutathione-dependent drug efflux. Mol Pharmacol 2006;69:1499–505. [DOI] [PubMed] [Google Scholar]
- Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, Buck E, Foreman K, Landfair D, et al. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med Chem 2009;1:1153–71. [DOI] [PubMed] [Google Scholar]
- Nicolle E, Boumendjel A, Macalou S, Genoux E, Ahmed-Belkacem A, Carrupt PA, et al. QSAR analysis and molecular modeling of ABCG2-specific inhibitors. Adv Drug Deliv Rev 2009;61:34–46. [DOI] [PubMed] [Google Scholar]
- Patel A, Tiwari AK, Chufan EE, Sodani K, Anreddy N, Singh S, et al. PD173074, a selective FGFR inhibitor, reverses ABCB1-mediated drug resistance in cancer cells. Cancer Chemother Pharmacol 2013;72:189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer 2003;89:1971–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scagliotti GV, Novello S. The role of the insulin-like growth factor signaling pathway in non-small cell lung cancer and other solid tumors. Cancer Treat Rev 2012;38:292–302. [DOI] [PubMed] [Google Scholar]
- Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics 2008;9:105–27. [DOI] [PubMed] [Google Scholar]
- Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos 2009;37:359–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodani K, Patel A, Anreddy N, Singh S, Yang DH, Kathawala RJl, et al. Telatinib reverses chemotherapeutic multidrug resistance mediated by ABCG2 efflux transporter in vitro and in vivo. Biochem Pharmacol 2014, Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Sodani K, Patel A, Kathawala RJ, Chen ZS. Multidrug resistance associated proteins in multidrug resistance. Chin J Cancer 2012a;31:58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodani K, Tiwari AK, Singh S, Patel A, Xiao ZJ, Chen JJ, et al. GW583340 and GW2974, human EGFR and HER-2 inhibitors, reverse ABCG2- and ABCB1-mediated drug resistance. Biochem Pharmacol 2012b;83:1613–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YL, Chen JJ, Kumar P, Chen K, Sodani K, Patel A, et al. Reversal of MRP7 (ABCC10)-mediated multidrug resistance by tariquidar. PLoS ONE 2013;8:e55576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YL, Kathawala RJ, Singh S, Zheng K, Talele TT, Jiang WQ, et al. Zafirlukast antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance. Anticancer Drugs 2012;23:865–73. [DOI] [PubMed] [Google Scholar]
- Tiwari AK, Sodani K, Dai CL, Abuznait AH, Singh S, Xiao ZJ, et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett 2013;328:307–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Kathawala RJ, Chufan EE, Patel A, Ambudkar SV, Chen ZS, et al. Tivozanib reverses multidrug resistance mediated by ABCB1 (P-glycoprotein) and ABCG2 (BCRP). Future Oncol 2013, Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Zhao XQ, Xie JD, Chen XG, Sim HM, Zhang X, Liang YJ, et al. Neratinib reverses ATP-binding cassette B1-mediated chemotherapeutic drug resistance in vitro, in vivo, and ex vivo. Mol Pharmacol 2012;82:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.