

Abstract
Abstract
To identify novel adenosine receptor (AR) ligands based on the chalcone scaffold, herein the synthesis, characterization and in vitro and in silico evaluation of 33 chalcones (15–36 and 37–41) and structurally related compounds (42–47) are reported. These compounds were characterized by radioligand binding and GTP shift assays to determine the degree and type of binding affinity, respectively, against rat (r) A1 and A2A ARs. The chalcone derivatives 24, 29, 37 and 38 possessed selective A1 affinity below 10 µM, and thus, are the most active compounds of the present series; compound 38 was the most potent selective A1 AR antagonist (Ki (r) = 1.6 µM). The structure–affinity relationships (SAR) revealed that the NH2-group at position C3 of ring A of the chalcone scaffold played a key role in affinity, and also, the Br-atom at position C3′ on benzylidene ring B. Upon in vitro and in silico evaluation, the novel C3 amino-substituted chalcone derivative 38—that contains an α,ß-unsaturated carbonyl system and easily allows structural modification—may possibly be a synthon in future drug discovery.
Graphic abstract
C3 amino-substituted chalcone derivative (38) with C3′ Br substitution on benzylidene ring B possesses selective adenosine rA1 receptor affinity in micromolar range.
Electronic supplementary material
The online version of this article (10.1007/s11696-020-01414-9) contains supplementary material, which is available to authorized users.
Keywords: Chalcone, Schiff base, Base- and acid-catalysed Claisen–Schmidt condensation reactions, Selective adenosine A1 receptor antagonist, Neurological conditions
Introduction
In the human body, adenosine takes part in both physiologic and pathophysiologic processes (Boison 2018). Most, if not all, of these actions are mediated by four cell surface receptors, denoted A1, A2A, A2B and A3 (Fredholm et al. 1994, 2001, 2011). Ensuing almost 100 years of research on adenosine and its receptors as well as the ligands that bind to these adenosine receptors (ARs), the potential of the adenosine system as a drug discovery target is evident (de Lera Ruiz et al. 2013).
The A1 AR has been cloned from different mammalian species, including humans (Müller 2001) and show structural homology among diverse mammalian species (Ralevic and Burnstock 1998) (although lower affinity for human than rat receptors have been reported) (Klotz et al. 1997). A member of the seven transmembrane-spanning G-protein-coupled receptor superfamily (that preferentially couples to inhibitory Gi/o heterotrimeric G-proteins) (Freissmuth et al. 1991; Munshi et al. 1991; Ralevic and Burnstock 1998), the A1 AR inhibits adenylate cyclase and decreases cyclic adenosine monophosphate (Londos et al. 1980; Van Calker et al. 1978). Other second messenger systems may also be coupled to the A1 AR, such as phospholipase C and several types of calcium and potassium channels (Ralevic and Burnstock 1998). A1 ARs are widely distributed in different mammalian species (Ralevic and Burnstock 1998) and ubiquitous within the central nervous system, with the highest density in the cerebral cortex, cerebellum, hippocampus, thalamus, brainstem and spinal cord (Dixon et al. 1996; Reppert et al. 1991). These receptors are localized in the active zone of the presynaptic nerve terminal (Rebola et al. 2003; Swanson et al. 1995), a highly specialized region of the cytoplasm that directly faces the synaptic cleft (Sankaranarayanan and Ryan 2007).
At presynaptic nerve terminals, A1 ARs play a role in the release of neurotransmitters (Dunwiddie 1985). Adenosine acts by inhibiting cholinergic transmission, among other, via A1 ARs (Phillis 1991). The cholinergic system has been associated with a number of cognitive functions, for example, learning and memory as well as emotion (Jackson 2011).
Interaction between the adenosine and cholinergic system is regrettably controversial; on the one hand, administration of a selective A1 AR agonist (e.g. CPA) caused learning and memory deficits, which may be prevented by a selective A1 AR antagonist (e.g. DPCPX) (Normile and Barraco 1991), and on the other hand, the said antagonist also caused learning and memory deficits (Vollert et al. 2013)! Moreover, it was found that chronic treatment with A1 AR modulators result in behavioural effects different from those observed following acute administration; therefore, particular caution is required in the development of adenosine-based strategies for the chronic treatment of neurodegenerative or cognitive disorders (Von Lubitz et al. 1993). Other studies advocated the use of A1 AR antagonists and considered the A1 AR an interesting drug target for the development of cognitive enhancers based on the observation that A1 AR antagonism positively modulated memory process (Maemoto et al. 2004; Normile and Barraco 1991; Pitsikas and Borsini 1997; Suzuki et al. 1993).
Apart from learning and memory, adenosine and its receptors—specifically the A1 AR—also modulate anxiety where blockade of the A1 AR results in anxiolytic actions (Maemoto et al. 2004). The A1 AR agonist R-PIA exacerbated the effects of ethanol withdrawal, whereas the A1 AR antagonist CPT improved these anxiogenic effects in the elevated plus-maze and light/dark test, relevant behavioural animal models of anxiety (Gatch et al. 1999). These results suggest that A1 AR antagonists, at some doses, may be useful for ameliorating the anxiogenic effects produced by ethanol withdrawal, although it does not appear useful for reducing ethanol consumption (Gatch et al. 1999). Furthermore, the widely used anxiolytic agents, benzodiazepines, block adenosine uptake (Noji et al. 2004) and decreased A1 AR binding capacity in vivo at doses of clinical relevance (Kaplan et al. 1992). As with the adenosine system and its effects on learning and memory, there is controversy: DPCPX did not affect the anxiety state of mice in the elevated plus-maze test (Jain et al. 1995) and A1 AR knockout mice showed signs of increased anxiety in the light/dark test (Johansson et al. 2001).
Adenosine and its analogues have been known to cause behavioural despair in animal models relevant to depression. The A2A, rather than the A1, AR is involved in depression; based on evidence from pharmacology and A2A AR knockout mice (Yacoubi et al. 2001). (Additionally, the selective A2A AR antagonist istradefylline not only improves motor fluctuations but also some non-motor symptoms of Parkinson’s disease (PD); such as the mood disorder depression—the most common non-motor symptom of PD (Aarsland et al. 2012; Nagayama et al. 2019) This is yet to be confirmed by a double-blind placebo-controlled trial (Nagayama et al. 2019). Interestingly, the selective A1 AR antagonist DPCPX enhanced the anti-depressant effects of selective serotonin re-uptake inhibitors (SSRIs) imipramine, escitalopram and reboxetine in mice behavioural tests (Szopa et al. 2018); nevertheless, the effects of atypical antidepressants agomelatine and tianeptine were increased, not by DPCPX, but by the selective A2A AR antagonist DMPX (Szopa et al. 2019).
The orally active and brain penetrable pyrazolopyridine derivative FR194921 (1) exhibits potent affinity for the A1 AR (A1 (h) Ki = 2.9 nM) without affinity for the A2A and A3 ARs (Fig. 1). In addition, no species differences among human, rat and mouse were observed in the binding affinity profile of this compound (Maemoto et al. 2004). After oral administration of FR194921 (1), hypolocomotion in rats caused by the A1 AR agonist CPA was bettered demonstrating A1 AR antagonism in vivo (Maemoto et al. 2004). Additionally, scopolamine-induced memory deficits (passive avoidance test) as well as anxiety (social interaction test and elevated plus-maze test) were also bettered by the said compound (1), without influencing general behaviour (Maemoto et al. 2004). FR194921 (1) showed no anti-depressant activity (forced swim test)—even at high doses. It may be said that FR194921 (1) shows promise in the treatment of memory deficits and anxiety (Maemoto et al. 2004).
Fig. 1.

The chemical structure and A1Ki (h) value (nM) of FR194921 (1)
The role of A1 AR antagonists in brain functioning is not yet fully understood, and at times, debatable—yet, selective A1 AR antagonists have potential for the treatment of neurological and psychiatric conditions, as seen with the potent and selective A1 AR antagonist FR184921 (1).
While diverse classes of A1 AR antagonists have been documented since the discovery of the receptor and specific radiolabelled ligands, most of these compounds were derived from xanthine analogues which show poor water solubility and oral bioavailability, central nervous system penetration and considerable species differences in terms of receptor binding affinity (Maemoto et al. 1997). Due to these undesirable physiochemical properties of xanthine derivatives, the focus has shifted from xanthine to non-xanthine based heterocycles as A1 AR antagonists (de Lera Ruiz et al. 2013).
The chemical structure 1,3-diphenyl-2-propen-1-one—perhaps better known by the general term “chalcone”—has attracted attention from both chemical (biosynthesis and synthesis) and biological standpoints; due to the wide-ranging pharmacological properties exhibited by these compounds (Mathew et al. 2019; Zhuang et al. 2017). These properties include anticancer, antileishmanial, antimalarial, antimicrobial, antiviral, antifungal, antioxidant, antihypertensive and antidiabetic biological activities—to name a few (Batovska and Todorova 2010; Sahu et al. 2012; Karthikeyan et al. 2015; Singh et al. 2014; Zhou and Xing 2015). Additionally, chalcones show potential in the treatment of neurological conditions by acting as anxiolytics and antidepressants, as well as the modulation of gamma-aminobutyric acid (GABA) receptors and the inhibition of acetylcholinesterase (AChE), butyrylcholinesterase (BChE) and monoamine oxidase (MAO) (Mathew et al. 2019). Of note, the chalcone–coumarin hybrid (2) possesses selective A3 AR affinity (A3Ki (h) = 5.2 µM) (Vazquez‐Rodriguez et al. 2013), the blockade of A3 ARs may be advantageous in brain ischemia (Fig. 2) (Jacobson and Gao 2006).
Fig. 2.

The chemical structure and A1, A2A and A3Ki (h) values of 2
The term chalcone generally refers to chemicals with an α,ß-unsaturated carbonyl system; thus, the chalcone family has extensive structural diversity (Zhuang et al. 2017). For example, the chalcone-based benzocycloalkanone derivatives are hybrid chalcones with the core scaffold of 1,3-diaryl-2-propen-1-one (Fig. 3). Some of these compounds structurally related to chalcones include aurone derivatives, which also possess selective A1 affinity [such as hispidol (3)] (Jacobson et al. 2002) and served as inspiration for the structurally related 2-benzylidene-1-tetralone (4–8) (Janse van Rensburg et al. 2017; Legoabe et al. 2018) and 2-benzylidene-1-indanone derivatives (9–12) (Janse van Rensburg et al. 2019a, b), leading to compounds with selective affinity for the A1 AR in the micromolar range.
Fig. 3.

The chemical structure and A1Ki (r) values of aurone (3), 2-benzylidene-1-tetralone (4–8) and 2-benzylidene-1-indanone (9–12) derivatives
The chemistry of chalcones is as attractive today as it was years ago; due to the open-chain model and the feature of skeletal modification to produce a new class of organic compounds such as isoxazole-, pyrazole- and indole-based chalcones. Heterocycles occupy a central position in medicinal chemistry and a number of both approved and potential drugs contain at least one heterocyclic nucleus (Khanam 2015; McGrath et al. 2010). Based on the above and in continuation of the efforts to obtain heterocycles as A1 and/or A2A AR antagonists, simple chalcone derivatives (15–36) with either ring A or ring B substitutions as well as a series of novel C3 amino-substituted chalcone derivatives (37–41) with halogen (Br-, Cl- and F-atoms) substitutions on ring B and their regioisomers (42–47) were designed containing the basic chalcone scaffold as structural core. Herein the synthesis, characterization and evaluation, both in vitro and in silico, will be discussed to ascertain the structure–affinity relationships (SAR) that govern A1 and/or A2A AR affinity as well as the future of the said compounds as synthons by which a range of analogues may be designed for future drug discovery.
Results and discussion
Chemistry
The synthesis of the chalcone derivatives 15–36 and 37–41 was done by a Claisen–Schmidt condensation reaction (Claisen and Claparède 1881; Schmidt 1881) of acetophenone and benzaldehyde using either a base or an acid catalyst in a polar solvent (Fig. 4a, b)—this well-known synthetic route is deemed a classical organic chemistry reaction. As stated, the condensing agents are either strong bases or acids (generally, basic conditions are more common in chalcone synthesis) (Gaonkar and Vignesh 2017; Gomes et al. 2017; Rammohan et al. 2020; Zhuang et al. 2017); in the case of base catalysts (15–36), the chalcone is generated from the aldol product via dehydration in an enolate mechanism, while in the case of acid catalysts (37–41), the chalcone is generated via an enol mechanism (Nielsen and Houlihan 2004; Noyce and Pryor 1955). The Claisen–Schmidt condensation reaction is widespread in the literature because of its experimental simplicity and highly efficient formation of the carbon–carbon double bond.
Fig. 4.

Synthesis of 15–36, 37–41 and 42–47 (R substituents are identified in Tables 2, 3, 4). Reagents and conditions: a EtOH, KOH (10% (w/v) aqueous solution), room temperature; b MeOH, HCl (32 wt. % in H2O, FCC), 120 °C
The synthesis of the Schiff base or imine derivatives 42–47 by condensation of 3-aminoacetophenone and different substituted benzaldehydes using a base catalyst in a polar solvent (Fig. 4a)—analogous to the reaction conditions of chalcone derivatives 15–36—was not planned. Initially, it was thought that the said reaction conditions would yield compounds 37–41 and not the regioisomers of these C3 amino-substituted chalcone derivatives comprised of two aromatic rings linked via an azomethine (C=N) group. Schiff base or imine compounds are formed by the reaction of a primary amine (such as 3-aminoacetophenone) and either aldehydes (such as the different substituted benzaldehydes) or ketones with the simultaneous removal of water (Furniss et al. 1989). When one, or both, of the reactants are aromatic, the imine is quite stable and usually known as a Schiff base, additionally, in the case of wholly aliphatic reactants the imines tend to decompose or polymerise (Furniss et al. 1989).
Under the adopted synthetic routes, the test compounds 15–36, 37–41 and 42–47 were obtained in fair to good yields, purified by recrystallization from a suitable solvent (EtOH), and in the instance of compounds 37–41 and 42–47, the structure, molecular mass and/or purity of these compounds were verified by 1H NMR, 13C NMR, MS and/or HPLC (Supplementary data). The known chalcone derivatives 15–36 were characterized by 1H NMR only, melting point (DSC) and HPLC and are in accordance with the literature values.
Taking the novel 3-amino-substituted chalcone derivative 38 (R2 = NH2; R2′ = Br) as a representative case, the 1H NMR spectrum displayed two characteristic signals for the ethylenic protons Hα (nearer the carbonyl group) and Hß (next to the Hα proton) at 7.73 and 7.97 ppm, respectively, as doublets (JHα,Hß = 15.7 Hz). Protons on the NH2-group (similar to the proton on the OH-group) are not always visible on a 1H NMR spectrum as protons attached to a N-atom (or O-atom) are acidic, and thus, exchangeable. Also, the 13C NMR spectrum displayed three characteristic signals: a prominent signal for the carbonyl group at 188.36 ppm and two other signals typical of Cα (121.39 ppm) and Cß (142.81 ppm).
From the coupling constant value of the ethylenic protons Hα and Hß (JHα,Hß = 15.7 Hz), it may be deduced that compound 38 exists in the trans (E) configuration (Aksöz and Ertan 2012; Barros et al. 2004; Jung et al. 2008; Opletalova et al. 2000; Rao et al. 2001, 2004). As a result of the diamagnetic anisotropy (deshielding of proton due to local diamagnetic current) of the carbonyl functional group, the ethylenic proton of the (E) isomer gave a signal at a greater chemical shift than the ethylenic proton of the (Z) isomer, as the latter is more remote from the carbonyl functional group (Bayer et al. 1991). Stereochemically, a chalcone might exist as either a cis (Z)- or trans (E)-isomer, having two aromatic rings linked via a three carbon α,ß-unsaturated carbonyl system (Gomes et al. 2017; Hallgas et al. 2005). The cis (Z)-conformer is thermodynamically less stable than the trans (E)-conformer due to the steric effects between the carbonyl group and ring A (Hallgas et al. 2005; Van der Werten et al. 1995). The more stable trans (E)-isomer is the predominant configuration among chalcones (Gomes et al. 2017).
The mass spectrum of compound 38 displayed an intense [M + H]+ molecular ion peak at 302.0164 (79Br isotope) as well as an intense [M + H]+2 peak at 304.0152 (81Br isotope)—in accordance with the formulation depicted (C15H13BrNO). Since Bromine has two isotopes (in a ratio of approximately 1:1), a compound containing a Br-atom (such as compounds 37–38) will have two peaks of similar height in the molecular ion region depending on which bromine isotope the molecular ion contains. Additionally, the two M+ molecular ion peaks for compound 38 (C15H12BrNO) may be seen at 301.0096 (79Br isotope) and 303.0095 (81Br isotope). Of note, chlorine also has two isotopes, namely 35Cl and 37Cl (in a ratio of 3:1) meaning a similar effect may be observed on the mass spectrum of an organic compound containing a Cl-atom (for example, compounds 39–40). The purity of compound 38 determined by HPLC was 98.0549%. The melting point of compound 38 determined by DSC was 176.78 °C.
The word chalcone is derived from the Greek word “chalcos”, meaning “bronze”, which results from the colours of most natural chalcones (Sahu et al. 2012). The bronze colour of chalcones is most likely due to the reactive keto-ethylenic group CO–CH=CH–; a chromophore responsible for the colour of chalcones depending on the presence of other auxochromes (Gaonkar and Vignesh 2017). This bronze colour scheme was also observed with most of the synthesised chalcones, for example, the light brown colour of 38.
As stated, the 3-amino-substituted chalcones and the Schiff base derivatives 42–47 are regioisomers (molecules with the same molecular formula, but different bonding patterns and atomic organisations), and as such, dissimilarities were detected on the 1H NMR and 13C NMR spectra when comparing these compounds. The absence of the ethylenic protons Hα and Hß may be observed on the 1H NMR spectrum and the signals typical of Cα and Cß, on the 13C NMR. In the place of these signals, the proton adjacent to the azomethine group was visible on the 1H NMR spectrum and the carbon of the azomethine group on the 13C NMR spectrum. Additionally, on both the 1H NMR and 13C NMR spectra the methyl group from the acetophenone moiety was clearly visible downfield of all other signals.
Biology
In vitro evaluation
Radioligand binding assays
The degree of binding affinity that the test compounds showed toward rat (r) A1 and A2A ARs were determined via radioligand binding assays in either duplicate [specific binding (%)] or triplicate [inhibition constant (Ki, µM)] and expressed as either mean or mean ± standard error of the mean (SEM), respectively. Only compounds 24, 29, 37 and 38—which displayed specific binding values < 20% at a maximum tested concentration of 100 µM—justified the determination of inhibition constant values (Ki, µM), unlike compounds 15–23, 25–28, 30–36, 39–41 and 42–47 (specific binding values > 20%). The radioligand binding assays were validated with CPA (A1 agonist), DPCPX (A1 antagonist) and istradefylline (A2A antagonist) as reference compounds and results were in accordance with literature values (Table 1).
Table 1.
Ki values for the binding affinity of reference compounds against rat (r) A1 and A2A ARs
| # | Ki ± SEM (µM)a | SIg | GTP shifth | ||
|---|---|---|---|---|---|
| rA1b vs [3H]DPCPXd | rA2Ac vs [3H]NECAe | rA1c + GTPf vs [3H]DPCPXd | |||
| CPA (A1 agonist) |
0.0057 ± 0.0015 (0.0068)i (0.015)j (0.0079)k |
0.40 ± 0.17 (0.16)i (0.33)j |
0.099 ± 0.015 (0.099)i (0.099)j |
70 (24) (22) |
17 (15)i (14)j |
| DPCPX (A1 antagonist) |
0.0005 ± 0.00003 (0.0004)i (0.0005)j (0.0003)l |
0.23 ± 0.03 (0.55)i (0.53)j (0.34)l |
0.0006 ± 0.00003 (0.0004)i (0.0004)j |
468 (1362) (1060) (1133) |
1.2 (1)i (1.3)j |
| Istradefylline (A2A antagonist) | 0.19 ± 0.01 (0.23)m | 0.0014 ± 0.0003 (0.0022)m | 0.15 ± 0.02 | 7.3 (0.0096) | 0.79 |
aAll inhibition constant (Ki) values were determined in triplicate and expressed as mean ± standard error of the mean (SEM) in µM
bRat receptors were used (rA1: rat whole brain membranes)
cRat receptors were used (rA2A: rat striatal membranes)
d0.1 nM [3H]DPCPX
e4 nM [3H]NECA
fAddition of 100 µM GTP to A1 AR radioligand binding assay
gSelectivity index (SI) for the A1 AR isoform calculated as a ratio of A2AKi/A1Ki
hGTP shift calculated by dividing Ki value in the presence of 100 µM GTP by Ki value in the absence of 100 µM GTP
iLiterature value obtained from (Janse van Rensburg et al. 2017)
jLiterature value obtained from (Van der Walt and Terre’Blanche 2015)
kLiterature value obtained from (Bruns et al. 1987)
lLiterature value obtained from (Lohse et al. 1987)
mLiterature value obtained from (Shimada et al. 1997)
Structure–affinity relationships (SAR)
As depicted in Tables 2, 3 and 4, most of the test compounds showed poor A1 and/or A2A AR affinity upon in vitro evaluation—making it difficult to determine SAR; however, some broad conclusions may be drawn from these results. In brief, the test compounds were noticeably more active against the A1 AR than A2A AR and the chalcone derivatives 24, 29, 37 and 38 showed selective A1 AR affinity below 10 µM—of these compounds, 38 had the best A1 AR affinity (A1Ki (r) = 1.6 µM). The type of substitution on ring A of the chalcone scaffold played a key role in affinity, and also, the type and position of the substitution on benzylidene ring B.
Table 2.
Ki values for the binding affinity of chalcone derivatives (15–36) against rat (r) A1 and A2A ARs
|
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| # | Ring A | Ring B | Ki ± SEM (µM)a [specific binding (%)]b | |||||||
| 2 | 3 | 4 | 2′ | 3′ | 4′ | 5′ | 6′ | rA1c vs [3H]DPCPXe | rA2Ad vs [3H]NECAf | |
| R1 | R2 | R3 | R1′ | R2′ | R3′ | R4′ | R5′ | |||
| Structural modification of ring A | ||||||||||
| 15 | H | H | H | H | H | H | H | H | (35) | (79) |
| 16 | OH | H | H | H | H | H | H | H | (65) | (129) |
| 17 | H | H | OH | H | H | H | H | H | (56) | (83) |
| 18 | H | H | OCH3 | H | H | H | H | H | (141) | (106) |
| 19 | H | H | Br | H | H | H | H | H | (117) | (117) |
| Structural modification of ring B | ||||||||||
| 20 | H | H | H | H | OH | H | H | H | (32) | (40) |
| 21 | H | H | H | H | OCH3 | H | H | H | (33) | (106) |
| 22 | H | H | H | H | H | OCH3 | H | H | (45) | (78) |
| 23 | H | H | H | OCH3 | H | OCH3 | H | H | (30) | (50) |
| 24 | H | H | H | OCH3 | H | OCH3 | OCH3 | H | 5.3 ± 1 | (28) |
| 25 | H | H | H | OCH3 | H | OCH3 | H | OCH3 | (27) | (28) |
| 26 | H | H | H | Br | H | H | H | H | (24) | (50) |
| 27 | H | H | H | H | Br | H | H | H | (28) | (42) |
| 28 | H | H | H | H | H | Br | H | H | (110) | (91) |
| 29 | H | H | H | Cl | H | H | H | H | 6.1 ± 1.1 | (42) |
| 30 | H | H | H | H | Cl | H | H | H | (23) | (88) |
| 31 | H | H | H | H | Cl | Cl | H | H | (114) | (77) |
| 32 | H | H | H | H | H | F | H | H | (49) | (98) |
| 33 | H | H | H | H | H | CF3 | H | H | (80) | (156) |
| 34 | H | H | H | H | CN | H | H | H | (52) | (155) |
| 35 | H | H | H | – | – | – | – | – | (55) | (75) |
| 36 | H | OCH3 | H | – | – | – | – | – | (43) | (73) |
aAll Ki values were determined in triplicate and expressed as mean ± standard error of the mean (SEM) in µM
bSpecific binding (%) of the radioligand at a maximum tested concentration of 100 µM were determined in duplicate and expressed as the mean in %
cRat receptors were used (rA1: rat whole brain membranes)
dRat receptors were used (rA2A: rat striatal membranes)
e0.1 nM [3H]DPCPX
f4 nM [3H]NECA
Table 3.
Ki values for the binding affinity of chalcone derivatives (37–41) against rat (r) A1 and A2A ARs
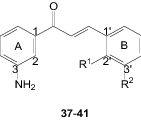
| ||||
|---|---|---|---|---|
| # | Ring B | Ki ± SEM (µM)a [specific binding (%)]b | ||
| 2′ | 3′ | rA1c vs [3H]DPCPXe | rA2Ad vs [3H]NECAf | |
| R1 | R2 | |||
| 37 | Br | H | 7.1 ± 0.57a | (30)b |
| 38 | H | Br | 1.6 ± 0.02a | (75)b |
| 39 | Cl | H | (26)b | (23)b |
| 40 | H | Cl | (30)b | (72)b |
| 41 | H | F | (26)b | (71)b |
aAll Ki values were determined in triplicate and expressed as mean ± standard error of the mean (SEM) in µM
bSpecific binding (%) of the radioligand at a maximum tested concentration of 100 µM were determined in duplicate and expressed as the mean in %
cRat receptors were used (rA1: rat whole brain membranes)
dRat receptors were used (rA2A: rat striatal membranes)
e0.1 nM [3H]DPCPX
f4 nM [3H]NECA
Table 4.
Ki values for the binding affinity of Schiff base derivatives (42–47) against rat (r) A1 and A2A ARs
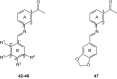
| ||||||
|---|---|---|---|---|---|---|
| # | Ring B | Ki ± SEM (µM)a [specific binding (%)]b | ||||
| 2′ | 3′ | 4′ | 5′ | rA1c vs [3H]DPCPXe | rA2Ad vs [3H]NECAf | |
| R1′ | R2′ | R3′ | R4′ | |||
| 42 | OH | H | OCH3 | H | (91)b | (197)b |
| 43 | OH | H | H | OCH3 | (70)b | (75)b |
| 44 | OH | Br | H | Cl | (101)b | (95)b |
| 45 | OH | H | H | Cl | (44)b | (43)b |
| 46 | OH | H | N(CH2CH3)2 | H | (74)b | (69)b |
| 47 | – | – | – | – | (47)b | (66)b |
aAll Ki values were determined in triplicate and expressed as mean ± standard error of the mean (SEM) in µM
bSpecific binding (%) of the radioligand at a maximum tested concentration of 100 µM were determined in duplicate and expressed as the mean in %
cRat receptors were used (rA1: rat whole brain membranes)
dRat receptors were used (rA2A: rat striatal membranes)
e0.1 nM [3H]DPCPX
f4 nM [3H]NECA
As stated, the chalcone derivatives 24 (A1Ki (r) = 5.3 µM) and 29 (A1Ki (r) = 6.1 µM) (Table 2) showed selective A1 AR affinity below 10 µM, and although, the Ki (r) values of these compounds were quite similar the substitutions on ring B were not; 24 was C2′, C4′, C5′-trimethoxy substituted and 29 was C2′-chloro substituted. In the past, the antimicrobial (Karaman et al. 2010; Ramyashree et al. 2017), anticancer (Juvale et al. 2012; Mellado et al. 2018) and/or antioxidant (Shenvi et al. 2013) activities of compounds 24 and 29 were also explored, and now, selective A1 AR affinity may be added.
With regard to compound 24 and A1 AR affinity, it seems that the C2′, C4′, C5′ substitution pattern on ring B was preferred to that of compound 25 ([3H]DPCPX-specific binding (r) = 27%)—interestingly, these compounds differ only at the C5′ (24: –OCH3; 25: –H) and C6′ (24: –H; 25: –OCH3) positions. However, the radioligand-specific binding against the A2A AR of these compounds were the same (24–25: [3H]NECA-specific binding (r) = 28%). Comparison of 24 to the structurally related 2-benzylidene-1-indanone compound 13 (Fig. 5) consisting of a fused 6- and 5-membered ring system (ring A and ring C, respectively) along with C4′-methoxy substitution on ring A had poor A1 and A2A AR affinity (Janse van Rensburg et al. 2019b). The decreased affinity may be due to the increased number of C-, H- and O atoms in the form of the C4 OCH3-group substitution on ring A and the CH2-group present in ring C, increasing the molecular weight from 298.34 g/mol (24) to 340.38 g/mol (13), and perhaps, the resultant steric hindrance (of the CH2-group present in ring C and C2′ OCH3-group on ring B) also decreased binding affinity (Liu et al. 2014; Vásquez-Martínez et al. 2019). Interestingly, according to the physiochemical properties calculated by the free web-tool SwissADME, compound 24 will not be orally bioavailable, but compound 13 will be. Both of these compounds were, unfortunately, not lead-like based on the said parameters. (See “In silico evaluation” for the complete results and discussion).
Fig. 5.

The structure–affinity relationships (SAR) of compound 24 versus 13
Comparison of 29 (A1Ki (r) = 6.1 µM) to 30–31 ([3H]DPCPX-specific binding > 20%) demonstrated the importance of the position of the Cl-atom on ring B for A1 AR affinity; the C2′ (ortho) position (29) is preferred to the C3′ (meta) and/or C4′ (para) positions (30–31). Additionally, at the C2′ (ortho) position on ring B the more electronegative Cl-atom (29) is favoured over the less electronegative Br-atom (26). (It must be noted that compounds 26–27 and 30 retained some affinity against the A1 AR based on the specific binding of less than 30%).
These trends were not observed with the C3 amino-substituted chalcone derivatives 37–41 (Table 3). Compound 29 paralleled to its counterpart 39 showed decreased A1 AR affinity (39: [3H]DPCPX-specific binding = 26%); this may be credited to the combination of the NH2-group at position C3 on ring A and the Cl-atom at position C2′. Furthermore, C2′ (ortho) Br-group substitution on ring B led to A1 AR affinity below 10 µM; for example, compound 37 (A1Ki (r) = 7.1 µM) versus 39 ([3H]DPCPX-specific binding = 26%). The A1 AR affinity was enhanced almost five-fold when the Br-atom was moved from the C2′ position (37) to the C3′ position (38) on ring B. The importance of the C3 NH2-group substitution on ring A to attain A1 AR affinity was demonstrated by comparison of compound 38 to its unsubstituted counterpart 27 ([3H]DPCPX-specific binding = 28%). Compound 38 had a Ki value of 1.6 µM against the A1 AR—the best of the evaluated compounds Therefore, it may be said that the A1 AR favours C3 NH2-group substitution on ring A in combination with C3′ Br-atom substitution on ring B; based on these chalcones (15–41). The SAR of the chalcone derivatives 27 and 29 and 37–39 are summarized in Fig. 6.
Fig. 6.

The structure–affinity relationships (SAR) of compounds 27 and 29 and 37–39; highlighting the importance of C3 NH2-substitution on ring A in combination with C3′ (meta) Br substitution on ring B for selective rA1 AR affinity
Interestingly, the C3 amino-substituted chalcones 37 ([3H]NECA-specific binding = 30%) and 39 ([3H]NECA-specific binding = 23%) with a C2′ substituent [either a Br- (37) or Cl-atom (39)] on ring B retained some affinity against the A2A AR, unlike compounds 38 ([3H]NECA-specific binding = 75%), 40 ([3H]NECA-specific binding = 72%) and 41 ([3H]NECA-specific binding = 71%) with a C3′ substituent [either a Br- (38), Cl- (40) or F-atom (41)]. (Again, it must be noted that compounds 39–41 retained some affinity against the A1 AR based on the radioligand-specific binding of less than 30%). The importance of not only the type of substituent, but also the position of the substituent on ring B is clear from these comparisons, for example, a compound with selective A1 AR affinity may be obtained when a C3 NH2-group substitution on ring A is combined with a C3′ halogen (Br > Cl > F) substitution on ring B of the chalcone scaffold (e.g. 38, 40–41), whereas a C2′ halogen-substituted ring B could yield a compound with dual A1 and/or A2A AR affinity (37 and 39). Previously, it was found that a coplanar geometry between the two ring systems (namely, ring A and benzylidene ring B) in the flavonols (notably, chalcones may be considered open-chain flavonols) is not required for residual A1 and/or A2A AR affinity; however, the C2′ position on ring B of compounds 37 and 39 was substituted (like the flavonol MRS 1067 with a planar geometry), and thus, there is likely also steric hindrance to free rotation of the benzylidene ring B of these compounds (37 and 39) (Karton et al. 1996). Notably, the structurally related 2-benzylidene-1-indanone 14 (Janse van Rensburg et al. 2019a) that is also C3′ bromo-substituted on benzylidene ring B has selective A2A AR affinity, unlike compound 38 (Fig. 7). Therefore, the benzylidene indanone scaffold with C4 hydroxy-substitution on ring A may be essential to selective A2A AR affinity when compared to the C3 amino-substituted chalcone scaffold. Compound 14 is structurally related to chromone hits with selective A2A AR affinity based on virtual screening, and perhaps, the conformation of these chromones binding to the A2A AR is similar to that of compound 14—leading to selectivity of the A2A AR versus the A1 AR (Langmead et al. 2012).
Fig. 7.

The structure–affinity relationships (SAR) of compound 38 versus 14
Additionally, SAR demonstrated the importance of, among other, the phenol functionality in triazine virtual screening hits for selective A2A AR affinity; possibly due to the hydrogen bonding between the phenol functionality and Asn2536.55 (Langmead et al. 2012). This key hydrogen bonding residue (Asn2536.55) is situated centrally and capable of high-quality interactions with diverse heterocyclic compounds (Langmead et al. 2012). Therefore, the phenol functionality (present in compound 14) may also be responsible for the selectivity of the said compound (as seen with the triazine virtual screening hits).
As seen in Table 4 the Schiff base containing compounds 42–47 showed poor A1 and A2A AR affinity. The best radioligand-specific binding at a concentration of 100 µM against A1 and/or A2A AR was demonstrated by compounds 45 and 47; displacing more than 50% of the radioligand at A1 (45 and 47) and A2A (45) ARs. Comparison of 44 to 45 showed that the inclusion of a bromine atom at position C3′ (44), rather than a hydrogen atom (45), in combination with C2′–OH and C5′–Cl substitution on ring B was detrimental to both A1 and A2A AR affinity as the radioligand-specific binding increased more than twofold. The A1 AR seemed to be more tolerable toward the said substitution. Schiff base or imine derivatives are important synthetic intermediates (Furniss et al. 1989); therefore, the synthesised and evaluated compounds 42–47 may be of use in future studies.
GTP shift assays
The type of binding affinity that test compounds 24 and 38 exhibited at the rat A1 AR was determined via a GTP shift assay, as described previously (Lohse et al. 1987; Van der Walt and Terre’Blanche 2015; Van der Werten et al. 1995). GTP shifts were calculated by dividing the Ki values of compounds reported in the presence of GTP by the Ki values obtained in the absence of GTP and the results are summarized in Table 5.
Table 5.
A1Ki values (in the absence and presence of GTP) and calculated GTP shifts of 24 and 38
| # | Ki ± SEM (µM)a | GTP shifte | |
|---|---|---|---|
| rA1b vs [3H]DPCPXc | rA1b + GTPd vs [3H]DPCPXc | ||
| 24 | 5.3 ± 1a | 7.8 ± 0.68 | 1.5 |
| 38 | 1.6 ± 0.02a | 1.7 ± 1.3 | 1.1 |
aKi values were determined in triplicate and expressed as mean ± standard error of the mean (SEM) in µM
bRat receptors were used (A1: rat whole brain membranes)
c0.1 nM [3H]DPCPX
dAddition of 100 µM GTP to A1 AR radioligand binding assay
eGTP shift calculated by dividing Ki value in the presence of 100 µM GTP by Ki value in the absence of 100 µM GTP
Test compounds 24 and 38 were selected as they possessed the highest A1 AR affinity in the three respective series (Tables 2, 3, 4). The results suggested that compounds 24 and 38 acted as A1 AR antagonists, as the binding curves in the presence of GTP were almost unaffected and the calculated GTP shifts were approximately 1 (Table 5; Fig. 8) (Van der Werten et al. 1995; Gütschow et al. 2012). On account of the structural similarity of the test compounds (15–23, 25–36, 37 and 39–41), it may be supposed that these chalcone derivatives were all A1 AR antagonists.
Fig. 8.

The binding curves of compounds 24 (a) and 38 (b) are examples of A1 AR antagonistic action determined via a GTP shift assay performed in triplicate (with and without 100 μM GTP) using rat whole brain membranes expressing A1 ARs with [3H]DPCPX as radioligand. Calculated GTP shift of: 1.5 (24) and 1.1 (38)
In silico evaluation
The physiochemical, pharmacokinetic as well as drug-likeness and medicinal chemistry friendliness of compounds 24, 29, 37 and 38 (the only test compounds with selective A1 AR affinity) were computed via SwissADME and these results are summarized in Tables 6, 7 and 8, respectively.
Table 6.
Physiochemical properties of compounds 24, 29, 37 and 38
| # | Physiochemical properties | Lipophilicity | Water solubility | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Molecular formula | Molecular weight (g/mol) | Fraction Csp3 | Num. rotatable bonds | Num. H-bond acceptors | Num. H-bond donors | Molar refractivity | TPSA (Å2) | Consensus logPo/w | Consensus logS | |
| 24 | C18H18O4 | 298.33 | 0.17 | 6 | 4 | 0 | 85.72 | 44.76 | 3.36 | − 4.51 |
| 29 | C15H13lO | 242.70 | 0 | 3 | 1 | 0 | 71.26 | 17.07 | 3.98 | − 4.89 |
| 37 | C15H12BrNO | 302.17 | 0 | 3 | 1 | 1 | 78.35 | 43.09 | 3.46 | − 4.69 |
| 38 | C15H12BrNO | 302.17 | 0 | 3 | 1 | 1 | 78.35 | 43.09 | 3.53 | − 4.86 |
Table 7.
Pharmacokinetic properties of compounds 24, 29, 37 and 38
| # | Pharmacokinetic properties | |||||||
|---|---|---|---|---|---|---|---|---|
| GI absorption | BBB permeation | Pgp substrate | CYP12 inhibitor | CYP2C19 inhibitor | CYP2C9 inhibitor | CYP2D6 inhibitor | CYP3A4 inhibitor | |
| 24 | High | Yes | No | Yes | Yes | Yes | Yes | Yes |
| 29 | High | Yes | No | Yes | Yes | Yes | No | No |
| 37 | High | Yes | No | Yes | Yes | Yes | No | Yes |
| 38 | High | Yes | No | Yes | Yes | Yes | No | Yes |
Table 8.
Drug-likeness and medicinal chemistry friendliness of compounds 24, 29, 37 and 38
| # | Drug-likeness | Lead-likeness | ||||||
|---|---|---|---|---|---|---|---|---|
| Num. violations | ||||||||
| Lipinskia | Ghoseb | Veberc | Egand | Mueggee | PAINSf | Brenkg | Teagueh | |
| 24 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| 29 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 2 |
| 37 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| 38 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
aLipinski: MW < 500, MLOGP < 4.15, N or O < 10, NH or OH < 5 (Lipinski et al. 1997)
bGhose: 160 < MW < 480, − 04 < WLOGP < 5.6, 40 < MR < 130, 20 < atoms < 70 (Ghose et al. 1999)
cVeber: Num. rotatable bonds < 10, TPSA < 140 (Veber et al. 2002)
dEgan: WLOGP < 5.88, TPSA < 131.6 (Egan et al. 2000)
eMuegge: 200 < MW < 600, − 2 < XLOGP < 5, TPSA < 150, num. rings < 7, num. carbon > 4, num. heteroatoms > 1, num. rotatable bonds < 15, num. H-bond acceptors < 10, num. H-bond donors < 5 (Muegge et al. 2001)
fPan assay interference compounds (PAINS) implemented from Baell and Holloway (2010)
gStructural alert implemented from Brenk et al. (2008)
hTeague: 250 < MW < 350, num. rotatable bonds < 7, XLOGP3 < 3.5 (Teague et al. 1999)
The bioavailability radar (Fig. 9) gave a first glance at the drug-likeness of these compounds; compounds 24, 29, 37 and 38 fall within the optimal range for the parameters lipophilicity, size, polarity, solubility and flexibility (see Table 6), except for saturation (Csp3 > 0.25). The fraction of carbon atoms in the sp3 hybridization of compounds 24 (0.17), 29 (0), 37 (0) and 38 (0) were smaller than 0.25 and, thus, out of range. Consequently, compounds 24, 29, 37 and 38 were predicted not orally bioavailable.
Fig. 9.

The pink area represents the optimal range for lipophilcity (LIPO: − 0.7 < XLOGP3 < + 5.0), size (SIZE: 150 < MW < 500), polarity (POLAR: 20 < TPSA < 130), solubility (INSOLU: logS < 6), saturation (INSATU: fraction Csp3 > 0.25) and flexibility (FLEX: num. rotatable bonds < 9). The red lines represent the said parameters of compounds 24, 29, 37 and 38. The red lines must fall completely within the pink area for a compound to be considered drug-like; therefore, compounds 24, 29, 37 and 38 are predicted not orally bioavailable
The qualitative solubility classes of compounds 24, 29, 37 and 38 were moderately soluble to soluble in water [the predicted values are the decimal logarithm of the molar solubility in water (logS)]. This was encouraging given the poor solubility of most chalcone-based compounds [most probably the reason for poor in vivo efficacy in preclinical studies (Zhuang et al. 2017)]. Seeing as solubility influences GI absorption (Ottaviani et al. 2010), a soluble molecule will most probably facilitate drug development (Ritchie et al. 2013).
The BOILED-Egg model (Fig. 10) predicted passive human gastrointestinal absorption (HIA) and blood–brain-barrier (BBB) permeation as a function of the position of the compound in the WLOGP-versus-TPSA referential (Daina and Zoete 2016). Compounds 24, 29, 37 and 38 were predicted to have high GI absorption and permeate the BBB (Table 7). Additionally, these compounds were not substrates for the permeability glycoprotein (Pgp) responsible for efflux through biological membranes, for instance from the gastrointestinal wall to the lumen or from the brain (Montanari and Ecker 2015). Pgp also protects the CNS from xenobiotics (Szakács et al. 2008).
Fig. 10.

Compounds 24, 29, 37 and 38, which are not a substrate for Pgp (PGP-), is represented by the red circles in the yellow region. The white region is for high probability of passive absorption by the gastrointestinal tract (HIA), and the yellow region (yolk) is for high probability of brain penetration (BBB). White and yellow (yolk) regions are not mutually exclusive
The interaction of a compound with cytochromes P450 (CYP) is important; seeing as these isoenzymes modulate drug elimination through metabolic transformation (Testa and Kraemer 2007); probably causing pharmacokinetics-related drug–drug interactions (Hollenberg 2002; Huang et al. 2008), leading to adverse or even toxic effects due to the lower clearance and accumulation of the drug or its metabolites (Kirchmair et al. 2015). Compounds 24, 29, 37 and 38 were predicted to inhibit the major CYP isoforms (CYP12, CYP2C19, CYP2C9, CYP2D6 and CYP3A4), with the exception of 24 and/or 37–38 that did not inhibit CYP2D6 (29 and 37–38) and CYP3A4 (29).
The drug-likeness of compounds 24, 29, 37 and 38 was assessed by various rule-based filters (Egan et al. 2000; Ghose et al. 1999; Lipinski et al. 1997; Muegge et al. 2001; Veber et al. 2002) (Table 8). Compound 29 violated a rule-based filter from Muegge (Muegge et al. 2001); namely that a compound should contain more than one heteroatom and the only atom other than carbon and hydrogen that 29 contained was a chloro atom. Additionally, compounds 24, 29, 37 and 38 all had a bioavailability score of 0.55 (the probability that a compound will have > 10% bioavailability in rat or measurable Caco-2 permeability) (Martin 2005).
The medicinal chemistry friendliness of compounds 24, 29, 37 and 38 was assessed by the identification of potentially problematic fragments; first, pan assay interference compounds (PAINS) (Baell and Holloway 2010) and, second, structural alerts (Brenk et al. 2008). Compounds 24, 29, 37 and 38 contained no PAINS; however, Brenk violations were present (Table 8). All these compounds contained an α,ß-unsaturated carbonyl system perceived as a potential Michael acceptor. Additionally, compounds 37–38 contained a potentially unwanted aniline group; as aniline groups are perhaps inclined toward problems with genetic toxicity (Clayson 1981).
As compounds 24, 29, 37 and 38 did not have pronounced affinity toward the A1 AR, the present structures need to be optimised, and here, lead-likeness [a molecular entity suitable for optimization, most probably increasing size and lipophilicity (Hann and Keserü 2012)] plays a role (Teague et al. 1999). Accordingly, compounds 24, 37 and 38 (but not compound 29; due to molecular weight value smaller than 250 g/mol and consensus logPo/w value larger than 3.5) may be considered lead-like (Tables 6, 8).
Therefore, the optimization of the physiochemical properties of chalcones and, thus, the pharmacokinetic properties as well as drug-likeness and medicinal chemistry friendliness is of great importance for medicinal chemistry research on chalcone-based compounds.
Experimental
Chemistry
Materials
Unless otherwise noted, all starting materials and solvents were purchased from commercial vendors and used without further purification. Thin layer chromatography on TLC silica gel 60 F254 aluminium sheets from Merck was used to monitor reaction progress. Melting points were determined by means of differential scanning calorimetry (DSC) with a Mettler DSC 3 Star System (Mettler Toledo, Greifensee, Switzerland). Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III 600 spectrometer at frequencies of 600 and 151 MHz, respectively, using either CDCl3 or DMSO-d6 as solvent and TMS as reference. Chemical shifts were reported in parts per million (ppm) in relation to the solvent peak (CDCl3: residual CH at 7.26 ppm and DMSO-d6: residual CH3 at 2.50 ppm for 1H NMR). Spin multiplicities were indicated as follows: singlet (s), doublet (d), triplet (t), quartet (q), doublet of doublets (dd), triplet of doublets (td), double double doublet (ddd) and multiplet (m). Coupling constant (J) values were reported in Hertz (Hz). High-resolution mass spectra (HRMS) were recorded on a Bruker micrOTOF-Q II mass spectrometer in atmospheric chemical ionisation (APCI) mode. High-performance liquid chromatography (HPLC) analyses were done on an Agilent 1100 HPLC system.
Synthesis of 15–36
(2E)-1,3-Diphenylprop-2-en-1-one (15)
A solution of acetophenone (0.50 g, 4.16 mmol) and benzaldehyde (0.44 g, 4.16 mmol) in EtOH (5 mL) was mechanically stirred at room temperature for approximately 5 min before the dropwise addition of KOH (10% (w/v) aqueous solution, 5 mL). The subsequent reaction mixture was mechanically stirred at room temperature and continuously monitored by TLC. Upon completion, the reaction mixture was quenched with crushed ice (15 g) and acidified to pH 2 with HCl (32 wt. % in H2O, FCC). The subsequent precipitate was collected by vacuum filtration, dried (30 °C) and recrystallized from EtOH to yield the title compound 15 as light yellow crystals (0.43 g, 49%): mp: 57.87 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.16 (d, J = 7.7 Hz, 2H), 7.95 (d, J = 15.6 Hz, 1H), 7.89 (d, J = 4.8 Hz, 2H), 7.76 (d, J = 15.6 Hz, 1H), 7.68 (t, J = 7.3 Hz, 1H), 7.58 (t, J = 7.5 Hz, 2H), 7.46 (d, J = 4.4 Hz, 3H) (Xie et al. 2017). Purity (HPLC): 99.8%.
(2E)-1-(2-Hydroxyphenyl)-3-phenylprop-2-en-1-one (16)
Prepared as for 15 from 2′-hydroxyacetophenone (0.50 g, 3.67 mmol) and benzaldehyde (0.39 g, 3.67 mmol) to yield the title compound 16 as dark yellow crystals (0.05 g, 6%): mp: 87.21 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 12.82 (s, 1H), 7.97–7.90 (m, 2H), 7.71–7.64 (m, 3H), 7.51 (t, J = 7.7 Hz, 1H), 7.45 (d, J = 2.6 Hz, 3H), 7.04 (d, J = 8.3 Hz, 1H), 6.95 (t, J = 7.5 Hz, 1H) (Xie et al. 2017). Purity (HPLC): 99%.
(2E)-1-(4-Hydroxyphenyl)-3-phenylprop-2-en-1-one (17)
Prepared as for 15 from 4′-hydroxyacetophenone (0.50 g, 3.67 mmol) and benzaldehyde (0.39 g, 3.67 mmol) to yield the title compound 17 as beige crystals (0.15 g, 18%): mp: 171.29 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 10.46 (d, J = 7.9 Hz, 1H), 8.10–8.04 (m, 2H), 7.90 (d, J = 15.6 Hz, 1H), 7.86 (dd, J = 7.5, 1.7 Hz, 2H), 7.68 (d, J = 15.6 Hz, 1H), 7.45 (d, J = 6.9 Hz, 3H), 6.93–6.89 (m, 2H) (Xie et al. 2017). Purity (HPLC): 98.9%.
(2E)-1-(4-Methoxyphenyl)-3-phenylprop-2-en-1-one (18)
Prepared as for 15 from 4′-methoxyacetophenone (0.50 g, 3.33 mmol) and benzaldehyde (0.35 g, 3.33 mmol) to yield the title compound 18 as white crystals (0.79 g, 52%): mp: 105.91 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.17 (d, J = 7.7 Hz, 2H), 7.94 (d, J = 15.6 Hz, 1H), 7.88 (d, J = 6.8 Hz, 2H), 7.71 (d, J = 15.6 Hz, 1H), 7.45 (d, J = 5.5 Hz, 3H), 7.09 (d, J = 7.7 Hz, 2H), 3.87 (s, 3H) (Zhang et al. 2015). Purity (HPLC): 97.2%.
(2E)-1-(4-Bromophenyl)-3-phenylprop-2-en-1-one (19)
Prepared as for 15 from 4′-bromoacetophenone (0.50 g, 2.51 mmol) and benzaldehyde (0.27 g, 2.51 mmol) to yield the title compound 19 as light yellow crystals (0.72 g, 63%): mp: 103.26 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 7.92–7.87 (m, 2H), 7.82 (d, J = 15.7 Hz, 1H), 7.69–7.60 (m, 4H), 7.48 (d, J = 15.7 Hz, 1H), 7.44–7.40 (m, 3H) (Zhou et al. 2016). Purity (HPLC): 98%.
(2E)-3-(3-Hydroxyphenyl)-1-phenylprop-2-en-1-one (20)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 3-hydroxybenzaldehyde (0.51 g, 4.16 mmol) to yield the title compound 20 as light yellow crystals (0.51 g, 55%): mp: 163.81 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 9.63 (s, 1H), 8.17–8.09 (m, 2H), 7.83 (d, J = 15.6 Hz, 1H), 7.69–7.63 (m, 2H), 7.57 (t, J = 7.7 Hz, 2H), 7.31 (d, J = 7.7 Hz, 1H), 7.26 (t, J = 7.8 Hz, 1H), 7.23 (s, 1H), 6.88 (dd, J = 7.9, 1.7 Hz, 1H) (Xie et al. 2017). Purity (HPLC): 99.9%.
(2E)-3-(3-Methoxyphenyl)-1-phenylprop-2-en-1-one (21)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 3-methoxybenzaldehyde (0.57 g, 4.16 mmol) to yield the title compound 21 as dark yellow crystals (0.19 g, 19%): mp: 60.43 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 8.14–8.10 (m, 2H), 7.88 (d, J = 15.7 Hz, 1H), 7.69 (t, J = 7.4 Hz, 1H), 7.64–7.58 (m, 3H), 7.44 (t, J = 7.9 Hz, 1H), 7.38–7.32 (m, 1H), 7.26 (d, J = 1.7 Hz, 1H), 7.07 (dd, J = 8.2, 2.4 Hz, 1H), 3.96 (s, 3H) (Unoh et al. 2013). Purity (HPLC): 98.2%.
(2E)-3-(4-Methoxyphenyl)-1-phenylprop-2-en-1-one (22)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 4-methoxybenzaldehyde (0.57 g, 4.16 mmol) to yield the title compound 22 as light yellow crystals (0.06 g, 6%): mp: 72.02 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.17–8.08 (m, 2H), 7.88–7.83 (m, 2H), 7.80 (d, J = 15.6 Hz, 1H), 7.72 (d, J = 15.6 Hz, 1H), 7.66 (t, J = 7.4 Hz, 1H), 7.57 (t, J = 7.7 Hz, 2H), 7.06–6.97 (m, 2H), 3.83 (s, 3H) (Zhang et al. 2015). Purity (HPLC): 100%.
(2E)-3-(2,4-Dimethoxyphenyl)-1-phenylprop-2-en-1-one (23)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 2,4-dimethoxybenzaldehyde (0.69 g, 4.16 mmol) to yield the title compound 23 as dark yellow crystals (0.18 g, 16%): mp: 69.63 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.08 (d, J = 7.5 Hz, 2H), 7.99 (d, J = 15.7 Hz, 1H), 7.91 (d, J = 8.6 Hz, 1H), 7.74 (d, J = 15.7 Hz, 1H), 7.64 (t, J = 7.3 Hz, 1H), 7.56 (t, J = 7.6 Hz, 2H), 6.67–6.60 (m, 2H), 3.90 (s, 3H), 3.84 (s, 3H) (Suwito et al. 2014). Purity (HPLC): 94.8%.
(2E)-1-Phenyl-3-(2,4,5-trimethoxyphenyl)prop-2-en-1-one (24)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 2,4,5-trimethoxybenzaldehyde (0.82 g, 4.16 mmol) to yield the title compound 24 as dark yellow crystals (0.95 g, 79%): mp: 102.97 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 8.09 (d, J = 15.8 Hz, 1H), 8.03–7.97 (m, 2H), 7.55 (t, J = 7.3 Hz, 1H), 7.47 (dd, J = 20.6, 11.8 Hz, 3H), 7.13 (s, 1H), 6.52 (s, 1H), 3.94 (s, 3H), 3.90 (s, 6H) (Shenvi et al. 2013). Purity (HPLC): 97.6%.
(2E)-1-Phenyl-3-(2,4,6-trimethoxyphenyl)prop-2-en-1-one (25)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 2,4,6-trimethoxybenzaldehyde (0.82 g, 4.16 mmol) to yield the title compound 25 as bright yellow crystals (0.43 g, 35%): mp: 109.17 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 8.26 (d, J = 15.9 Hz, 1H), 8.03–7.98 (m, 2H), 7.88 (d, J = 15.9 Hz, 1H), 7.53 (t, J = 7.3 Hz, 1H), 7.47 (t, J = 7.5 Hz, 2H), 6.14 (s, 2H), 3.90 (s, 6H), 3.86 (s, 3H) (Sawle et al. 2008). Purity (HPLC): 88%.
(2E)-3-(2-Bromophenyl)-1-phenylprop-2-en-1-one (26)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 2-bromobenzaldehyde (0.77 g, 4.16 mmol) to yield the title compound 26 as dark yellow crystals (0.90 g, 75%): mp: 47.76 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.24–8.12 (m, 3H), 8.01 (d, J = 15.5 Hz, 1H), 7.95 (d, J = 15.5 Hz, 1H), 7.75 (dd, J = 8.0, 0.9 Hz, 1H), 7.69 (t, J = 7.4 Hz, 1H), 7.59 (t, J = 7.7 Hz, 2H), 7.50 (t, J = 7.5 Hz, 1H), 7.40 (td, J = 7.9, 1.6 Hz, 1H) (Wu et al. 2017). Purity (HPLC): 97.1%.
(2E)-3-(3-Bromophenyl)-1-phenylprop-2-en-1-one (27)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 3-bromobenzaldehyde (0.77 g, 4.16 mmol) to yield the title compound 27 as light yellow crystals (0.62 g, 52%): mp: 84.50 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.24–8.16 (m, 3H), 8.03 (d, J = 15.6 Hz, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.74–7.62 (m, 3H), 7.58 (dd, J = 10.7, 4.7 Hz, 2H), 7.42 (t, J = 7.8 Hz, 1H) (Wang et al. 2019). Purity (HPLC): 86.5%.
(2E)-3-(4-Bromophenyl)-1-phenylprop-2-en-1-one (28)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 4-bromobenzaldehyde (0.77 g, 4.16 mmol) to yield the title compound 28 as light yellow crystals (0.58 g, 49%): mp: 119.66 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.19–8.12 (m, 2H), 7.98 (d, J = 15.7 Hz, 1H), 7.87 (d, J = 8.5 Hz, 2H), 7.72 (d, J = 15.7 Hz, 1H), 7.69–7.64 (m, 3H), 7.58 (t, J = 7.7 Hz, 2H) (Wu et al. 2017). Purity (HPLC): 98.3%.
(2E)-3-(2-Chlorophenyl)-1-phenylprop-2-en-1-one (29)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 2-chlorobenzaldehyde (0.58 g, 4.16 mmol) to yield the title compound 29 as light yellow crystals (0.32 g, 32%): mp: 50.30 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.23 (dd, J = 7.5, 1.9 Hz, 1H), 8.20–8.15 (m, 2H), 8.03 (q, J = 15.6 Hz, 2H), 7.72–7.66 (m, 1H), 7.62–7.55 (m, 3H), 7.52–7.43 (m, 2H) (Wu et al. 2017). Purity (HPLC): 97.5%.
(2E)-3-(3-Chlorophenyl)-1-phenylprop-2-en-1-one (30)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 3-chlorobenzaldehyde (0.58 g, 4.16 mmol) to yield the title compound 30 as light yellow crystals (0.32 g, 32%): mp: 75.30 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.19 (d, J = 7.3 Hz, 2H), 8.09 (s, 1H), 8.04 (d, J = 15.7 Hz, 1H), 7.83 (d, J = 7.2 Hz, 1H), 7.73 (d, J = 15.7 Hz, 1H), 7.69 (t, J = 7.4 Hz, 1H), 7.58 (t, J = 7.7 Hz, 2H), 7.53–7.46 (m, 2H) (Robinson et al. 2015). Purity (HPLC): 96%.
(2E)-3-(3,4-Dichlorophenyl)-1-phenylprop-2-en-1-one (31)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 3,4-dichlorobenzaldehyde (0.73 g, 4.16 mmol) to yield the title compound 31 as light yellow crystals (0.35 g, 30%): mp: 108.62 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.30 (d, J = 2.0 Hz, 1H), 8.21–8.17 (m, 2H), 8.07 (d, J = 15.7 Hz, 1H), 7.89 (dd, J = 8.4, 1.9 Hz, 1H), 7.75–7.66 (m, 3H), 7.59 (t, J = 7.7 Hz, 2H) (Choi et al. 2016). Purity (HPLC): 95.9%.
(2E)-3-(4-Fluorophenyl)-1-phenylprop-2-en-1-one (32)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 4-fluorobenzaldehyde (0.52 g, 4.16 mmol) to yield the title compound 32 as light yellow crystals (0.32 g, 34%): mp: 86.71 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.15 (dd, J = 8.2, 1.1 Hz, 2H), 8.03–7.94 (m, 2H), 7.91 (d, J = 15.6 Hz, 1H), 7.75 (d, J = 15.7 Hz, 1H), 7.70–7.64 (m, 1H), 7.57 (dd, J = 10.7, 4.7 Hz, 2H), 7.34–7.26 (m, 2H) (Stroba et al. 2009). Purity (HPLC): 100%.
(2E)-1-Phenyl-3-[4-(trifluoromethyl)phenyl]prop-2-en-1-one (33)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 4-(trifluoromethyl)benzaldehyde (0.72 g, 4.16 mmol) to yield the title compound 33 as light yellow crystals (0.41 g, 36%): mp: 126.59 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.20–8.16 (m, 2H), 8.11 (dd, J = 21.5, 11.9 Hz, 3H), 7.81 (dd, J = 11.9, 8.8 Hz, 3H), 7.73–7.66 (m, 1H), 7.59 (dd, J = 10.6, 4.8 Hz, 2H) (Downey et al. 2018). Purity (HPLC): 94.4%.
3-[(1E)-3-Oxo-3-phenylprop-1-en-1-yl]benzonitrile (34)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 3-cyanobenzaldehyde (0.55 g, 4.16 mmol) to yield the title compound 34 as light yellow crystals (0.63 g, 65%): mp: 113.82 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.51 (s, 1H), 8.20 (dd, J = 8.1, 1.0 Hz, 3H), 8.12 (d, J = 15.7 Hz, 1H), 7.90 (d, J = 7.7 Hz, 1H), 7.77 (d, J = 15.7 Hz, 1H), 7.67 (dd, J = 12.7, 4.7 Hz, 2H), 7.59 (t, J = 7.7 Hz, 2H) (Nagarajan and Shechter 1984). Purity (HPLC): 94.7%.
(2E)-3-[4-(Morpholin-4-yl)phenyl]-1-phenylprop-2-en-1-one (35)
Prepared as for 15 from acetophenone (0.50 g, 4.16 mmol) and 4-(4-morpholinyl)benzaldehyde (0.80 g, 4.16 mmol) to yield the title compound 35 as bright yellow crystals (0.15 g, 13%): mp: 149.99 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.14–8.09 (m, 2H), 7.78–7.61 (m, 5H), 7.55 (t, J = 7.7 Hz, 2H), 6.99 (d, J = 8.9 Hz, 2H), 3.77–3.71 (m, 4H), 3.28–3.22 (m, 4H) (Li et al. 2017). Purity (HPLC): 99.2%.
(2E)-3-(2H-1,3-benzodioxol-5-yl)-1-(3-methoxyphenyl)prop-2-en-1-one (36)
Prepared as for 15 from 3-methoxyacetophenone (0.50 g, 3.33 mmol) and 3,4-(methylenedioxy)benzaldehyde (0.50 g, 3.33 mmol) to yield the title compound 36 as bright yellow crystals (0.92 g, 98%): mp: 77.10 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 7.73 (d, J = 15.5 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.53 (s, 1H), 7.43–7.31 (m, 2H), 7.16 (s, 1H), 7.12 (d, J = 7.9 Hz, 2H), 6.84 (d, J = 7.5 Hz, 1H), 6.02 (s, 2H), 3.88 (s, 3H) (Ruparelia et al. 2018). Purity (HPLC): 99.1%.
Synthesis of 37–41
(2E)-1-(3-Aminophenyl)-3-(2-bromophenyl)prop-2-en-1-one (37)
3′-Aminoacetophenone (0.50 g, 3.70 mmol) and 2-bromobenzaldehyde (0.68 g, 3.70 mmol) were suspended in MeOH (4 mL) and HCl (32 wt.% in H2O, FCC, 6 mL). The subsequent reaction mixture was mechanically stirred at 120 °C under reflux while being continuously monitored by TLC. Upon completion, the reaction mixture was cooled to room temperature, ice (15 g) was added and the resulting precipitate was filtered, dried (30 °C) and recrystallized from a suitable solvent to yield the title compound 37 as dark brown powder (1.10 g, 99%): mp: 189.86 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.21–8.12 (m, 2H), 8.01 (d, J = 15.5 Hz, 1H), 7.95 (s, 1H), 7.91 (d, J = 15.5 Hz, 1H), 7.75 (d, J = 7.9 Hz, 1H), 7.64 (t, J = 7.8 Hz, 1H), 7.58 (dd, J = 8.1, 0.8 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.44–7.37 (m, 1H); 13C NMR (151 MHz, DMSO) δ 188.30, 141.78, 138.30, 133.80, 133.34, 132.35, 130.19, 128.76, 128.26, 126.55, 126.49, 125.44, 124.70, 121.31. APCI-HRMS m/z calculated for C15H13BrNO [M + H] + : 302.0175, found: 302.0176. Purity (HPLC): 99.8%.
(2E)-1-(3-Aminophenyl)-3-(3-bromophenyl)prop-2-en-1-one (38)
Prepared as for 37 from 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 3-bromobenzaldehyde (0.68 g, 3.70 mmol) to yield the title compound 38 as light brown powder (0.80 g, 72%): mp: 176.78 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.26–8.13 (m, 2H), 7.97 (d, J = 15.7 Hz, 1H), 7.95 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 15.7 Hz, 1H), 7.64 (t, J = 7.8 Hz, 2H), 7.58 (d, J = 7.8 Hz, 1H), 7.43 (t, J = 7.8 Hz, 1H); 13C NMR (151 MHz, DMSO) δ 188.36, 142.82, 138.42, 137.04, 135.23, 133.22, 130.98, 130.91, 130.17, 128.27, 126.72, 126.60, 123.32, 122.40, 121.45. APCI-HRMS m/z calculated for C15H13BrNO [M + H] + : 302.0175, found: 302.0164. Purity (HPLC): 98.1%.
(2E)-1-(3-Aminophenyl)-3-(2-chlorophenyl)prop-2-en-1-one (39)
Prepared as for 37 from 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 2-chlorobenzaldehyde (0.52 g, 3.70 mmol) to yield the title compound 39 as light brown powder (0.94 g, 99%): mp: 188.04 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.23–8.13 (m, 2H), 8.05 (d, J = 15.6 Hz, 1H), 7.95 (d, J = 15.6 Hz, 2H), 7.65 (t, J = 7.8 Hz, 1H), 7.62–7.56 (m, 2H), 7.53–7.43 (m, 2H); 13C NMR (151 MHz, DMSO) δ 188.30, 139.05, 138.31, 134.43, 132.20, 132.12, 130.20, 130.08, 128.61, 127.73, 126.63, 126.58, 124.56, 121.39. APCI-HRMS m/z calculated for C15H13ClNO [M + H] + : 258.0680, found: 258.0673. Purity (HPLC): 99.8%.
(2E)-1-(3-Aminophenyl)-3-(3-chlorophenyl)prop-2-en-1-one (40)
Prepared as for 37 from 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 3-chlorobenzaldehyde (0.52 g, 3.70 mmol) to yield the title compound 40 as light brown powder (0.89 g, 93%): mp: 175.73 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.17 (d, J = 7.6 Hz, 1H), 8.06 (s, 1H), 7.97 (d, J = 15.7 Hz, 1H), 7.95 (s, 1H), 7.83 (d, J = 7.3 Hz, 1H), 7.74 (d, J = 15.7 Hz, 1H), 7.64 (t, J = 7.8 Hz, 1H), 7.58 (d, J = 7.9 Hz, 1H), 7.54–7.45 (m, 2H); 13C NMR (151 MHz, DMSO) δ 188.39, 142.86, 138.41, 136.78, 133.81, 130.73, 130.32, 130.16, 128.05, 127.90, 126.61, 126.56, 123.36, 121.40. APCI-HRMS m/z calculated for C15H13ClNO [M + H] + : 258.0680, found: 258.0658. Purity (HPLC): 98.1%.
(2E)-1-(3-Aminophenyl)-3-(3-fluorophenyl)prop-2-en-1-one (41)
Prepared as for 37 from 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 3-fluorobenzaldehyde (0.46 g, 3.70 mmol) to yield the title compound 41 as dark brown powder (0.19 g, 21%): mp: 206.07 °C (EtOH); 1H NMR (600 MHz, DMSO) δ 8.17 (d, J = 7.7 Hz, 1H), 7.99–7.93 (m, 2H), 7.85 (d, J = 10.3 Hz, 1H), 7.76 (d, J = 15.7 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.63–7.57 (m, 1H), 7.55–7.47 (m, J = 7.9, 6.3 Hz, 1H), 7.30 (td, J = 8.5, 2.4 Hz, 1H); 13C NMR (151 MHz, DMSO) δ 188.43, 163.28, 161.67, 143.14, 138.43, 137.12, 137.07, 135.17, 130.93, 130.88, 130.19, 126.74, 126.66, 125.61, 123.29, 121.54, 117.54, 117.40, 114.83, 114.69. APCI-HRMS m/z calculated for C15H13FNO [M + H] + : 242.0976, found: 242.0956. Purity (HPLC): 96.4%.
Synthesis of 42–47
1-(3-{(E)-[(2-Hydroxy-4-methoxyphenyl)methylidene]amino}phenyl)ethan-1-one (42)
A solution of 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 2-hydroxy-4-methoxybenzaldehyde (0.56 g, 3.70 mmol) in EtOH (5 mL) was mechanically stirred at room temperature for approximately 5 min before the dropwise addition of KOH (10% (w/v) aqueous solution, 5 mL). The subsequent reaction mixture was mechanically stirred at room temperature and continuously monitored by TLC. Upon completion, the reaction mixture was quenched with crushed ice (15 g) and acidified to pH 2 with HCl (32 wt. % in H2O, FCC). The subsequent precipitate was collected by vacuum filtration, dried (30 °C) and recrystallized from EtOH to yield the title compound 42 as dark green crystals (0.08 g, 8%): mp: 94.63 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 13.47 (d, J = 2.0 Hz, 1H), 8.58 (s, 1H), 7.83 (s, 2H), 7.54–7.41 (m, 2H), 7.30 (d, J = 8.3 Hz, 1H), 6.51 (d, J = 8.0 Hz, 2H), 3.85 (s, 3H), 2.64 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 197.84, 164.45, 163.95, 162.75, 149.21, 138.47, 133.96, 129.76, 126.32, 126.21, 120.41, 113.11, 107.54, 101.24, 55.65, 26.85. APCI-HRMS m/z calculated for C16H16NO3 [M + H] + : 270.1125, found: 270.1116. Purity (HPLC): 99.8%.
1-(3-{(Z)-[(2-Hydroxy-5-methoxyphenyl)methylidene]amino}phenyl)ethan-1-one (43)
Prepared as for 42 from 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 2-hydroxy-5-methoxybenzaldehyde (0.56 g, 3.70 mmol) to yield compound 43 as dark red crystals (0.59 g, 59%): mp: 92.81 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 12.53 (s, 1H), 8.63 (s, 1H), 7.86 (d, J = 11.7 Hz, 2H), 7.52 (t, J = 7.6 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.02 (d, J = 9.0 Hz, 1H), 6.97 (d, J = 8.9 Hz, 1H), 6.91 (s, 1H), 3.81 (s, 3H), 2.65 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 197.73, 163.63, 155.57, 152.50, 149.20, 138.48, 129.86, 126.84, 126.37, 121.20, 120.44, 118.63, 118.32, 115.52, 56.08, 26.91. APCI-HRMS m/z calculated for C16H16NO3 [M + H] + : 270.1125, found: 270.1112. Purity (HPLC): 94.4%.
1-(3-{(E)-[(3-Bromo-5-chlorophenyl)methylidene]amino}phenyl)ethan-1-one (44)
Prepared as for 42 from 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 3-bromo-5-chlorosalicylaldehyde (0.87 g, 3.70 mmol) to yield the title compound 44 as light orange crystals (0.42 g, 32%): mp: 183.31 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 14.07 (s, 1H), 8.61 (s, 1H), 7.95–7.84 (m, 2H), 7.64 (s, 1H), 7.56 (t, J = 7.7 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.38 (s, 1H), 2.65 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 197.38, 161.58, 156.92, 147.57, 138.65, 136.12, 130.85, 130.11, 127.76, 126.39, 124.20, 120.40, 120.01, 112.03, 26.90. APCI-HRMS m/z calculated for C15H13rClNO2 [M+ H]+: 351.9734, found: 351.9714. Purity (HPLC): 99.7%.
1-(3-{(E)-[(5-Chloro-2-hydroxyphenyl)methylidene]amino}phenyl)ethan-1-one (45)
Prepared as for 42 from 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 5-chlorosalicylaldehyde (0.58 g, 3.70 mmol) to yield the title compound 45 as light orange crystals (0.18 g, 18%): mp: 118.02 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 12.97 (s, 1H), 8.61 (s, 1H), 7.92–7.82 (m, 2H), 7.54 (t, J = 7.7 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.39 (s, 1H), 7.34 (d, J = 8.8 Hz, 1H), 6.99 (d, J = 8.8 Hz, 1H), 2.65 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 197.56, 162.59, 159.84, 148.66, 138.57, 133.51, 131.60, 129.94, 127.23, 126.22, 124.01, 120.58, 119.86, 119.08, 26.91. APCI-HRMS m/z calculated for C15H13ClNO2 [M+ H]+: 274.0629, found: 274.0629. Purity (HPLC): 100%.
1-{3-[(E)-{[4-(Diethylamino)-2-hydroxyphenyl]methylidene}amino]phenyl}ethan-1-one (46)
Prepared as for 42 from 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 4-(diethylamino)salicylaldehyde (0.71 g, 3.70 mmol) to yield the title compound 46 as gold crystals (0.46 g, 42%): mp: 117.85 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 13.52 (s, 1H), 8.47 (s, 1H), 7.84–7.73 (m, 2H), 7.50–7.38 (m, 2H), 7.18 (d, J = 8.7 Hz, 1H), 6.26 (d, J = 8.8 Hz, 1H), 6.20 (s, 1H), 3.41 (q, J = 7.0 Hz, 4H), 2.63 (s, 3H), 1.21 (t, J = 7.0 Hz, 6H); 13C NMR (151 MHz, CDCl3) δ 197.89, 163.87, 161.38, 152.00, 149.49, 138.15, 133.95, 129.39, 125.85, 125.17, 120.03, 108.90, 103.90, 97.61, 44.55, 26.72, 12.63. APCI-HRMS m/z calculated for C19H23N2O2 [M + H] + : 311.1754, found: 311.1746. Purity (HPLC): 100%.
1-(3-{(E)-[(2H-1,3-benzodioxol-5-yl)methylidene]amino}phenyl)ethan-1-one (47)
Prepared as for 42 from 3′-aminoacetophenone (0.50 g, 3.70 mmol) and 3,4-(methylenedioxy)benzaldehyde (0.56 g, 3.70 mmol) to yield the title compound 47 as light green crystals (0.98 g, 99%): mp: 127.64 °C (EtOH); 1H NMR (600 MHz, CDCl3) δ 7.81 (d, J = 15.6 Hz, 1H), 7.49–7.32 (m, 4H), 7.21 (d, J = 7.9 Hz, 1H), 6.99 (d, J = 8.0 Hz, 1H), 6.94 (d, J = 7.7 Hz, 1H), 6.13 (s, 2H), 3.93 (s, 2H), 3.82 (q, J = 6.9 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ 190.69, 149.97, 148.53, 146.93, 144.50, 139.67, 129.59, 129.54, 125.28, 120.54, 119.41, 118.91, 114.52, 108.79, 106.79, 101.75, 58.61, 18.58. APCI-HRMS m/z calculated for C16H14NO3 [M+ H]+: 268.0968, found: 268.0951. Purity (HPLC): 90.4%.
Biology
In vitro evaluation
Materials
All reagents were commercially available and purchased from various manufacturers. Radioligands [3H]DPCPX (specific activity 120 Ci/mmol) and [3H]NECA (specific activity 21.1 Ci/mmol) were obtained from PerkinElmer. Radioactivity was counted by a Packard Tri-CARB 2810 liquid scintillation counter.
Ethics
The collection of tissue samples for the A1 and A2A AR radioligand binding assays were approved by the Health Sciences Ethics Office for Research, Training and Support, North-West University (application number NWU-00260-17-A5) and were performed in accordance with the guidelines of the South African National Standard (SANS) document (The care and use of animals for scientific purposes).
Tissue samples
Male Sprague–Dawley rats were dissected to attain rat whole brain membranes (excluding cerebellum and brain stem) and rat striatal membranes for the A1 and A2A AR radioligand binding assays, respectively. Tissue samples were prepared and stored as described in literature (Van der Walt and Terre’Blanche 2015). Protein concentrations were determined according to the Bradford assay, using bovine serum albumin as reference standard (Bradford 1976).
Adenosine A1 and A2A receptor radioligand binding assays
The degree of binding affinity the test compounds possess towards rat A1 and A2A ARs were determined via radioligand binding assays, as described previously (Bradford 1976; Bruns 1987; Bruns and Watson 2012; Van der Walt and Terre’Blanche 2015). The A1 AR radioligand binding assay used rat whole brain membranes (expressing A1 ARs) and 0.1 nM 1,3-[3H]-dipropyl-8-cyclopentylxanthine ([3H]DPCPX) as radioligand (Bruns 1987) and, in turn, the A2A AR radioligand binding assay used rat striatal membranes (expressing A2A ARs) and 4 nM 5′-N-ethylcarboxamido[3H]adenosine ([3H]NECA) as radioligand (Bruns et al. 1986). N6-Cyclopentyladenosine (CPA) was also added to the A2A AR radioligand binding assay (in order to eliminate the A1 component of binding exhibited by the non-selective [3H]NECA) as well as MgCl2 (in order to increase radioligand binding and decrease non-specific binding) (Bruns et al. 1986). The final volume of all incubations contained 1 mL 50 mM Tris.HCl buffer and 1% DMSO (Van der Walt and Terre’Blanche 2015). Non-specific binding of [3H]DPCPX and [3H]NECA for the radioligand binding assays were defined as binding in the presence of 100 µM CPA (Bruns et al. 1986; Van der Walt and Terre’Blanche 2015). Specific binding was defined as the total binding minus the non-specific binding (Van der Walt and Terre’Blanche 2015).
Guanosine triphosphate (GTP) shift assays
The type of binding affinity test compounds exhibited at the rat A1 AR was determined via a guanosine triphosphate (GTP) shift assay, as described previously (Lohse et al. 1987; Van der Walt and Terre’Blanche 2015; Van der Werten et al. 1995). A GTP shift assay resembles the A1 AR radioligand binding assay. However, it requires the addition of 100 µM GTP. Non-specific binding was defined as binding in the presence of 10 µM DPCPX (Van der Werten et al. 1995; Lohse et al. 1984).
Statistical data analyses
In short, all statistical data analyses were done using Microsoft Excel and GraphPad Prism Software. Sigmoidal dose response curves, from which IC50 values were calculated, were obtained by plotting the specific binding against the logarithm of the test compounds’ concentrations. Subsequently, the IC50 values were used to calculate the Ki values for the competitive inhibition of [3H]DPCPX (Kd = 0.36 nM) against rat whole brain membranes and [3H]NECA (Kd = 15.3 nM) against rat striatal membranes by the test compounds by means of the Cheng–Prusoff equation. All calculated Ki values were determined in triplicate and given as mean ± standard error of the mean (SEM). Specific binding (%) of the radioligand at a maximum tested concentration of 100 µM were determined in duplicate and expressed as the mean in %. SI index values were calculated as a ratio of the A1 and A2A AR Ki values of test compounds. GTP shifts were calculated by dividing the Ki values of compounds reported in the presence of GTP by the Ki values obtained in the absence of GTP.
In silico evaluation
SwissADME (https://www.swissadme.ch), a free web tool, was used to evaluate key parameters of small molecules; such as pharmacokinetics, drug-likeness and medicinal chemistry friendliness. Pharmacokinetic parameters (absorption, distribution, metabolism and excretion), among others, were predicted from molecular structures using the most relevant computational methods, and thus, do not focus on just one specific property or model (Daina et al. 2017).
Conclusions
A total of 33 chalcones (15–36 and 37–41) and structurally related compounds (42–47) with different substitutions on ring A and/or benzylidene ring B were synthesised, characterized and evaluated in vitro to determine the degree and type of A1 and A2A receptor binding. While most compounds were not novel, the application of all compounds is original. The chalcone derivatives 24, 29, 37 and 38 possessed selective A1 affinity below 10 µM; compound 38 was the most potent selective A1 AR antagonist (Ki (r) = 1.6 µM). Most of the test compounds showed poor A1 and/or A2A AR affinity upon in vitro evaluation—making it difficult to determine SARs; however, some broad conclusions were drawn from these results. The type of substitution on ring A of the chalcone scaffold played a key role in affinity, and the position of the substitution on benzylidene ring B; the NH2-group at position C3 of ring A of the chalcone scaffold, and the Br-atom at position C3′ on benzylidene ring B were essential to selective A1 AR affinity (exemplified by compound 27 versus 38 and 37 versus 38). Selective A1 AR affinity and antagonistic activity may be added to the known biological activities of compounds 24 and 29. The physiochemical and pharmacokinetic properties (based on in silico evaluation) proved the chalcone derivatives 24, 29, 37 and 38, although drug-like, not at all lead-like (29). Thus far, the chalcone chemical structure was a promising scaffold in medicinal chemistry; due to the diverse chemistry and biology of this α,ß-unsaturated carbonyl system. The said system may be troublesome (it is perceived as a potential Michael acceptor); however, it easily allows structural modification and introduction of a heterocyclic ring system (such as isoxazole, pyrazole, pyrrole etcetera) which may just be beneficial to A1 and/or A2A AR binding affinity; thus, leading to the discovery of new pharmacophores from this classes of compounds.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
This study was funded by the National Research Foundation (NRF) of South Africa (Grant number 111814) and the North-West University (NWU). The authors wish to thank Dr D. Otto for NMR analyses and Dr J. Jordaan for MS analyses both from Chemical Research Beneficiation at NWU, as well as Prof. W. Liebenberg for DSC analyses, Prof F. Van der Kooy for HPLC analyses and Ms S. Lowe for assistance with biological assays from the Centre of Excellence for Pharmaceutical Sciences (Pharmacen), NWU.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Aarsland D, Påhlhagen S, Ballard CG, Ehrt U, Svenningsson P. Depression in Parkinson disease—epidemiology, mechanisms and management. Nat Rev Neurol. 2012;8:35. doi: 10.1038/nrneurol.2011.189. [DOI] [PubMed] [Google Scholar]
- Aksöz BE, Ertan R (2012) Spectral properties of chalcones II. Fabad J Pharm Sci 37:205–216. https://www.semanticscholar.org/paper/Spectral-Properties-of-Chalcones-II-Aks%C3%B6z-Ertan/9c63104669fa8cd865bd5c65da7a1d78c8cce36c?p2df
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Barros AI, Silva AM, Alkorta I, Elguero J. Synthesis, experimental and theoretical NMR study of 2′-hydroxychalcones bearing a nitro substituent on their B ring. Tetrahedron. 2004;60:6513–6521. doi: 10.1016/j.tet.2004.06.005. [DOI] [Google Scholar]
- Batovska DI, Todorova IT. Trends in utilization of the pharmacological potential of chalcones. Curr Clin Pharmacol. 2010;5:1–29. doi: 10.2174/157488410790410579. [DOI] [PubMed] [Google Scholar]
- Bayer H, Batzl C, Hartman RW, Mannschreck A. New aromatase inhibitors. Synthesis and biological activity of pyridyl-substituted tetralone derivatives. J Med Chem. 1991;34:2685–2691. doi: 10.1021/jm00113a004. [DOI] [PubMed] [Google Scholar]
- Boison D. Regulation of extracellular adenosine. In: Borea PA, Varani K, Gessi S, Merighi S, Vincenzi F, editors. The adenosine receptors, vol 34. The receptors. Totowa: Humana Press; 2018. pp. 13–58. [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Brenk R, Schipani A, James D, Krasowski A, Gilbert IH, Frearson J, Wyatt PG. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem Chem Enabling Drug Discov. 2008;3:435–444. doi: 10.1002/cmdc.200700139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns RF et al. (1987) Binding of the A 1-selective adenosine antagonist 8-cyclopentyl-1, 3-dipropylxanthine to rat brain membranes. Naunyn Schmiedeberg's Arch Pharmacol 335:59–63. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Bruns+RF+et+al.+%281987%29+Binding+of+the+A+1-selective+adenosine+antagonist+8-cyclopentyl-1%2C+3-dipropylxanthine+to+rat+brain+membranes+Naunyn-Schmiedeberg%27s+archives+of+pharmacology+335%3A59-63&btnG= [DOI] [PubMed]
- Bruns RF, Watson IA. Rules for identifying potentially reactive or promiscuous compounds. J Med Chem. 2012;55:9763–9772. doi: 10.1021/jm301008n. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Lu GH, Pugsley TA (1986) Characterization of the A2 adenosine receptor labeled by [3H] NECA in rat striatal membranes. Mol Pharmacol 29:331–34. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Bruns+RF%2C+Lu+GH%2C+Pugsley+TA+%281986%29+Characterization+of+the+A2+adenosine+receptor+labeled+by+%5B3H%5D+NECA+in+rat+striatal+membranes+Molecular+Pharmacology+29%3A331-346&btnG= [PubMed]
- Choi D, Yu S, Baek SH, Kang Y-H, Chang Y-C, Cho H. Synthesis and algicidal activity of new dichlorobenzylamine derivatives against harmful red tides. Biotechnol Bioprocess Eng. 2016;21:463–476. doi: 10.1007/s12257-016-0175-8. [DOI] [Google Scholar]
- Claisen L, Claparède A. Condensationen von Ketonen mit Aldehyden. Ber Dtsch Chem Ges. 1881;14:2460–2468. doi: 10.1002/cber.18810140182. [DOI] [Google Scholar]
- Clayson DB (1981) Aromatic amines: an assessment of the biological and environmental effects. National Academies Press, Washington D.C. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Clayson+DB+%281981%29+Aromatic+amines%3A+an+assessment+of+the+biological+and+environmental+effects.+National+Academies+Press%2C+&btnG=
- Daina A, Zoete V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. ChemMedChem. 2016;11:1117–1121. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lera Ruiz M, Lim Y-H, Zheng J. Adenosine A2A receptor as a drug discovery target. J Med Chem. 2013;57:3623–3650. doi: 10.1021/jm4011669. [DOI] [PubMed] [Google Scholar]
- Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ, Freeman TC. Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol. 1996;118:1461–1468. doi: 10.1111/j.1476-5381.1996.tb15561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey CW, Glist HM, Takashima A, Bottum SR, Dixon GJ. Chalcone and cinnamate synthesis via one-pot enol silane formation-Mukaiyama aldol reactions of ketones and acetate esters. Tetrahedron Lett. 2018;59:3080–3083. doi: 10.1016/j.tetlet.2018.06.066. [DOI] [Google Scholar]
- Dunwiddie TV. International review of neurobiology. Amsterdam: Elsevier; 1985. The physiological role of adenosine in the central nervous system; pp. 63–139. [DOI] [PubMed] [Google Scholar]
- Egan WJ, Merz KM, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43:3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- Fredholm BB et al. (1994) VI. Nomenclature and classification of purinoceptors. Pharmacol Rev 46:143. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Fredholm+BB+et+al.+%281994%29+VI.+Nomenclature+and+classification+of+purinoceptors+Pharmacological+reviews+46%3A143&btnG= [PMC free article] [PubMed]
- Fredholm BB, IJzerman AP, Jacobson KA, Klotz K-N, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53:527–552. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Fredholm+BB%2C+IJzerman+AP%2C+Jacobson+KA%2C+Klotz+K-N%2C+Linden+J+%282001%29+International+Union+of+Pharmacology.+XXV.+Nomenclature+and+classification+of+adenosine+receptors+Pharmacological+reviews+53%3A527-552&btnG= [PMC free article] [PubMed]
- Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE, International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freissmuth M, Schütz W, Linder M (1991) Interactions of the bovine brain A1-adenosine receptor with recombinant G protein alpha-subunits. Selectivity for rGi alpha-3. J Biol Chem 266:17778–17783. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Freissmuth+M%2C+Sch%C3%BCtz+W%2C+Linder+M+%281991%29+Interactions+of+the+bovine+brain+A1-adenosine+receptor+with+recombinant+G+protein+alpha-subunits.+Selectivity+for+rGi+alpha-3+Journal+of+Biological+Chemistry+266%3A17778-17783&btnG= [PubMed]
- Furniss B, Hannaford A, Smith P, Tatchell A (1989) Vogel’s textbook of practical organic chemistry, 5th edn. Longman Group UK Limited. https://fac.ksu.edu.sa/sites/default/files/vogel_-_practical_organic_chemistry_5th_edition.pdf
- Gaonkar SL, Vignesh U. Synthesis and pharmacological properties of chalcones: a review. Res Chem Intermed. 2017;43:6043–6077. doi: 10.1007/s11164-017-2977-5. [DOI] [Google Scholar]
- Gatch MB, Wallis CJ, Lal H. The effects of adenosine ligands R-PIA and CPT on ethanol withdrawal. Alcohol. 1999;19:9–14. doi: 10.1016/S0741-8329(99)00009-9. [DOI] [PubMed] [Google Scholar]
- Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1 A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- Gomes MN, et al. Chalcone derivatives: promising starting points for drug design. Molecules. 2017;22:1210. doi: 10.3390/molecules22081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gütschow M, et al. Benzothiazinones: a novel class of adenosine receptor antagonists structurally unrelated to xanthine and adenine derivatives. J Med Chem. 2012;55:3331–3341. doi: 10.1021/jm300029s. [DOI] [PubMed] [Google Scholar]
- Hallgas B, et al. Characterization of lipophilicity and antiproliferative activity of E-2-arylmethylene-1-tetralones and their heteroanalogues. J Chromatogr B. 2005;819:283–291. doi: 10.1016/j.jchromb.2005.02.014. [DOI] [PubMed] [Google Scholar]
- Hann MM, Keserü GM. Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat Rev Drug Discov. 2012;11:355. doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34:17–35. doi: 10.1081/DMR-120001387. [DOI] [PubMed] [Google Scholar]
- Huang SM, et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48:662–670. doi: 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- Jackson CE. Cholinergic system. In: Kreutzer JS, DeLuca J, Caplan B, editors. Encyclopedia of clinical neuropsychology. New York: Springer; 2011. pp. 562–564. [Google Scholar]
- Jacobson KA, Gao Z-G. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Moro S, Manthey JA, West PL, Ji X-d. Flavonoids in cell function. Berlin: Springer; 2002. Interactions of flavones and other phytochemicals with adenosine receptors; pp. 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain N, Kemp N, Adeyemo O, Buchanan P, Stone TW. Anxiolytic activity of adenosine receptor activation in mice. Br J Pharmacol. 1995;116:2127–2133. doi: 10.1111/j.1476-5381.1995.tb16421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse van Rensburg H, Terre'Blanche G, van der Walt M, Legoabe L. 5-Substituted 2-benzylidene-1-tetralone analogues as A1 and/or A2A antagonists for the potential treatment of neurological conditions. Bioorg Chem. 2017;74:251–259. doi: 10.1016/j.bioorg.2017.08.013. [DOI] [PubMed] [Google Scholar]
- Janse van Rensburg HD, Legoabe LJ, Terre'Blanche G, Van der Walt MM. 2-benzylidene-1-indanone analogues as dual adenosine A1/A2a receptor antagonists for the potential treatment of neurological conditions. Drug Res. 2019;69(7):382–391. doi: 10.1055/a-0808-3993. [DOI] [PubMed] [Google Scholar]
- Janse van Rensburg HD, Legoabe LJ, Terre'Blanche G, Van der Walt MM. Methoxy substituted 2-benzylidene-1-indanone derivatives as A1 and/or A2A AR antagonists for the potential treatment of neurological conditions. Medchemcomm. 2019;10(2):300–309. doi: 10.1039/C8MD00540K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson B, et al. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y, Son K-I, Oh YE, Noh D-Y. Ferrocenyl chalcones containing anthracenyl group: synthesis, X-ray crystal structures and electrochemical properties. Polyhedron. 2008;27:861–867. doi: 10.1016/j.poly.2007.11.015. [DOI] [Google Scholar]
- Juvale K, Pape VF, Wiese M. Investigation of chalcones and benzochalcones as inhibitors of breast cancer resistance protein. Bioorg Med Chem. 2012;20:346–355. doi: 10.1016/j.bmc.2011.10.074. [DOI] [PubMed] [Google Scholar]
- Kaplan GB, Cotreau MM, Greenblatt DJ. Effects of benzodiazepine administration on A1 adenosine receptor binding in-vivo and ex-vivo. J Pharm Pharmacol. 1992;44:700–703. doi: 10.1111/j.2042-7158.1992.tb05502.x. [DOI] [PubMed] [Google Scholar]
- Karaman İ, Gezegen H, Gürdere MB, Dingil A, Ceylan M. Screening of biological activities of a series of chalcone derivatives against human pathogenic microorganisms. Chem Biodivers. 2010;7:400–408. doi: 10.1002/cbdv.200900027. [DOI] [PubMed] [Google Scholar]
- Karthikeyan C, SH Narayana Moorthy N, Ramasamy S, Vanam U, Manivannan E, Karunagaran D, Trivedi P (2015) Advances in chalcones with anticancer activities. In: Recent patents on anti-cancer drug discovery, vol 10. pp 97–115. https://www.ingentaconnect.com/content/ben/pra/2015/00000010/00000001/art00006 [DOI] [PubMed]
- Karton Y, Jiang J-l, Ji X-D, Melman N, Olah ME, Stiles GL, Jacobson KA. Synthesis and biological activities of flavonoid derivatives as A3 adenosine receptor antagonists. J Med Chem. 1996;39:2293–2301. doi: 10.1021/jm950923i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanam H. Bioactive Benzofuran derivatives: a review. Eur J Med Chem. 2015;97:483–504. doi: 10.1016/j.ejmech.2014.11.039. [DOI] [PubMed] [Google Scholar]
- Kirchmair J, et al. Predicting drug metabolism: experiment and/or computation? Nat Rev Drug Discov. 2015;14:387. doi: 10.1038/nrd4581. [DOI] [PubMed] [Google Scholar]
- Klotz K-N, Hessling J, Hegler J, Owman C, Kull B, Fredholm B, Lohse M (1997) Comparative pharmacology of human adenosine receptor subtypes–characterization of stably transfected receptors in CHO cells. Naunyn Schmiedeberg's Arch Pharmacol 357:1–9. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Klotz+K-N%2C+Hessling+J%2C+Hegler+J%2C+Owman+C%2C+Kull+B%2C+Fredholm+B%2C+Lohse+M+%281997%29+Comparative+pharmacology+of+human+adenosine+receptor+subtypes%E2%80%93characterization+of+stably+transfected+receptors+in+CHO+cells+Naunyn-Schmiedeberg%27s+archives+of+pharmacology+357%3A1-9&btnG= [DOI] [PubMed]
- Langmead CJ, et al. Identification of novel adenosine A2A receptor antagonists by virtual screening. J Med Chem. 2012;55:1904–1909. doi: 10.1021/jm201455y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legoabe LJ, Van der Walt MM, Terre'Blanche G. Evaluation of 2-benzylidene-1-tetralone derivatives as antagonists of A1 and A2A adenosine receptors. Chem Biol Drug Design. 2018;91:234–244. doi: 10.1111/cbdd.13074. [DOI] [PubMed] [Google Scholar]
- Li M, Fang D, Geng F, Dai X. Silver-catalyzed efficient synthesis of enaminones from propargyl alcohols and amines. Tetrahedron Lett. 2017;58:4747–4749. doi: 10.1016/j.tetlet.2017.09.054. [DOI] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Liu S, Guo C, Guo Y, Yu H, Greenaway F, Sun M-Z (2014) Comparative binding affinities of flavonoid phytochemicals with bovine serum albumin. Iran J Pharm Res IJPR 13:1019. https://scholar.google.com/scholar?q=Liu+S,+Guo+C,+Guo+Y,+Yu+H,+Greenaway+F,+Sun+M-Z+(2014)+Comparative+binding+affinities+of+flavonoid+phytochemicals+with+bovine+serum+albumin+Iranian+journal+of+pharmaceutical+research:+IJPR+13:1019&hl=en&as_sdt=0,5 [PMC free article] [PubMed]
- Lohse MJ, Lenschow V, Schwabe U (1984) Interaction of barbiturates with adenosine receptors in rat brain. Naunyn Schmiedeberg's Arch Pharmacol 326:69–74. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Lohse+MJ%2C+Lenschow+V%2C+Schwabe+U+%281984%29+Interaction+of+barbiturates+with+adenosine+receptors+in+rat+brain+Naunyn-Schmiedeberg%27s+archives+of+pharmacology+326%3A69-74&btnG= [DOI] [PubMed]
- Lohse MJ, Klotz K-N, Lindenborn-Fotinos J, Reddington M, Schwabe U, Olsson RA. 8-Cyclopentyl-1, 3-dipropylxanthine (DPCPX)—a selective high affinity antagonist radioligand for A 1 adenosine receptors. Naunyn Schmiedeberg's Arch Pharmacol. 1987;336:204–210. doi: 10.1007/BF00165806. [DOI] [PubMed] [Google Scholar]
- Londos C, Cooper D, Wolff J. Subclasses of external adenosine receptors. Proc Natl Acad Sci. 1980;77:2551–2554. doi: 10.1073/pnas.77.5.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maemoto T, Finlayson K, Olverman HJ, Akahane A, Horton RW, Butcher SP. Species differences in brain adenosine A1 receptor pharmacology revealed by use of xanthine and pyrazolopyridine based antagonists. Br J Pharmacol. 1997;122:1202–1208. doi: 10.1038/sj.bjp.0701465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maemoto T, et al. Pharmacological characterization of FR194921, a new potent, selective, and orally active antagonist for central adenosine A1 receptors. J Pharmacol Sci. 2004;96:42–52. doi: 10.1254/jphs.FP0040359. [DOI] [PubMed] [Google Scholar]
- Martin YC. A bioavailability score. J Med Chem. 2005;48:3164–3170. doi: 10.1021/jm0492002. [DOI] [PubMed] [Google Scholar]
- Mathew B, et al. Perspective design of chalcones for the management of CNS disorders: a mini-review. CNS Neurol Disord Drug Targets. 2019 doi: 10.2174/1871527318666190610111246. [DOI] [PubMed] [Google Scholar]
- McGrath NA, Brichacek M, Njardarson JT. A graphical journey of innovative organic architectures that have improved our lives. J Chem Educ. 2010;87:1348–1349. doi: 10.1021/ed1003806. [DOI] [Google Scholar]
- Mellado M, et al. Synthesis of chalcones with antiproliferative activity on the SH-SY5Y neuroblastoma cell line: quantitative structure–activity relationship models. Med Chem Res. 2018;27:2414–2425. doi: 10.1007/s00044-018-2245-2. [DOI] [Google Scholar]
- Montanari F, Ecker GF. Prediction of drug–ABC-transporter interaction—recent advances and future challenges. Adv Drug Deliv Rev. 2015;86:17–26. doi: 10.1016/j.addr.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muegge I, Heald SL, Brittelli D. Simple selection criteria for drug-like chemical matter. J Med Chem. 2001;44:1841–1846. doi: 10.1021/jm015507e. [DOI] [PubMed] [Google Scholar]
- Müller CE. A1 adenosine receptors and their ligands: overview and recent developments. Il Farmaco. 2001;56:77–80. doi: 10.1016/S0014-827X(01)01005-9. [DOI] [PubMed] [Google Scholar]
- Munshi R, Pang I-H, Sternweis PC, Linden J (1991) A1 adenosine receptors of bovine brain couple to guanine nucleotide-binding proteins Gi1, Gi2, and Go. J Biol Chem 266:22285–22289. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Munshi+R%2C+Pang+I-H%2C+Sternweis+PC%2C+Linden+J+%281991%29+A1+adenosine+receptors+of+bovine+brain+couple+to+guanine+nucleotide-binding+proteins+Gi1%2C+Gi2%2C+and+Go+Journal+of+Biological+Chemistry+266%3A22285-22289&btnG= [PubMed]
- Nagarajan M, Shechter H (1984) The diverse carbenic and cationic chemistry of 3-diazo-2, 5-diphenylpyrrole. J Org Chem 49:62–74. https://scholar.google.com/scholar?q=Nagarajan+M,+Shechter+H+(1984)+The+diverse+carbenic+and+cationic+chemistry+of+3-diazo-2,+5-diphenylpyrrole+The+Journal+of+Organic+Chemistry+49:62-74&hl=en&as_sdt=0,5
- Nagayama H, et al. Effect of istradefylline on mood disorders in Parkinson's disease. J Neurol Sci. 2019;396:78–83. doi: 10.1016/j.jns.2018.11.005. [DOI] [PubMed] [Google Scholar]
- Nielsen AT, Houlihan WJ. The aldol condensation. Org React. 2004;16:1–438. doi: 10.1002/0471264180.or016.01. [DOI] [Google Scholar]
- Noji T, Karasawa A, Kusaka H. Adenosine uptake inhibitors. Eur J Pharmacol. 2004;495:1–16. doi: 10.1016/j.ejphar.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Normile H, Barraco RA. N6-cyclopentyladenosine impairs passive avoidance retention by selective action at A1 receptors. Brain Res Bull. 1991;27:101–104. doi: 10.1016/0361-9230(91)90288-U. [DOI] [PubMed] [Google Scholar]
- Noyce DS, Pryor WA. Carbonyl reactions. I. Kinetics and mechanism of the acid-catalyzed aldol condensation of benzaldehyde and acetophenone1. J Am Chem Soc. 1955;77:1397–1401. doi: 10.1021/ja01611a001. [DOI] [Google Scholar]
- Opletalova V, Hartl J, Palát K, Jr, Patel A. Conformational analysis of 2-hydroxy-2′, 5′-diazachalcones. J Pharm Biomed Anal. 2000;23:55–59. doi: 10.1016/S0731-7085(00)00263-6. [DOI] [PubMed] [Google Scholar]
- Ottaviani G, Gosling DJ, Patissier C, Rodde S, Zhou L, Faller B. What is modulating solubility in simulated intestinal fluids? Eur J Pharm Sci. 2010;41:452–457. doi: 10.1016/j.ejps.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Phillis JW (1991) Adenosine and adenine nucleotides as regulators of cellular function. CRC Press Incorporated. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Phillis+JW+%281991%29+Adenosine+and+adenine+nucleotides+as+regulators+of+cellular+function.+CRC+press%2C+&btnG=
- Pitsikas N, Borsini F. The adenosine A1 receptor antagonist BIIP 20 counteracts scopolamine-induced behavioral deficits in the passive avoidance task in the rat. Eur J Pharmacol. 1997;328:19–22. doi: 10.1016/S0014-2999(97)83021-X. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. doi: 10.1007/978-3-642-28863-0_5. [DOI] [PubMed] [Google Scholar]
- Rammohan A, Reddy JS, Sravya G, Rao CN, Zyryanov GV. Chalcone synthesis, properties and medicinal applications: a review. Environ Chem Lett. 2020;18:1–26. doi: 10.1007/s10311-019-00959-w. [DOI] [Google Scholar]
- Ramyashree D, Raghavendra K, Kumar AD, Vagish C, Kumar KA (2017) Synthesis, characterization and antimicrobial activities of chalcones and their post transformation to pyrazole derivatives. Asian J Chem 29:1538–1542. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Ramyashree+D%2C+Raghavendra+K%2C+Kumar+AD%2C+Vagish+C%2C+Kumar+KA+%282017%29+Synthesis%2C+characterization+and+antimicrobial+activities+of+chalcones+and+their+post+transformation+to+pyrazole+derivatives+Asian+J+Chem+29%3A1538-1542&btnG=
- Rao ML, Houjou H, Hiratani K. Novel synthesis of macrocycles with chalcone moieties through mixed aldol reaction. Tetrahedron Lett. 2001;42:8351–8355. doi: 10.1016/S0040-4039(01)01793-2. [DOI] [Google Scholar]
- Rao YK, Fang S-H, Tzeng Y-M. Differential effects of synthesized 2′-oxygenated chalcone derivatives: modulation of human cell cycle phase distribution. Bioorg Med Chem. 2004;12:2679–2686. doi: 10.1016/j.bmc.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Rebola N, Pinheiro PC, Oliveira CR, Malva JO, Cunha RA. Subcellular localization of adenosine A1 receptors in nerve terminals and synapses of the rat hippocampus. Brain Res. 2003;987:49–58. doi: 10.1016/S0006-8993(03)03247-5. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR, Stehle JH, Rivkees SA. Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Mol Endocrinol. 1991;5:1037–1048. doi: 10.1210/mend-5-8-1037. [DOI] [PubMed] [Google Scholar]
- Ritchie TJ, Macdonald SJ, Peace S, Pickett SD, Luscombe CN. Increasing small molecule drug developability in sub-optimal chemical space. MedChemComm. 2013;4:673–680. doi: 10.1039/C3MD00003F. [DOI] [Google Scholar]
- Robinson SJ, Petzer JP, Terre’Blanche G, Petzer A, Van der Walt MM, Bergh JJ, Lourens AC. 2-Aminopyrimidines as dual adenosine A1/A2A antagonists. Eur J Med Chem. 2015;104:177–188. doi: 10.1016/j.ejmech.2015.09.035. [DOI] [PubMed] [Google Scholar]
- Ruparelia KC, et al. The synthesis of chalcones as anticancer prodrugs and their bioactivation in CYP1 expressing breast cancer cells. Med Chem. 2018;14:322–332. doi: 10.2174/1573406414666180112120134. [DOI] [PubMed] [Google Scholar]
- Sahu NK, Balbhadra S, Choudhary J, Kohli DV. Exploring pharmacological significance of chalcone scaffold: a review. Curr Med Chem. 2012;19:209–225. doi: 10.2174/092986712803414132. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Ryan TA. Protein trafficking in neurons. Amsterdam: Elsevier; 2007. Neuronal exocytosis; pp. 97–124. [Google Scholar]
- Sawle P, Moulton BE, Jarzykowska M, Green CJ, Foresti R, Fairlamb IJ, Motterlini R. Structure−activity relationships of methoxychalcones as inducers of heme oxygenase-1. Chem Res Toxicol. 2008;21:1484–1494. doi: 10.1021/tx800115g. [DOI] [PubMed] [Google Scholar]
- Schmidt JG. Ueber die Einwirkung von Aceton auf Furfurol und auf Bittermandelöl bei Gegenwart von Alkalilauge. Ber Dtsch Chem Ges. 1881;14:1459–1461. doi: 10.1002/cber.188101401306. [DOI] [Google Scholar]
- Shenvi S, Kumar K, Hatti KS, Rijesh K, Diwakar L, Reddy GC. Synthesis, anticancer and antioxidant activities of 2, 4, 5-trimethoxy chalcones and analogues from asaronaldehyde: structure–activity relationship. Eur J Med Chem. 2013;62:435–442. doi: 10.1016/j.ejmech.2013.01.018. [DOI] [PubMed] [Google Scholar]
- Shimada J, et al. Adenosine A2A antagonists with potent anti-cataleptic activity. Bioorg Med Chem Lett. 1997;7:2349–2352. doi: 10.1016/S0960-894X(97)00440-X. [DOI] [Google Scholar]
- Singh P, Anand A, Kumar V. Recent developments in biological activities of chalcones: a mini review. Eur J Med Chem. 2014;85:758–777. doi: 10.1016/j.ejmech.2014.08.033. [DOI] [PubMed] [Google Scholar]
- Stroba A, et al. 3, 5-Diphenylpent-2-enoic acids as allosteric activators of the protein kinase PDK1: structure−activity relationships and thermodynamic characterization of binding as paradigms for PIF-binding pocket-targeting compounds. J Med Chem. 2009;52:4683–4693. doi: 10.1021/jm9001499. [DOI] [PubMed] [Google Scholar]
- Suwito H, et al. Design and synthesis of chalcone derivatives as inhibitors of the ferredoxin—ferredoxin-NADP+ reductase interaction of plasmodium falciparum: pursuing new antimalarial agents. Molecules. 2014;19:21473–21488. doi: 10.3390/molecules191221473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki F et al. (1993) Adenosine A1 antagonists. 3. Structure-activity relationships on amelioration against scopolamine-or N6-[(R)-phenylisopropyl] adenosine-induced cognitive disturbance. J Med Chem 36:2508–2518. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Suzuki+F+et+al.+%281993%29+Adenosine+A1+antagonists.+3.+Structure-activity+relationships+on+amelioration+against+scopolamine-or+N6-%5B%28R%29-phenylisopropyl%5D+adenosine-induced+cognitive+disturbance+Journal+of+medicinal+chemistry+36%3A2508-2518&btnG= [DOI] [PubMed]
- Swanson TH, Drazba JA, Rivkees SA. Adenosine A1 receptors are located predominantly on axons in the rat hippocampal formation. J Comp Neurol. 1995;363:517–531. doi: 10.1002/cne.903630402. [DOI] [PubMed] [Google Scholar]
- Szakács G, Váradi A, Özvegy-Laczka C, Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME–Tox) Drug Discov Today. 2008;13:379–393. doi: 10.1016/j.drudis.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Szopa A, et al. DPCPX, a selective adenosine A1 receptor antagonist, enhances the antidepressant-like effects of imipramine, escitalopram, and reboxetine in mice behavioral tests. Naunyn Schmiedeberg's Arch Pharmacol. 2018;391:1361–1371. doi: 10.1007/s00210-018-1551-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szopa A, et al. Agomelatine and tianeptine antidepressant activity in mice behavioral despair tests is enhanced by DMPX, a selective adenosine A2A receptor antagonist, but not DPCPX, a selective adenosine a1 receptor antagonist. Pharmacol Rep. 2019;71:676–681. doi: 10.1016/j.pharep.2019.03.007. [DOI] [PubMed] [Google Scholar]
- Teague SJ, Davis AM, Leeson PD, Oprea T. The design of leadlike combinatorial libraries. Angew Chem Int Ed. 1999;38:3743–3748. doi: 10.1002/(SICI)1521-3773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Testa B, Kraemer S. The biochemistry of drug metabolism—an introduction-testa-2007-chemistry and biodiversity-Wiley Online Library. Chem Biodivers. 2007 doi: 10.1002/cbdv.200890199. [DOI] [PubMed] [Google Scholar]
- Unoh Y, Hirano K, Satoh T, Miura M. Palladium-catalyzed decarboxylative arylation of benzoylacrylic acids toward the synthesis of chalcones. J Org Chem. 2013;78:5096–5102. doi: 10.1021/jo400716e. [DOI] [PubMed] [Google Scholar]
- Van Calker D, Müller M, Hamprecht B (1978) Adenosine inhibits the accumulation of cyclic AMP in cultured brain cells. Nature 276:839. https://scholar.google.com/scholar?hl=en&as_sdt=0%2C5&q=Van+Calker+D%2C+M%C3%BCller+M%2C+Hamprecht+B+%281978%29+Adenosine+inhibits+the+accumulation+of+cyclic+AMP+in+cultured+brain+cells+Nature+276%3A839&btnG= [DOI] [PubMed]
- Van der Walt MM, Terre’Blanche G. 1, 3, 7-Triethyl-substituted xanthines—possess nanomolar affinity for the adenosine A1 receptor. Bioorg Med Chem. 2015;23:6641–6649. doi: 10.1016/j.bmc.2015.09.012. [DOI] [PubMed] [Google Scholar]
- Van der Werten EM, et al. 8-Substituted adenosine and theophylline-7-riboside analogues as potential partial agonists for the adenosine A1 receptor. Eur J Pharmacol Mol Pharmacol. 1995;290:189–199. doi: 10.1016/0922-4106(95)00064-X. [DOI] [PubMed] [Google Scholar]
- Vásquez-Martínez YA, et al. Antimicrobial, anti-inflammatory and antioxidant activities of polyoxygenated chalcones. J Braz Chem Soc. 2019;30:286–304. doi: 10.21577/0103-5053.20180177. [DOI] [Google Scholar]
- Vazquez-Rodriguez S, Matos MJ, Santana L, Uriarte E, Borges F, Kachler S, Klotz KN. Chalcone-based derivatives as new scaffolds for h A 3 adenosine receptor antagonists. J Pharm Pharmacol. 2013;65:697–703. doi: 10.1111/jphp.12028. [DOI] [PubMed] [Google Scholar]
- Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Vollert C, Forkuo GS, Bond RA, Eriksen JL. Chronic treatment with DCPCX, an adenosine A1 antagonist, worsens long-term memory. Neurosci Lett. 2013;548:296–300. doi: 10.1016/j.neulet.2013.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Lubitz DK, Paul IA, Bartus RT, Jacobson KA. Effects of chronic administration of adenosine A1 receptor agonist and antagonist on spatial learning and memory. Eur J Pharmacol. 1993;249:271. doi: 10.1016/0014-2999(93)90522-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Chen G, Lu YJ, Chen Q, Huo Y, Li X. Intermolecular multiple dehydrogenative cross-couplings of ketones with boronic acids and amines via copper catalysis. Adv Synth Catal. 2019;361:3886–3892. doi: 10.1002/adsc.201900419. [DOI] [Google Scholar]
- Wu S, Yu H, Hu Q, Yang Q, Xu S, Liu T. Silver-catalyzed decarboxylative cross-coupling of α-keto acids with alkenes giving approach to chalcones. Tetrahedron Lett. 2017;58:4763–4765. doi: 10.1016/j.tetlet.2017.11.005. [DOI] [Google Scholar]
- Xie Z, et al. Synthesis and evaluation of hydroxychalcones as multifunctional non-purine xanthine oxidase inhibitors for the treatment of hyperuricemia. Bioorg Med Chem Lett. 2017;27:3602–3606. doi: 10.1016/j.bmcl.2017.01.053. [DOI] [PubMed] [Google Scholar]
- Yacoubi ME, Ledent C, Parmentier M, Bertorelli R, Ongini E, Costentin J, Vaugeois JM. Adenosine A2A receptor antagonists are potential antidepressants: evidence based on pharmacology and A2A receptor knockout mice. Br J Pharmacol. 2001;134:68–77. doi: 10.1038/sj.bjp.0704240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Yang D, Wei W, Yuan L, Nie F, Tian L, Wang H. Silver-catalyzed double-decarboxylative cross-coupling of α-keto acids with cinnamic acids in water: a strategy for the preparation of chalcones. J Org Chem. 2015;80:3258–3263. doi: 10.1021/jo502642n. [DOI] [PubMed] [Google Scholar]
- Zhou B, Xing C. Diverse molecular targets for chalcones with varied bioactivities. Med Chem. 2015;5:388. doi: 10.4172/2161-0444.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Li Z, Yang X, Chen X, Li M, Chen T, Yin S-F. Phosphorous acid promoted hydration–condensation of aromatic alkynes with aldehydes affording chalcones in an oil/water two-phase system. Synthesis. 2016;48:231–237. doi: 10.1055/s-0035-1560832. [DOI] [Google Scholar]
- Zhuang C, Zhang W, Sheng C, Zhang W, Xing C, Miao Z. Chalcone: a privileged structure in medicinal chemistry. Chem Rev. 2017;117:7762–7810. doi: 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.