

Abstract
A major factor in the progression to heart failure in humans is the inability of the adult heart to repair itself after injury. We recently demonstrated that the early postnatal mammalian heart is capable of regeneration following injury through proliferation of preexisting cardiomyocytes1,2 and that Meis1, a three amino acid loop extension (TALE) family homeodomain transcription factor, translocates to cardiomyocyte nuclei shortly after birth and mediates postnatal cell cycle arrest3. Here we report that Hoxb13 acts as a cofactor of Meis1 in postnatal cardiomyocytes. Cardiomyocyte-specific deletion of Hoxb13 can extend the postnatal window of cardiomyocyte proliferation and reactivate the cardiomyocyte cell cycle in the adult heart. Moreover, adult Meis1-Hoxb13 double-knockout hearts display widespread cardiomyocyte mitosis, sarcomere disassembly and improved left ventricular systolic function following myocardial infarction, as demonstrated by echocardiography and magnetic resonance imaging. Chromatin immunoprecipitation with sequencing demonstrates that Meis1 and Hoxb13 act cooperatively to regulate cardiomyocyte maturation and cell cycle. Finally, we show that the calcium-activated protein phosphatase calcineurin dephosphorylates Hoxb13 at serine-204, resulting in its nuclear localization and cell cycle arrest. These results demonstrate that Meis1 and Hoxb13 act cooperatively to regulate cardiomyocyte maturation and proliferation and provide mechanistic insights into the link between hyperplastic and hypertrophic growth of cardiomyocytes.
Heart failure affects more than 26 million people worldwide1, and thus finding effective therapies for heart failure is a major public health goal. A key underlying cause of heart failure is the inability of the adult human myocardium to regenerate following injury. Although both the adult mouse2,3 and human4 hearts have a limited capacity for cardiomyocyte turnover5, this low basal rate of cardiomyogenesis is not therapeutically relevant from a regeneration perspective. By contrast, newborn mammals are capable of myocardial regeneration for a short time after birth, mediated by proliferation of preexisting cardiomyocytes6,7. Several regulators of postnatal cardiomyocyte cell cycle arrest have been described thus far8–13, highlighting the complexity of cardiomyocyte cell cycle regulation and the need for better understanding of the interplay between these redundant mechanisms.
Meis1, a homeobox transcription factor belonging to the TALE family, was identified as a regulator of the cardiomyocyte cell cycle9. Deletion of Meis1 prolongs the postnatal window of myocyte proliferation, and reactivates cardiomyocyte mitosis in adult mouse heart. However, cardiomyocyte proliferation following Meis1 deletion is transient, and cell cycle arrest eventually ensues9. Indeed, although conditional deletion of Meis1 prevented continued deterioration of left ventricular function following proximal left anterior descending coronary artery (LAD) ligation, there was minimal improvement in function compared with immediately after myocardial infarction (MI) values (Extended Fig.1a). Therefore, it is plausible that other additional redundant mechanisms are activated following Meis1 deletion. Meis1 is known to associate with a number of proteins that regulate its cellular localization and transcriptional activity14–16; however, their role in cardiomyocytes remains unknown. We set out to identify regulators of Meis1 expression and function in the postnatal heart and to define their role in regulation of cardiomyocyte cell cycle.
Hoxb13 associates with Meis1 postnatally.
Among the Hox family of transcription factors that are upregulated in the early postnatal window9, Hoxb13 was the only one with an expression pattern similar to that of Meis1 (Fig.1a–b, Extended Fig.1b, c). Hoxb13 localized to cardiomyocyte nuclei at the time of cell cycle arrest on postnatal day (P)7, and Hoxb13 levels were low in P7 hearts that had been subjected to LAD ligation at P1, a procedure that extends cardiomyocyte proliferation seven days after injury, and Hoxb13 levels increased as the mouse heart switched from hyperplastic (P1) to hypertrophic growth (P7). Moreover, coimmunoprecipitation and proximity ligation assays (PLA) revealed the association of Hoxb13 and Meis1 at P7 (Fig.1c–e, Extended Fig.1d–g).
Figure 1: Identification of Hoxb13 as a Meis1-interacting transcription factor in postnatal cardiomyocytes.
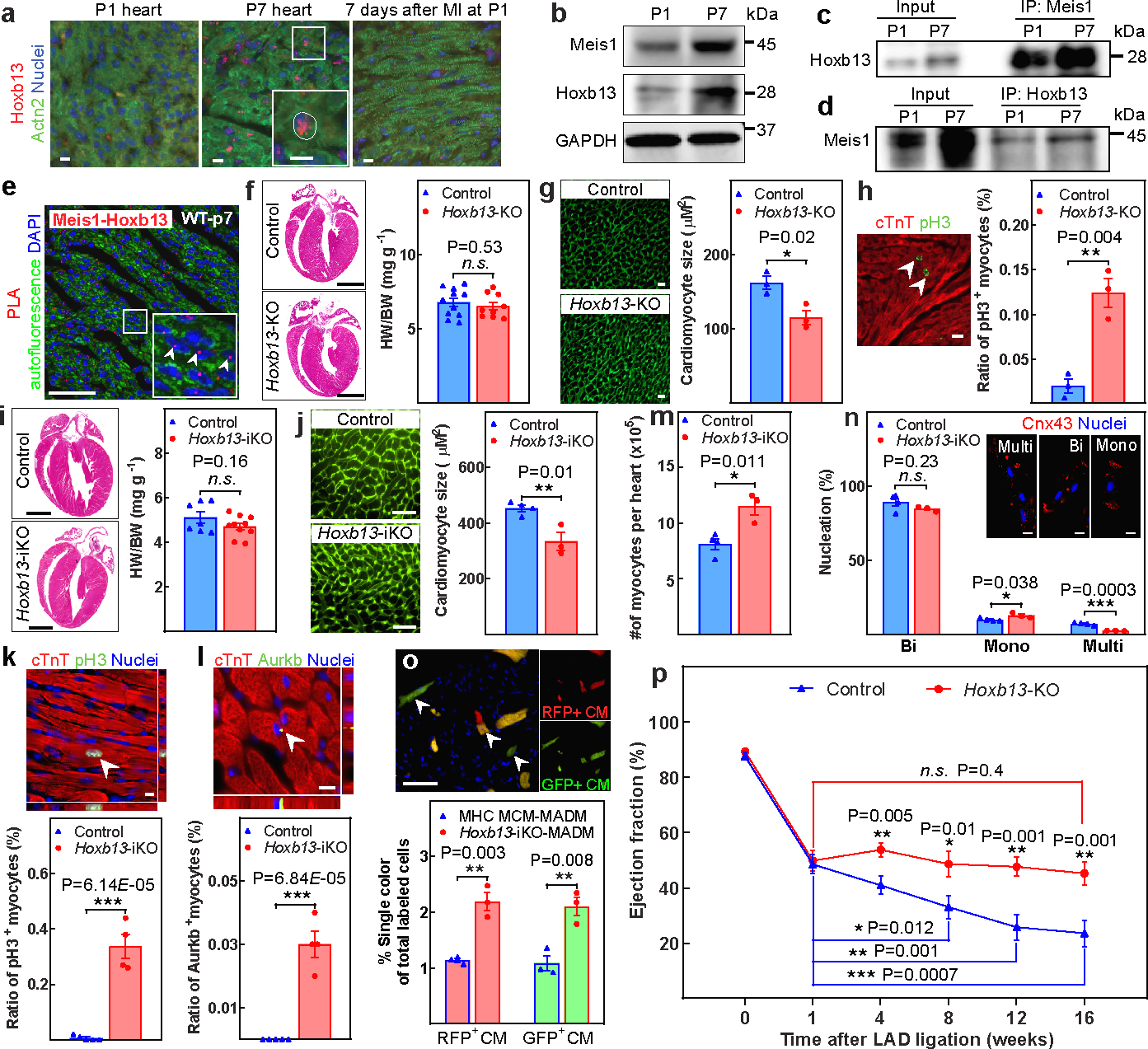
a, Immunostaining of Hoxb13 (red), sarcomeric-α-actinin (Actn2; green) and nucleus (blue) at P1, P7 and seven days after P1 MI. The inset shows a higher magnification of the outlined region. b, Western blot analysis of Meis1 and Hoxb13 proteins during regenerative stages. c, d, Coimmunoprecipitation (IP) using Meis1 (c) and Hoxb13 (d) antibodies from total heart extracts at P1 and P7. e, PLA of Meis1-Hoxb13 (red dots) in P7 heart. f, Haematoxylin and eosin-stained heart sections and heart weight/body weight (HW/BW) in Hoxb13-KO mice at P14. g, Wheat germ agglutinin (WGA) staining and CSA quantification in Hoxb13-KO hearts. h, Immunostaining for PH3 (green) and cardiac troponin T (cTnT, red) showing the percentage of mitotic cardiomyocytes (arrowheads). i–n, Quantification of Hoxb13-iKO hearts at 14 days after deletion. i, Heart weight/body weight and H&E-stained heart sections. j, WGA staining and CSA quantification. k, l, Immunostaining for PH3 or aurora B kinase (AurkB) (green), cardiac troponin T (red) and nucleus (blue), showing the percentage of mitotic (k) and cytokinetic (l) cardiomyocytes. m, n, Total number (m) and nucleation (n) of isolated cardiomyocytes. Cx43, connexin 43. o, Representative immunofluorescence and quantification of single-labelled (red or green, arrowheads) cardiomyocytes (CM) in Hoxb13-iKO MADM mice. p, Serial echocardiographic measurements of control and Hoxb13-KO MI mice. Data are mean ± s.e.m.; unpaired two-sided t-test. Data in a–e were independently repeated three times with similar results. *P < 0.05, **P < 0.01, ***P < 0.001. NS, not significant. For gel source data, see Supplementary Fig. 1. For sample numbers, see Methods. Scale bars, 10 μm (a, g, h, k, l, n), 50 μm (e, j, o), 1 mm (f, i). See also Extended Data Figs. 1, 2.
Genetic deletion of Hoxb13.
As previously noted, Hoxb13 might have similar a role as Meis1 in postnatal cardiomyocyte proliferation. We therefore generated conditional cardiomyocyte-specific Myh6cre:Hoxb13fl/fl (hereafter, Hoxb13-KO) mice (Extended Fig.1h, i). Western blot analysis revealed a reduction of Hoxb13 protein in Hoxb13-KO hearts (Extended Fig.1j, left). Meis1 was cytoplasmic in cardiomyocytes of Hoxb13-KO hearts at P14, although Meis1 was localized to the nucleus in non-cardiomyocyte cells and in wild-type cardiomyocytes (Extended Fig.1k). Compared with wild-type hearts, despite the absence of differences in cardiac morphology, heart weight to body weight ratio (Fig.1f), and left ventricle (LV) function (Extended Fig.1l), we observed a significant reduction in individual cardiomyocyte cross-sectional area (CSA) (Fig.1g) and an increase in phosphorylated histone H3 (PH3)-positive mitotic cardiomyocytes (Fig.1h) in Hoxb13-KO hearts at P14, but not in older hearts (data not shown). Thus, prolongation of the postnatal window of cardiomyocyte proliferation secondary to Hoxb13 deletion is not sustained, similar to the phenotype resulting from Meis1 deletion. To test whether Hoxb13 deletion is sufficient to induce postmitotic cardiomyocyte cycle re-entry, we generated a tamoxifen-inducible cardiomyocyte-specific Myh6mERcremERHoxb13fl/fl mouse (hereafter, Hoxb13-iKO). Hoxb13 protein reduction in adult heart was confirmed by western blot two weeks after tamoxifen injection (Extended Fig.1j, right). Cardiac morphology, heart weight/body weight (Fig.1i), and fibrosis (Extended Fig.1m) were unaltered in the Hoxb13-iKO hearts. However, cardiomyocyte CSA was significantly reduced in the Hoxb13-iKO hearts (Fig.1j). There was also a marked increase in PH3+ (Fig.1k) and aurora B kinase-positive (Aurkb+) cardiomyocytes (Fig.1l). Quantification of adult cardiomyocyte number following collagenase digestion showed a significant increase in the number of cardiomyocytes in the Hoxb13-iKO hearts (Fig.1m). There was also a significant increase in the fraction of mononucleated cardiomyocytes, and a decrease in multinucleated cardiomyocytes (Fig.1n). In addition, we used a lineage-tracing system in which we crossed cardiomyocyte-specific Myh6mERcremER and Mosaic Analysis with Double Markers (MADM) mice17 with the Hoxb13fl/fl mice to examine cardiomyocyte division. Assessment of single-coloured green or red cardiomyocytes confirmed that Hoxb13 deletion resulted in more than twofold induction of new cardiomyocyte formation (Fig.1o). Finally, there were no significant changes in cell death, as measured using TUNEL assay (Extended Fig. 1n). The evidence thus far suggested that cardiomyocyte-specific deletion of Hoxb13 is sufficient to induce measurable cardiomyocyte mitosis in adult hearts.
To determine whether Hoxb13 deletion leads to meaningful cardiomyocyte regeneration, we induced MI in adult Hoxb13-KO mice (Extended Fig.2a). Although the initial reduction in left-ventricle ejection fraction (LVEF) was comparable between the two groups at one week after MI (Fig. 1p), serial echocardiographic assessment revealed a progressive deterioration of LVEF in the controls, but not in Hoxb13-KO mice, such that at 8 to 16 weeks after the MI, the Hoxb13-KO hearts had significantly higher LVEF compared to controls (Fig. 1p, Extended Data Fig. 2b). There was no significant difference in heart weight/body weight, wet-to-dry lung ratio or scar formation (Extended Fig.2c–e). Similarly, inducible knockout of Hoxb13 in adult mice also prevented the progressive decline in LVEF (Extended Fig.2f–i). However, there was no improvement in LV systolic function in the MI hearts following deletion of Hoxb13 over time, compared with immediate post-injury values. Taken together, these findings suggest that loss of Hoxb13 prevents progressive deterioration of systolic function after MI, but does not induce substantial functional recovery. This may be owing to the transient nature of activation of cardiomyocyte mitosis following Hoxb13 deletion.
Combined loss of both Meis1 and Hoxb13
Next we generated cardiomyocyte-specific Myh6cre:Meis1-Hoxb13 double-knockout mice (hereafter, DKO) to examine cardiomyocyte proliferation at P28. The Meis1-KO and Hoxb13-KO mice do not show significant cardiomyocyte division at this time point. We observed a modest but significant decrease in heart weight/body weight (Extended Fig.3a) in DKO mice, despite a significant decrease in cardiomyocyte CSA in the DKO hearts compared with wild-type hearts (Extended Fig.3b). Of note, there was a marked increase PH3+ and aurora B kinase-positive cardiomyocytes in the DKO hearts (Extended Fig.3c, d respectively), and some cardiomyocytes displayed sarcomere disassembly (Extended Fig.3c), which is characterized by loss of striations and localization of the sarcomeres to the periphery of cardiomyocytes. This is a hallmark of cardiomyocyte proliferation in neonatal hearts and is seldom seen in the adult heart, even when mitotic indices are present. In support of the increased mitosis, the total cardiomyocyte number in DKO hearts was significantly greater than in controls, with an associated increase in mononucleated cardiomyocytes and a decrease in binucleated and multinucleated cardiomyocytes (Extended Fig.3e, f). Cardiac morphology and function (Extended Fig.3a, g) revealed no differences between DKO and control mice. Cardiomyocyte proliferation remained active in six-month-old DKO hearts (Extended Fig.3h, i). Although the cardiomyocyte CSA in the six-month-old DKO mice was still significantly smaller, the LVEF remained similar to that of controls (Extended Fig.3j, k).
Given that DKO hearts consistently displayed a smaller cell size, we performed further studies to assess the effect of deletion of Meis1 and Hoxb13 on the cardiomyocyte hypertrophy. We detected a blunted response to pathological hypertrophy following pressure overload induced by transverse aortic constriction (TAC) in DKO hearts, in which DKO hearts had better LVEF, and smaller absolute heart weight, heart weight/body weight, heart weight/tibia length and cardiomyocyte CSA than controls at three weeks after TAC (Extended Fig.4a–h). There was no difference in tibia length and myocardial interstitial fibrosis (Extended Fig.4d, g). In addition, the DKO hearts exhibited an attenuated exercise-induced physiological hypertrophic response (Extended Fig.4i–p).
Heart regeneration in the DiKO
We next examined whether conditional inducible deletion of Meis1 and Hoxb13 in the adult heart (DiKO) could promote cardiomyocyte cell cycle re-entry. Inducible deletion of both genes did not alter cardiac morphology or fibrosis (Fig.2a, Extended Fig.5a). Notably, however, heart weight/body weight increased (Fig.2b); the DiKO hearts also showed diminished cardiomyocyte CSA by about 30% one week after tamoxifen injection (Fig.2c). There was also a significant increase in the number of ventricular cardiomyocytes with disassembled sarcomeres at this time point (Fig.2d, Extended Fig.5b, c). Because sarcomere disassembly is an unusual event in adult hearts, we examined cardiomyocyte death and found a significant increase in TUNEL+ cardiomyocytes one week after deletion, but not at a later time point (two months after deletion) (Fig.2e, Extended Fig.5d). Of note, the number of cardiomyocytes undergoing mitosis and cytokinesis were also markedly increased in DiKO hearts one week after deletion. However, the number of mitotic cardiomyocytes in DiKO mice decreased by two months after deletion compared with one week after deletion (Fig. 2f, Extended Fig. 5e–h). Assessment of DiKO-MADM mice showed over a threefold increase in single-labelled green or red cardiomyocytes (Fig. 2g). Consistently, 5-bromodeoxyuridine (BrdU) staining, coupled with cardiomyocyte nuclear staining using PCM-1 revealed labeling of approximately 7 % of cardiomyocyte nuclei in the DiKO hearts two weeks after deletion (Fig. 2h). Finally, we observed a significant increase in the total number of cardiomyocytes in the DiKO hearts (Fig.2i), with an increase in the number of mononucleated cardiomyocytes (Fig.2j). Together these results demonstrate that concomitant cardiomyocyte-specific loss of Meis1 and Hoxb13 induces proliferation of adult ventricular cardiomyocytes.
Figure 2: Loss of Meis1 and Hoxb13 induces cardiomyocyte proliferation and heart regeneration.
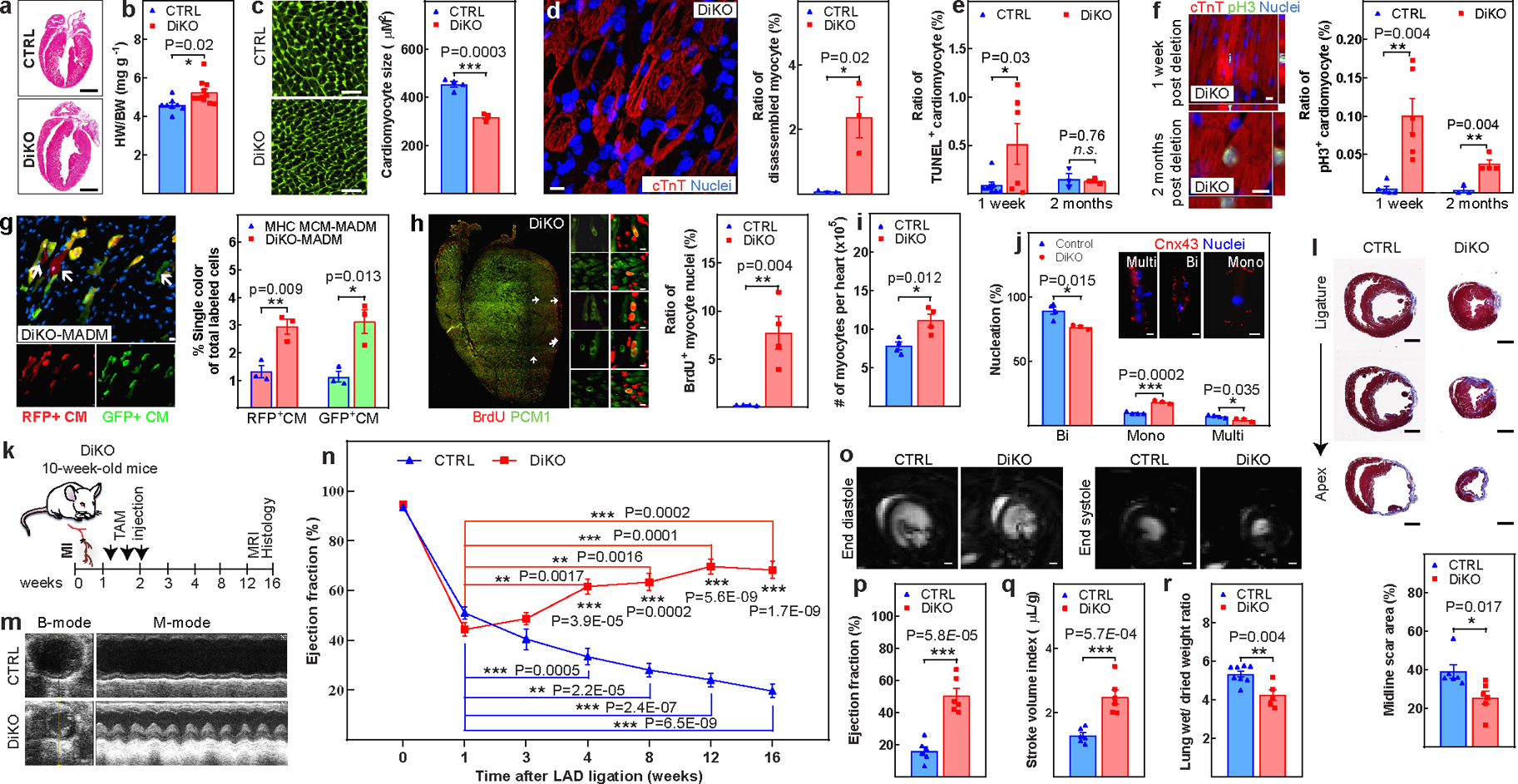
a, Representative haematoxylin and eosin-stained heart sections four weeks after inducible deletion of Meis1 and Hoxb13 (DiKO). b–d, Heart weight/body weight (b), WGA staining and CSA quantification (c), and representative image of sarcomere disassembly and quantification (d) in DiKO hearts at one week after deletion. WGA (green), cardiac troponin T (red) and nucleus (blue). e, Quantification of TUNEL+ cardiomyocytes in DiKO hearts at one week and two months after deletion. f, Immunostaining for PH3 (green) and cardiac troponin T (red) showing the percentage of mitotic cardiomyocytes. g, Representative immunofluorescence images and quantification of single-labelled (red or green, arrows) cardiomyocytes in DiKO-MADM mice. h, Representative images and quantification of proliferating cardiomyocytes (PCM+BrdU+ cells, arrows) from BrdU pulse–chase experiments from cryosections of an entire heart. The smaller panels show selected regions at higher magnification. i, j, Total number of cardiomyocytes (i) and nucleation quantification (j) in DiKO hearts. k, Schematic of MI model in DiKO mice. Tam, tamoxifen. l, m, Representative serial Masson’s trichrome-stained transverse sections and quantification of fibrotic scars (l), and representative echocardiography of control and DiKO MI mice at 16 weeks after injury (m). n, Serial echocardiographic measurement of control and DiKO MI mice. o–q, Representative images of MRI (o), LVEF (p) and stroke volume index (q). r, Wet-to-dry lung weight ratio in DiKO mice 16 weeks after MI. Data are mean ± s.e.m.; unpaired two-sided t-test. *P < 0.05, **P < 0.01, ***P < 0.001. For sample numbers, see Methods. Scale bars, 10 μm (d, f, g, h), 50 μm (c), 1 mm (a, l, o). See also Extended Data Figs. 3–6.
To test whether the increase in aforementioned cardiomyocyte proliferation can contribute to consequential cardiac repair in the adult heart, we first examined the expression of cell cycle inhibitors two weeks after MI by western blot analysis, revealing a post-MI decrease in p21 and p15/16 proteins in the DKO hearts (Extended Fig.6a–c). Next, we subjected DiKO mice to MI one week before gene deletion (Fig.2k). The results revealed that the infarcted DiKO hearts had smaller fibrotic scars, and lower heart weight/body weight and absolute heart weight values at 16 weeks after MI compared with controls, although their body weight was comparable (Fig.2l, Extended Fig.6d, ,e, f, h). None of the DiKO mice displayed left atrial dilatation (Extended Fig.6g). Although both groups showed a similar reduction in LVEF one week after MI, starting from week 4, the DiKO mice displayed a gradual and significant improvement in LVEF (Fig.2m, n). DiKO mice also showed a marked increase in the infarcted anterior wall thickness and a significant decrease in ventricular dilation (Extended Fig.6k–q). Consistent with these results, magnetic resonance imaging (MRI) at 16 weeks after MI showed a significant increase in LVEF, stroke volume, and cardiac output indexes (Fig.2o–q, Extended Data Fig 6i), with a similar heart rate (Extended Data Fig 6j), in the DiKO mice compared with controls. In addition, the wet-to-dry lung weight ratio in the DiKO was smaller than in the control group, indicating prevention of pulmonary oedema (Fig.2r). These results suggest that the induction of cardiomyocyte proliferation following loss of both Meis1 and Hoxb13 can improve LV systolic function after MI.
Next, we used chromatin immunoprecipitation with next-generation sequencing (ChIP-Seq) to identify the in vivo targets of Meis1 and Hoxb13. Three distinct sets of genes were identified; 272 genes that are targets for both Meis1 and Hoxb13, 1,647 genes that are targets for Meis1 only and 1,326 genes that are targets Hoxb13 only (Extended Fig.7). Pathway analysis suggested that Meis1 and Hoxb13 synergistically regulate cardiomyocyte differentiation and cell cycle.
Calcineurin regulates Hoxb13
Concurrent with the postnatal loss of proliferative capacity, cardiomyocytes switch to hypertrophic growth. Calcineurin-dependent signalling is known to be a fundamental mediator of hypertrophic growth. Therefore, we examined postnatal changes in calcineurin abundance. Western blot analyses showed an increase in protein levels of the catalytic subunit of calcineurin (CnA) at P7, similar to Meis1 and Hoxb13 (Fig.3a). Coimmunoprecipitation using CnA, Meis1 and Hoxb13 antibodies revealed that these three proteins interact in the postnatal heart (Fig.3b–d, Extended Fig.8a–c). Notably, although PLA confirmed the association of CnA with both Meis1 and Hoxb13, the frequency and intensity of the foci for Hoxb13-CnA was higher than for Meis1-CnA (Fig.3e, Extended Fig.8d–f). We next examined whether Meis1 and Hoxb13 are direct targets of calcineurin by scanning for calcineurin-docking motifs. A sequence in Hoxb13, ITIWFQ (aa 262–267) was identified as a potential PxIxIT site that, similar to the motif in AKAP79, contains an isoleucine in the first position (Extended Fig.8h)18. To determine whether CnA interacts with this sequence in Hoxb13, we performed an in vitro pull-down assay for CnA-His6 with glutathione S-transferase (GST) fused to a 16-amino-acid peptide containing either the ITIWFQ sequence, or a mutant peptide containing alanine substitutions in this putative motif (Fig.3f). The results showed that CnA co-purified with the GST-Hoxb13 peptide, but not the mutated form, indicating a PxIxIT-mediated interaction (Fig.3g), suggesting that Hoxb13 directly interacts with, and may be dephosphorylated by, calcineurin.
Figure 3: Calcineurin regulates Hoxb13 expression in the postnatal hearts.
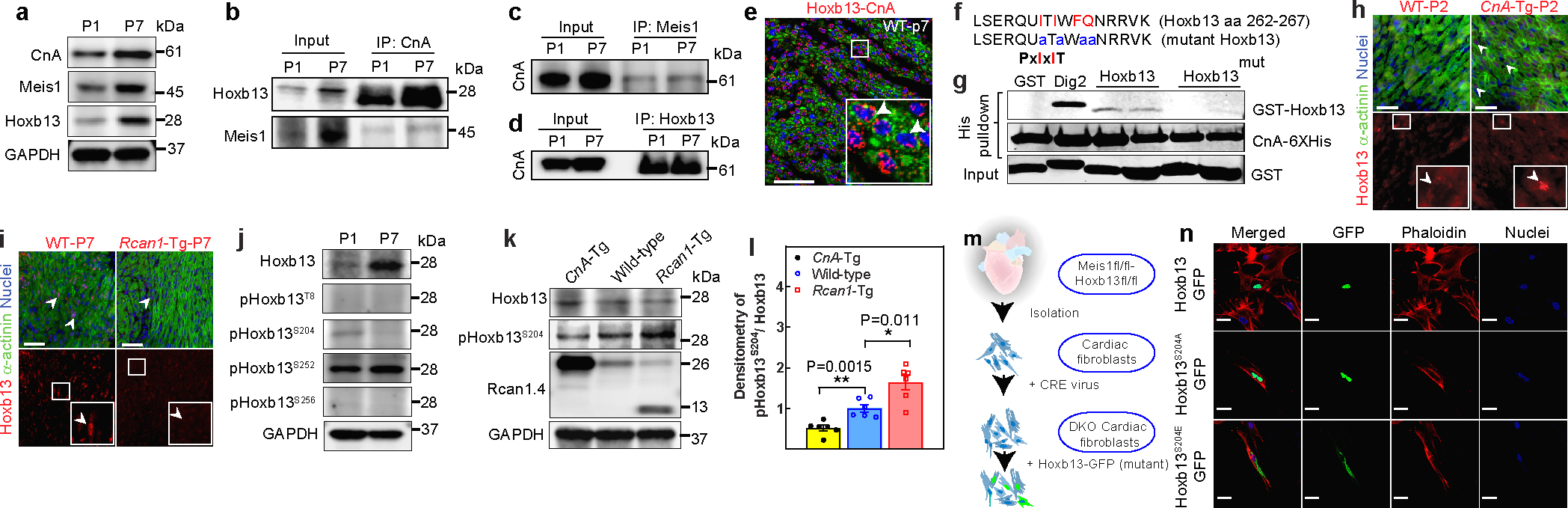
a, Western blot analysis of Meis1, Hoxb13 and CnA proteins during regenerative stages. b–d, Coimmunoprecipitation of CnA (b), Meis1 (c) and Hoxb13 (d) from total heart lysates at P1 and P7. e, PLA showing Hoxb13–calcineurin (red dots) in P7 heart. f, Schematic of Hoxb13 mutant at the predicated PxIxIT site. aa, amino acid. g, Quantitative pull-down assay with purified recombinant His6-tagged CnA and GST-tagged Hoxb13 peptide or mutant (Mut) Hoxb13 peptides. Dig2–GST serves as positive control. h, i, Top, merged images of immunostaining for Hoxb13 (red, arrow, bottom) and sarcomeric α-actinin (green) showing the subcellular localization of Hoxb13 in P2 CnA-Tg (h) or P7 Rcan1-Tg (i) hearts. DAPI (blue) indicates nucleus. j, Western blot analysis of phosphorylated Hoxb13 proteins during regenerative stages. k, l, Western blot analysis of phosphorylated Hoxb13S204 in CnA-Tg and Rcan1-Tg P2 hearts (k) and densitometry quantification (l). Rcan1.4 (26 kDa) indicates CnA-Tg heart; truncated Rcan1.4 (13 kDa) indicates Rcan1-Tg heart. GAPDH serves as loading controls (a, j, k). m, Schematic showing generation of primary cardiac DKO fibroblasts for assessing S204 phosphorylation-dependent nuclear localization of Hoxb13. n, Representative images of subcellular localization of Hoxb13–GFP mutants in cardiac DKO fibroblasts. Data are mean ± s.e.m.; unpaired two-sided t-test. Data in a–e, g, j, n were independently repeated three to six times with similar results. *P < 0.05, **P < 0.01. For gel source data, see Supplementary Fig. 1. For sample numbers, see Methods. Scale bars, 50 μm (e, h, i, n). See also Extended Data Fig. 8.
Hoxb13 immunostaining in hearts with cardiomyocyte-specific expression of either a constitutively active transgene of Ppp3ca (the gene encoding CnA) (CnA-Tg), or Rcan1, an endogenous inhibitor of calcineurin (Rcan1-Tg), showed that Hoxb13 was prematurely localized to nuclei in CnA-Tg hearts at P2 (Fig.3h). In contrast to wild-type hearts, which show a strong nuclear Hoxb13 signal at P7, Rcan1-Tg hearts at P7 showed only a faint Hoxb13 signal (Fig.3i). We hypothesized that calcineurin-mediated dephosphorylation controls nuclear localization of Meis1 and Hoxb13. We generated custom polyclonal phospho-specific antibodies against four predicted sites in Hoxb13 and six sites in Meis1 (Extended Fig.8g, h) and used them to compare the Meis1 or Hoxb13 phosphorylation status in postnatal hearts. We found that only the antibody directed against Hoxb13 S204 detected a band that was more intense at P1 than at P7 (Fig.3j, Extended Fig.8i, j). None of the Meis1 phospho antibodies showed evidence of differential phosphorylation (Extended Fig.8k). Of note, Hoxb13 S204 phosphorylation was decreased in the CnA-Tg P2 hearts (a timepoint when S204-phosphorylation can be detected in wild-type). Conversely, the Rcan1-Tg P2 hearts showed an increase in S204 phosphorylation (Fig. 3k, l). Thus, this previously unidentified phosphosite at S204 in Hoxb13 appears to be dephosphorylated by calcineurin in the postnatal heart at the time of cardiomyocyte cycle arrest.
To better understand the effect of S204 phosphorylation on Hoxb13, we constructed plasmids expressing wild-type and mutant GFP-Hoxb13 fusion proteins and examined their spatial expression in primary cultured DKO cardiac fibroblasts (Fig. 3m, Extended Fig.8l). Transient transfection with wild-type Hoxb13 or dephosphorylation-mimetic Hoxb13(S204A) constructs resulted in nuclear localization of GFP-Hoxb13, whereas the phospho-mimetic Hoxb13 constructs (Hoxb13(S204D) and Hoxb13(S204E)) completely eliminated Hoxb13-GFP nuclear localization (Fig. 3n). These results suggest that calcineurin-dependent dephosphorylation of Hoxb13 at S204 promotes nuclear translocation of Hoxb13 and facilitates cell cycle arrest.
Calcineurin regulates cardiomyocyte proliferation
Previous reports indicate that CnA overexpression in cardiomyocytes results in profound pathological hypertrophy19. Consistently, we observed an increase in heart weight/body weight and cardiomyocyte CSA as early as P2 and subsequently, at P21 (Fig.4a–b, Extended Fig.9a–d). In addition, CnA-Tg hearts contained significantly fewer PH3+ cardiomyocytes at P2 (Fig.4c, Extended Fig.9e). Conversely, in Rcan1-Tg hearts, there were more PH3+ cardiomyocytes compared to wild type at P21 (Fig.4d, Extended Fig.9f). This suggests that forced expression of activated calcineurin causes a premature switch from hyperplasic to hypertrophic growth, whereas inhibition of calcineurin prolongs the window of cardiomyocyte proliferation.
Figure 4: Increased calcineurin activity suppresses postnatal cardiomyocyte proliferation.
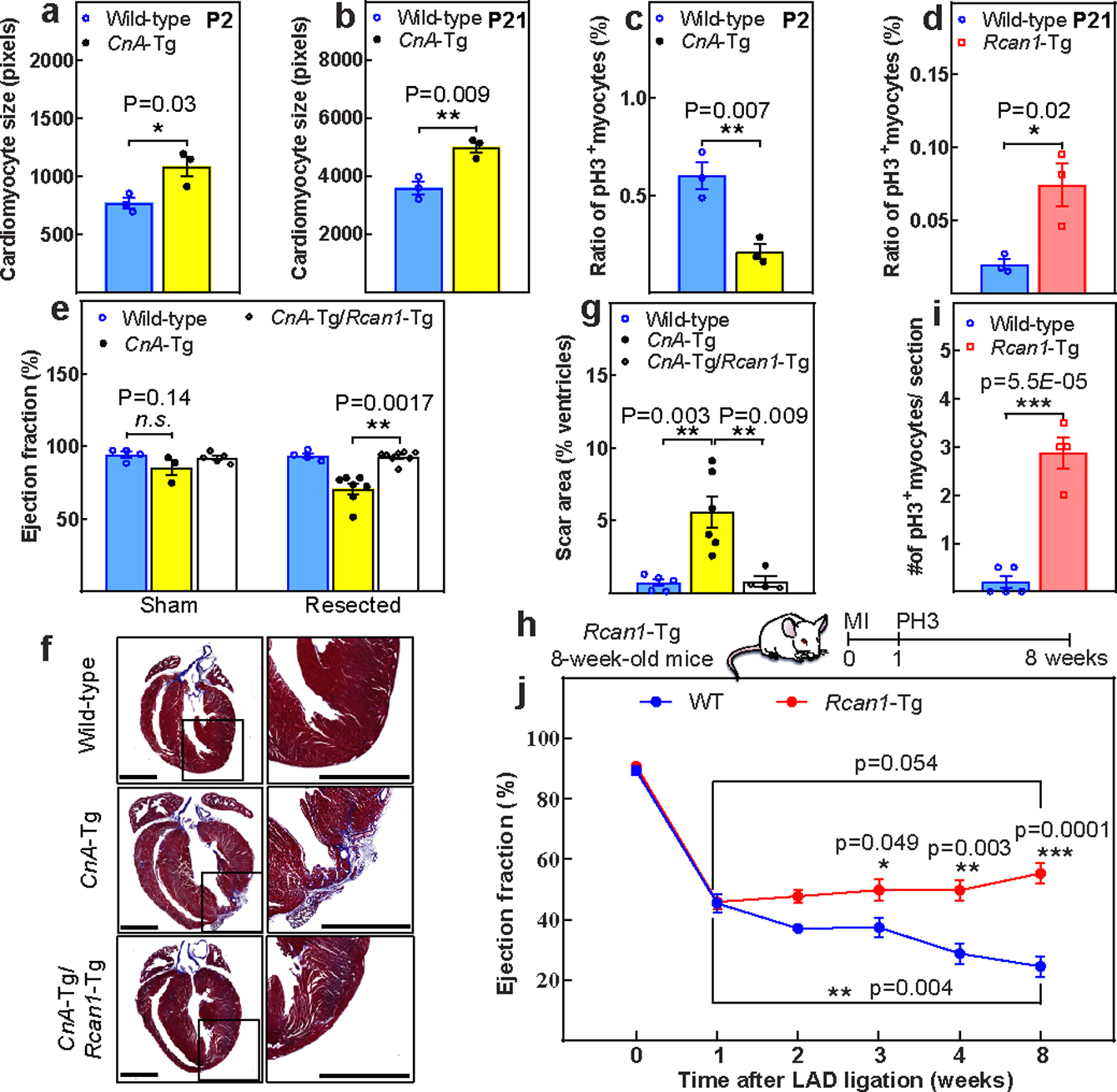
a, b, CSA quantification in wild-type and CnA-Tg in P2 (a) and P21 hearts (b). c, d, Percentage of mitotic cardiomyocytes in CnA-Tg P2 (c) or Rcan1-Tg P21 (d) hearts. e, Echocardiographic measurements of cardiac function 21 days after resection. f, g, Representative Masson’s trichrome staining (f; scale bars, 1 mm) and scar area quantification (g) of apical resected hearts 21 days after resection. h, Schematic of MI model in Rcan1-Tg mice. i, Quantification of PH3+ cardiomyocytes in wild-type and Rcan1-Tg heart sections one week after MI induction. j, Serial echocardiographic measurement of wild-type and Rcan1-Tg MI mice. Data are mean ± s.e.m.; unpaired two-sided t-test. *P < 0.05, **P < 0.01, ***P < 0.001. For sample numbers, see Methods. See also Extended Data Fig. 9.
Next, we examined the regenerative capacity of CnA-Tg hearts following apical resection at P1 (Extended Fig.9g). Twenty-one days after resection, CnA-Tg hearts displayed a significantly reduced LVEF (Fig.4e) with prominent scars (Fig.4f, g) compared with controls. Moreover, similar to wild-type hearts, CnA-Tg/Rcan1-Tg double-transgenic hearts recovered fully by P21. Consistently, Rcan1 overexpression-inhibited CnA activity enhanced cardiomyocyte proliferation at one week after MI (Fig4.h–i), and improved LVEF over two months compared with wild-type mice. Although LVEF of the Rcan1-Tg mice showed a trend towards improvement compared to post-MI values, this did not reach statistical significance (Fig4.j).
In this study, we identify Hoxb13 as a Meis1 cofactor in postnatal cardiomyocytes. Whereas Hoxb13 deletion resulted in transient induction of cardiomyocyte proliferation, loss of both Hoxb13 and Meis1 resulted in blunting of both physiological and pathological hypertrophic growth, and a robust and sustained induction of cardiomyocyte mitosis. The morphological changes in some DKO cardiomyocytes are reminiscent of neonatal cardiomyocytes, with sarcomere disassembly and localization of sarcomeres to the cellular cortex. We also report robust improvement in LVEF when Meis1 and Hoxb13 are conditionally deleted following injury. This is supported by results of ChIP-seq, which demonstrate that the targets of Meis1 and Hoxb13 in the mouse heart include a diverse set of pathways involved in cardiomyocyte proliferation, differentiation and sarcomere organization, in addition to genes involved in regulation of cardiomyocyte hypertrophy.
We further show how suppression of hyperplastic growth by Hoxb13-Meis1 is integrated with activation of hypertrophic growth through calcineurin-dependent signalling. The mammalian heart undergoes substantial circulatory remodelling after birth, resulting in structural and pressure changes that result in a significant and persistent increase in pressure load on the left ventricle20. This increase in load coincides with cell cycle withdrawal of cardiomyocytes, and is associated with activation of a number of pathways that mediate the postnatal hypertrophic growth. In particular, the phosphatase calcineurin is thought to have a critical role in induction of cardiomyocyte hypertrophy in response to load; however, whether it simultaneously has a role in suppressing cell cycle progression—thereby coordinating the switch from hyperplastic to hypertrophic cardiac growth—is not known.
Calcineurin is a Ca2+/calmodulin-activated phosphatase which has been extensively studied for its role in a wide array of biological functions, including cardiac hypertrophy19,21–24. Calcineurin-dependent hypertrophic growth occurs when there is a sufficient rise in diastolic Ca2+ levels, can occur on a broad array of substrates and mediates diverse signalling cascades25. Relative to cardiac hypertrophy, many studies have focused on the ability of calcineurin to activate transcription via the nuclear factor of activated T cell (NFAT) family of transcription factors19,26,27. Although most studies have focused on the role of calcineurin in pathological hypertrophic remodelling, there is substantial evidence that it is involved in normal postnatal cardiac growth as well as physiological hypertrophy28,29. Our results demonstrating that Meis1 and Hoxb13 are involved in induction of both pathological and physiological cardiomyocyte hypertrophy as well as cardiomyocyte cell cycle downstream of calcineurin highlight the complex role that calcineurin plays in cardiomyocyte maturation in the postnatal heart.
We provide several lines of evidence that support the role of calcineurin in postnatal cell cycle arrest of cardiomyocytes through its regulation of Hoxb13. First, we show that calcineurin associates with Hoxb13 in postnatal cardiomyocytes at the time of cell cycle arrest. Second, we identify a calcineurin PxIxIT site in Hoxb13, which mediates binding of Hoxb13 to calcineurin. Third, we show that calcineurin gain and loss of function in cardiomyocytes regulates nuclear localization of Hoxb13. Fourth, we show that Hoxb13 S204, which is dephosphorylated at P7 compared with P1 in the postnatal heart, is differentially dephosphorylated by calcineurin gain and loss of function, and regulates nuclear localization of Hoxb13. Finally, we show that gain and loss of function of calcineurin regulates postnatal cardiomyocyte proliferation and heart regeneration.
In summary, our results indicate that flux through calcineurin signalling provides a coordinating link between the postnatal increase in cardiac load and suppression of the regenerative capacity of the mammalian heart, and identifies Hoxb13 as an important regulator of cardiomyocyte differentiation and proliferation. The Meis1 Hoxb13 DKO cardiomyocyte mitosis phenotype outlined in this study indicates that reversion to a more immature state of cardiomyocytes is feasible, and suggests that this pathway might be a viable target for therapeutic interventions. Overall, these findings support the notion that adaptation to environmental cues of the postnatal life is an important determinant of postnatal cardiomyocyte cell cycle arrest.
METHODS
Mice
All animal experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Texas Southwestern Medical Center (UTSW), and were in compliance with the relevant ethical regulations regarding animal research. Mice were housed in a 12-h light/dark cycle in a temperature-controlled room in the Animal Research Center of the UTSW, with free access to water and food. In each experiment, the age of the mouse is indicated in the text and in the figure. Littermate controls were used whenever possible. Both male and female mice were used on age-matched and gender-matched mice. No statistical analysis was performed to predetermine sample size. All surgeries, echocardiographic, and MRI studies were carried out blinded to the genotype of the mice during the experiments and outcome assessments. CD1 mice (Charles River Laboratories) were used for wild-type studies. The Myh6Cre (αMHCCre); Myh6Cre-ERT2 (αMHCmERCremER); Myh6Cre/activated Calcineurin (CnA-Tg); Myh6cre/Rcan1 (Rcan1-Tg) transgenic lines, cardiac-specific inducible deletion of Meis1fl/fl (Meis1-iKO) have been described previously9,29. The Hoxb13 floxed (Hoxb13fl/fl) mice were generated by The KOMP Repository (Hoxb13tm1a(KOMP)Wtsi https://www.komp.org). Cardiac-specific deletion of Hoxb13 (Hoxb13-KO or Hoxb13-iKO) was achieved by crossing Hoxb13fl/fl mice with αMHCcre or αMHCmERcremER mice respectively. The Myh6cre/CnA-Tg/Rcan1-Tg (CnA-Tg/Rcan1-Tg); Myh6cre/Meis1fl/fl Hoxb13fl/fl (DKO) and Myh6mERcremER/Meis1fl/fl Hoxb13fl/fl mice were generated by cross-breeding. Combinations of littermate floxed (fl/fl) were used as controls for conditional deletion studies. Combinations of littermate fl/fl and age- and gender-matched αMHCmERcremER were used as controls for inducible deletion studies. MADM-ML-11GT/TG mice (JAX #030578) were obtained from Jackson Laboratory. Lineage analysis of Hoxb13-KO or DKO cardiomyocytes was performed by crossing Hoxb13-iKO or Meis1/Hoxb13-iKO with MADM TG/TG and MADM GT/GT mice to obtain Myh6mERcremER/Hoxb13-MADM GT/TG or Meis1-Hoxb13-MADM GT/TG mice. Age- and gender-matched Myh6mERcremER/ MADM GT/TG mice were used as control for MADM study.
Drug administration
Tamoxifen (Sigma) was prepared by dissolving in sesame oil (Sigma) to a concentration of 10 mg ml−1. Standard tamoxifen induction was done by injecting 3 times every other day by intraperitoneal (IP) injection at a dosage of 100 mg kg−1 per day per mouse. For the MADM clonal analysis, 14 consecutive injections of tamoxifen were administered and mice were euthanized at two weeks after the last injection. To monitor proliferation of endogenous cardiomyocytes, mice were administered daily with 0.25 mg ml−1 BrdU (Sigma) in the drinking water for up to 3 weeks after the first dose of tamoxifen injection. The BrdU mixture was prepared freshly and changed daily. Mice were euthanized at the same time of day to limit circadian variability.
Mouse model of MI
Induction of adult anterior wall MI was performed as previously described30. In brief, ten-week-old mice were subjected to MI by ligation of the proximal aspect of the LAD coronary artery. Mice were anesthetized in an airtight chamber using 4% isoflurane, endotracheally intubated, and ventilated using a volume control ventilator with 100% O2, supplemented with 2% vaporized isoflurane (Harvard Apparatus). Following lateral thoracotomy and pericardiectomy, the LAD coronary artery was identified along the anterior wall of the left ventricle. Prolene sutures (6–0 non-absorbable) was used to ligate the LAD. Proper occlusion of the LAD artery was noted by three criteria: immediate blanching of the left ventricle anterior wall myocardium below the ligature, echocardiographic assessment 7 days post-surgery showing an EF in the range of 35–65%, and histological analysis. Vicryl sutures (6–0 absorbable) were used to close the thoracic cavity. Tamoxifen was administrated one week after MI. Mice were subjected to echocardiography at serial time points and then euthanized at 4 months after ligation. Hearts were collected and processed for histology and immunohistochemistry. Fibrotic scar size was measured using MIQuant31. Neonatal MI of P1 mice was performed as described elsewhere32. Prolene sutures (8–0 non-absorbable) was used to occlude the LAD. Hearts were collected 7 days after MI and processed for immunohistochemistry.
Cardiac apical resection
Apical resection of P1 mice was performed as described previously6. Sham procedures excluded apex amputation. Mice were subjected to echocardiography and then euthanized 21 days after resection. Dissected hearts were processed for histology. Fibrotic scar size was measured using ImageJ (National Institutes of Health).
Mouse model of TAC
To perform TAC, eight-week-old DKO mice and corresponding CTL littermate mice were anesthetized, restrained in a supine position, intubated and ventilated using a Harvard Apparatus respirator. Following dissection through the intercostal muscles, the aorta was identified and freed by blunt dissection. 7.0 silk suture was placed around the great vessel, tied around a blunt 27-gauge needle. Sham-operated mice and wild-type littermates were used as controls. The surgical incision was closed and the mice were recovered on a warmer until they were returned to their cages. Cardiac function was evaluated by echocardiography for up to three weeks after surgery. The mice were euthanized, postmortem and histological studies were performed as described below.
Voluntary wheel running
Eight-week-old DKO mice and corresponding control littermates were randomly assigned to housing in individual cages with or without a running wheel for a total of four weeks. The mice were acclimatized with the running wheel for a week before the experiment. Rotation recordings are made every 5 minute and integrated into the computer program. Echocardiography was performed to access left ventricular systolic function. Run distances were determined at the end of the study.
Transthoracic echocardiography
Assessment of in vivo heart function on conscious, non-sedated mice was performed using a Vevo2100 micro-ultrasound system, MS400C probe (VisualSonics) at baseline, 1 week after injury, 1 week after tamoxifen-induced gene deletion, and 4, 8, 12, 16 weeks after occlusion. Echocardiographic M-mode images were obtained from a parasternal short-axis view at the level of the papillary muscles. Left ventricular internal diameters at end diastole (LVIDd) and end systole (LVIDs) were measured from M-mode recordings. Six representative contraction cycles were selected for analysis, and average indexes (LVIDd, LVIDs, fractional shortening) were calculated for each mouse. All echocardiography measurements were performed in a blinded manner.
Magnetic Resonance Imaging
The cardiac function of mice was evaluated by cardiac MRI using a 7T small animal MRI scanner (Bruker, Rheinstetten, German) equipped with a 40 mm quadrature RF coil (ExtendMR LLC, Milpitas, CA). Under anesthesia by inhalation of 1.5 – 3% isoflurane mixed in with medical-grade oxygen via nose-cone, the animals were placed supine on a mouse holder, with a pneumatic respiratory sensor and ECG electrodes for cardiac sensing, head first with the heart centered with respect to the center of the RF coil. The mice’s chests were shaved and conducting hydrogels were applied to optimize ECG contact between electrodes and mouse. All MRI acquisitions were gated using both cardiac and respiratory triggering. The bore temperature was kept at 33 ± 2 °C to assure adequate and constant heart rate. Two-dimensional (2D) gradient echo images on three orthogonal planes (transverse, coronal and sagittal) were acquired to determine the long-axis of the heart in each mouse. Axial images perpendicular to the long axis of the heart was chosen for Cine-imaging. Cine images at 12 phases per cardiac cycle was obtained with an echo time of 1.5 ms, repetition time = ECG R-R interval / 12 (typically, RR ≈ 120–150 ms, which means TR ≈ 10–12.5 ms), flip angle of 45°, and NEX= 6 (Number of Average). Each scan consisted of five to nine slices from apex to LV outflow with 1 mm thickness, no-gap between slices, a matrix size of 128 × 128, and a field of view of 40 × 40 mm. Endocardial borders were manually traced for calculation of left ventricular end systolic and end diastolic volumes (LVESV and LVEDV) using NIH ImageJ (v1.47j) software. Total LV volumes were calculated as the sum of all slice volumes. Stroke volume was calculated by the equation, LVEDV - LVESV. Cardiac output was calculated by the equation, stroke volume × heart rate. The left ventricular ejection fraction (LVEF) was calculated by the equation, (LVEDV - LVESV) / LVEDV × 100%. Investigators performing MRI acquisition and analysis were blinded to the assignment of mice group.
Histology
Hearts were harvested and fixed in 4% paraformaldehyde (PFA)/PBS solution overnight at 4°C and then processed for either paraffin or cryo embedding. Haematoxylin and eosin and Masson’s trichrome staining were performed according to standard procedures at UTSW core histology facility on paraffin sections.
Immunofluorescence staining
Immunostaining was performed according to previous descriptions30,33. In brief, heart cryosections were equilibrated with antigen retrieval buffer with 1 mM EDTA or epitope retrieval buffer (IHC World). Samples were permeabilized and blocked with 0.3% Triton X-100 and 10% serum from the host animal of secondary antibodies in PBS for 20 min at room temperature. Then, samples were incubated overnight at 4°C with the primary antibodies. After three times washed with PBS, samples were incubated for 1 hour at room temperature with corresponding fluorescence secondary antibodies conjugated to Alexa Fluor 488 or 555 (Invitrogen) at 1:400. The slides were mounted in Vectashield Antifade Mounting Medium (Vector Laboratories). Primary antibodies used: phospho histone H3 Ser10 (EMD Millipore, 06-570; 1:100), aurora B kinase (Sigma, A5102; 1:25) to analyse cell cycle re-entry; troponin T, cardiac isoform Ab-1, clone 13-11 (Thermo Scientific, MS-295-P1; 1:200), sarcomeric α-actinin (Abcam, ab68167; 1:200) to identify cardiomyocytes. Other antibodies used in the studies included Meis1/2(C-17) (Santa Cruz, sc-10599; 1:25), Hoxb13(F-9) (Santa Cruz, sc-28333; 1:10), Hoxa10(N-20) (Santa Cruz, sc-17158; 1:50), Hoxa11 (Abcam, ab28699; 1:50), Hoxd12(G-13) (Santa Cruz, sc-82922; 1:50), BrdU (Abcam, ab92837; 1:400), PCM1 (Sigma-Aldrich, HPA023370; 1:1,000). DAPI or Hoechst was used for nuclear staining. Images were obtained using a Nikon Eclipse Ni or Nikon A1 laser scanning confocal microscopes.
WGA staining and cardiomyocyte size quantification
WGA staining and quantification was performed as previously described30. In brief, the slides were incubated with WGA conjugated to Alexa Fluor 488 (50 mg mL−1, Life Technologies) for 1 h at room temperature following washing with PBS. To quantify the cross-sectional cell size, three to five independent hearts per group with three different views and positions, each from left and right ventricles, and septum were captured at 40X magnification. ImageJ was used to quantify the size of cardiomyocytes that were round and contained a nucleus. At least 500 cells per sample were quantified.
Cardiomyocyte isolation
Adult hearts were freshly collected and fixed in 4% PFA at 4°C overnight. The hearts were minced to smaller pieces and subsequently incubated with collagenase D (2.4 mg mL−1, Roche) and B (1.8 mg mL−1, Roche) for 12 hours at 37°C using an end-over-end shaker. The supernatants were collected via 160-μm nylon mesh filter, and the procedure was repeated until no more cardiomyocytes were dissociated from the tissue. The isolated cardiomyocytes were co-stained with connexin 43 (IHCWorld, IW-PA1026, 1:100) and DAPI (Sigma, D9542, 1:10000) for further quantification. For nucleation counts, at least 300 cardiomyocytes per sample were quantified.
Generation of phospho-specific Meis1 and Hoxb13 antibodies
Prediction servers (https://www.rcsb.org/pdb/home/hom.do and http://www.cbs.dtu.dk/services/NetPhos/) were used to predict phosphorylation sites in Meis1 and Hoxb13. We found that phosphorylation may occur at 9 phosphosites in Meis1 (S21, T22, Y24, S196, S222, T223, T260, T278) and 6 sites in Hoxb13 (T8, S204, S252, S256, K272, K279). pMeis1 and pHoxb13 (4 μg/ml, polyclonal) antibodies were raised (Abmart). Antibodies against 4 sites in Hoxb13 and 6 sites in Meis1 were successfully produced. The sequences of peptides are indicated in Extended Data Table 1.
Western blotting
Ventricles were collected and lysed in RIPA buffer with the addition of Complete protease inhibitor cocktail (Roche). Protein concentration was quantified using Pierce BCA protein assay kit (Pierce Biotechnology), with three biological replicates. After separation via SDS-PAGE, proteins were transferred to nitrocellulose membranes (Bio-Rad), blocked in 5% skim milk/TBS and incubated with appropriate primary antibodies: Hoxb13 (Santa Cruz, sc-28333; 1:250), Hoxb13X (Santa Cruz, sc-28333X; 1:1000), Meis1/2(C-17) (Santa Cruz, sc-10599; 1:1,500), Meis1/2(C-17)X (Santa Cruz, sc-10599X; 1:2,000), calcineurin A (Abcam, ab71149; 1:2,000), Rcan1 (C-terminal) (Sigma, D6694; 1:1,500), GAPDH (EMD Milipore, AB2302; 1:5,000), Hoxa10(N-20) (Santa Cruz, sc-17158; 1:1,000), Hoxa11(5A3) (Abcam, ab28699; 1:1,000), Hoxd12(G-13) (Santa Cruz, sc-82922,;1:1,000), phospho-Meis1 (Abmart; 1:500), phospho-Hoxb13 (Abmart; 1:500), p21 Waf1/Cip1 (12D1) (Cell Signaling, 2947; 1:1,000), p15/16 (Santa Cruz, sc-377412; 1:1,000), α-tubulin (Milipore, 04-1117; 1:2,000). Horseradish peroxidase (HRP)-conjugated peroxidase anti-mouse, anti-rabbit, anti-chicken, or anti-goat antibodies (Jackson ImmunoResearch, 115-035-166, 111-035-144, 703-035-155, 705-035-147, 1:25000–1:50000) were used as secondary antibodies. The membranes were explored using Licor Odyssey Fc system and quantified by Image Studio Lite Ver.5.2 software.
Site-directed mutagenesis of serine 204 of Hoxb13
Phospho-mimetic (S204A) and non-phosphorylated (S204D, S204E) Hoxb13 mutants were constructed from pCMV-murine Hoxb13-EGFP using GeneArt Site-Directed Mutagenesis Plus Kit (Life Technologies, cat# A14604). The point mutations were produced by overlap extension PCR with primers bearing mutations as follows:
Hoxb13_S204A_F: GCGTTTGCAGAGCCCGCCGTCCAGCACCCTCCT
Hoxb13_S204A_R: AGGAGGGTGCTGGACGGCGGGCTCTGCAAACGC
Hoxb13_S204D_F: GCGTTTGCAGAGCCCGACGTCCAGCACCCTCCT
Hoxb13_S204D_R: AGGAGGGTGCTGGACGTCGGGCTCTGCAAACGC
Hoxb13_S204E_F: GCGTTTGCAGAGCCCGAGGTCCAGCACCCTCCT
Hoxb13_S204E_R: AGGAGGGTGCTGGACCTCGGGCTCTGCAAACGC
All mutations were verified by DNA sequencing.
CRE lentiviral production
Host 293T cells were plated on 10-cm plates until 70% confluence, at which point they were transfected with 5 μg of Cre-ires-puro vector (Addgene Plasmid no. 30205) along with 3 μg pAX2 and 2 μg VSVG packaging plasmids using Lipofectamine 3000 Reagent (Invitrogen). Fourty-eight hours later, the supernatant was collected and spun at 300g for 5 min to remove cells. The resulting lentivirus-containing supernatant was filtered through a 0.45-μm polyethersulfone membrane filter (Thermo Scientific, cat no. 725–2545), and stored at −80 °C or used immediately.
Generation of DKO cardiac fibroblasts
Adult murine cardiac fibroblasts (CF) were isolated from eight-week-old male Meis1fl/fl Hoxb13fl/fl hearts. Hearts were collected and rinsed in Gerard isotonic solution. Subsequently, hearts were minced and submitted to digestion with Gerard solution containing 1 mg/mL type II collagenase (Sigma-Aldrich) at 37°C for 1 hours with gentle shaking. Repeated digestions and centrifugations were performed, and CF were obtained by plating the cells in 10% FBS, DMEM-F12 (ThermoFisher Scientific), and 20 ng/mL FGF (Merck Millipore). Fresh medium containing FGF was added every 2 days. After reaching 80–100% confluence (3 weeks), CFs were passaged to a 6-well plate to perform further experiments. For Cre lentivirus infection, in one well of the 6-well plate, when cells reached about 60% confluence, the media was removed and replaced with One millilitre Optimem supplemented with 16 μg ml−1 polybrene. One millilitre of Cre lentivirus was added dropwise to the plate. Lentivirus without Cre was used as control. After 48 h, puromyocin (ThermoFisher Scientific) selection with media containing 1 μg ml−1 puromycin was added for up to two weeks to select for lentiviral-infected cells.
Cell culture, transfection, and immunostaining
Culture cells were maintained at 37 °C in a humidified incubator containing 5% CO2. 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% (v/v) fetal bovine serum (FBS; Hyclone) and 1% penicillin/streptomycin. Cardiac fibroblasts (CFs) were cultured in 10% FBS, DMEM-F12. CFs were grown on a Poly-L-lysin-coated covers slips 24 h before transfection. When the cells were 60% confluent, transient transfection was performed with Lipofectamine 3000 Reagent (ThermoFisher Scientific) according to the manufacturer’s protocol. After 48 h, CFs were fixed and permeabilized with ice-cold acetone for 15 min at −20°C, then blocked with PBS containing 0.03% Triton X-100 and 3% bovine serum albumin (BSA) for 20 min at room temperature. CFs were then washed three times with PBS and stained for 1 h at room temperature with Rhodamine-Phalloidin (Cytoskeleton; 1:400). The cells were mounted by Prolong gold containing DAPI (Invitrogen) and viewed under a Nikon fluorescence microscope.
Coimmunoprecipitation
Mouse hearts at P1 and P7 (n=3 for each) were collected and lysed in immunoprecipitation lysis buffer: 2.5mM Tris, pH7.4, 150 mM NaCl, 1 mM EDTA, 1% NP40, 5% glycerol. Proteinase inhibitor cocktail and phosphatase inhibitor were included at all times. 5 μg primary antibodies were used for binding with 1 mg digested heart samples with gentle rocking at 4°C overnight. Protein A agarose beads (Calbiochem) were added to the mixture and incubation was continued with gentle rocking for 4 h at 4°C. After centrifugation for 30 seconds at 4°C, the pellets were washed three times with 1 ml immunoprecipitation lysis buffer. Final pellets were resuspended with 25 μl 2XSDS sample buffer. Negative control IgG is used in place of a primary antibodies to evaluate nonspecific binding. The samples were then analysed by western blotting. The same procedure was done using DKO heart lysate as negative control for protein binding.
Calcineurin binding assay
N-terminally His6-tagged human calcineurin A (α isoform, truncated at residue 400), was expressed in tandem with the calcineurin B subunit in E.coli BL21 (DE3) cells (Invitrogen) and affinity purified using Ni-NTA-agarose with methods identical to the purification of His-tagged yeast calcineurin34. A 16-amino-acid peptide corresponding to HoxB13 residues 255–270, or a mutant version with residues 260, 262, 264 and 265 changed to alanine, or a positive control (Saccharomyces cerevisiae Dig2 residues 95–116 containing a PxIxIT site) were fused to GST in vector pGX4T-3. Cell lysates from bacteria expressing the were obtained as in a previous study34. Bacterial cell lysate (50–100 μg) was used to test co-purification with 2μg of purified His6-calcineurin as described34. Co-purifying proteins were observed by western blotting with mouse GST (Bio Legend) and anti-His monoclonal primary antibodies and an IRDye680RD goat anti-mouse secondary antibody (LiCor) and imaged with the Li-Cor Odyssey imaging system.
Proximity ligation assay
Proximity ligation assay (Duolink, no. DUO92101, Sigma) was performed according to the manufacturer’s instructions. Cryo-sections at P7 were used for all studies. DKO hearts were used as negative control for PLA proteins. Antigen retrieval and permeabilization steps were the same as per standard immunostaining procedure.
TUNEL assay
Cryo-sections underwent immunofluorescent staining for cardiac troponin T as abovementioned. Following incubation with corresponding secondary antibody conjugated to Alexa Fluor 555 (Invitrogen), TUNEL staining was performed according to manufacturer’s guideline (In-Situ Cell Death Detection Kit, Fluorescein, Roche). All staining was performed on 3 hearts per group, with 3 sections per heart.
ChIP-seq
Ventricles from three P15 mouse hearts were used as biological triplicates for the ChIP-seq experiments. Chromatin immunoprecipitation was performed as previously described35,36, using the following antibodies: Meis1/2 (Santa Cruz, sc-10599X; 1:200) or Hoxb13 (GeneTex, GTX129245; 1:100). ChIP-Seq libraries were generated using NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, E7645L) according to manufacturer’s protocol. Final ChIP-Seq libraries were pooled and sequenced on an Illumina Nextseq 500 system using the 75bp high output sequencing kit for single-end sequencing. Reads were quality trimmed and mapped to mouse genome (mm10) by bowtie2 (v.2.2.3)37 with parameter “--sensitive”. Uniquely mapped reads were filtered by removing alignments with mapping quality less than 10, and removing duplicate reads identified by Picard MarkDuplicates (v1.127) (http://broadinstitute.github.io/picard). Enriched regions (peaks) were identified using the IDR pipeline38 and spp. package39. Peak regions were annotated by HOMER40. Genome-wide core motifs were found for Meis1 and Hoxb13 using HOMER (scanMotifGenomeWide.pl). Input samples were used as reference controls for background correction. Genes with peaks in their promoters and also including the core motif were used to generate the 3 categories: Meis1, Hoxb13, and Meis1-Hoxb13 (Vene diagram). Gene ontology analysis for ChIP-Seq peak target genes was performed using Metascape41.
Statistics and Reproducibility
Differences between groups were examined for statistical significance using unpaired two-sided Student’s t-test. All bar graphs represent mean ± s.e.m. Statistical significance was assessed as *P < 0.05, **P < 0.01 and *** P < 0.001. P values are shown in graphs. All ChIP–seq samples were derived from n = 3 biologically independent samples. The sample sizes were as follows: Fig. 1f, n = 11 mice per Hoxb13fl/fl group, n = 9 mice per Hoxb13-KO group; Fig. 1g, h, n = 3 biologically independent samples per group; Fig. 1i, n = 7 mice per MHC MCM group, n = 10 mice per Hoxb13-iKO group; Fig. 1j n = 4 biologically independent samples per control group (3 MHC MCM, 1 Hoxb13fl/fl), n = 3 biologically independent samples per Hoxb13-iKO group; Fig. 1k, l, n = 5 biologically independent samples per control group (3 MHC MCM, 2 Hoxb13fl/fl), n = 4 biologically independent samples per Hoxb13-iKO group; Fig. 1m, n, n = 4 biologically independent samples per control group, n = 3 biologically independent samples per Hoxb13-iKO group; Fig. 1o, n = 3 biologically independent samples per group; Fig. 1p, n = 11 mice per Hoxb13fl/fl group, n = 12 mice per Hoxb13-KO group; Fig. 2b, n = 8 mice per control group (5 MHC MCM, 3 Meis1fl/fl Hoxb13fl/fl), n = 11 mice per DiKO group; Fig. 2c, n = 4 biologically independent samples per control group, n = 3 biologically independent samples per DiKO group; Fig. 2d, n = 3 biologically independent samples per group; Fig. 2e, n = 8 biologically independent samples per control group and n = 6 biologically independent samples per DiKO group at one week post-deletion, n = 3 biologically independent samples per group at two months post-deletion; Fig. 2f, n = 5 biologically independent samples per control group and n = 6 biologically independent samples per DiKO group at one week post-deletion, n = 3 biologically independent samples per control group and n = 4 biologically independent samples per DiKO group at two months post-deletion; Fig. 2g, n = 3 biologically independent samples per group; Fig. 2h, i, n = 4 biologically independent samples per group; Fig. 2j, n = 4 mice per control group, n = 3 mice per DiKO group; Fig. 2l, n = 6 biologically independent samples per group; Fig. 2n, n = 13 mice per control group (10 MHC MCM, 3 Meis1fl/fl Hoxb13fl/fl), n = 8 mice per DiKO group; Fig. 2p, q, n = 6 mice per group; Fig. 2r, n = 8 mice per control group, n = 5 mice per DiKO group; Fig. 3l, n = 6 biologically independent samples per group; Fig. 4a–d, n = 3 biologically independent samples per group; Fig. 4e, for sham cohort: n = 4 mice per wild-type group, n = 3 mice per CnA-Tg group, n = 5 per CnA-Tg/Rcan1-Tg group, for resected cohort: n = 4 mice per wild-type group, n = 7 mice per CnA-Tg group, n = 8 per CnA-Tg/Rcan1-Tg group; Fig. 4g, n = 5 biologically independent samples per wild-type group, n = 6 biologically independent samples per CnA-Tg group, n = 4 biologically independent samples per CnA-Tg/Rcan1-Tg group; Fig. 4i, n = 5 biologically independent samples per wild-type group, n = 4 biologically independent samples per Rcan1-Tg group; Fig. 4j, n = 5 mice per wild-type group, n = 8 mice per Rcan1-Tg group; Extended Data Fig. 1a, n = 9 mice per MHC MCM group, n = 6 mice per Meis1-iKO group; Extended Data Fig. 1j, n = 3 biologically independent experiments; Extended Data Fig. 1l, n = 11 mice per group; Extended Data Fig. 1n, n = 5 mice per control group (3 MHC MCM, 2 Hoxb13f/f), n = 6 mice per Hoxb13-iKO group; Extended Data Fig. 2c, n = 13 mice per control group, n = 11 mice per Hoxb13-KO group; Extended Data Fig. 2d, n = 10 mice per control group, n = 6 mice per Hoxb13-KO group; Extended Data Fig. 2e, n = 6 biologically independent samples per group; Extended Data Fig. 2g, for MI cohort: n = 14 mice per control group, n = 8 mice per Hoxb13-iKO group, for sham cohort: n = 10 mice per control group, n = 6 mice per Hoxb13-iKO group; Extended Data Fig. 2h, n = 8 biologically independent samples per group; Extended Data Fig. 2i, for MI cohort: n = 14 mice per control group, n = 8 mice per Hoxb13-iKO group, for sham cohort: n = 10 mice per control group, n = 10 mice per Hoxb13-iKO group; Extended Data Fig. 3a, n = 9 mice per Meis1fl/fl Hoxb13fl/fl group, n = 7 mice per DKO group; Extended Data Fig. 3b, n = 3 biologically independent samples per group; Extended Data Fig. 3c, n = 3 biologically independent samples per Meis1fl/fl Hoxb13fl/fl group, n = 4 biologically independent samples per DKO group; Extended Data Fig. 3d, n = 5 biologically independent samples per group; Extended Data Fig. 3e, n = 4 biologically independent samples per group; Extended Data Fig. 3f, n = 3 biologically independent samples per Meis1fl/fl Hoxb13fl/fl group, n = 4 biologically independent samples per DKO group; Extended Data Fig. 3g, n = 4 mice per Meis1fl/fl Hoxb13fl/fl group, n = 5 mice per DKO group; Extended Data Fig. 3h, n = 3 biologically independent samples per Meis1fl/fl Hoxb13fl/fl group, n = 5 biologically independent samples per DKO group; Extended Data Fig. 3i, n = 4 biologically independent samples per Meis1fl/fl Hoxb13fl/fl group, n = 4 biologically independent samples per DKO group; Extended Data Fig. 3j, n = 3 biologically independent samples per group; Extended Data Fig. 3k, n = 3 mice per Meis1fl/fl Hoxb13fl/fl group, n = 5 mice per DKO group; Extended Data Fig. 4b–f, for sham cohort: n = 6 mice per Meis1fl/fl Hoxb13fl/fl group, n = 10 mice per DKO group, for TAC cohort: n = 7 mice per Meis1fl/fl Hoxb13fl/fl group, n = 11 mice per DKO group; Extended Data Fig. 4h, for sham cohort: n = 5 biologically independent samples per Meis1fl/fl Hoxb13fl/fl group, n = 6 biologically independent samples per DKO group, for TAC cohort: n = 4 biologically independent samples per Meis1fl/fl Hoxb13fl/fl group, n = 6 biologically independent samples per DKO group; Extended Data Fig. 4j–n, for sham cohort: n = 6 mice per Meis1fl/fl Hoxb13fl/fl group, n = 10 mice per DKO group, for exercise cohort: n = 6 mice per group; Extended Data Fig. 4p, for sham cohort: n = 5 biologically independent samples per Meis1fl/fl Hoxb13fl/fl group, n = 6 biologically independent samples per DKO group, for exercise cohort: n = 6 biologically independent samples per group; Extended Data Fig. 5g, n = 5 biologically independent samples per control group, n = 3 biologically independent samples per DiKO; Extended Data Fig. 6c, n = 4 biologically independent samples per group; Extended Data Fig. 6d, n = 13 mice per control group (10 MHC MCM, 3 Meis1fl/fl Hoxb13fl/fl), n = 8 mice per DiKO group; Extended Data Fig. 6e, f, for sham cohort: n = 9 mice per group, for MI cohort: n = 13 mice per control group (10 MHC MCM, 3 Meis1fl/fl Hoxb13fl/fl), n = 8 mice per DiKO group; Extended Data Fig. 6i, j, n = 6 mice per group; Extended Data Fig. 6k, n = 9 mice per group; Extended Data Fig. 6l–q, n = 13 mice per control group (10 MHC MCM, 3 Meis1fl/fl Hoxb13fl/fl), n = 8 mice per DiKO group; Extended Data Fig. 8i, j, n = 3 biologically independent samples per group; Extended Data Fig. 9a, n = 18 mice per wild-type group, n = 11 mice per CnA-Tg group, n = 14 mice per Rcan1-Tg group, n = 6 per CnA-Tg/Rcan1-Tg group; Extended Data Fig. 9b, n = 7 mice per wild-type group, n = 6 mice per CnA-Tg group, n = 16 mice per Rcan1-Tg group, n = 4 per CnA-Tg/Rcan1-Tg group.
DATA AVAILABILITY
The data that support the findings of this study are available within the paper and its Supplementary Information file. Source Data for Figs. 1–4 and Extended Data Figs. 1–6, 8, 9 are provided with the online version of the paper. ChIP-seq data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE141769. Data or other materials are available from the corresponding author upon reasonable request.
Extended Data
Extended Data Fig. 1. Additional characterization of Meis1-iKO, Hoxb13-KO, and Hoxb13-iKO mice.
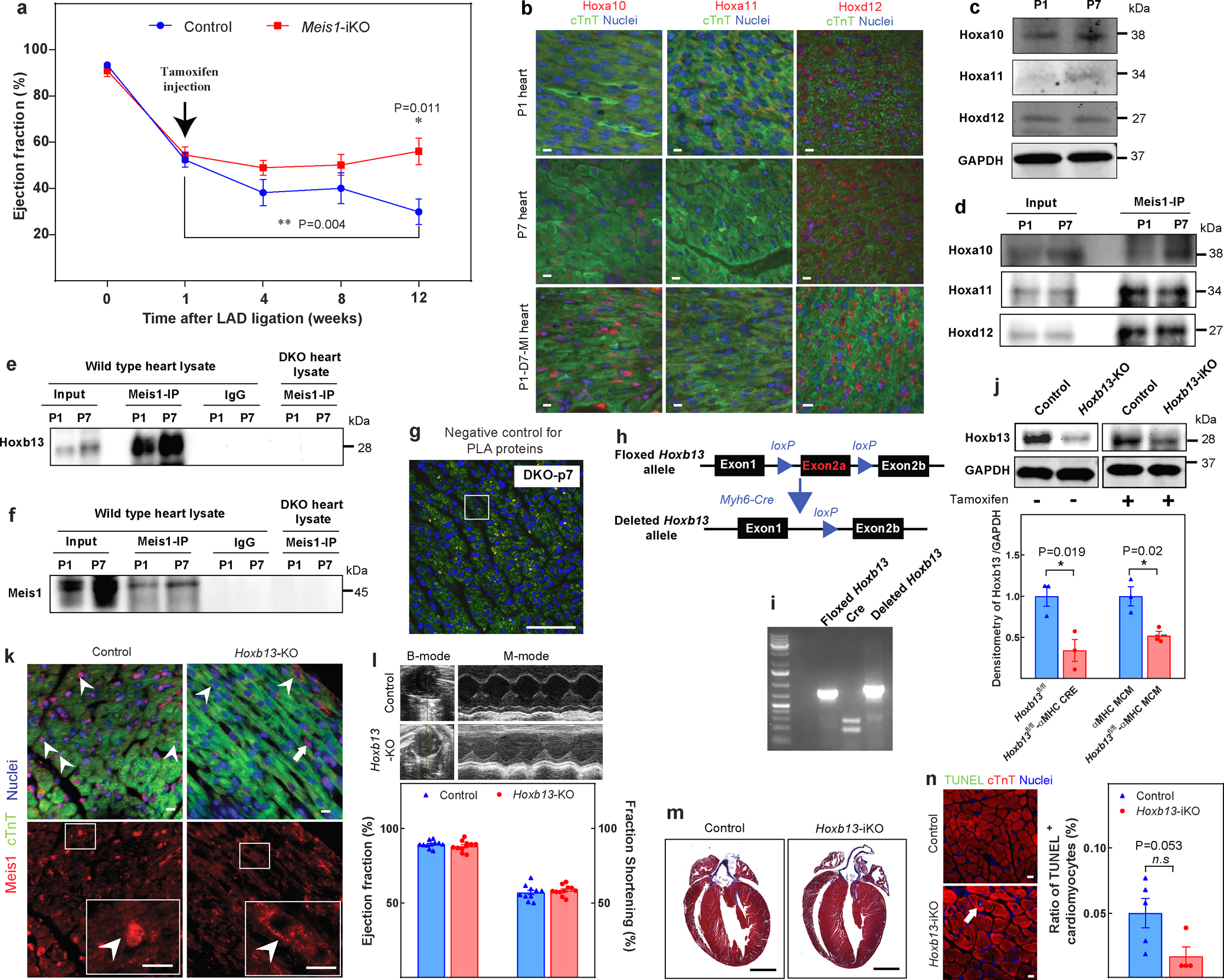
a, Echocardiographic assessment of LVEF in Meis1-iKO hearts at the indicated time points post-MI. b, Immunostaining of Meis1-interacting Hox proteins (red), cardiac troponin T (green), and nucleus (blue) at P1, P7 and 7 days after P1 MI. c, Western blot analysis of Meis1 and Hox proteins during regenerative stages. GAPDH serves as loading control. d, Coimmunoprecipitation of Meis1 from total heart extracts at P1 and P7. e, Coimmunoprecipitation from Fig. 1c, with negative controls (IgG and DKO heart). f, Coimmunoprecipitation from Fig. 1d, with negative controls (IgG and DKO heart). g, PLA of Meis1 and Hoxb13 in P7 DKO heart; serves as negative control for PLA assay in Fig. 1e. h, Schematic of targeting strategy for generation of cardiomyocyte-specific Hoxb13 mutants. Partial map of the Hoxb13 floxed allele (top), and of the knockout allele (bottom) following cardiomyocyte-specific Cre-mediated excision. Exons and loxP sites are indicated as rectangles and triangles, respectively. i, PCR analysis of heart DNA obtained by crossing Hoxb13loxP/loxP (female) mutants with MHC-cre (male) transgenic mice. Left lane shows Hoxb13loxP/loxP. Middle lane indicates cardiomyocyte-specific Cre. Right lane indicates that the deleted allele is present in cardiomyocyte DNA of this offspring. j, Top, western blot of protein extracts from hearts of constitutive (left) or inducible (right) Hoxb13 knockout and Hoxb13fl/fl littermate mice. GAPDH serves as loading control. Bottom, densitometry quantification of the above western blot. k, Control and Hoxb13-KO P14 hearts stained for Meis1 (red), cardiac troponin T (green), and nucleus (blue). Arrowheads indicated Meis1-expressing cardiomyocytes, and arrows indicate Meis1-expressing non-cardiomyocytes. l, Representative echocardiography, ejection fraction and fraction shortening in Hoxb13-KO mice P14 hearts. m, Representative images of Masson’s trichrome staining of control and Hoxb13-iKO hearts at 14 days post-deletion. n, Representative images of immunostaining for TUNEL (green), cardiac troponin T (red) and nucleus (blue) (left) and quantification (right) 14 days post-deletion. Arrow indicates TUNEL-positive cardiomyocytes. Data are mean ± s.e.m.; unpaired two-sided t-test. Data in b±f, g, i, k, m were independently repeated three times with similar results. *P < 0.05, **P < 0.01. For gel source data, see Supplementary Fig. 1. For n values, see Methods. Scale bars, 10 μm (b, k, n), 50 μm; (g), 1 mm (m).
Extended Data Fig. 2. Assessment of Hoxb13-KO and Hoxb13-iKO hearts post-MI.
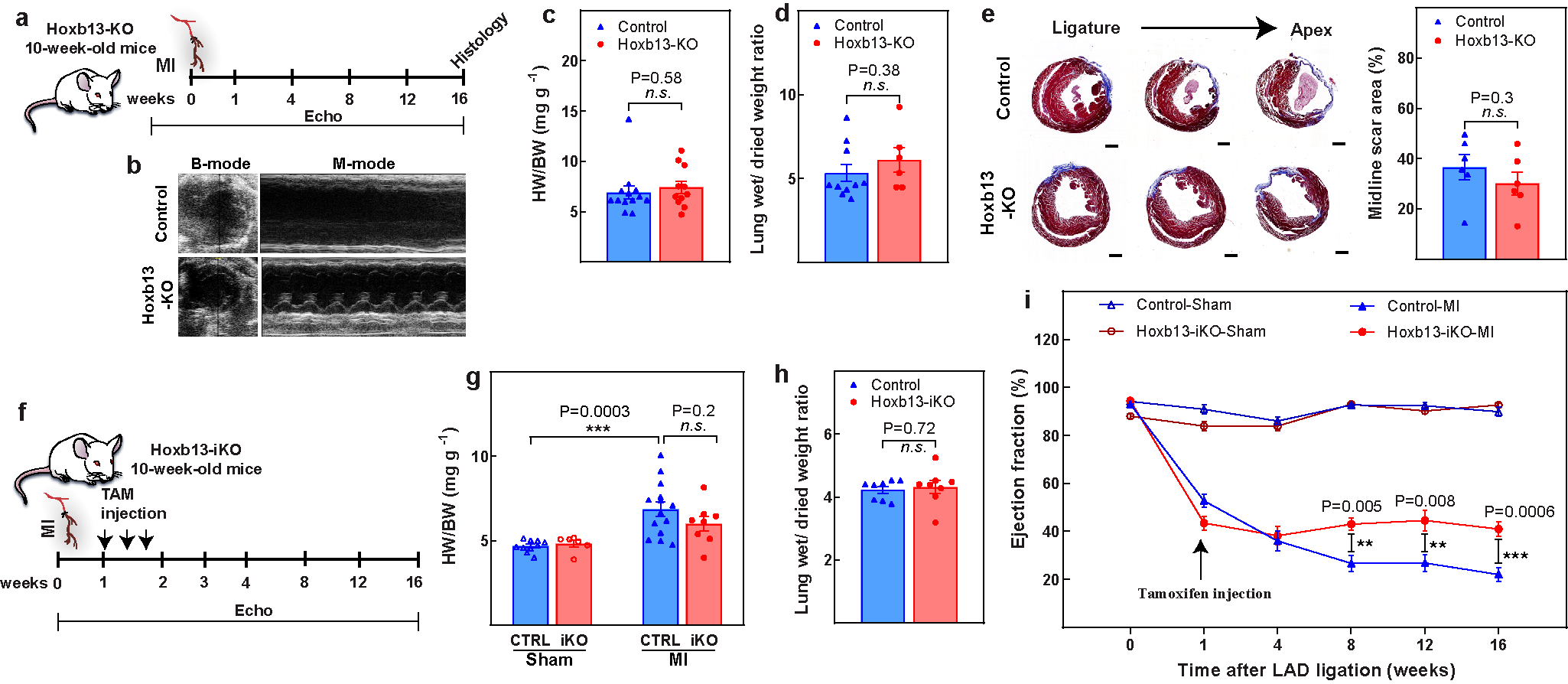
a, Schematic of MI model in Hoxb13-KO mice. b–d, Representative echocardiography (b), heart weight/body weight (c), and wet-to-dry lung weight ratio (d) in Hoxb13-KO mice 16 weeks after MI. e, Representative serial Masson’s trichrome staining in transversal sections (left) and quantification of fibrotic scars (right) in Hoxb13-KO MI hearts at 16 weeks after injury. f, Schematic of MI model in Hoxb13-iKO mice. g, h, Heart weight/body weight (g) and wet-to-dry lung weight ratio (h) in Hoxb13-iKO mice 16 weeks after MI. i, Serial echocardiography of Hoxb13-iKO MI and sham mice. Data are mean ± s.e.m.; unpaired two-sided t-test. **P < 0.01, ***P < 0.001; n.s., not significant. For n values, see Methods. Scale bar 1 mm (e).
Extended Data Fig. 3. Concurrent knockout of Meis1 and Hoxb13 preserves cardiomyocyte proliferative capacity.
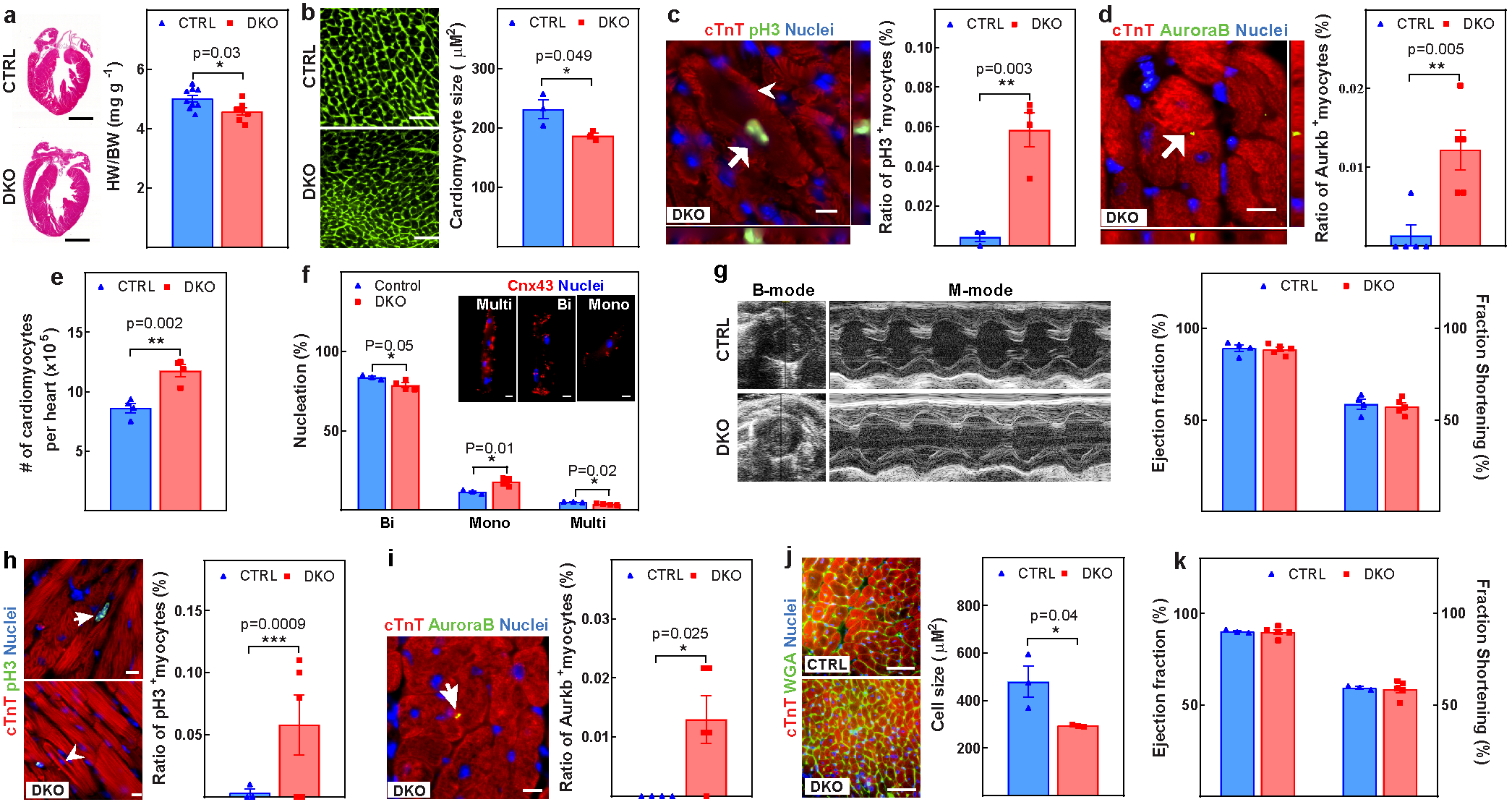
a–g, Assessment of control and DKO hearts at P28. a, Representative images of haematoxylin and eosin-stained heart sections and heart weight/body weight ratio. b, Representative images of WGA staining and cardiomyocyte cross-sectional area quantification. c, Representative images of immunostaining for PH3 (green), cardiac troponin T (red) and nucleus (blue) (left) and the percentage (right) of mitotic cardiomyocytes (arrow). Arrowhead indicates sarcomere disassembly. d, Representative images of immunostaining for aurora B kinase (green), cardiac troponin T (red) and nucleus (blue) (left) showing the percentage (right) of cardiomyocytes undergoing cytokinesis (arrow). e, f, Total number of cardiomyocytes (e) and quantification of nucleation (f). Cnx43, connexin-43. g, Representative echocardiography and LV systolic function. h–k, Assessment of control and DKO hearts at 6 months of age. h, Representative images of immunostaining for PH3 (green), cardiac troponin T (red), and nucleus (blue) (left) and the percentage (right) of mitotic cardiomyocytes (arrow). Arrowhead indicates cardiomyocytes with sarcomere disassembly. i, Representative images of immunostaining for aurora B kinase (green), cardiac troponin T (red) and nucleus (blue) (left) and the percentage (right) of cardiomyocytes undergoing cytokinesis (arrow). j, Representative images of WGA (green), cardiac troponin T (red) and nucleus (blue) staining (left) and cardiomyocyte CSA quantification (right). k, LV systolic function quantified by ejection fraction in 6-month-old DKO mice. Data are mean ± s.e.m.; unpaired two-sided t-test. *P < 0.05, **P < 0.01, ***P < 0.001. For n values, see Methods. Scale bars, 10 μm (c, d, f, h, i), 50 μm (b, j), 1 mm (a).
Extended Data Fig. 4. Loss of Meis1 and Hoxb13 attenuates both pathological and physiological hypertrophic responses.
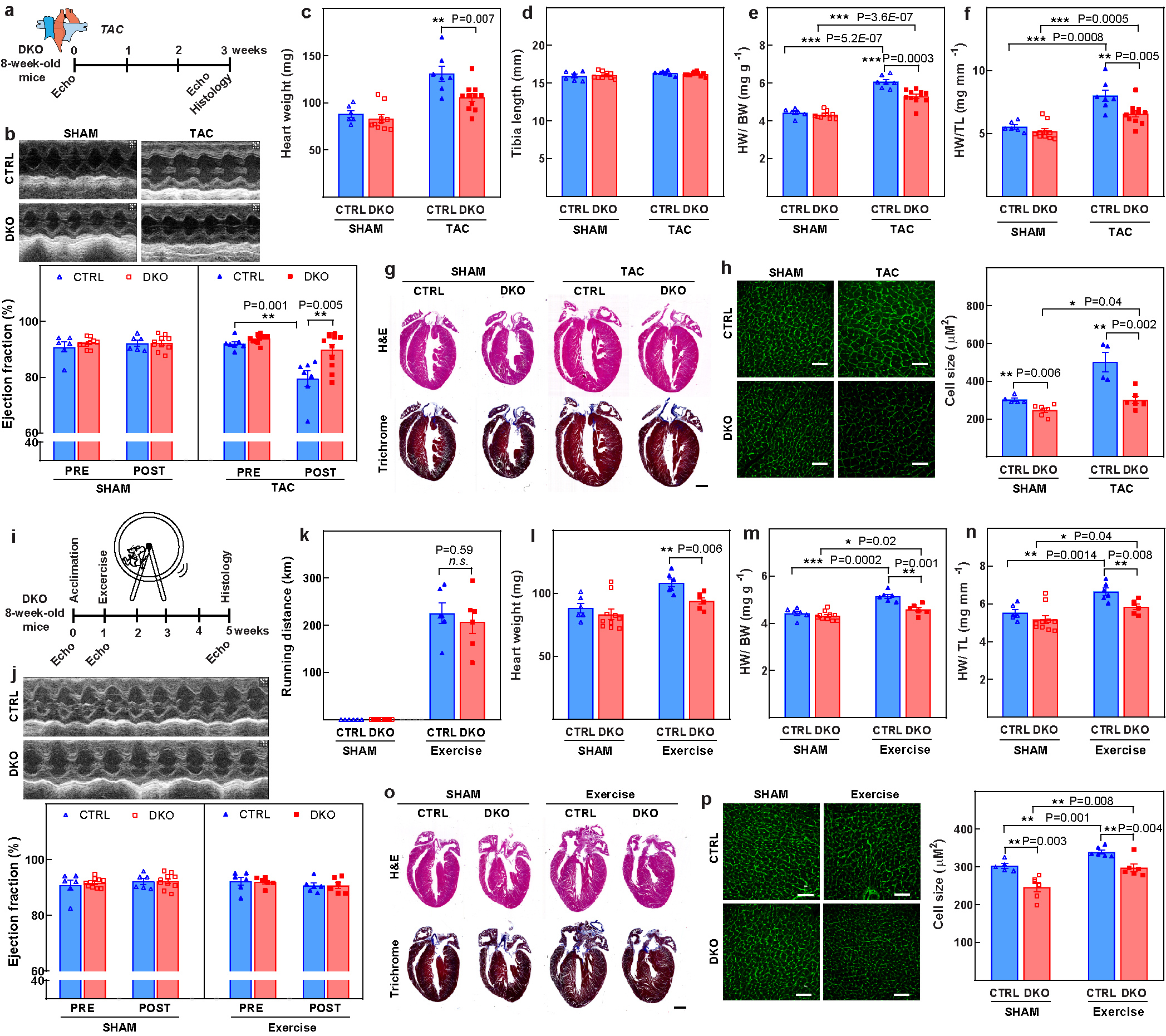
a, Schematic representing the TAC model. b, Representative images of M-mode echocardiography (top) and quantification of ejection fraction (bottom), demonstrating improved LVEF in the DKO mice compared with wild-type mice three weeks after TAC. c, Heart weights, showing significantly higher heart weight in the wild-type group. d, Tibia length was similar in both groups. e, f, The ratio of heart weight/body weight (e) and heart weight/tibial length (f) indicate that the DKO mice have a blunted response to pressure overload. g, Representative images of mouse heart sections stained with haematoxylin and eosin (H&E; top) or Masson’s trichrome (bottom) three weeks after surgery. h, Representative confocal images of cardiomyocyte WGA staining (left) and CSA quantification (right), confirming the blunted hypertrophic response in the DKO hearts in response to pressure overload. i, Schematic representation of the exercise protocol. j, Representative images of echocardiography (top) and ejection fraction quantification (bottom), demonstrating no difference in ejection fraction at baseline and after exercise. k, Running distance of control and DKO mice over four weeks, demonstrating no difference between the two groups. l, Heart weight. m, The ratio of heart weight/body weight suggests that both control and DKO hearts respond to exercise-induced cardiac hypertrophy. n, The ratio of heart weight/tibia length also indicates cardiac hypertrophy in both groups in response to exercise. o, Representative images of mouse heart sections stained with either haematoxylin and eosin (top) or Masson’s trichrome (bottom) four weeks after exercise. p, Confocal images of cardiomyocyte WGA staining and CSA quantification. Data are mean ± s.e.m.; unpaired two-sided t-test. Images in g, o are representative of six independently performed experiments with similar results. *P < 0.05, **P < 0.01, ***P < 0.001. For n values, see Methods. Scale bars, 50 μm (h, p), 1 mm (g, o).
Extended Data Fig. 5. Inducible knockout of Meis1 and Hoxb13 promotes sarcomere disassembly and cardiomyocyte division.
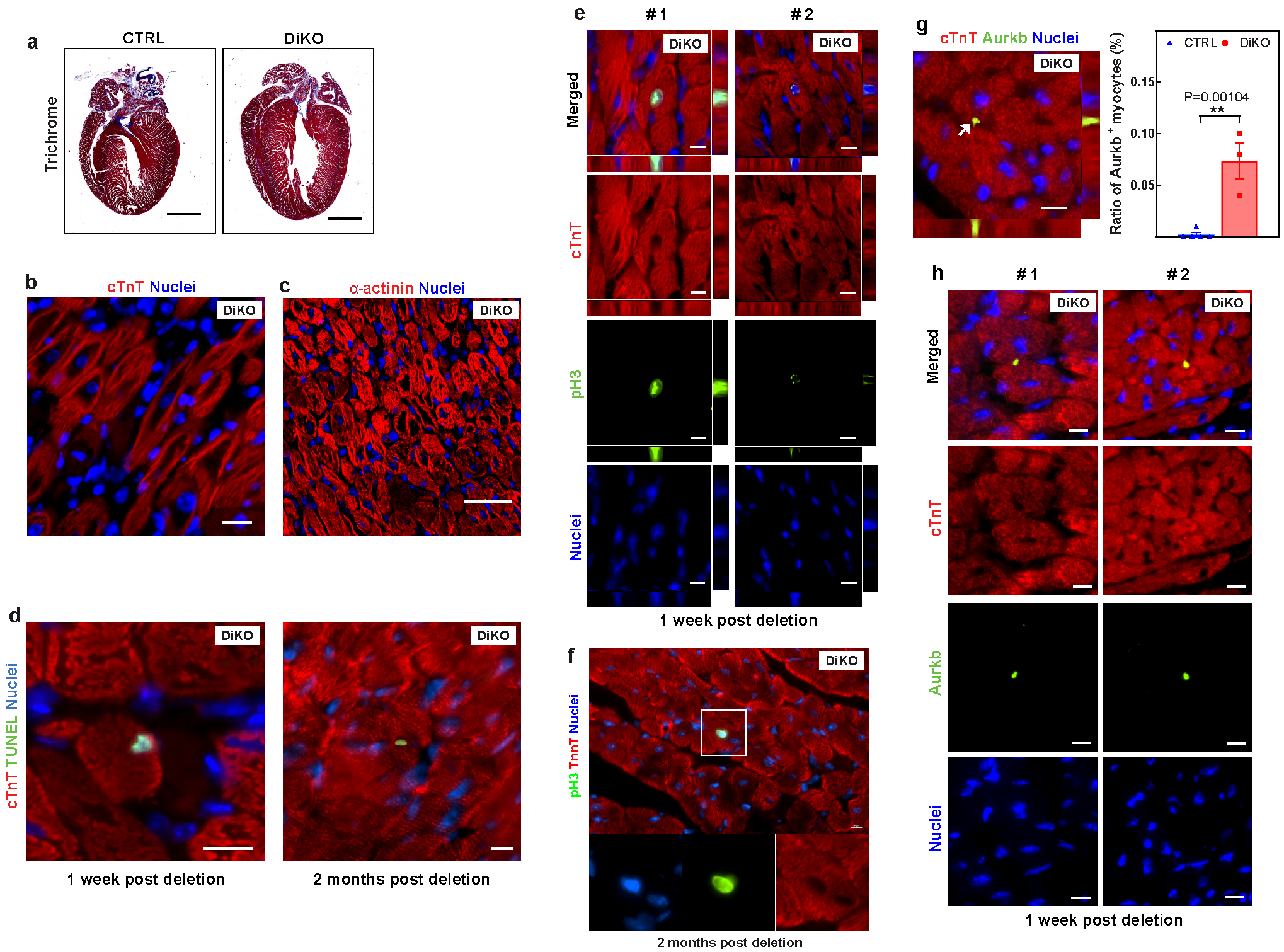
a, Representative images of Masson’s trichrome staining of DiKO hearts four weeks after deletion. b, c, Disassembled sarcomere cardiomyocytes in DiKO hearts one week after deletion, demonstrated by cardiac troponin T (red) (b) or sarcomeric α-actinin (red) (c). d, Representative images of immunostaining for TUNEL (green) and cardiac troponin T (red) in DiKO hearts one week and two months after deletion, as shown in Fig. 2e. e, f, Representative images of immunostaining for PH3 (green), cardiac troponin T (red) and nucleus (blue), showing additional examples of proliferating cardiomyocytes in DiKO hearts at (e) one week and (f) two months after gene deletion. g, Representative images of immunostaining for aurora B kinase (green), cardiac troponin T (red) and nucleus (blue) (left) and the percentage (right) of proliferating cardiomyocytes (arrow) one week after gene deletion. h, Additional examples of cytokinetic cardiomyocytes in DiKO hearts one week after gene deletion. Images in a–c are representative of three independently performed experiments with similar results. Scale bars, 10 μm (b, d–h), 20 μm (c), 1 mm (a).
Extended Data Fig. 6. LV function of DKO hearts after MI.
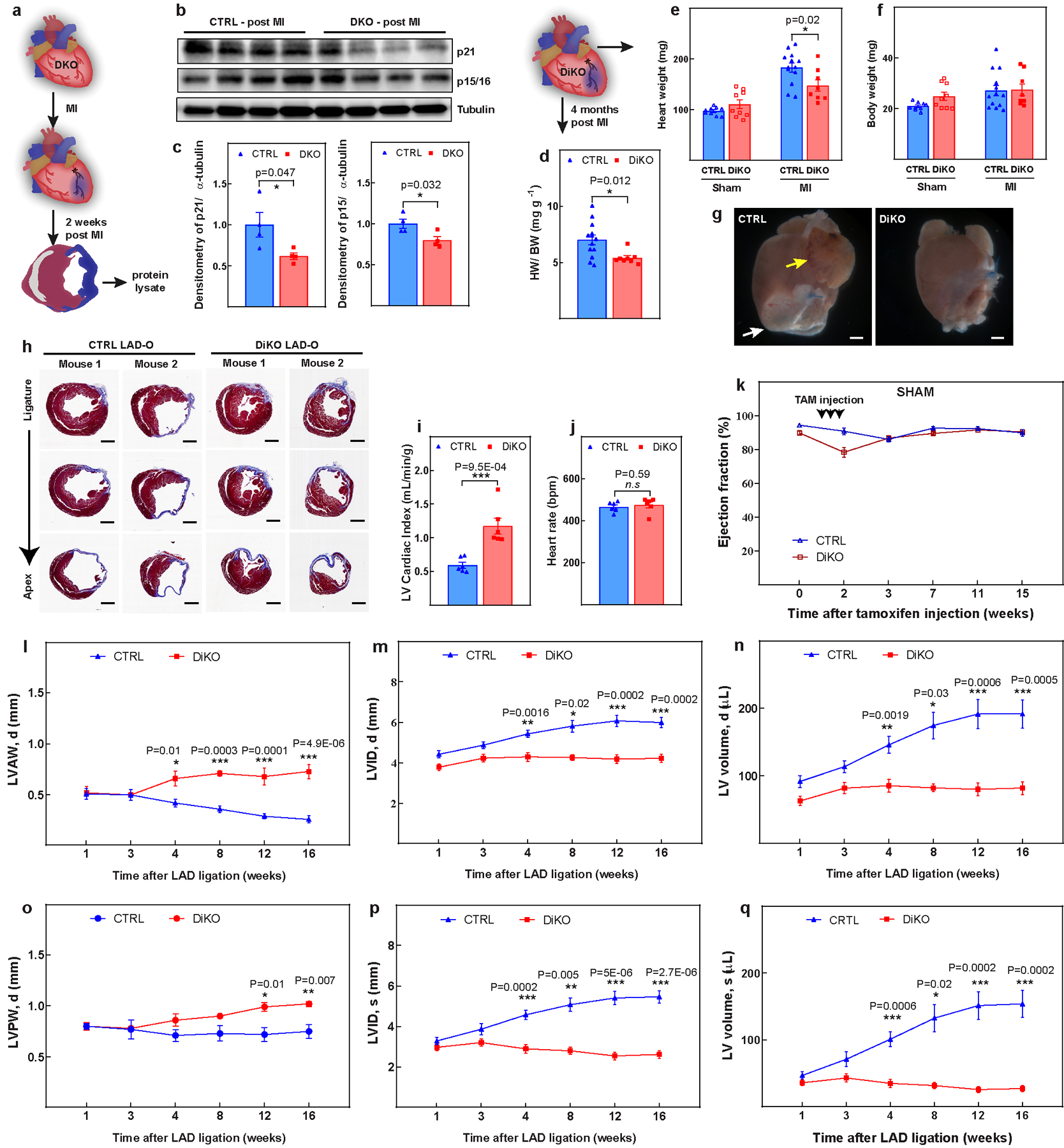
a, Schematic of MI model in DKO hearts 2 weeks post-MI. b, c, Western blot analysis (b) and quantification (c), showing decrease in expression of p21, p15/p16 in DKO hearts. d–f, Heart weight/body weight (d), heart weight (e) and body weight (f) in control and DiKO mice 16 weeks after injury. g, Representative images of the whole hearts at 16 weeks post-MI. The white arrow indicates a region with cell death and the yellow arrow indicates the dilated left atrial appendage. h, Additional representative serial Masson’s trichrome-stained transversal sections, as in Fig. 2l. i, j, LV cardiac index (i) and heart rate (j) in DiKO mice 16 weeks after MI measured by MRI. k, LVEF for sham groups. l–q, Cardiac function analysis for left-ventricular end-diastolic anterior wall thickness (l), left-ventricular end-diastolic internal diameter (m), left-ventricular end-diastolic volume (n), left-ventricular end-diastolic posterior wall thickness (o), left-ventricular end-systolic internal diameter (p), and left ventricular end-systolic volume (q) in DiKO and control mice after MI. Data are mean ± s.e.m.; unpaired two-sided t-test. *P < 0.05, **P < 0.01, ***P < 0.001. For gel source data, see Supplementary Fig. 1. For n values, see Methods. Scale bars, 1 mm (g, h).
Extended Data Fig. 7. Genome-wide identification of Meis1 and Hoxb13 binding sites in heart using ChIP–seq.
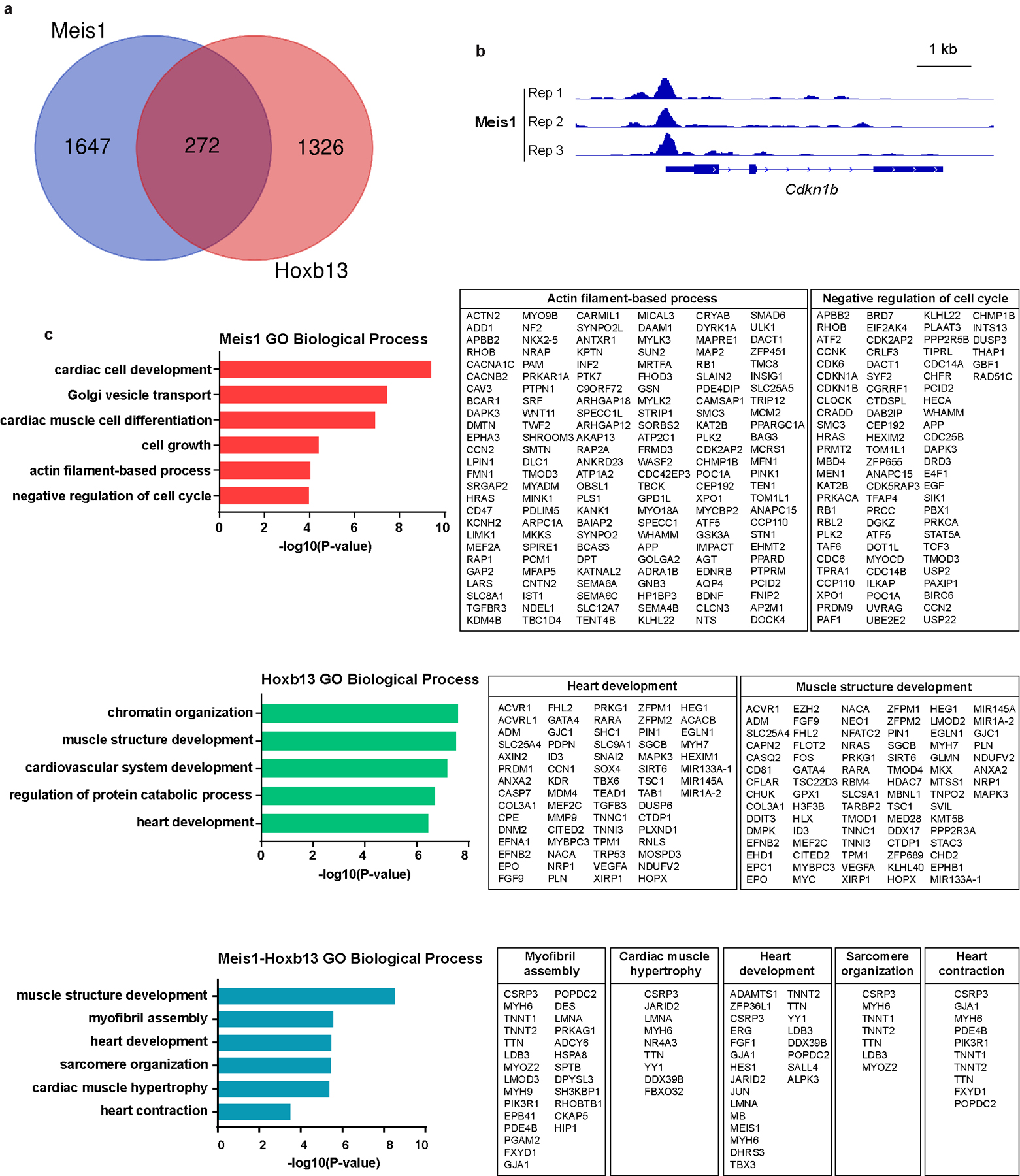
a, Venn diagram showing Meis1 and Hoxb13 binding-site identification. Three replicate sequencing runs were performed for each factor, and the enriched regions were selected only if they were detected in three biological replicates. b, An example of a Meis1 binding site in the cell cycle arrest gene Cdkn1b. c, Gene ontology terms enriched in Meis1-, Hoxb13- and Meis1–Hoxb13-bound peak regions identified from a.
Extended Data Fig. 8. Interaction and dephosphorylation status of Meis1 and Hoxb13 with calcineurin.
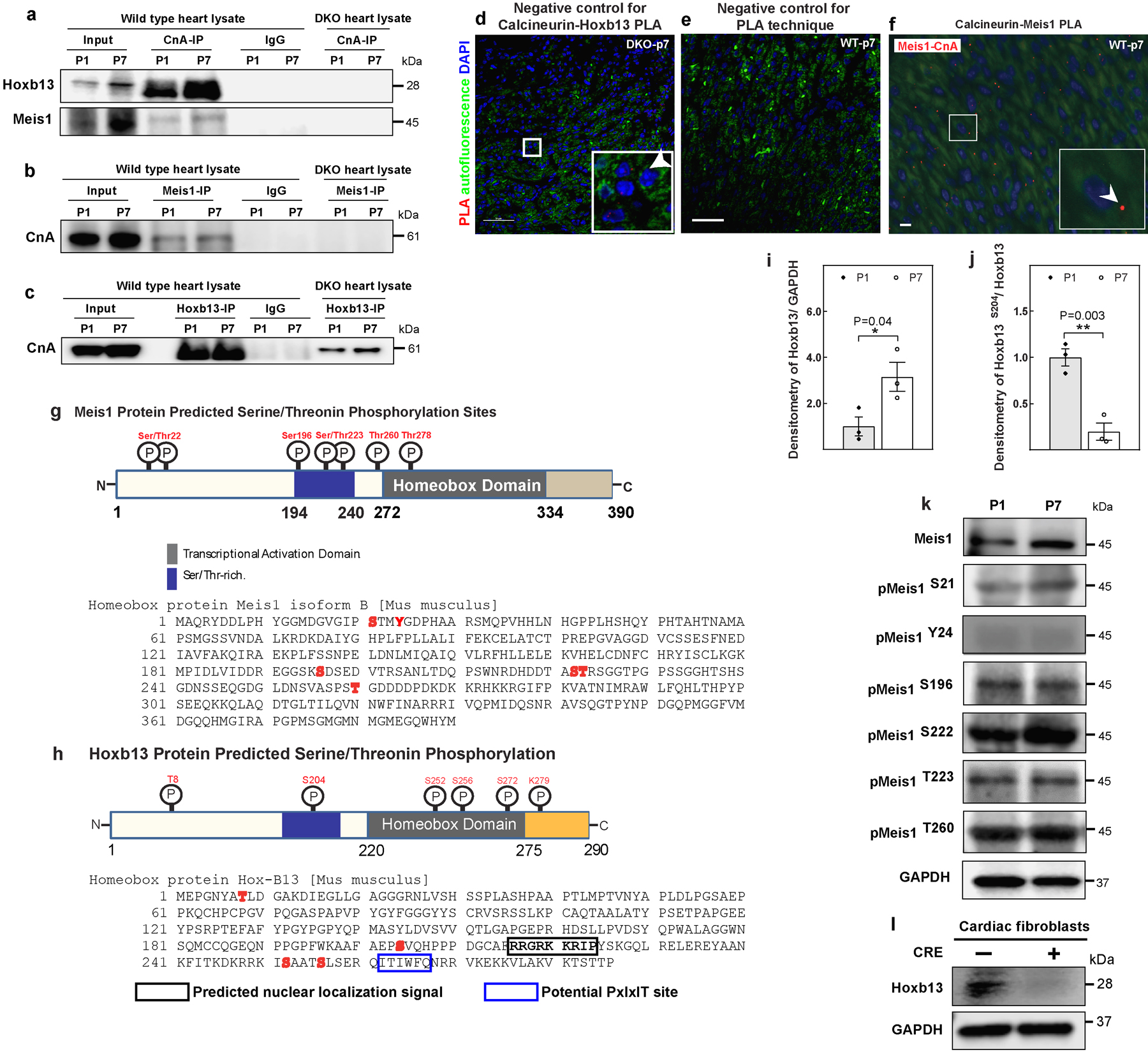
a–c, Coimmunoprecipitation of Fig. 3b–d with negative controls (IgG and DKO heart). d, PLA of CnA and Hoxb13 at P7 of DKO heart; serves as negative control for PLA assay in Fig. 3e. e, PLA assay using only one antibody; serves as negative control for the assay. f, PLA of CnA and Meis1 at P7 heart showed Meis1–CnA association (red dot). g, h, Schematic representation of Meis1 (g, top) and Hoxb13 (h, top) proteins showing the phosphorylatable serine and threonine residues, amino acid sequence of Meis1 (g, bottom) and Hoxb13 (h, bottom) with potentially phosphorylatable serine and threonine residues in red. Box indicates the predicted nuclear localization signal. Numbers indicate amino acid positions. i, j, Densitometry quantification of western blot analysis in Fig. 3j for Hoxb13 (i) and phosphorylated Hoxb13 S204 (j). k, Western blot analysis of phosphorylated Meis1 protein at P1 and P7. GAPDH serves as loading control. l, Western blot analysis from stable DKO cardiac fibroblasts confirms the loss of Hoxb13 in Cre-treated cells. Data are mean ± s.e.m.; unpaired two-sided t-test. Experiments in a–f, k–m were repeated independently three times with similar results. *P < 0.05, **P < 0.01. For gel source data, see Supplementary Fig. 1. For n values, see Methods. Scale bars, 10 μm (f), 50 μm (d, e).
Extended Data Fig. 9. Increased calcineurin activity suppresses postnatal cardiomyocyte proliferation.
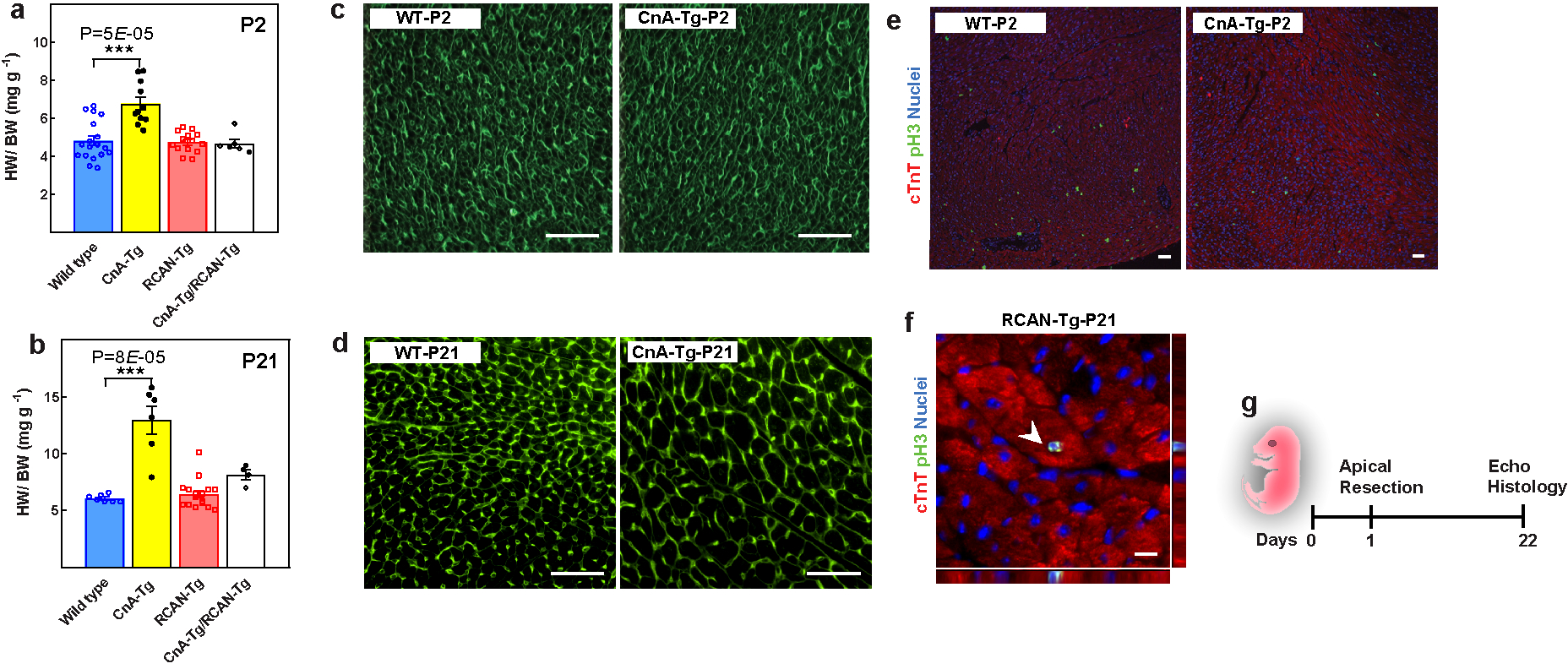
a, Heart weight/body weight in wild-type, CnA-Tg, Rcan1-Tg, and double-Tg mice at P2 (a) and P21 (b). c, d, Representative images of WGA staining for CSA quantification in Fig. 4a (c) and Fig. 4b (d). e, f, Representative images of immunostaining for PH3 (green) and cardiac troponin T (red) for mitotic cardiomyocytes quantification in Fig. 4c (e) and Fig. 4d (f). g, Schematic of apical resection model in P1 mice. Scale bars, 10 μm (f), 50 μm (c, d). Data are mean ± s.e.m.; unpaired two-sided t-test. ***P < 0.001. For n values, see Methods.
Extended Data Table 1.
| TARGET PHOSPHO PROTEIN | PRODUCT NAME | PEPTIDE SEQUENCE |
|---|---|---|
| MEIS1-S21 | 20592-1NB-5 | Ac-CGVGIPS (P) TMY-NH2 |
| MEIS1-Y24 | 26328 | Ac-C-IPSTMY (P) GDP-NH2 |
| MEIS1-S196 | 28112 | Ac-C-EGGSKS (P) DSE-NH2 |
| MEIS1-S222 | 20592-1NB-17 | Ac-CHDDTAS (P) TRS-NH2 |
| MEIS1-T223 | 28113 | Ac-CDDTAST (P) RSG-NH2 |
| MEIS1-T260 | 28114 | Ac-C-VASPST (P) GDD-NH2 |
| TARGET PHOSPHO PROTEIN | PRODUCT NAME | PEPTIDE SEQUENCE |
| HOXB13-T8 | 28115 | AC-C-PGNYAT (P) LDG-NH2 |
| HOXB13-S252 | 28118 | Ac-C-KRRKIS (P) AAT-NH2 |
| HOXB13-S256 | 28119 | Ac-C-ISAATS (P) LSE-NH2 |
| HOXB13-S204 | 28117 | Ac-C-AFAEPS (P) VQH-NH2 |
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Zhang for MRI studies at the Mouse MRI Core, Advanced Imaging Research Center, University of Texas Southwestern Medical Center; J. Shelton for assistance with histology; H. I. May for conducting mouse surgeries; J. Heinrich for helping with voluntary exercise; J. Xu, X. Liu and Y. Jung Kim from the Sequencing Core Facility in Children’s Research Institute in University of Texas Southwestern Medical Center for performing the Illumina sequencing. The UTSW mouse MRI facility is supported by an NIH shared instrumentation grant 1S10OD023552-01. H.A.S. is supported by grants from the NIH (1R01HL115275 and 5R01H2131778), National Aeronautics and Space Administration (NNX-15AE06G), American Heart Association (16EIA27740034), Cancer Prevention and Research Institute of Texas (RP160520), Hamon Center for Regenerative Science and Medicine, and Fondation Leducq (Redox Regulation of Cardiomyocyte Renewal). M.S.C. and J.R. are funded by NIH R01GM119336. B.A.R. was supported by grants from the NIH (R01 HD101006, R01 HL072016) and Wellstone (U54HD087351). J.A.H. is supported by grants from NIH (R01 HL120732, R01 HL128215, R01 HL126012). N.U.N.N. is supported by AHA Postdoctoral Fellowship 19POST34450039. Z.W. was supported by a predoctoral fellowship from the American Heart Association and the Harry S. Moss Heart Trust (19PRE34380436). N.T.L. is supported by a Haberecht Wildhare-Idea Research Grant.
Footnotes
COMPETING INTEREST DECLARATION
The authors declare no competing interests.
Supplementary Figures
Uncropped western blot images with molecular weight markers and indication of how the gels were cropped.
REFERENCES
- 1.Bui AL, Horwich TB & Fonarow GC Epidemiology and risk profile of heart failure. Nature reviews. Cardiology 8, 30–41, doi: 10.1038/nrcardio.2010.165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soonpaa MH & Field LJ Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol 272, H220–226 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Senyo SE et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 493, 433–436, doi: 10.1038/nature11682 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bergmann O et al. Evidence for cardiomyocyte renewal in humans. Science 324, 98–102, doi: 10.1126/science.1164680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eschenhagen T et al. Cardiomyocyte Regeneration: A Consensus Statement. Circulation 136, 680–686, doi: 10.1161/CIRCULATIONAHA.117.029343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porrello ER et al. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080, doi: 10.1126/science.1200708 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porrello ER et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A 110, 187–192, doi: 10.1073/pnas.1208863110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soonpaa MH et al. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest 99, 2644–2654, doi: 10.1172/JCI119453 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahmoud AI et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 497, 249–253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eulalio A et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492, 376–381, doi: 10.1038/nature11739 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Puente BN et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157, 565–579, doi: 10.1016/j.cell.2014.03.032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heallen T et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461, doi: 10.1126/science.1199010 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J et al. mir-17–92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res 112, 1557–1566, doi: 10.1161/CIRCRESAHA.112.300658 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen WF et al. HOXA9 forms triple complexes with PBX2 and MEIS1 in myeloid cells. Mol Cell Biol 19, 3051–3061 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen WF et al. AbdB-like Hox proteins stabilize DNA binding by the Meis1 homeodomain proteins. Mol Cell Biol 17, 6448–6458 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berthelsen J, Kilstrup-Nielsen C, Blasi F, Mavilio F & Zappavigna V The subcellular localization of PBX1 and EXD proteins depends on nuclear import and export signals and is modulated by association with PREP1 and HTH. Genes Dev 13, 946–953 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ali SR et al. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A 111, 8850–8855, doi: 10.1073/pnas.1408233111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H et al. Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat Struct Mol Biol 19, 337–345, doi: 10.1038/nsmb.2238 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molkentin JD et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228, doi: 10.1016/s0092-8674(00)81573-1 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiserud T Physiology of the fetal circulation. Semin Fetal Neonatal Med 10, 493–503, doi: 10.1016/j.siny.2005.08.007 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Fiedler B & Wollert KC Targeting calcineurin and associated pathways in cardiac hypertrophy and failure. Expert Opin Ther Targets 9, 963–973, doi: 10.1517/14728222.9.5.963 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Heineke J & Ritter O Cardiomyocyte calcineurin signaling in subcellular domains: from the sarcolemma to the nucleus and beyond. J Mol Cell Cardiol 52, 62–73, doi: 10.1016/j.yjmcc.2011.10.018 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Wilkins BJ & Molkentin JD Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun 322, 1178–1191, doi: 10.1016/j.bbrc.2004.07.121 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Colella M et al. Ca2+ oscillation frequency decoding in cardiac cell hypertrophy: role of calcineurin/NFAT as Ca2+ signal integrators. Proc Natl Acad Sci U S A 105, 2859–2864, doi: 10.1073/pnas.0712316105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parra V & Rothermel BA Calcineurin signaling in the heart: The importance of time and place. J Mol Cell Cardiol 103, 121–136, doi: 10.1016/j.yjmcc.2016.12.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Rooij E et al. Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J Biol Chem 277, 48617–48626, doi: 10.1074/jbc.M206532200 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Hogan PG, Chen L, Nardone J & Rao A Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 17, 2205–2232, doi: 10.1101/gad.1102703 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Schaeffer PJ et al. Impaired contractile function and calcium handling in hearts of cardiac-specific calcineurin b1-deficient mice. Am J Physiol Heart Circ Physiol 297, H1263–1273, doi: 10.1152/ajpheart.00152.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothermel BA et al. Myocyte-enriched calcineurin-interacting protein, MCIP1, inhibits cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A 98, 3328–3333, doi: 10.1073/pnas.041614798 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakada Y et al. Hypoxia induces heart regeneration in adult mice. Nature 541, 222–227 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Nascimento DS et al. MIQuant—semi-automation of infarct size assessment in models of cardiac ischemic injury. PLoS ONE 6, e25045 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahmoud AI, Porrello ER, Kimura W, Olson EN & Sadek HA Surgical models for cardiac regeneration in neonatal mice. Nat. Protoc 9, 305–311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura W et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 523, 226–230 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Goldman A et al. The calcineurin signaling network evolves via conserved kinase-phosphatase modules that transcend substrate identity. Mol. Cell 55, 422–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z et al. Mechanistic basis of neonatal heart regeneration revealed by transcriptome and histone modification profiling. Proc. Natl Acad. Sci. USA 116, 18455–18465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashimoto H et al. Cardiac reprogramming factors synergistically activate genome-wide cardiogenic stage-specific enhancers. Cell Stem Cell 25, 69–86 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Q, Brown JB, Huang H & Bickel PJ Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat 5, 1752–1779 (2011). [Google Scholar]
- 39.Kharchenko PV, Tolstorukov MY & Park PJ Design and analysis of ChIP–seq experiments for DNA-binding proteins. Nat. Biotechnol 26, 1351–1359 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities.. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available within the paper and its Supplementary Information file. Source Data for Figs. 1–4 and Extended Data Figs. 1–6, 8, 9 are provided with the online version of the paper. ChIP-seq data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE141769. Data or other materials are available from the corresponding author upon reasonable request.