

Abstract
Numerous bacteriophages—viruses of bacteria, also known as phages—have been described for hundreds of bacterial species. The Gram-negative Shigella species are close relatives of Escherichia coli, yet relatively few previously described phages appear to exclusively infect this genus. Recent efforts to isolate Shigella phages have indicated these viruses are surprisingly abundant in the environment and have distinct genomic and structural properties. In addition, at least one model system used for experimental evolution studies has revealed a unique mechanism for developing faster infection cycles. Differences between these bacteriophages and other well-described model systems may mirror differences between their hosts’ ecology and defense mechanisms. In this review, we discuss the history of Shigella phages and recent developments in their isolation and characterization and the structural information available for three model systems, Sf6, Sf14, and HRP29; we also provide an overview of potential selective pressures guiding both Shigella phage and host evolution.
Keywords: Omps, lipopolysaccharide, phage biology, podoviruses, myoviruses, Shigella
INTRODUCTION
Bacteriophages have been instrumental in both fundamental and applied research, including molecular biology, microbial ecology and evolution, phage display, and phage therapy. These viruses have also been used to manipulate their hosts and to develop bacteria as model organisms. Despite their close relationship to Escherichia coli based on genomic data, bacteria in the genus Shigella—comprising the four species S. boydii, S. dysenteriae, S. flexneri, and S. sonnei—are phenotypically distinct (1–4) and have had comparatively few bacteriophages described.
Although few Shigella phages have been fully characterized, they are surprisingly easy to isolate from the environment (5). While the ecology of these phages is not well characterized, many appear to be specific for Shigella and are abundant regardless of local shigellosis outbreaks (6, 7). Part of this abundance may be related to aspects of Shigella physiology. One major difference between E. coli and Shigella is the clustered regularly interspaced palindromic repeats (CRISPRs) encoded in their genomes. CRISPR is an acquired defense mechanism that protects the host against phage infection by cleaving foreign DNA While most strains of E. coli described thus far encode at least one functional CRISPR system, Shigella has none (8, 9). While remnants of CRISPR-1 and CRISPR-3 systems have been detected for some Shigella species, these are interrupted by deletions, insertion elements, frameshifts, and truncations, rendering them nonfunctional (9). Besides CRISPR, bacteria have innate immunity via restriction modification (RM) systems. These systems use enzymes that can recognize self versus nonself DNA. Compared with E. coli, Shigella also has relatively few RM systems (10). Overall, without the ability to differentiate between native versus foreign DNA, Shigella appears to have been parasitized by numerous mobile genetic elements—or selfish genes—including phage genes and transposons (11). This could explain the ongoing degradation of Shigella genomes, which is affecting the genus in numerous ways. For example, large deletions, or genomic black holes, small deletions, and pseudogenes are responsible for enhancing its virulence (12–14). Many species lack functional flagella and fimbriae (13), and others have deleted numerous outer membrane proteins (Omps), some of which likely serve as receptors for bacteriophages (11).
Some of these differences between E. coli and Shigella may help explain why the latter is parasitized by so many phages. In this review, we discuss Shigella phage history and ecology, the development and current understanding of a few model systems, and studies addressing the evolution of both host and phage. We also address numerous unknown aspects of Shigella phages throughout the review and highlight areas in critical need of research.
HISTORY OF SHIGELLA PHAGES
One of the first bacteriophages ever discovered was by Felix d’Herelle in 1917, which was subsequently claimed to cure children suffering from Shigella infection (15). Before about the year 2000, most research regarding Shigella phages was in their use for typing—i.e., identifying specific Shigella strains—or in the ability of temperate phages to alter the serotype of their host (16–18). Aside from host range, there is little information on the properties of these phage. To the best of our knowledge, these historic isolates have since been lost, although the strains from which they were induced may exist in public or private collections.
Part of the problem with identifying and maintaining historic Shigella phages may have been their origin from clinical samples collected from shigellosis patients. The first modern Shigella phages were released from lysogenized clinical strains of virulent S. flexneri. These phages—SfI, SfII, SfIV, SfV, Sf6, SfX, Sf101, and SfMu—could alter the surface properties of their host bacterium when integrated into its genome (19–26). This phenomenon of serotype conversion can now be investigated more directly by modern techniques and is still an active area of research. The serotype refers to surface antigens on a bacterium, facilitating classification of strains within a species. For Gram-negative bacteria, this is often determined by the lipopolysaccharide (LPS) structure on the bacterial cell surface, which is discussed in more detail below (see the section titled Interactions Between Shigella and Its Phages). By studying these phages, it was determined that phage-encoded O-antigen modifying enzymes were responsible for altering the host’s LPS structure. If multiple phages coinfect the same host cell, they have the potential to produce completely new serotypes (19), with significant implications for the pathogenicity and epidemiology of S. flexneri.
In more recent years, Shigella phage isolation has not been limited to induction of or spontaneous release from clinical isolates, which biases toward isolation of temperate phage. Numerous lytic phages have now been isolated from environmental samples. Many of these isolates were described in terms of their usefulness for clinical control of shigellosis, but they also illustrate the prevalence and diversity of Shigella phages. A comprehensive list of characterized Shigella phages is summarized in Table 1, including some basic characteristics. Thus far, the 78 known Shigella phages fall into a few main groups. The largest group is the T4-like myovirus subfamily Teven-virinae, with genome sizes of 164.0–176.0 kbp. The next largest groups are the T1-like siphovirus subfamily Tunavirinae, with genome sizes of 47.7–51.9 kbp, and the FelixO1-like myovirus subfamily Ounavirinae, with genome sizes of 85.9–90.4 kbp. Relatively few phages are short-tailed podoviruses, but these span across Autographivirinae, Sepvirinae, Gamaleyaviruses, Kuraviruses, and Lederbergviruses. Given the recent development of isolating environmental Shigella phages, a brief overview of what is known about their ecology is presented next.
Table 1.
Basic properties of Shigella phages that have been described and/or deposited into the National Center for Biotechnology Information GenBank with isolation source and geographic location, taxonomic family, genome length (in kbp), GenBank accession number, and reference if available
| Name | Isolation source | Familya | Genome | Accession | Reference(s) |
|---|---|---|---|---|---|
| JK32 | Glencar Waterfall, Ireland | Myoviridae | 176.0 | MK962753 | 89 |
| JK45 | Sewage, Ireland | Myoviridae | 170.7 | MK962757 | 89 |
| SHFML-11 | Intesti bacteriophage lot #010504, Eliava Institute, Tbilisi, Georgia | Myoviridae | 170.7 | NC_030953 | 90, 91 |
| SP18 | Gap River, Korea | Myoviridae | 170.6 | NC_014595 | 92 |
| SSE1 | China | Myoviridae | 169.7 | MK639187 | NA |
| SHSML-52-1 | Park surface water, MD, USA | Myoviridae | 169.6 | NC_031090 | 90, 91 |
| Shfl125875 | Wastewater, Charleston, SC, USA | Myoviridae | 169.1 | NC_025437 | 93 |
| SHFML-26 | Ses D-90 lot #010104, Eliava Institute, Tbilisi, Georgia | Myoviridae | 169.0 | NC_031011 | 90, 91 |
| JK36 | Sewage, Ireland | Myoviridae | 168.9 | MK962754 | 89 |
| Sf25 | Red Cedar River, Lansing, MI, USA | Myoviridae | 168.6 | MF327009 | 6 |
| JK23 | Stream from Connemara National Park, Ireland | Myoviridae | 168.3 | MK962752 | 89 |
| JK42 | Sewage, Ireland | Myoviridae | 168.3 | MK962756 | 89 |
| Sf24 | Red Cedar River, Lansing, MI, USA | Myoviridae | 168.1 | NC_042078 | 6 |
| SfPhi01 | Wastewater, Japan | Myoviridae | 168.0 | LC465543 | 94 |
| JK38 | Sewage, Ireland | Myoviridae | 167.9 | MK962755 | 89 |
| Sf23 | Grand River, East Lansing, MI, USA | Myoviridae | 167.7 | MF158046 | 6 |
| KRT47 | Red Cedar River, East Lansing, MI, USA | Myoviridae | 167.5 | MN781580 | NA |
| phi25–307 | Not known | Myoviridae | 167.5 | MG589383 | NA |
| CM8 | Chicken meat | Myoviridae | 167.2 | MK962750 | 89 |
| SHBML-50–1 | Park surface water, MD, USA | Myoviridae | 166.6 | NC_031085 | 90, 91 |
| Sf22 | Grand River, Lansing, MI, USA | Myoviridae | 166.3 | NC_042039 | 6 |
| vB_SdyM_006 | China | Myoviridae | 166.1 | MK295204 | NA |
| Sf21 | Red Cedar River, East Lansing, MI, USA | Myoviridae | 166.0 | MF327007 | 6 |
| Shfl2 | Sewage water, Brazil | Myoviridae | 166.0 | NC_015457 | NA |
| pSs-1 | Surface water, South Korea | Myoviridae | 165.0 | NC_025829 | 95 |
| SH7 | Water in Chotrana, Tunisia | Myoviridae | 164.9 | KX828711 | 96 |
| Sf20 | Red Cedar River, East Lansing, MI, USA | Myoviridae | 164.0 | MF327006 | 6 |
| MK-13 | Surface water, Dhaka City, Bangladesh | Ackermannviridae | 158.8 | MK509462 | 97 |
| phiSboM-AG3 | Sewage, Guelph, ON, Canada | Ackermannviridae | 158.0 | NC_013693 | 98 |
| CM1 | Chicken meat | Myoviridae | 140.0 | MK962749 | 89 |
| SSP1 | Not known | Siphoviridae | 113.3 | KY963424 | NA |
| SHSML-45 | Encophagum D-90, lot #140704, Eliava Institute, Tbilisi, Georgia | Siphoviridae | 108.0 | NC_031022 | 90, 91 |
| Sf19 | Red Cedar River, East Lansing, MI, USA | Myoviridae | 90.4 | MF327005 | 6 |
| Sf18 | Grand River, Lansing, MI, USA | Myoviridae | 90.3 | MF158044 | 6 |
| KPS64 | Surface water, Lincoln, NE, USA | Myoviridae | 90.2 | MK562502 | 7 |
| Sf17 | Red Cedar River, East Lansing, MI, USA | Myoviridae | 90.1 | NC_042076 | 6 |
| Silverhawkium | Lincoln, NE, USA | Myoviridae | 88.8 | MK562505 | 7 |
| Sf16 | Grand River, Lansing, MI, USA | Myoviridae | 88.6 | MF158043 | 6 |
| Sf15 | Red Cedar River, East Lansing, MI, USA | Myoviridae | 88.5 | MF158041 | 6 |
| Sf14 | Red Cedar River, East Lansing, MI, USA | Myoviridae | 87.6 | NC_042075 | 6 |
| Sf13 | Red Cedar River, East Lansing, MI, USA | Myoviridae | 87.6 | NC_042017 | 6 |
| JK55 | Sewage, Belgium | Myoviridae | 86.2 | MK962758 | 89 |
| vB_SflM_004 | China | Myoviridae | 85.9 | MK295205 | NA |
| SGF2 | Water, China | Podoviridae | 77.0 | MN148435 | NA |
| pSb-1 | Dorimcheon stream, Seoul, South Korea | Podoviridae | 71.6 | NC_023589 | 99 |
| Ss-VASD | Induced from clinical isolate (S. sonnei from 2014) | Podoviridae | 62.9 | NC_028685 | 100 |
| POCJ13 | Induced from clinical isolate (S. flexneri strain BS937) | Podoviridae | 62.7 | NC_025434 | 101 |
| 75/02 Stx | Induced from clinical isolate (S. sonnei strain 75/02) | Podoviridae | 60.9 | NC_029120 | 102 |
| JK16 | Surface water, Cork City, Ireland | Siphoviridae | 51.9 | MK962751 | 89 |
| pSf-1 | Han River, South Korea | Siphoviridae | 51.8 | NC_021331 | 103 |
| Shfl1 | Sewage water, Brazil | Siphoviridae | 50.7 | NC_015456 | NA |
| vB_SsoS-ISF002 | Wastewater treatment plant, Isfahan, Iran | Siphoviridae | 50.6 | NC_041995 | 104 |
| SH6 | Water in Chotrana, Tunisia | Siphoviridae | 50.6 | KX828710 | 96 |
| vB_SflS-ISF001 | Wastewater treatment plant, Isfahan, Iran | Siphoviridae | 50.6 | MG049919 | 105 |
| vB_SsoS_008 | China | Siphoviridae | 50.4 | MK335533 | NA |
| Sfin-5 | Environmental surface water, India | Siphoviridae | 50.4 | MN342247 | NA |
| Sfin-4 | India | Siphoviridae | 50.4 | MN337573 | NA |
| Sfin-1 | Ganga River, West Bengal, India | Siphoviridae | 50.4 | MF468274 | 106 |
| phi2457T | Intesti Bacteriophage cocktail, Eliava Institute, Tbilisi, Georgia | Siphoviridae | 50.2 | MH917278 | 107 |
| pSf-2 | Geolpocheon stream, South Korea | Siphoviridae | 50.1 | NC_026010 | 108 |
| Sd1 | Grand River, Lansing, MI, USA | Siphoviridae | 48.3 | MF158042 | 6 |
| Sf12 | Grand River, Lansing, MI, USA | Siphoviridae | 47.7 | MF158039 | 6 |
| DS8 | China | Siphoviridae | 44.6 | MK759854 | NA |
| EP23 | Gap River, South Korea | Siphoviridae | 44.1 | NC_016566 | 109 |
| HRP29 | Surface water, Lincoln, NE, USA | Podoviridae | 43.6 | MK562503 | 7 |
| SFN6B | Reservoir water, Malaysia | Podoviridae | 43.0 | KY684082 | NA |
| SfII | Induced from clinical isolate NCTC 4 | Myoviridae | 41.5 | NC_021857 | 110 |
| SfX | Induced from clinical isolate 2002017 | Podoviridae | 41.2 | Not available | 111 |
| SFPH2 | Hospital sewage, Beijing, China | Podoviridae | 40.4 | MH464253 | 112 |
| vB_Ship_A7 | China | Podoviridae | 40.1 | MK685668 | NA |
| SfIV | Induced from clinical isolate SFL1522, International Center for Diarrheal Disease Research, Bangladesh | Myoviridae | 39.8 | NC_022749 | 21 |
| Sf6 | Spontaneous release from strain 3-19 | Podoviridae | 39.0 | NC_005344 | 34 |
| Sf101 | Induced from clinical isolate SFL1683 | Podoviridae | 38.7 | NC_027398 | 25 |
| SfI | Induced from clinical isolate ICDC 019 | Myoviridae | 38.4 | NC_027339 | 19 |
| SfV | Induced from clinical isolate EW 595/52 | Myoviridae | 37.1 | NC_003444 | 22 |
| SfMu | Induced from clinical isolate (SFL2241, serotype 4a) | Myoviridae | 37.1 | NC_027382 | 26 |
| R18C | Rabbit feces, Hungary | Myoviridae | 31.8 | MN016939 | 113 |
| SGF3 | Unknown | Microviridae | 5.4 | MN266305 | NA |
Conventional from Reference 114.
Abbreviation: NA, not applicable.
ECOLOGY OF SHIGELLA AND ITS PHAGES
Most Shigella phages isolated from the environment originated from wastewater, rivers, or streams. While the latter two may seem surprising, Connor et al. (27) describe the persistence of S. flexneri throughout the world. The authors hypothesize that contaminated environmental water serves as a reservoir for the species, similar to Vibrio cholerae. Despite these findings, Shigella has thus far not been isolated from samples in which phage are found. This could be an effect of sampling: Shigella is likely dilute in the source waters tested, as calculated previously (28). Alternatively, because many environmental samples undergo an enrichment process after collection, lytic bacteriophages that were also collected in the sample would have a chance to amplify and kill any susceptible hosts (29). Finally, if Shigella is similar to V. cholerae, an environmental stage might be important for the infection cycle, where it goes through a nonculturable phase depending on environmental conditions (30). Nonculturability has been described for S. dysenteriae when in environmental water samples (31) and for other species of Shigella during sewage sludge treatment (32). In the latter study, the authors collected samples and determined the number of culturable cells (via colony plating), viable cells (via transcriptional activity measured by reverse transcriptase PCR), and total cells (via number of genomes measured by quantitative PCR).
A nonculturable phase may protect cells during stressful abiotic conditions, but whether those cells are still susceptible to phages has not been directly tested. Additional studies on the abundance of Shigella in the environment and its physiology in these conditions would be informative. The prevalence of Shigella and its phages is also largely unknown for nonwater environments. Although most phages were isolated from water samples, it has not been determined whether they are limited to this environment or why they are more prevalent in water. Future studies designed to directly compare Shigella phage ecology with phages infecting other types of enteric bacteria could provide information about the dynamics between Shigella and its phages during its residence in the environment and how these dynamics are affected by both abiotic and biotic factors.
Thus far, both old and new Shigella phages have been excellent systems to study numerous aspects of phage biology. Isolated in 1975, Sf6 and its host S. flexneri have since provided a wealth of information on genomic and structural aspects of podoviruses, phage-host interactions, and novel mechanisms for how phage evolve faster replication rates (see the section titled Evolution of Shigella Phages). The environmental isolates Sf14 and HRP29, discovered in the past few years, are two new emerging systems with unusual genomic and structural properties and relatively little information on their family members. The genomes and initial structural analyses of these two phages are shown in Figure 1 compared with Sf6. Characterizing Sf14 and HRP29 further will begin to fill critical gaps in our knowledge of phage structure, mechanisms of infection, and diversity.
Figure 1.
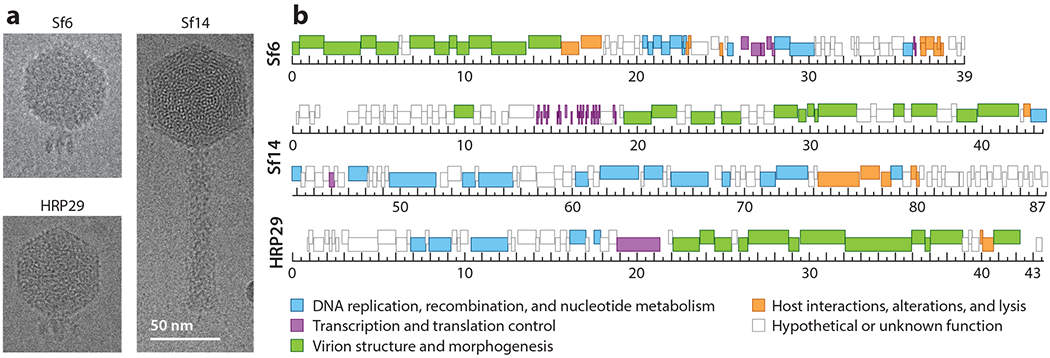
Characteristics of the developing Shigella phage model systems Sf14 and HRP29 compared with the established model system Sf6. (a) Representative images from cryo-electron micrographs. (b) Genome maps, with genes colored according to function, as indicated. The ruler is in kilo-base pairs. The cluster of short, dark-colored bars at 14–18 kbp in the Sf14 genome represents transfer RNA genes.
CURRENT AND DEVELOPING MODEL SYSTEMS
Established Model System Sf6
Sf6 was initially isolated from a clinical isolate of S. flexneri via spontaneous lysis, where it had persisted as a lysogen before coming into the hands of Gemski et al. (23). It is a member of the short-tailed Podoviridae family, where it can be further classified as a P22virus with the capsid exhibiting T = 7 icosahedral symmetry (33). Detailed analyses of its genome revealed extensive mosaicism relative to related phages and how this mosaicism influences function (34). The overall architecture of the Sf6 genome is similar to the lambdoid phages (34), defined here as phages with genomes that closely resemble phage λ in terms of gene order and organization (35, 36). Sf6 encodes 68 predicted open reading frames, nine of which have no known phage homologs. One key feature is the nin region, which—in addition to ten open reading frames common to other nin regions—contains two open reading frames with no known phage homologs, two nonfunctional open reading frames, and an IS911 transposon element. In most other lambdoid phages, this region encodes a variable number of intact open reading frames involved in transcriptional regulation, recombination, or an unknown function; these are largely nonessential in the laboratory. The IS911 transposon of Sf6 is the most striking feature, as it shares 98.6% of nucleotide identity to the Shigella IS911 element. Although many phage genomes contain small mobile elements, harboring a large, functional transposon is uncommon. Because Sf6 can lysogenize S. flexneri by integrating into the host’s argW transfer RNA (tRNA) gene, and because Shigella genomes are particularly susceptible to mobile genetic elements, this could explain how Sf6 acquired its own transposon. Although bacteriophage genomes are notoriously mosaic, harboring an IS911 sequence is not a common feature (34).
During a lytic infection cycle, Sf6 assembles into virions that can initiate subsequent infection after host cell lysis. Like other P22viruses, all double-stranded DNA–containing bacteriophages and herpesvirus assembly begins with procapsid formation. The procapsid is a metastable assembly intermediate composed of the portal, scaffolding, and coat proteins. The procapsid gets filled with DNA through the portal via ATPase activity of the terminase complex, triggering release of the scaffolding protein and subsequent expansion of the procapsid (37). Head completion proteins, so-called plug proteins, and the tail apparatus are subsequently added (38). Finally, addition of the tail needle trimer and six tailspike trimers completes the virion assembly process (39). Based on protein sequence comparisons, mosaicism of the tailspike protein suggests the N-terminal domain of this protein is responsible for binding to the capsid via the P22 gp10 homolog (34). This N-terminal domain is conserved within the genus, as P22, Sf6, and CUS-3 share similar sequences in this region, but their C-terminal domains vary in both sequence and the substrates they degrade (40). This is discussed in greater detail below.
Numerous structures of Sf6 complexes and proteins have been resolved. These include the procapsid, capsid, and virion resolved by cryo–electron microscopy (39,41) (Figure 2) and structures of the small terminase subunit gp1 (42), large terminase subunit gp2 (43), tail adaptor protein gp7 (44), DNA-injection protein gp12 (45), tail needle knob gp9 (46), and tailspike gp14 (47) resolved by X-ray crystallography (Figure 3). These can be used as tools to understand capsid assembly, stability, and the infection process. For example, although the structural genes are closely related to P22, the initiation of DNA packaging varies significantly: Sf6 can initiate packaging anywhere within a 1,800-bp region around the packaging (or pac) site, in contrast to the 120-bp region of P22 (34). This results in Sf6 virions with highly diverse genomic content in a population due to headful packaging of DNA.
Figure 2.
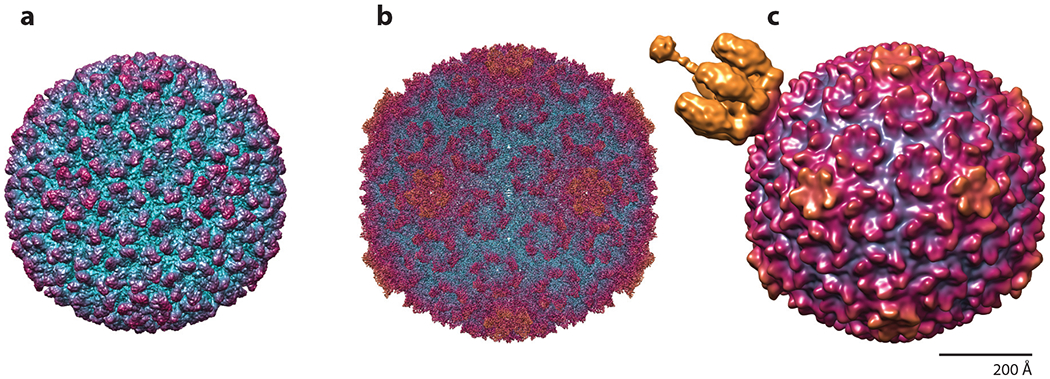
Structures of Sf6 macromolecular complexes, colored according to radial distance. (a) Procapsid at 7.8 Å, EMDB 5724. (b) Capsid at 2.9 Å, EMDB 8314. (c) Virion at 16.0 Å, EMDB 5730. Like other precursor procapsids, it has the smallest diameter and the hexons are skewed with twofold symmetry. Upon expansion, the capsid walls become thinner, the hexons isomerize, and the internal volume increases by ~10%. The mature virion includes the tail apparatus, shown in gold, attached to the portal vertex.
Figure 3.
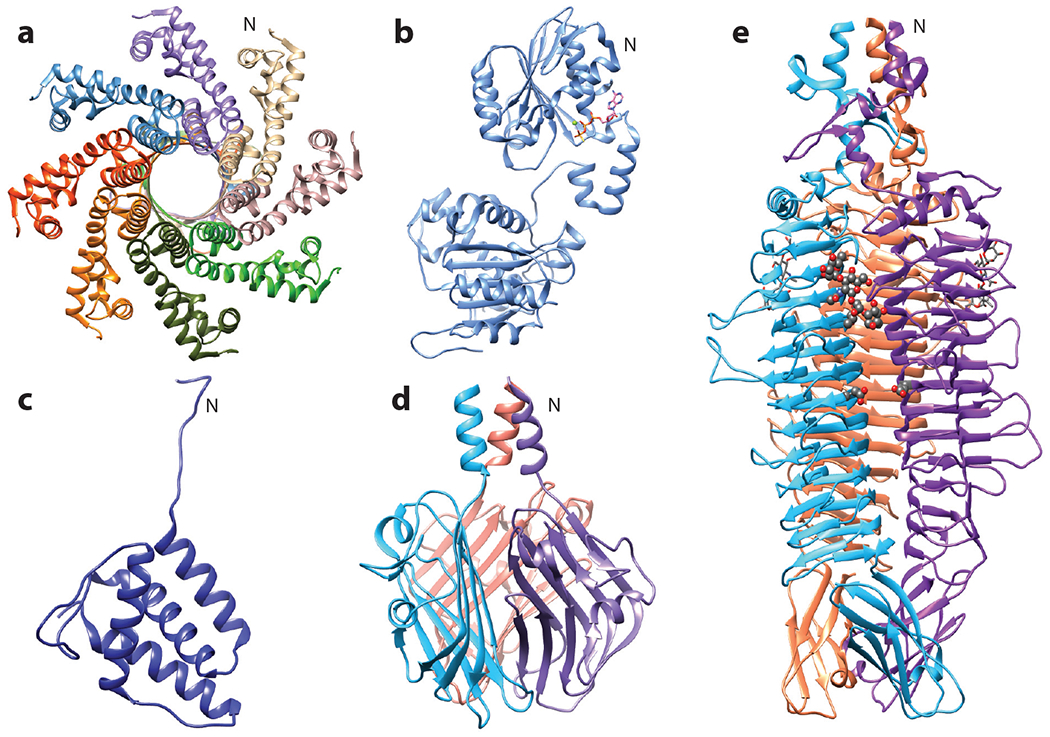
Structures of Sf6 proteins. (a) Small terminase octamer at 1.65 Å, with the N terminus forming the body facing the viewer and the neck facing away, PDB 3HEF. (b) Large terminase monomer bound to ATPγS at 1.89 Å, PDB 4IEE. (c) Tail adaptor monomer at 1.77 Å, PDB 5VGT. (d) Tail needle knob trimer showing the jellyroll fold at 1 Å, PDB 3RWN. (e) Tailspike trimer with one tetrasaccharide molecule and the catalytic active site residues Asp 399 and Glu 366 shown in gray ball-stick model, resolved to 2 Å, PDB 2VBM. For multimeric proteins, individual subunits are shown in different colors. The N indicates the N terminus for one monomer.
The structures of the small (gp1) and large (gp2) terminase subunits responsible for DNA packaging into the procapsid have been resolved individually, with gp1 in an octameric state and gp2 in a monomeric state (42, 43) (Figure 3a,b). Details on the assembly of terminase proteins onto the portal protein of Sf6 are not yet known. Like its P22 homolog, Sf6 gp1 recognizes the pac site via specific interactions with the N terminus. This region also appears to be responsible for the extreme fuzziness of the Sf6 packaging initiation region, as chimeric P22 small terminase with the Sf6 N terminus exhibits an expanded initiation region similar to Sf6; however, the molecular mechanisms determining this difference are not yet clear (48). The C terminus of gp1 interacts with the large terminase gp2, which packages the genome via its N-terminal ATPase activity (43, 48). Specific residues in Sf6 gp1 likely determine DNA specificity, ultimately affecting whether the Sf6 genome or foreign DNA gets packaged into the procapsid, although it is not clear which residues serve this function (49, 50). Once the procapsid senses it is full—hence the term headful packaging—the gp2 C-terminal nuclease domain cleaves and releases the DNA. During packaging, the gp2 nuclease domain is inactive until the capsid is full, signaling the nuclease to cleave DNA. Like other P22viruses, the icosahedrally averaged Sf6 capsid structure contains densely packed DNA, but unlike P22, the Sf6 coat protein forms distinct contacts with the outermost shell of packaged DNA (39, 41). The significance of this has not been directly determined, but other phages such as ϕ29 also share this feature (51), suggesting these interactions may be important in particle stabilization or some other unknown aspect of phage biology. More detailed analyses on these characteristics and structural differences between Sf6, P22, and CUS-3 capsids can be found in References 39 and 41.
The complete virion structure of Sf6 shows details on the architecture and organization of the tail machine. Several crystal structures could be fit into this asymmetric model, including the Sf6 portal gp4, P22 tail adaptor gp10, Sf6 homology model of the tail needle gp9, and Sf6 tailspike gp14. Assembly of the tail onto the capsid begins with the P22 tail adaptor protein homolog, denoted gp7 in Sf6, which connects the portal and tail proteins (Figure 3c). Gp7 binds to the portal via a flexible C-terminal domain and then undergoes conformational changes to recruit additional gp7 proteins. Formation of a dodecameric complex with the portal produces a binding site for the tail nozzle gp8, to which the tail needle gp9 subsequently binds (44). The Sf6 tail needle is unique when compared with other characterized members of P22-like family, as this protein forms a C-terminal globular—or knob—domain (Figure 3d). The function of the tail needle knob is unknown, but it may act extracellularly as a pressure sensor or fulcrum for reorienting the particle prior to membrane penetration; alternatively, it may directly penetrate the Shigella membrane, perhaps governing kinetics of DNA delivery or interacting with inner membrane proteins during infection.
Although added during the last stages of assembly, the tailspike gp14 is the first protein to contact the host cell. For both P22 and Sf6, the tailspike degrades the host LPS via its endorhamnosidase activity, ultimately bringing the phage closer to the membrane and enabling the tail to interact with its secondary receptors. Through cocrystallization of the tailspike with a tetrasaccharide derived from LPS—the product of the O-antigen degradation—the protein’s catalytic site was identified (Figure 3e). Residues in the tetrasaccharide binding pocket were mutagenized, and the ability for the tailspike to bind or cleave substrate was subsequently measured. From these experiments, it was determined that two acidic glutamate and aspartate residues were responsible for cleavage, with two additional glutamate and aspartate residues playing an auxiliary role, perhaps in stabilizing the longer O-antigen chain (52). Mutational studies confirmed this active site was located between two β-strand subunits of tailspike, rather than in a single β-strand subunit seen in P22 and other enzymes involved in degrading polysaccharides. This may suggest potential convergent evolution of P22 and Sf6 tailspikes from different ancestral origins. Whether the Sf6 tailspike also interacts directly with secondary receptors or serves another function during infection is not yet known.
Even though Sf6 has been biophysically characterized, several questions remain unanswered. First, the location and binding mechanisms of the scaffolding protein to the capsid protein during morphogenesis are unclear. The scaffolding protein drives capsid morphogenesis by specifically interacting with the coat protein, catalyzing procapsid formation and stabilizing the structure while the capsid is being polymerized (53). Although the scaffolding protein has been studied in the context of P22, it is unknown whether the same principles hold true for Sf6 and whether the mechanisms are common to all P22viruses. In addition, whether the scaffolding protein is cleaved—as is the case for many viruses—or whether it leaves the capsid to be recycled is unknown. Finally, the structure and location of ejection proteins in the procapsid and/or virion have not been determined. These proteins are likely involved in genome delivery, potentially forming a channel to protect DNA as it travels through the bacterial periplasm. However, very little is known about their role in assembly or infection, their function, or the mechanisms they use during these putative roles (38). During the infection process, some major questions are: How do the phage tail proteins interact with the secondary receptor(s)? How does the structure of the virion change to facilitate genome ejection into the host? And how do ejection proteins function throughout this process, i.e., what is their location and role in the virion during genome ejection? Do they form a tube to protect the DNA during translocation or use some other mechanism? While Sf6 may follow an infection process similar to P22, key differences between these phages suggest these mechanisms still warrant exploration. As mentioned, these two phages use different types of active sites on their tailspikes to bind and cleave their LPS receptors, suggesting potentially different mechanisms of interaction. In addition, they exhibit different requirements and kinetics for genome ejection in vitro (54, 55), suggesting differences in vivo as well. Finally, the presence of the C-terminal tail needle knob in Sf6 may affect infection kinetics and/or interaction with host receptors, although the latter does not seem to be true for other P22-like phages (56).
Developing Model Systems Sf14 and HRP29
Sf14 is one of seven FelixO1-like Shigella bacteriophages isolated from the environment in Michigan during 2016, being closely related to Citrobacter phage Moogle in terms of genome content and nucleotide identity (6). This isolate was chosen for further characterization because of its unusual genome size and uncommon T = 9 capsid geometry. Sf14 and the other Moogle-like bacteriophages of S. flexneri have genomes between 85.0 and 95.0 kbp, which at the time of isolation was representative of only 2% of the deposited phage genomes. The genome also encodes 26 tRNAs, which is significantly more than most phages. The biological significance of both the uncommon genome size and the number of tRNAs is not yet known. Furthermore, the Moogle-like Shigella bacteriophages have been the predominant species isolated from two different environmental locations, suggesting they warrant further study (6, 7).
Structurally, the capsid geometry of Sf14 has been seen in only two other bacteriophages, N4 and Basilisk (57, 58). The Sf14 capsid also possesses decoration proteins, which bind to the external surface of the capsid and, although they do not have a clear function in all systems, can be involved in virion assembly, stability, or infection in others (59–61). The decoration protein of Sf14 consists of an N-terminal α-helical capsid binding domain and two C-terminal Ig-like domains. This arrangement is similar to the T5 pb10 decoration protein, although pb10 has only one C-terminal Ig-like domain (61). Both the location and domain architecture of the Sf14 decoration protein are therefore unique compared with other decoration proteins found in bacteriophage capsids (59–64).
A thorough characterization of Sf14 is ongoing, and various aspects of the Sf14 life cycle are currently unresolved. First, the assembly pathway of Sf14 may contain unique aspects, as morphogenesis of a T = 9 capsid has not been well characterized. Second, the function of the decoration protein has not been tested, and the residues involved in capsid binding are unknown. Third, Sf14 has been shown to infect numerous serotypes of S. flexneri, suggesting it may have a semibroad host range. Understanding how the tail interacts with its host could begin to identify the mechanisms behind this type of host range. Finally, structures of the tail apparatus before and after genome ejection into the host will be informative for the infection process, as this phage uses short tail fibers as opposed to the long and short tail fibers that have previously been characterized for T4. Taken together, Sf14 is poised for further biophysical characterization and studies in bacteriophage diversity.
While Moogle-like viruses were found to be abundant in the environment in these studies, short-tailed podoviruses were surprisingly rare. HRP29—isolated from a phage hunt in Lincoln, Nebraska—was the only podovirus isolated from phage hunts across multiple years (6, 7). Podoviruses of S. flexneri in general are underrepresented when compared with myoviruses and siphoviruses, at least in the types of environments currently sampled (65). Moreover, the genome of HRP29 has only 10% average nucleotide identity (calculated as percent identity multiplied by percent coverage) to any publicly available phage genome, with its closest relative being KP34 of the Drulisvirus genus. Although rare, efforts to isolate podoviruses will significantly contribute to our understanding of phage diversity, as these have thus far been much more diverse that the commonly isolated myoviruses. Morphological analysis of HRP29 indicates it is structurally similar to both T7 and P22, as the central tail structure resembles T7 but possesses tailspikes rather than tail fibers; thus, this phage may possess a hybrid tail. Mass spectrometry analysis of virions further confirmed the presence of predicted structural proteins, supporting the hybrid tail hypothesis (7). To our knowledge, no other phage with this type of tail has been biophysically characterized to date. Determining the three-dimensional structure and performing more comprehensive biological characterization of HRP29 may indicate whether Drulisviruses use a unique mechanism of host recognition and/or infection. Because HRP29 has similar host specificity to Sf6, infecting only S. flexneri serotype Y, this may suggest similar initial binding but divergent genome ejection strategies.
INTERACTIONS BETWEEN SHIGELLA AND ITS PHAGES
Primary and Secondary Phage Receptors
To successfully infect a host cell, a phage must first recognize and bind to it. After initial contact with the host, many phages of Gram-negative bacteria typically use a two-step process to initiate infection (66). First, they bind weakly to a primary receptor, typically LPS, flagella, or pili. This interaction is reversible, with the phage able to detach and reattach elsewhere on the cell or onto a different one altogether. If this first interaction is favorable, the phage will move closer to the cell membrane actively via enzymatic action or passively during flagellar rotation or pilus retraction. Once at the membrane, the phage then binds irreversibly to a secondary receptor and initiates movement of the genome across the bacterial membranes.
Most of the work regarding Shigella phages has investigated the role of LPS as a primary receptor. LPS is a complex molecule comprising multiple regions, which differ both within and between species of Shigella; of these regions, the O-antigen is the most variable (67, 68) (Figure 4a). Many phages studied thus far specifically use the O-antigen for host identification, while specific receptors for other phages have yet to be determined. The O-antigen is a repeating unit of polysaccharides that is located farthest from the cell surface. Therefore, it is often the first molecule a phage contacts. For S. flexneri, the most basic O-antigen structure is serotype Y, with the repeating unit formed by an N-acetylglucosamine (GlcNAc) sugar followed by three rhamnose sugars. Other serotypes are determined by the pattern of glucosyl, acetyl, or phosphoethanolamine groups attached to this basic repeating unit (19) (Figure 4b). The LPS layer can present a significant physical barrier to phage infection due to strong, stabilizing interactions between molecules (69). Therefore, many phages encode polysaccharide depolymerases (e.g., rhamnosidases, sialidases, lyases) or virion-associated lysins (e.g., glycosidases, amidases, endopeptidases) to penetrate the LPS layer and access the outer membrane (70). This subsequently allows the tail proteins to interact with the secondary protein receptor. For Sf6, this protein receptor can be either OmpA or OmpC (54) (Figure 4c).
Figure 4.
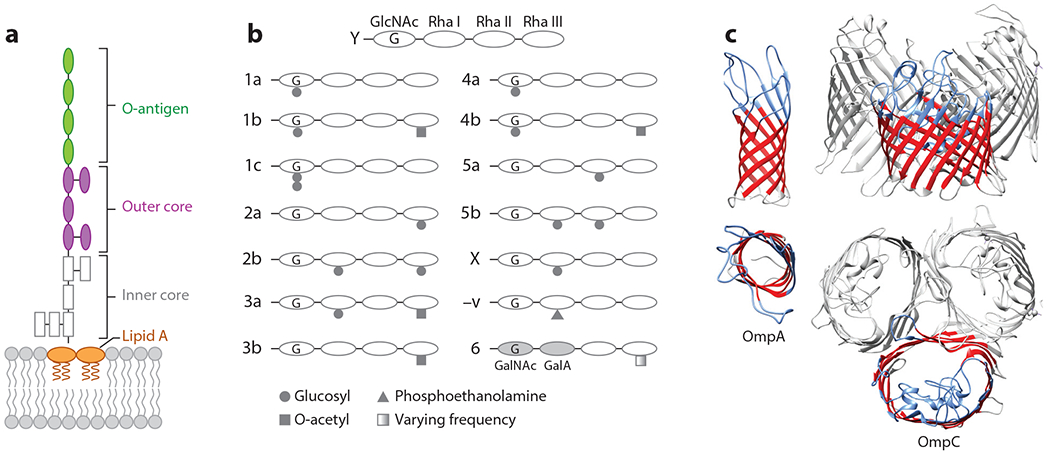
Primary and secondary phage receptors in Shigella flexneri. Primary receptors: (a) simplified diagram of smooth lipopolysaccharide, with (b) illustrating modifications to the O-antigen, producing serotypes indicated to the left. Secondary receptors: (c) outer membrane proteins (Omps) A and C, PDB 3NB3 and 2J1N, respectively.
Upon binding to the secondary receptor, phage virions undergo extensive rearrangements to translocate their genomes into the host cell (71–73). Because this process is irreversible, the interactions between a phage and its secondary receptor are often highly specific to ensure the phage is truly infecting a susceptible host. Numerous Omps are known to act as receptors for bacteriophages, e.g., LamB and FhuA (66). Some bacteriophages have been shown to release their genomes in vitro in the presence of LPS alone (74–77) or LPS plus the purified protein receptor (54, 55, 78). This can act as proxy to understand factors required for bacteriophage host recognition. The inability of LPS alone to trigger Sf6 genome release emphasizes that secondary receptors are necessary for this recognition process.
Sf6 and Its Secondary Receptors
During routine growth, Sf6 typically associates with outer membrane vesicles—which are shed from the host—even after cesium chloride purification (54). Based on sodium dodecyl sulfate-polyacrylamide gel electrophoresis, two host proteins were predominantly found in these purified samples, which were identified by mass spectrometry as OmpA and OmpC and characterized further in the context of phage infection (41). Quantitative plaque assays were used to compare Sf6 growth on a S. flexneri mutant lacking both OmpA and OmpC with the wild-type host. These assays showed that plaque formation was reduced by 90% but only when both proteins were absent—i.e., hosts with individual knockouts of either ompA or ompC did not significantly affect plaque formation. However, because even the double knockout did not have complete immunity against Sf6, this phage can likely use at least one additional receptor, the identity of which is not known.
In vitro, a combination of purified LPS and OmpA was sufficient to trigger Sf6 genome release (54). This result supported OmpA’s role as a secondary receptor, and additional experiments were conducted to determine which residues were necessary for Sf6 infection (55). Although the amino acid identity between E. coli OmpA and S. flexneri OmpA is 99.6%, all differences map to extracellular loops. These differences appear to be sufficient for blocking infection, as E. coli OmpA cannot trigger Sf6 genome ejection. Conversely, OmpA from S. typhimirium, which has identical sequence to S. flexneri OmpA in loops 2 and 4, can induce Sf6 genome release. Efficiency of plating assays with cells expressing mutants of OmpA revealed loops 2 and 4 were indeed important for recognition by Sf6, although no single mutation completely blocked infection. Because loops 2 and 4 are on opposite sides of the OmpA barrel, this suggests Sf6 does not preferentially bind to a single portion of the receptor. Instead, it may recognize the overall surface of the protein. This mode of binding may make it more difficult for the host to acquire simple point mutations to escape phage infection.
Mechanisms of Host Resistance
Because Shigella are unable to defend against phage via CRISPR-mediated immunity or RM systems, there may be extra selective pressure on Omps to avoid phage interactions. If a receptor is not required for growth of the host, complete deletions of phage receptors—e.g., flagella and other Omps—can also be an effective defense against phage. These genes have been deleted in many Shigella genomes (12). This type of scorched earth policy at least thus far seems to be effective, as Shigella can persist despite the apparent abundance of these phages in the environment. To date, no ecological study on Shigella phage abundance has been performed; however, from studies of phage isolation using the enteric bacteria E. coli, Salmonella, and Shigella, phages infecting Shigella have been the most frequently isolated (6, 7). In addition, a majority of the known Shigella phages have come from environmental sources (Table 1). Because some phages such as Sf6 are capable of using more than one secondary receptor, this could be facilitating the continued degradation of Shigella genomes via genomic deletions. Alternatively, another mechanism of defense is dependent on phage itself because many temperate S. flexneri phages encode proteins leading to serotype conversion, as discussed previously (79). While Sf6 can use multiple Omps as secondary receptors, it can infect only one serotype of S. flexneri (23). In many species of bacteria, temperate phages are known to confer immunity to subsequent infection via numerous methods including host cell surface modification, superinfection exclusion, repression, restriction, or prevention of cell growth (80, 81). Although these mechanisms have not been identified in Shigella specifically, uncharacterized genes in Shigella prophages may provide a similar benefit.
Overall, Shigella’s dynamic genomes—whether phage mediated or not—may also explain some of the diversity of Shigella phages. With quickly changing cell surface properties, it may be more advantageous for phage to rely on multiple receptors or to recognize regions of the LPS besides O-antigen in the case of S. flexneri. This leads to several challenges and questions in the field. Despite the apparent similarity to E. coli and instability of its genome, S. flexneri has been surprisingly difficult to manipulate genetically. Some groups have had success with lambda red recombineering (82), transposon mutagenesis (83), or P1 transduction (84), but the success rate is generally several orders of magnitude lower than for its close relative E. coli. Because of this, targeted mutagenesis has been difficult, and studies using high-throughput mutagenesis such as Transposon insertion sequencing or single-gene deletion libraries are even rarer (3). Establishing genetic tools will be essential in furthering Shigella as a model system, where many questions regarding phage-host interactions can be addressed. A thorough characterization of LPS biosynthesis and modification by phages is an active area of research, but much remains to be investigated. In addition, because Shigella genomes are riddled with prophages and appear to be parasitized by numerous temperate phages, the effect of these prophage or lysogenic phage on subsequent infection by lytic phage is largely unknown.
EVOLUTION OF SHIGELLA PHAGES
Few studies have been conducted on the ecology and evolutionary dynamics of Shigella phages. Although characterizing phages from various sources is useful in understanding the environments and conditions in which phage are found, long-term studies have yet to be conducted. Given the differences between Shigella and other model organisms, Shigella phages may employ alternative strategies to infect, persist, and adapt to their hosts. Experimental evolution of Shigella and its phages could be used to investigate some of these. One study done by Dover et al. (85) in 2016 examined the evolution of phage Sf6 when serially passaged on a host strain lacking two of its secondary receptors, OmpA and OmpC. The authors started ten independent lineages of Sf6 growing in an ompA−C− host, with each lineage serially passed for 20 rounds of growth in shaking culture flasks. This corresponded to over a hundred generations for each lineage, from which 82 progeny were isolated for further testing. These contained at least one of three changes: (a) a mutation in gene 61, which encodes holin, a protein important for cell lysis at the end of the phage life cycle; (b) a mutation in the nonprotein coding region corresponding to the major promoter of early genes, PR; and/or (c) a deletion of the nin/IS911 region, with a range of 1.2–4.0 kbp being deleted. Rather than affecting rate of phage adsorption to the host, which would suggest better usage of an additional receptor, these mutations all resulted in faster life cycles of the phage. The first two had been seen in previous evolution experiments (86, 87), but the third had not. The mutations were also additive. Some phage had a combination of the nin/IS911 deletion and a single mutation in either the holin or promoter regions. These mutants exhibited population lysis times up to 50 min earlier than the ancestor, which is significant given the approximately 20-min doubling time of the host under those experimental conditions. The mutation in holin can reasonably explain this faster lysis time. Because Sf6 uses a headful packaging mechanism, where the length of DNA packaged into the capsid is a fixed value, large deletions did not have an obvious explainable benefit.
For Sf6, the nin region is involved in transcriptional regulation of late genes, although two of these genes are nonfunctional due to interruption by the IS911 mobile genetic element. Despite several large deletions in the nin region during evolution, there was no significant difference observed in transcriptional kinetics of late genes between the mutants and the ancestor. Due to Sf6’s headful packaging mechanism, the same length of DNA is packaged regardless of genome size; however, in the case of the deletion mutants, each phage particle contained more than one full-length genome. This results in increased length of the terminal redundancy in packaged genomes, potentially increasing the rate or efficiency of circularization of the phage genome after infection. Alternatively, removing this large region could effectively increase the efficiency of transcribing phage genes by removing extraneous information, reducing competition or interference of RNA polymerase activity, or increasing the concentration of essential genes in the infected cell. Experiments to differentiate between these mechanisms have not yet been conducted; however, none of these mutations were what the authors expected. The original experiment was designed to elucidate structural mechanisms of phage-host interactions (for example, as in Reference 88), yet the primary selective pressure suggested intracellular infection kinetics were more critical than kinetics of phage adsorption to the host.
Because Shigella phage ecology and evolution is largely unexplored, numerous questions remain, and any future studies would likely be informative. Specifically, follow-up studies regarding phage host range expansion and receptor usage may indicate other mechanisms by which Shigella phage evolve in unexpected ways (S.M. Doore et al., unpublished article). Alternatively, these may address the original questions intended in the Sf6 ompA−C− work: How do phage evolve faster and/or more efficient adsorption and genome ejection? Which phage structures are responsible for recognizing secondary receptors, and how do they interact? In addition to these questions, experiments in which the host and phage are allowed to coevolve would indicate how S. flexneri escapes phage infection and how phage persist under a variety of environmental conditions. Finally, because the ecology of Shigella phages is largely unknown despite their apparent abundance, controlled studies would be immensely informative for our understanding of Shigella phage diversity, composition, and prevalence, and how these properties change over time. Since the isolation of Shigella phages from the environment is relatively recent, the relationship between shigellosis incidence, wastewater treatment methods, clinical phage composition, and environmental phage composition is unknown. It appears that Shigella phages are present in the environment regardless of shigellosis outbreaks, but isolation from clinical environments—not induced from Shigella directly—has not been common practice.
CONCLUSION
In the past few decades, a total of 79 Shigella phages have been isolated and characterized. Despite the low apparent prevalence of Shigella in the environment, a majority of these phages originated from environmental samples, with comparatively few induced from clinical isolates. Most phages belong to the Myoviridae family (total: 44), followed by Siphoviridae (16), Podoviridae (10), Ackermannviridae (2), and Microviridae (1), plus a few as-of-yet-unclassified phages. Three model systems are discussed in this review: Sf6, a member of the P22viruses, is well characterized in terms of genomic content and viral structures; Sf14 andHRP29, two new emerging model systems, present opportunities to understand phage diversity and other aspects of Shigella phage biology. For example, the uncommon capsid structure of Sf14 can be used to address interesting questions regarding virion assembly and biophysical characteristics. Similarly, the overall dearth of information regarding relatives of HRP29 makes this an excellent system for future research in phage tail structures and genome evolution. For both Sf14 and HRP29, receptor binding proteins and mechanisms of host interaction are unknown and need further characterization, as these may present novel infection mechanisms. On the bacterial side, identifying the specific host genes required for successful phage propagation would be informative. This has been carried out for Salmonella and its phage P22, but follow-up studies with Shigella and Sf6, Sf14, or HRP29 may identify some commonly used genes as well as different sets of genes. Further exploring these dynamics via experimental evolution could indicate how bacteria might escape phage infection and how phage can either expand their host ranges or overcome resistance—all important aspects when considering phage for applications, whether medical, agricultural, or biotechnological. With the rise of antibiotic resistance and renewed interest in bacteriophage therapy, it is important to understand both phage and host in terms of structure, infection mechanisms, and long-term dynamics of these interactions. Finally, characterizing where Shigella phages are found in the environment, how abundant and diverse they are, and how these aspects change over time would provide information on biotic and abiotic factors that affect phage biology. Since phage have been used as indicators for bacterial presence, these studies may also indicate how and where their Shigella hosts survive in the environment, which could be used to identify and/or prevent outbreaks in the future.
Acknowledgments
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Pupo GM, Lan R, Reeves PR. 2000. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. PNAS 97:10567–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y, Lin K.2012. A phylogenomic analysis of Escherichia coli/Shigella group: implications of genomic features associated with pathogenicity and ecological adaptation. BMC Evol. Biol. 12:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freed NE, Bumann D, Silander OK. 2016. Combining Shigella Tn-seq data with gold-standard E. coli gene deletion data suggests rare transitions between essential and non-essential gene functionality. BMC Microbiol. 16:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rautureau GJP, Palama TL, Canard I, Mirande C, Chatellier S,et al. 2019. Discrimination of Escherichia coli and Shigella spp. by nuclear magnetic resonance based metabolomic characterization of culture media. ACS Infect. Dis. 5:1879–86 [DOI] [PubMed] [Google Scholar]
- 5.Goodridge LD. 2013. Bacteriophages for managing Shigella in various clinical and non-clinical settings. Bacteriophage 3:e25098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doore SM, Schrad JR, Dean WF, Dover JA, Parent KN. 2018. Shigella phages isolated during a dysentery outbreak reveal uncommon structures and broad species diversity. J. Virol. 92(8):e02117–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doore SM, Schrad JR, Perrett HR, Schrad KP, Dean WF, Parent KN. 2019. A cornucopia of Shigella phages from the Cornhusker State. Virology 538:45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diez-Villasenor C, Almendros C, Garcia-Martinez J, Mojica FJ. 2010. Diversity of CRISPR loci in Escherichia coli. Microbiology 156:1351–61 [DOI] [PubMed] [Google Scholar]
- 9.Touchon M, Rocha EP. 2010. The small, slow and specialized CRISPR and anti-CRISPR of Escherichia and Salmonella. PLOS ONE 5:e11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee KF, Ling JM, Kam KM, Clark DR, Shaw PC. 1997. Restriction endonucleases in clinical isolates of Shigella spp. J. Med. Microbiol. 46:949–52 [DOI] [PubMed] [Google Scholar]
- 11.Yang F, Yang J, Zhang X, Chen L, Jiang Y, et al. 2005. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 33:6445–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maurelli AT, Fernandez RE, Bloch CA, Rode CK, Fasano A. 1998. “Black holes” and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. PNAS 95:3943–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakata N, Tobe T, Fukuda I, Suzuki T, Komatsu K, et al. 1993. The absence of a surface protease, OmpT, determines the intercellular spreading ability of Shigella: the relationship between the ompT and kcpA loci. Mol. Microbiol. 9:459–68 [DOI] [PubMed] [Google Scholar]
- 14.Day WA Jr., Fernandez RE, Maurelli AT. 2001. Pathoadaptive mutations that enhance virulence: genetic organization of the cadA regions of Shigella spp. Infect. Immun. 69:7471–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Herelle F 2007. On an invisible microbe antagonistic toward dysenteric bacilli: brief note by Mr. F. D’Herelle, presented by Mr. Roux. 1917. Res. Microbiol. 158:553–54 [DOI] [PubMed] [Google Scholar]
- 16.Hammarstrom E 1947. Bacteriophage classification of Shigella sonnei. Lancet 1:102. [PubMed] [Google Scholar]
- 17.Ketyi I 1974. Role in virulence of Shigella flexneri antigens derived from lysogenic conversion. Infect. Immun. 9:931–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Financsek I, Ketyi I, Sasak W, Jankowski W, Janczura E, Chojnacki T. 1976. Phage-dependent changes in Shigella flexneri type antigen synthesis. Infect. Immun. 14:1290–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Q, Lan R, Wang Y, Wang J, Luo X, et al. 2011. Genesis of a novel Shigella flexneri serotype by sequential infection of serotype-converting bacteriophages SfX and SfI. BMC Microbiol. 11:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mavris M, Manning PA,Morona R. 1997. Mechanism of bacteriophage SfII-mediated serotype conversion in Shigella flexneri. Mol. Microbiol. 26:939–50 [DOI] [PubMed] [Google Scholar]
- 21.Jakhetia R, Talukder KA, Verma NK. 2013. Isolation, characterization and comparative genomics of bacteriophage SfIV: a novel serotype converting phage from Shigella flexneri. BMC Genom. 14:677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huan PT, Whittle BL, Bastin DA, Lindberg AA, Verma NK. 1997. Shigella flexneri type-specific antigen V: cloning, sequencing and characterization of the glucosyl transferase gene of temperate bacteriophage SfV. Gene 195:207–16 [DOI] [PubMed] [Google Scholar]
- 23.Gemski P Jr., Koeltzow DE, Formal SB. 1975. Phage conversion of Shigella flexneri group antigens. Infect. Immun. 11:685–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verma NK, Verma DJ, Huan PT, Lindberg AA. 1993. Cloning and sequencing of the glucosyl transferase-encoding gene from converting bacteriophage X (SfX) of Shigella flexneri. Gene 129:99–101 [DOI] [PubMed] [Google Scholar]
- 25.Jakhetia R, Marri A, Stahle J, Widmalm G, Verma NK. 2014. Serotype-conversion in Shigella flexneri: identification of a novel bacteriophage, Sf101, from a serotype 7a strain. BMC Genom. 15:742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jakhetia R, Verma NK. 2015. Identification and molecular characterisation of a novel Mu-like bacteriophage, SfMu, of Shigella flexneri. PLOS ONE 10:e0124053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Connor TR, Barker CR, Baker KS, Weill FX, Talukder KA, et al. 2015. Species-wide whole genome sequencing reveals historical global spread and recentlocal persistence in Shigella flexneri. eLife 4:e07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang WL, Dunlop SG, Munson PS. 1966. Factors influencing the survival of Shigella in wastewater and irrigation water. J. Water Pollut. Control Fed. 38:1775–81 [PubMed] [Google Scholar]
- 29.Muniesa M, Blanch AR, Lucena F,Jofre J. 2005. Bacteriophages may bias outcome of bacterial enrichment cultures. Appl. Environ. Microbiol. 71:4269–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conner JG, Teschler JK, Jones CJ, Yildiz FH. 2016. Stayingalive: vibrio cholerae’s cycle of environmental survival, transmission, and dissemination In Virulence Mechanisms of Bacterial Pathogens, ed. Kudva IT, Cornick NA, Plummer PJ, Zhang Q, Nicholson TL, Bannantine JP, Bellaire BH, pp. 593–633. Washington, DC: ASM [Google Scholar]
- 31.Islam MS, Hasan MK, Miah MA, Sur GC, Felsenstein A, et al. 1993. Use of the polymerase chain reaction and fluorescent-antibody methods for detecting viable but nonculturable Shigella dysenteriae type 1 in laboratory microcosms. Appl. Environ. Microbiol. 59:536–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu B, Jiang Q, Liu HB, Liu H. 2015. Quantification of viable but nonculturable Salmonella spp. and Shigella spp. during sludge anaerobic digestion and their reactivation during cake storage. J. Appl. Microbiol. 119:1138–47 [DOI] [PubMed] [Google Scholar]
- 33.Lindberg AA, Wollin R, Gemski P, Wohlhieter JA. 1978. Interaction between bacteriophage Sf6 and Shigella flexneri. J. Virol. 27:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casjens S, Winn-Stapley DA, Gilcrease EB, Morona R, Kuhlewein C, et al. 2004. The chromosome of Shigella flexneri bacteriophage Sf6: complete nucleotide sequence, genetic mosaicism, and DNA packaging. J. Mol. Biol. 339:379–94 [DOI] [PubMed] [Google Scholar]
- 35.Hendrix RW, Casjens SR. 2006. Bacteriophage lambda and its genetic neighborhood In The Bacteriophages, ed. Calendar R, pp. 409–47. New York: Plenum; 2nd ed. [Google Scholar]
- 36.Casjens SR, Hendrix RW. 2015. Bacteriophage lambda: early pioneer and still relevant. Virology 479–80:310–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teschke CM, Parent KN. 2010. ‘Let the phage do the work’: using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology 401:119–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Casjens SR,Thuman-Commike PA. 2011Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology 411:393–415 [DOI] [PubMed] [Google Scholar]
- 39.Zhao H, Li K, Lynn AY, Aron KE, Yu G, et al. 2017. Structure of a headful DNA-packaging bacterial virus at 2.9 A resolution by electron cryo-microscopy. PNAS 114:3601–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stummeyer K, Schwarzer D, Claus H, Vogel U, Gerardy-Schahn R, Muhlenhoff M. 2006. Evolution of bacteriophages infecting encapsulated bacteria: lessons from Escherichia coli K1-specific phages. Mol. Microbiol. 60:1123–35 [DOI] [PubMed] [Google Scholar]
- 41.Parent KN, Gilcrease EB, Casjens SR, Baker TS. 2012. Structural evolution of the P22-like phages: comparison of Sf6 and P22 procapsid and virion architectures. Virology 427:177–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao H, Finch CJ, Sequeira RD, Johnson BA,Johnson JE, et al. 2010. Crystal structure of the DNA-recognition component of the bacterial virus Sf6 genome-packaging machine. PNAS 107:1971–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao H, Christensen TE, Kamau YN, Tang L. 2013. Structures of the phage Sf6 large terminase provide new insights into DNA translocation and cleavage. PNAS 110:8075–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang L, Zhao H, An B, Tang L. 2018. High-resolution structure of podovirus tail adaptor suggests repositioning of an octad motif that mediates the sequential tail assembly. PNAS 115:313–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao H, Speir JA, Matsui T, Lin Z, Liang L, et al. 2016. Structure of a bacterial virus DNA-injection protein complex reveals a decameric assembly with a constricted molecular channel. PLOS ONE 11:e0149337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhardwaj A, Molineux IJ, Casjens SR, Cingolani G. 2011. Atomic structure of bacteriophage Sf6 tail needle knob. J. Biol. Chem. 286:30867–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller JJ, Barbirz S, Heinle K, Freiberg A, Seckler R, Heinemann U. 2008. An intersubunit active site between supercoiled parallel β helices in the trimeric tailspike endorhamnosidase of Shigella flexneri phage Sf6. Structure 16:766–75 [DOI] [PubMed] [Google Scholar]
- 48.Leavitt JC, Gilcrease EB, Wilson K, Casjens SR. 2013. Function and horizontal transfer of the small terminase subunit of the tailed bacteriophage Sf6 DNA packaging nanomotor. Virology 440:117–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casjens S, Sampson L, Randall S, Eppler K, Wu H, et al. 1992. Molecular genetic analysis of bacteriophage P22 gene 3 product, a protein involved in the initiation of headful DNA packaging. J. Mol. Biol. 227:1086–99 [DOI] [PubMed] [Google Scholar]
- 50.Roy A, Bhardwaj A, Datta P, Lander GC, Cingolani G. 2012. Small terminase couples viral DNA binding to genome-packaging ATPase activity. Structure 20:1403–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang J, Olson N, Jardine PJ, Grimes S, Anderson DL, Baker TS. 2008. DNA poised for release in bacteriophage phi29. Structure 16:935–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kang Y, Gohlke U, Engstrom O,Hamark C, Scheidt T, et al. 2016. Bacteriophage tailspikes and bacterial O-antigens as a model system to study weak-affinity protein-polysaccharide interactions. J. Am. Chem. Soc. 138:9109–18 [DOI] [PubMed] [Google Scholar]
- 53.Prevelige PE, Fane BA. 2012. Building the machines: scaffolding protein functions during bacteriophage morphogenesis. Adv. Exp. Med. Biol. 726:325–50 [DOI] [PubMed] [Google Scholar]
- 54.Parent KN, Erb ML, Cardone G, Nguyen K, Gilcrease EB, et al. 2014. OmpA and OmpC are critical host factors for bacteriophage Sf6 entry in Shigella. Mol. Microbiol. 92:47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Porcek NB, Parent KN. 2015. Key residues of S. flexneri OmpA mediate infection by bacteriophage Sf6. J. Mol. Biol. 427:1964–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leavitt JC, Gogokhia L, Gilcrease EB, Bhardwaj A, Cingolani G, Casjens SR. 2013. The tip of the tail needle affects the rate of DNA delivery by bacteriophage P22. PLOS ONE 8:e70936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi KH, McPartland J, Kaganman I, Bowman VD, Rothman-Denes LB, Rossmann MG. 2008. Insight into DNA and protein transport in double-stranded DNA viruses: the structure of bacteriophage N4. J. Mol. Biol. 378:726–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grose JH, Belnap DM, Jensen JD, Mathis AD, Prince JT, et al. 2014. The genomes, proteomes, and structures of three novel phages that infect the Bacillus cereus group and carry putative virulence factors. J. Virol. 88:11846–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dokland T, Isaksen ML, Fuller SD, Lindqvist BH. 1993. Capsid localization of the bacteriophage P4 Psu protein. Virology 194:682–87 [DOI] [PubMed] [Google Scholar]
- 60.Lambert S, Yang Q, De Angeles R, Chang JR, Ortega M, et al. 2017. Molecular dissection of the forces responsible for viral capsid assembly and stabilization by decoration proteins. Biochemistry 56:767–78 [DOI] [PubMed] [Google Scholar]
- 61.Vernhes E, Renouard M, Gilquin B, Cuniasse P, Durand D, et al. 2017. High affinity anchoring of the decoration protein pb10 onto the bacteriophage T5 capsid. Sci. Rep. 7:41662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gilcrease EB, Winn-Stapley DA, Hewitt FC, Joss L, Casjens SR. 2005. Nucleotide sequence of the head assembly gene cluster of bacteriophage L and decoration protein characterization. J. Bacteriol. 187:2050–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang L, Gilcrease EB, Casjens SR, Johnson JE. 2006. Highly discriminatory binding of capsid-cementing proteins in bacteriophage L. Structure 14:837–45 [DOI] [PubMed] [Google Scholar]
- 64.Newcomer RL, Schrad JR, Gilcrease EB, Casjens SR, Feig M, et al. 2019. The phage L capsid decoration protein has a novel OB-fold and an unusual capsid binding strategy. eLife 8:e45345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Brien E, Munir M, Marsh T, Heran M, Lesage G, et al. 2017. Diversity of DNA viruses in effluents of membrane bioreactors in Traverse City, MI (USA) and La Grande Motte (France). Water Res. 111:338–45 [DOI] [PubMed] [Google Scholar]
- 66.Bertozzi Silva J, Storms Z, Sauvageau D. 2016. Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 363(4):fnw002. [DOI] [PubMed] [Google Scholar]
- 67.Knirel YA, Kondakova AN, Vinogradov E, Lindner B, Perepelov AV, Shashkov AS. 2011. Lipopolysaccharide core structures and their correlation with genetic groupings of Shigella strains. A novel core variant in Shigella boydii type 16. Glycobiology 21:1362–72 [DOI] [PubMed] [Google Scholar]
- 68.Sun Q, Knirel YA, Lan R, Wang J, Senchenkova SN, et al. 2014. Dissemination and serotype modification potential of pSFxv_2, an O-antigen PEtN modification plasmid in Shigella flexneri. Glycobiology 24:305–13 [DOI] [PubMed] [Google Scholar]
- 69.Frirdich E, Whitfield C. 2005. Lipopolysaccharide inner core oligosaccharide structure and outer membrane stability in human pathogens belonging to the Enterobacteriaceae. J. Endotoxin Res. 11:133–44 [DOI] [PubMed] [Google Scholar]
- 70.Latka A, Maciejewska B, Majkowska-Skrobek G, Briers Y, Drulis-Kawa Z. 2017. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 101:3103–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hu B, Margolin W, Molineux IJ, Liu J. 2013. The bacteriophage T7 virion undergoes extensive structural remodeling during infection. Science 339:576–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu B, Margolin W, Molineux IJ, Liu J. 2015. Structural remodeling of bacteriophage T4 and host membranes during infection initiation. PNAS 112:E4919–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang C, Tu J, Liu J, Molineux IJ. 2019. Structural dynamics of bacteriophage P22 infection initiation revealed by cryo-electron tomography. Nat. Microbiol. 4:1049–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andres D, Baxa U, Hanke C, Seckler R, Barbirz S. 2010. Carbohydrate binding of Salmonella phage P22 tailspike protein and its role during host cell infection. Biochem. Soc. Trans. 38:1386–89 [DOI] [PubMed] [Google Scholar]
- 75.Andres D, Hanke C, Baxa U, Seul A, Barbirz S, Seckler R. 2010. Tailspike interactions with lipopolysaccharide effect DNA ejection from phage P22 particles in vitro. J. Biol. Chem. 285:36768–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Andres D, Roske Y, Doering C, Heinemann U, Seckler R, Barbirz S. 2012. Tail morphology controls DNA release in two Salmonella phages with one lipopolysaccharide receptor recognition system. Mol. Microbiol. 83:1244–53 [DOI] [PubMed] [Google Scholar]
- 77.Broeker NK, Roske Y, Valleriani A, Stephan MS, Andres D, et al. 2019. Time-resolved DNA release from an O-antigen–specific Salmonella bacteriophage with a contractile tail. J. Biol. Chem. 294:11751–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jin Y, Sdao SM, Dover JA, Porcek NB, Knobler CM, et al. 2015. Bacteriophage P22 ejects all of its internal proteins before its genome. Virology 485:128–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Allison GE, Verma NK. 2000. Serotype-converting bacteriophages and O-antigen modification in Shigella flexneri. Trends Microbiol. 8:17–23 [DOI] [PubMed] [Google Scholar]
- 80.Bondy-Denomy J, Qian J, Westra ER, Buckling A, Guttman DS, et al. 2016. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 10:2854–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dedrick RM, Jacobs-Sera D, Bustamante CA, Garlena RA, Mavrich TN, et al. 2017. Prophage-mediated defence against viral attack and viral counter-defence. Nature Microbiol. 2:16251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ranallo RT, Barnoy S, Thakkar S, Urick T, Venkatesan MM. 2006. Developing live Shigella vaccines using λ Red recombineering. FEMS Immunol. Med. Microbiol. 47:462–69 [DOI] [PubMed] [Google Scholar]
- 83.Fisher CR, Davies NM, Wyckoff EE, Feng Z, Oaks EV, Payne SM. 2009. Genetics and virulence association of the Shigella flexneri sit iron transport system. Infect. Immun. 77:1992–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gore AL,Payne SM. 2010. CsrA and Cra influence Shigella flexneri pathogenesis. Infect. Immun. 78:4674–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dover JA, Burmeister AR, Molineux IJ, Parent KN. 2016. Evolved populations of Shigella flexneri phage Sf6 acquire large deletions, altered genomic architecture, and faster life cycles. Genome Biol. Evol. 8:2827–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lewis RJ, Brannigan JA, Offen WA, Smith I, Wilkinson AJ. 1998Anevolutionary link between sporulation and prophage induction in the structure of a repressor:anti-repressor complex. J. Mol. Biol. 283:907–12 [DOI] [PubMed] [Google Scholar]
- 87.Wang IN, Smith DL, Young R. 2000. Holins: the protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 54:799–825 [DOI] [PubMed] [Google Scholar]
- 88.Meyer JR, Dobias DT,Weitz JS, Barrick JE, Quick RT, Lenski RE. 2012. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 335:428–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kaczorowska J, Casey E, Neve H, Franz C, Noben JP, et al. 2019. A quest of great importance—developing a broad spectrum Escherichia coli phage collection. Viruses 11(10):899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mai V, Ukhanova M, Reinhard MK, Li M, Sulakvelidze A.2015. Bacteriophage administration signify-cantly reduces Shigella colonization and shedding by Shigella-challenged mice without deleterious side effects and distortions in the gut microbiota. Bacteriophage 5:e1088124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pasternack G, Sulakvelidze A. 2016. Novel Shigella bacteriophages and the uses thereof. US Patent 15/002,708
- 92.Kim KH, Chang HW, Nam YD, Roh SW, Bae JW. 2010. Phenotypic characterization and genomic analysis of the Shigella sonnei bacteriophage SP18. J. Microbiol. 48:213–22 [DOI] [PubMed] [Google Scholar]
- 93.Schofield DA, Wray DJ, Molineux IJ. 2015. Isolation and development of bioluminescent reporter phages for bacterial dysentery. Eur. J. Clin. Microbiol. Infect. Dis. 34:395–403 [DOI] [PubMed] [Google Scholar]
- 94.Kitajima M, Ishii S, Takagi T, Okabe S. 2019. Complete genome sequence of a novel Myoviridae phage, SfΦ01,infecting Shigella spp. Microbiol. Resour. Announc. 8(23):e00349–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jun JW, Giri SS, Kim HJ, Yun SK, Chi C, et al. 2016. Bacteriophage application to control the contaminated water with Shigella. Sci. Rep. 6:22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hamdi S, Rousseau GM, Labrie SJ, Tremblay DM, Kourda RS, et al. 2017. Characterization of two polyvalent phages infecting Enterobacteriaceae. Sci. Rep. 7:40349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Akter M, Brown N, Clokie M, Yeasmin M, Tareq TM, et al. 2019. Prevalence of Shigella boydii in Bangladesh: isolation and characterization of a rare phage MK-13 that can robustly identify shigellosis caused by Shigella boydii type 1. Front. Microbiol. 10:2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Anany H, Lingohr EJ, Villegas A, Ackermann HW, She YM, et al. 2011. Shigella boydii bacteriophage which resembles Salmonella phage ViI. Virol. J. 8:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jun JW, Yun SK, Kim HJ, Chai JY, Park SC. 2014. Characterization and complete genome sequence of a novel N4-like bacteriophage, pSb-1 infecting Shigella boydii. Res. Microbiol. 165:671–78 [DOI] [PubMed] [Google Scholar]
- 100.Carter CC, Fierer J, Chiu WW, Looney DJ, Strain M, Mehta SR. 2016. A novel shiga toxin 1a-converting bacteriophage of Shigella sonnei with close relationship to shiga toxin 2-converting pages of Escherichia coli. Open Forum Infect. Dis. 3 :ofw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gray MD, Lampel KA, Strockbine NA, Fernandez RE, Melton-Celsa AR,Maurelli AT. 2014. Clinical isolates of Shiga toxin 1a-producing Shigella flexneri with an epidemiological link to recent travel to Hispaniola. Emerg. Infect. Dis. 20:1669–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Toth I, Svab D, Balint B, Brown-Jaque M, Maroti G. 2016. Comparative analysis of the Shiga toxin converting bacteriophage first detected in Shigella sonnei. Infect. Genet. Evol. 37:150–57 [DOI] [PubMed] [Google Scholar]
- 103.Jun JW, Kim JH, Shin SP, Han JE, Chai JY, Park SC. 2013. Characterization and complete genome sequence of the Shigella bacteriophage pSf-1. Res. Microbiol. 164:979–86 [DOI] [PubMed] [Google Scholar]
- 104.Shahin K, Bouzari M, Wang R. 2018. Isolation, characterization and genomic analysis of a novel lytic bacteriophage vB_SsoS-ISF002 infecting Shigella sonnei and Shigella flexneri. J. Med. Microbiol. 67:376–86 [DOI] [PubMed] [Google Scholar]
- 105.Shahin K, Bouzari M. 2018. Bacteriophage application for biocontrolling Shigella flexneri in contaminated foods. J. Food Sci. Technol. 55:550–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ahamed ST, Roy B, Basu U, Dutta S, Ghosh AN, et al. 2019. Genomic and proteomic characterizations of Sfin-1, a novel lytic phage infecting multidrug-resistant Shigella spp. and Escherichia coli C. Front. Microbiol. 10:1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Llanos-Chea A, Citorik RJ, Nickerson KP, Ingano L, Serena G, et al. 2019. Bacteriophage therapy testing against Shigella flexneri in a novel human intestinal organoid-derived infection model. J. Pediatr. Gastroenterol. Nutr. 68:509–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jun JW, Kim HJ, Yun SK, Chai JY, Lee BC, Park SC. 2016. Isolation and comparative genomic analysis of T1-like Shigella bacteriophage pSf-2. Curr. Microbiol. 72:235–41 [DOI] [PubMed] [Google Scholar]
- 109.Chang HW, Kim KH. 2011. Comparative genomic analysis of bacteriophage EP23 infecting Shigella sonnei and Escherichia coli. J. Microbiol. 49:927–34 [DOI] [PubMed] [Google Scholar]
- 110.George DT, Stephenson DP, Tran E, Morona R, Verma NK. 2013. Complete genome sequence of SfII, a serotype-converting bacteriophage of the highly prevalent Shigella flexneri serotype 2a. Genome Announc. 1(5):e00626–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guan S, Bastin DA, Verma NK. 1999. Functional analysis of the O antigen glucosylation gene cluster of Shigella flexneri bacteriophage SfX. Microbiology 145(part 5):1263–73 [DOI] [PubMed] [Google Scholar]
- 112.Yang C, Wang H, Ma H, Bao R, Liu H, et al. 2018. Characterization and genomic analysis of SFPH2, a novel T7 virus infecting Shigella. Front. Microbiol. 9:3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Svab D, Horvath B, Rohde M, Maroti G, Toth I. 2019. R18C is a new viable P2-like bacteriophage of rabbit origin infecting Citrobacter rodentium and Shigella sonnei strains. Arch. Virol. 164:3157–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.ICTV (Int. Comm. Taxon. Viruses). 2018. Virus taxonomy: 2018b release, Washington, DC, updated July 2018. https://talk.ictvonline.org/taxonomy/ [Google Scholar]