

ABSTRACT
Widely metastatic cancers progress rapidly despite sharing genetic drivers with the primary tumor that seeds them. Our recent work indicates that metastatic pancreatic cancers evolve unique metabolic adaptations that are not genetically encoded. These adaptations harness niche-refined nutrients, such as hepatic glucose, to fuel malignant metaboloepigenetic programs that support widespread metastatic outgrowth.
KEYWORDS: Cancer, metabolism, metastasis, epigenetics, chromatin
By far the most common cause of cancer deaths is distant metastasis, which is a multi-step cascade that requires malignant cells to exit the primary tumor, disseminate in the circulation, seed foreign soils of other organs, and achieve successful metastatic outgrowth. Distant metastasis typically manifests as either limited (oligometastatic) or widely metastatic disease.1 The former progresses more indolently and is often treatable. The latter progresses rapidly and is nearly always lethal with few effective therapies. Unlike the rich genetic driver diversity of oligometastatic cancers, widely metastatic cancers possess surprisingly limited subclonal driver heterogeneity: genetic drivers are often shared by most or all primary tumor subclones and matched metastases alike within the same individual patient(s).1 This raises the possibility that additional mechanisms operate in parallel with preexisting genetic drivers to support or even accelerate widespread metastatic outgrowth.
Pancreatic ductal adenocarcinoma (PDAC) is one of the most striking examples of a widely metastatic cancer. PDAC genetic drivers are selected early in progression to activate unique metabolic adaptations that support survival within a densely fibrotic and nutrient-poor primary tumor stroma.2 In our recent study,3 we reported a more delicate stromal fibrosis pattern with high tumor cellularity in distant (liver, lung) metastases from PDAC patients with widely metastatic disease (for example, Figure 1). These clinical observations led us to hypothesize that widely metastatic PDACs might acquire additional nutrient dependencies beyond those that support tumor growth within densely fibrotic microenvironments.
Figure 1.
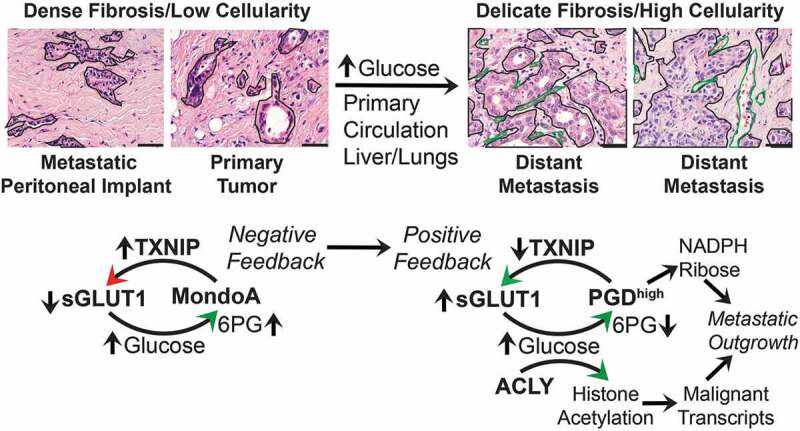
Pancreatic cancers suppress negative feedback of glucose transport to activate metaboloepigenetic programs that support distant metastasis. Left panels: hematoxylin and eosin (H&E) images show that primary pancreatic ductal adenocarcinomas (PDACs) and metastatic peritoneal implants are densely fibrotic with relatively low tumor cellularity. The invasive tumor glands (outlined) are embedded within a dense (scar-like) and hypovascular stroma. Genetic drivers are selected early in this microenvironment to activate scavenging pathways, such as autophagy and macropinocytosis, that promote tumor growth under these hypoglycemic conditions. In this context, MLX interacting protein (MLXIP, better known as MondoA) can appropriately sense glucose uptake if it occurs (possibly indirectly through 6-phosphogluconate (6PG) or other sugar metabolites) and prevent excessive consumption by activating expression of thioredoxin-interacting protein (TXNIP) for endocytosis of glucose transporter protein type 1 (GLUT1) off the cell surface (sGLUT1). Right panels: A subset of PDAC subclones are exposed to microenvironments that are more nutrient/glucose replete. This could occur within unusually well-vascularized regions of primary tumor, within the circulation, or at the metastatic site(s) itself (liver, lungs). This is depicted by typical H&E images from PDAC liver metastases, showing metastatic glands with higher tumor cellularity (outlined in black) growing in a more delicate (thin) fibrotic stroma with a rich microvascular network (green outlines). Such microenvironments may allow selection and clonal expansion of PDAC cells that acquire activation of phosphogluconate dehydrogenase (PGD), which suppresses the MondoA-TXNIP negative feedback loop. Mechanistically, high PGD catalysis (PGDhigh) (over)-consumes glucose-derived 6PG, which prevents MondoA-mediated activation of TXNIP. Suppression of TXNIP allows sGLUT1 to remain at the cell surface with corresponding increases in glucose import. The glucose fuels PGDhigh catalysis by replenishing depleted 6PG substrates. In parallel, glucose-derived citrate is provided to ATP citrate lyase (ACLY) for production of the bulk acetyl groups required to hyperacetylate histones within active chromatin. It is further possible that the PGD protein is itself acetylated into a more (hyper)-active conformation that helps facilitate rapid consumption of 6PG substrates. PGDhigh synthesizes ribulose/ribose (for nucleotides) and nicotinamide adenine dinucleotide phosphate (NADPH, for lipids and redox balance), while global histone hyperacetylation is permissive for full transcriptional activation of malignant gene transcripts. These two glucose-fueled tumorigenic activities synergize to support or even accelerate widespread metastatic outgrowth. H&E scale bars: 50 µm
We first detected abnormally high glucose consumption rates across distant metastases. This identified glucose as a nutrient that is both replete along metastatic routes and highly consumed by metastases. Examination of expression datasets from PDAC patients identified a potential mechanism for enhanced glucose uptake: down-regulation of thioredoxin-interacting protein (TXNIP). TXNIP encodes a multifunctional protein that promotes endocytosis of glucose transporters off the cell surface.4 This activity is tightly regulated by glucose-sensing negative feedback loops that employ TXNIP to prevent excessive glucose uptake when external supplies are replete (Figure 1, left panel).5 Loss of TXNIP might therefore cause glucose consumption rates to rise, as we observed in distant metastases.
We previously reported that catalytic rates of the glucose-metabolizing enzyme phosphogluconate dehydrogenase (PGD) were constitutively elevated in PDAC distant metastases (PGDhigh),6 and that the epigenomic landscape was reprogrammed to depend on PGD.7 Those studies and our new preliminary data raised intriguing questions. How is a negative feedback regulator of glucose (TXNIP) suppressed even when external glucose supplies are replete, and what metastatic fitness advantages are conferred? How is a pro-metastatic oncogene (PGD) activated without gain-of-function mutations, and how does this enzyme reprogram the metastatic epigenome? Because these are questions of glucose metabolism, we postulated that a glucose-fueled metastatic adaptation linking PGD with TXNIP might provide a unifying answer.3
To this end, we first discovered that PGDhigh was required to suppress TXNIP transcripts. That was because TXNIP is transcriptionally regulated by the glucose-sensing transcription factor MLX interacting protein (MLXIP, best known as MondoA5), and MondoA was highly sensitive to intracellular concentrations of the PGD substrate 6-phosphogluconate (6PG) in PGDhigh cells.3 Like other glucose-derived metabolites,5,8 MondoA may monitor 6PG as a surrogate of glucose uptake (6PG = glucose). Although 6PG may be well suited for this role normally, abnormally high PGD catalysis severely depletes steady state 6PG.7 PGDhigh would therefore cause MondoA to (mis)-interpret the resulting low 6PG as low glucose, even when glucose is replete and highly consumed. Indeed, MondoA failed to properly localize to the nucleus and activate TXNIP in PGDhigh PDACs, and this was dependent on high PGD catalysis and low 6PG. Thus, PGDhigh prevented MondoA-mediated transcriptional activation of TXNIP. The resulting failure to up-regulate TXNIP allowed glucose transporters to remain at the cell surface under glucose replete conditions, and glucose consumption rates rose accordingly. PGD-driven suppression of TXNIP was therefore responsible for the enhanced glucose uptake that we had initially observed in PDAC distant metastases.
We next asked if the excess glucose might be used to fuel selectable traits with metastatic fitness advantages. PGDhigh itself could be one such trait, since excess glucose can replenish rate-limiting 6PG substrates6 and the PGD reaction products themselves are highly tumorigenic (NADPH, ribulose, Figure 1).7 External glucose and intact surface transporters were indeed required to support high PGD catalysis, and restoring TXNIP normalized glucose consumption rates, lowered PGD catalysis, and phenocopied the effects of PGDhigh inactivation3,6,7 including impaired metastatic outgrowth in vivo.3
Another selectable trait that could be fueled by excess glucose is the PDAC epigenome. Chromatin is reprogrammed into a globally hyperacetylated state during distant metastasis, and this is permissive for activation of the metastatic transcriptome.7,9 Beyond 6PG, excess glucose can potentially supply the bulk acetyl groups required to hyperacetylate chromatin.10 Consistent with this, external glucose, surface glucose transporters, PGDhigh, mis-localized MondoA, low TXNIP, and ATP citrate lyase (ACLY)10 were all required to maintain hyperacetylated chromatin in PGDhigh PDACs.3 Thus, PGD-driven suppression of TXNIP allowed metastatic PDACs to consume the excess glucose required to both activate PGDhigh and reprogram the metastatic epigenome.
Mechanistically, our findings suggest positive feedback: PGDhigh stimulates excess glucose consumption, and the excess glucose reciprocally stimulates PGDhigh catalysis. This raises the possibility of pro-metastatic positive feedback loops (PGD-glucose) that eliminate negative feedback opposition (TXNIP) (Figure 1). We speculate that this represents a metabolic “feedback exchange” adaptation that does not require genetic hits to remain activated, so long as proper nutrient fuels are available. Because exchange of TXNIP for PGD is a glucose-fueled, self-reinforcing process that regulates global epigenetic state in the absence of a clear genetic underpinning,3 such feedback exchange adaptations are capable of providing malignant cells with heritable metaboloepigenetic programs that parlay niche-refined nutrient reservoirs into selectable traits without genetic constraints.
Funding Statement
The studies described in this commentary were funded by the National Institute of Health (NCI), grant (5R01CA222594), the Vanderbilt GI SPORE 5P50 CA95103, the Vanderbilt Digestive Diseases Research Center 5P30 DK058404-17, and the American Cancer Society IRG-58-009-54.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Iacobuzio-Donahue CA, Litchfield K, Swanton C.. Intratumor heterogeneity reflects clinical disease course. Nat Cancer. 2020;1:1–3. doi: 10.1038/s43018-019-0002-1. [DOI] [PubMed] [Google Scholar]
- 2.Halbrook CJ, Lyssiotis CA.. Employing metabolism to improve the diagnosis and treatment of pancreatic cancer. Cancer Cell. 2017;31:5–19. doi: 10.1016/j.ccell.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Bechard ME, Smalling R, Hayashi A, Zhong Y, Word AE, Campbell SL, Tran AV, Weiss VL, Iacobuzio-Donahue C, Wellen KE, McDonald OG. Pancreatic cancers suppress negative feedback of glucose transport to reprogram chromatin for metastasis. Nat Commun. 2020;11:4055. doi: 10.1038/s41467-020-17839-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, Kahn BB, Cantley LC. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucoseuptakeviaGLUT1.MolCell.2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoltzman CA, Peterson CW, Breen KT, Muoio DM, Billin AN, Ayer DE. Glucose sensing by MondoA:Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc Natl Acad Sci USA. 2008;105(19):6912–6917. doi: 10.1073/pnas.0712199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bechard ME, Word AE, Tran AV, Liu X, Locasale JW, McDonald OG. Pentose conversions support the tumorigenesis of pancreatic cancer distant metastases. Oncogene. 2018;37:5248–5256. doi: 10.1038/s41388-018-0346-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDonald OG, Zhang M, JG R, Bozic I, Allen B, Kundu D, Chatterjee K, Wong F, Jiao Y, ZA K, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017;49:367–376. doi: 10.1038/ng.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoltzman CA, Kaadige MR, Peterson CW, Ayer DE. MondoA senses non-glucose sugars: regulation of thioredoxin-interacting protein (TXNIP) and the hexose transport curb. J Biol Chem. 2011;286:38027–38034. doi: 10.1074/jbc.M111.275503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roe JS, Hwang C-I, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi K, et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell. 2017;170:875–888.e820. doi: 10.1016/j.cell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]