

ABSTRACT
We recently demonstrated that glioblastoma, the most lethal brain cancer, upregulates diacylglycerol O-acyltransferase 1 (DGAT1) to store excess fatty acids into triglycerides to prevent lipotoxicity and promote tumor growth. Targeting DGAT1 resulted in marked tumor cell death by triggering extensive oxidative stress, indicating that DGAT1 could be a promising target for cancer therapy.
KEYWORDS: DGAT1, triglycerides, lipotoxicity, oxidative stress, glioblastoma
Over the past decade, cancer cell metabolism has been extensively investigated and significant progress has been made from understanding glucose metabolism to identifying amino acids, nucleotides, lipids, and redox regulation.1-3 Nevertheless, translation of these exciting biological findings into the clinic has been limited. The hurdles include identification of metabolic targets that are also required for normal cell growth, or the quick adaptation to the metabolic inhibition by tumor cells that activate alternative metabolic pathways. In order to find effective metabolic targets for cancer therapy, identifying those that are unique to cancer cells and essential for tumor growth is required.
Recently, mounting evidence has shown that de novo fatty acid synthesis is greatly upregulated in various types of cancers, including breast, liver, colon and brain cancers, etc. 4,5 Many pharmaceutic companies have developed inhibitors that target the key enzymes regulating fatty acid synthesis, and some have already entered into Phase I/II clinical trials for cancer therapy.3 However, due to the activation of de novo fatty acid synthesis in many normal tissues and organs such as liver, it is highly conceivable that excessive toxicity could arise during the course of continuing treatment, resulting in adverse effects for patients. Therefore, there is a need to find better cancer therapy strategies to leverage the dependency of cancer cells on fatty acids.
Fatty acids are essential lipids that constitute the major components of the cell membrane phospholipids and also serve as important energy resources. Moreover, fatty acids can function as signaling molecules or bind to proteins to regulate cellular function.3 Therefore, it is reasonable to speculate that cancer cells activate de novo synthesis pathways in order to provide sufficient amount of fatty acids for tumor growth. Our previous study revealed that cancer cells depend on the activation of sterol regulatory element-binding protein 1 (SREBP-1), the master transcriptional factor of the fatty acid synthesis pathway, to promote tumor growth.4,6,7 However, in normal cells, it is known that excess free fatty acids (FFA) can disrupt lipid homeostasis and cause lipotoxicity, leading to cell damage. How cancer cells can maintain lipid homeostasis while avoiding lipotoxicity upon synthesis of high levels of fatty acids is poorly understood. Answering this question may lead to identification of effective strategies to treat cancer by disrupting the unique pathway that maintains lipid homeostasis in tumor cells.
Glioblastoma (GBM) is the most lethal brain tumor. Over the past two decades, the average survival time for GBM patients has remained about 12–15 months from diagnosis despite aggressive treatments. One of the main reasons for the failure to improve GBM therapy is our partial understanding of GBM biology. Our recent study published in Cell Metabolism8 demonstrated that GBM cells increase diacylglycerol O-acyltransferase-1 (DGAT1) expression to regulate fatty acid homeostasis, channeling excess FFA into triglycerides (TG) and lipid droplets (LDs) to prevent overactivation of fatty acid β-oxidation from inducing oxidative stress. We had previously shown that GBM tumors contain large amounts of LDs.9 Our current data showed that inhibition of DGAT1 blocks TG synthesis and LD formation, resulting in fatty acids overfeeding mitochondria and marked production of reactive oxygen species (ROS) that damage mitochondria and induce apoptosis to kill GBM cells. Moreover, inhibiting DGAT1 also significantly affected membrane structural lipids, increasing the flow from fatty acids to the major membrane lipids, phosphatidylcholine (PC) and phosphatidylethanolamine (PE), while reducing the flux toward phosphatidylserine (PS), phosphatidylinositol (PI), and phosphatidylglycerol (PG). Moreover, ceramide levels were also increased upon DGAT1 inhibition. Therefore, although it is clear that overactivation of fatty acid oxidation causing high levels of ROS and mitochondrial damage is sufficient to induce GBM cell apoptosis, we cannot rule out the contribution of membrane lipid dysregulation and endoplasmic reticulum (ER) stress to the induction of cell death by DGAT1 inhibition. Moreover, high levels of ROS can also cause lipid oxidation to trigger ferroptosis. Thus, dysregulated membrane lipids, ER stress, and lipid oxidation may strongly potentiate the toxic effects of oxidative stress upon DGAT1 inhibition to induce cancer cell death (Figure 1).
Figure 1.
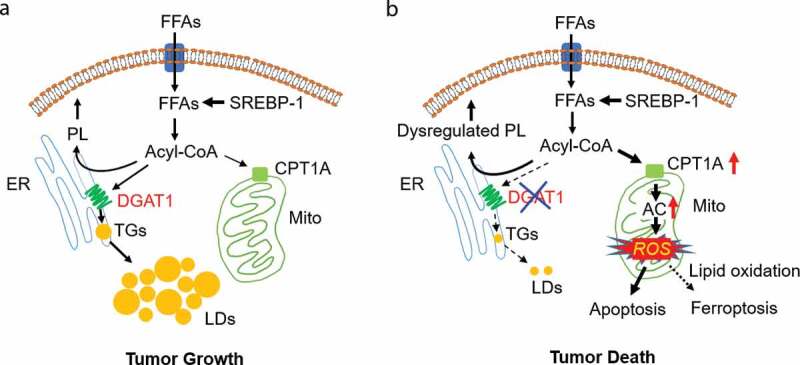
DGAT1: a promising metabolic target for cancer therapy. (a, b) Schematic model illustrating the function of diacylglycerol O-acyltransferase 1 (DGAT1) in regulating lipid homeostasis and lipotoxicity resulting from DGAT1 inhibition in tumor cells. Tumor cells acquire sufficient free fatty acids (FFA) for rapid growth via activation of sterol regulatory element-binding protein 1 (SREBP-1)-regulated de novo synthesis. In addition, FFA uptake also contribute to the pool of FFAs in tumor cells. FFA need to bind to coenzyme A to form acyl-CoA for subsequent synthesis of phospholipids (PL) in the endoplasmic reticulum (ER), or for shuttling to mitochondria for energy production. To prevent excess FFA from inducing toxicity, tumor cells upregulate the expression of DGAT1 to convert excess FFA to triglycerides (TGs) that form lipid droplets (LDs) for storage in the cytosol of cells (a). Targeting DGAT1 will cause the dysregulation of lipid homeostasis and the increase of carnitine palmitoyltransferase 1A (CPT1A) expression to convert excess FFA into acylcarnitines (AC) that enter into mitochondria to generate high levels of reactive oxygen species (ROS). ROS damage mitochondria, trigger apoptosis and can also increase lipid oxidation to induce ferroptosis, together killing tumor cells (b)
In humans, the major sites for TG synthesis are the adipose tissues and liver, where this process is regulated by the DGAT1 and DGAT2 enzymes. In the current study,8 we showed that expression of DGAT1 is much higher than that of DGAT2 in GBM. In contrast, DGAT1 expression is very low in normal brain tissues, where TG and LDs are rarely formed. Thus, targeting DGAT1 is likely to specifically exert toxic effects in GBM tumor tissues, while sparing normal brain and other organs. Through the Cancer Genome Atlas (TCGA) database analysis, we found that the transcription level of DGAT1 gene is very high in most types of tumors, including ovarian, prostate, breast, liver, lung, head and neck, melanoma, pancreas, sarcoma, cervical, thymoma, thyroid, and renal cancer.8 Importantly, LDs have also been observed in multiple types of cancers.10 Thus, blocking TG synthesis via DGAT1 targeting may have high therapeutic potential for a broad range of cancers. In addition, we found that GBM cells are more sensitive to DGAT1 pharmacological inhibition under hypoxia than normoxia conditions, and DGAT1 inhibition also induced mitochondrial damage and elevated ROS under hypoxia (data not shown), further supporting DGAT1 is a promising target for cancer therapy as hypoxia frequently occurs in growing tumors.
Future studies
Our laboratory will next focus on testing the currently available DGAT1 inhibitors in various types of cancers, aiming to move effective inhibitors to clinical trials for cancer therapy. We will also need to determine the impact of specific lipid changes, particularly those of structural lipids, caused by DGAT1 inhibition in tumor cells. In addition, further studies determining the underlying mechanisms upregulating DGAT1 in cancer cells are needed as these findings may lead to new avenues for cancer therapy.
Acknowledgments
We appreciate the support from OSUCCC-Pelotonia Idea grant to DG. We thank Dr. Martine Torres for her critical review and helpful comments on the manuscript.
Funding Statement
This work was supported by NINDS and NCI grants R01NS104332, R01NS112935, and R01CA240726 to DG, R01CA227874 to DG/AC and American Cancer Society Research Scholar Grant RSG-14-228-01–CSM to DG.
Disclosure of potential conflicts of interest
No potential conflicts of interest are disclosed.
References
- 1.Cheng C, Ru P, Geng F, Liu J, Yoo J, Wu X, Cheng X, Euthine V, Hu P, Guo J, et al. Glucose-mediated N-glycosylation of SCAP is essential for SREBP-1 activation and tumor growth. Cancer Cell. 2015;28(5):1–3. doi: 10.1016/j.ccell.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo D. SCAP links glucose to lipid metabolism in cancer cells. Mol Cell Oncol. 2016;3(2):e1132120. doi: 10.1080/23723556.2015.1132120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng C, Geng F, Cheng X, Guo D.. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). 2018;38(1):27. doi: 10.1186/s40880-018-0301-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo D, Bell E, Mischel P, Chakravarti A.. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des. 2014;20(15):2619–2626. doi: 10.2174/13816128113199990486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng X, Li J, Guo D. SCAP/SREBPs are central players in lipid metabolism and novel metabolic targets in cancer therapy. Curr Top Med Chem. 2018;18(6):484–493. doi: 10.2174/1568026618666180523104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo D, Hildebrandt IJ, Prins RM, Soto H, Mazzotta MM, Dang J, Czernin J, Shyy JY, Watson AD, Phelps M, et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc Natl Acad Sci USA. 2009;106(31):12932–12937. doi: 10.1073/pnas.0906606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1(5):442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng X, Geng F, Pan M, Wu X, Zhong Y, Wang C, Tian Z, Cheng C, Zhang R, Puduvalli V, et al. Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab. 2020;32(2):229-242. e8. doi: 10.1016/j.cmet.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geng F, Cheng X, Wu X, Yoo JY, Cheng C, Guo JY, Mo X, Ru P, Hurwitz B, Kim S-H, et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin Cancer Res. 2016;22(21):5337–5348. doi: 10.1158/1078-0432.CCR-15-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geng F, Guo D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern Med Rev (Wash D C). 2017;3(5). doi:10.18103/imr.v3i5.443 [DOI] [PMC free article] [PubMed] [Google Scholar]