

Abstract
We present G4-iM Grinder, a system for the localization, characterization and selection of potential G4s, i-Motifs and higher order structures. A robust and highly adaptable search engine identifies all structures that fit the user’s quadruplex definitions. Their biological relevance, in vitro formation probability and presence of known-to-form structures are then used as filters. The outcome is an efficient methodology that helps select the best candidates for a subsequent in vitro analysis or a macroscopic genomic quadruplex assessment. As proof of the analytical capabilities of G4-iM Grinder, the human genome was analyzed for potential G4s and i-Motifs. Many known-to-form structures were identified. New candidates were selected considering their score and appearance frequency. We also focused on locating Potential Higher Order Quadruplex Sequences (PHOQS). We developed a new methodology to predict the most probable subunits of these assemblies and applied it to a PHOQS candidate. Taking the human average density as reference, we examined the genomes of several etiological causes of disease. This first of its class comparative study found many organisms to be very dense in these potential quadruplexes. Many presented already known-to-form-G4s and i-Motifs. These findings suggest the potential quadruplexes have as therapeutic targets for these diseases that currently kill millions worldwide.
INTRODUCTION
Guanine rich nucleic acid sequences are capable of forming four-stranded structures called G-quadruplexes (G4), whilst cytosine-based assemblies can form i-motifs. These DNA and RNA conformations have been studied abundantly in the last few years due to the increasing evidence of their functional role in many living organisms (1,2), yet the natural properties by which they form and work are very much unknown. To identify new structures, in silico predictions are based on in vitro verified paradigms (3–5). Loops (6), tetrad number, run imperfections (7) and the flanking regions of the structures (5,8) all seem to play important roles in the topology and dynamics of these secondary structures.
Several tools for the identification of PQSs (putative G4 sequences) within a given DNA/RNA sequence are accessible to users nowadays. The first engines, such as Quadparser (9) and Quadfinder (10), were based on the folding rule that postulates that four perfect G-runs with shorter loops form the most stable G4s. Hence, results with these algorithms yield structures that usually fit the formula: (G-run {3:5} Loop {1:7})3G-run {3:5}, where the numbers inside the curly brackets are the range of acceptable lengths of the element.
However, many G4s have been identified that do not follow the folding rule. Loop range unconformity, G-run mismatches and bulges have been confirmed in several G4s (7), so a second generation of PQS search engines was designed to include them in the detection process.
QGRS Mapper (3,11) partially addressed these irregularities by relaxing the folding rule to accept G-runs of size 2 and loop lengths of up to 45. The likelihood of G4 formation for each result is defined here through a scoring system that favors short and equal loop lengths and higher quartet presence. Similarly, Quadbase2 (12), ImGQfinder (13) and PQSfinder (4) also follow the folding rule (or a similar regular expression model). Of these, Quadbase2 and ImGQfinder are the more basic search engines that heavily restrict user-defined variable configuration. Quadbase2 can detect a fixed number of bulges within the G-runs of predefined size (3) following a regular expression model, and ImGQfinder considers both mismatches and bulges within G-runs in variating G-run sizes. PQSfinder, to the contrary, grants greater parameter liberty and at the same time tolerates G-run defects, such as bulges and mismatches, in the detection process. Its scoring system has been proven to outmatch that of QGRS Mapper and is able to reduce false positive (PQS that are assumed to form G4 but do not) and false negative results (PQS that are assumed to be unable to form G4 but do). PQSfinder is also able to identify and resolve overlapping PQS, which is of utmost importance, as many G4 sequences overlap and compete for the common nucleotides to form the final structures (14).
Search engines that use the sliding window method and break with the folding rule have also been developed and used to detect potential G4s in a genome. Both G4 potential calculator (15) and G4Hunter (16) use this statistical analysis window that willingly defines neither individual PQS boundaries nor defect types. Hence, they can accommodate all G4-errors in the search at the expense of being unable to examine overlapping structures (as portions of nucleotides are analyzed instead of regular sequences). Results found with G4 potential calculator are then analyzed by their G-run density to determine G4-formation potential in a length independent manner. G4Hunter scoring system instead evaluates the result’s G-richness and C-skewness to also consider the experimental destabilization effect caused by nearby cytosine presence on the G-quadruplex (as C can base pair with G and ultimately hinder G-quartet formation (17)).
The newest approach in the field is the development and use of G4-potential scoring methods based on machine-learning algorithms. These avoid predefined motif definitions and minimize formation assumptions to improve the analytical accuracy on non-standard PQSs, at the cost of obscurity in their predictive features. G4NN, for example (18), employs an artificial neural network to classify the results of a sliding window model into forming and not-forming RNA G4 sequences. In a similar fashion, Quadron uses an artificial intelligence to classify folding rule abiding PQSs that return the results for all the possible nested and overlapping G4 sequences (5).
All quadruplex search models have several drawbacks and limitations despite the advances in the field. For the most part, variable configuration is usually heavily restricted meaning only the same kind of structures can be looked for (Table 1), excluding for example the detection of structures with more than four G-runs in the sequence. Even if only four G-runs can form the G4-tetrads, extra G-runs can also occur in G-quadruplexes (19,20) or as part of a fluctuating structure (21). Additionally, no current search engine considers or calculates genomic PQS frequency from the results. Even if a higher frequency of a PQS does not mean a stronger tendency of in vitro G4 formation, recurrent PQSs that potentially may form part of repetitive nucleotide segments, can be statistically more biologically important or less biologically problematic. Also, higher G4 frequency allows easier and more accessible targets for the current G4-ligands, which in general are not selective between G4s (22–24). As example, we recently published the results of a PQS search in several parasitic genomes whilst considering frequency, and identified numerous highly recurrent potential G4 candidates (25). Most of these had already been described in literature as G4-forming structures; yet other sequences were new, including EBR1 which is repeated 33 times in the genome of Trypanosoma brucei. Despite EBR1 being graded poorly by the engine employed, we confirmed that the recurrent parasitic PQS was able to form G4 in solution even in the absence of cations. Other examples of the use of frequency as a selection filter also exist (26).
Table 1.
Comparison of some of the search engines and analyzers available for use
| QGRS Mapper | G4Hunter | PQSfinder | G4RNA Screener | G4-iM Grinder | |
|---|---|---|---|---|---|
| Format | Web | R script/web | R package | Python script/web | R package |
| Search engine | |||||
| Model | F.R. | S.W. | F.F.R. | S.W. | F.F.R. |
| Run composition | G | G, C | G | G | G, C, T, U, A |
| Run imperfections | N | Y | Y | Y | Y |
| Modulable variables | 5 | 2 | 10 | 3 | 13 |
| Results analysis | |||||
| Structure analysis | Y | Y | Y | Y | Y |
| Structure frequency analysis | N | N | N | N | Y |
| High-order search and analysis | N | N | N | N | Y |
| Structure qualification | N | N | N | N | Y |
| PQS score system | G-Score | G4Hunter | PQSfinder | G4NN (A. N. N.) | Total Score |
| G4Hunter | Frequency | ||||
| cGcC | PQSfinder | ||||
| G4Hunter | |||||
| cGcC |
Structure qualification includes composition analysis and identification of sequences that are already known to form G4 in vitrowithin the results.
Abbreviations: A.N.N.: artificial neural network; F.R.: folding rule; F.F.R.: flexible folding rule; N: no; S.W.: sliding window; Y: yes.
To conclude, none of the search engines has been explicitly designed to detect, analyze and evaluate higher order sequences. These assemblies with great biological potential are the result of very rich genomic G-tracks that form consecutive G4s. These can be assembled into a higher order structure formed by several G4 subunits. The human telomere sequence (hTel) higher order assembly is currently the main focus of this new area of investigation (27–30). Although several different models exist regarding the interactions between the units, the supra-structure has been found to influence the interactions between the hTel G4s and the telomeric proteins compared to individual G4s.
MATERIALS AND METHODS
G4-iM Grinder
Our contribution to the field is focused on solving these limitations in an easy and fast manner for the user. G4-iM Grinder is an in silico tool designed as the starting step of the genomic quadruplex relevance study workflow. It is intended for researchers who want to detect quadruplex therapeutic targets (both known and new) on a genome, and efficiently filter and select the most interesting results to analyze in vitro. The final objective of the algorithm is to save time and resources in finding, evaluating, selecting and confirming these genomic structures.
Three distinct processes constitute G4-iM Grinder: the quadruplex search engine, the quadruplex qualification functions and the quantification functions. These processes tolerate parallelization over several cores to expedite the analysis.
Search engine: G4-iM Grinder’s search engine was developed to allow extensive freedom in the user’s definition of a quadruplex. These parameters are then applied to a fast, reliable and tolerant search motor capable of detecting even potential higher order structures (Supplementary Information S1: G4-iM Grinder’s search and analyzer algorithm). The result is a very flexible algorithm capable of detecting all structures that fit the user’s prerequisites, defined in 13 variables (Supplementary Information S2: Variables, predefined values and examples). These variables all have predefined values that conform to a functional yet broad definition of a flexible folding rule quadruplex disposition (Supplementary Information S3: G-quadruplex and G4-iM Grinder). They can, however, be easily modified if, for example, structures with longer loops, more run bulges and/or shorter run sizes are also to be detected.
Method 1 (M1), Method 2 (M2) and Method 3 (M3) sub-processes constitute the search engine (Figure 1A). M1 locates the runs (M1A) and finds their direct run-relationships (M1B), whilst M2 and M3 analyzes the potential quadruplex structure formation. M2 does this analysis in an overlapping size-restricted manner (to detect quadruplex structures), and M3 in a non-overlapping size unrestricted way (to detect higher order sequences). In each case, the genomic location (M2A and M3A) and the genomic appearance frequency of each sequence is returned (M2B and M3B).
Figure 1.

(A) G4-iM Grinder’s workflow when RunComposition = G to find PQS and PHOQS in a genome. Results are filtered by their scores, their frequency and the presence of known-to-form Quadruplex and known-NOT-to-form sequences. (B) G4-iM Grinder’s workflow results when applied to the human genome. Millions is abbreviated M.
Quadruplex qualification and quantification functions: Several qualification functions were created, updated or adapted to narrow the number of interesting sequences after a search. The traditional qualification approach is to apply scoring systems that link high scores with high in vitro probability of formation as a selection or filtering mechanism. G4-iM Grinder incorporates some of these systems (G4hunter and cGcC) to evaluate its results. PQSfinder scoring algorithm was upgraded using machine learning to overcome its current limitations and can also be used for this purpose. It is now capable of evaluating normal as well as irregular quadruplexes, i-Motifs and higher order sequences (Supplementary Information S5: Scoring models and their adaptations). A final score considering a weighted average formula of each scoring system and the frequency of the sequence (modulated by the variables ‘WeightedParameters’ and ‘FreqWeight’, respectively) can also be calculated as the sequence’s quantitative interest score.
Other functions have been integrated into G4-iM Grinder to complement the selection process. The quantification of a predefined pattern as a percentage of the sequence (for example ‘G’, ‘GGG’ or ‘TTA’) and the localization of in vitro known-to-form quadruplex and known-NOT-to-form sequences can further help select the most interesting sequences (Supplementary Information S6: Explanation of other variables).
Herein, we propose combining these G4-iM Grinder’s capabilities in an efficient workflow. All sequences found within G4-iM Grinder’s results with known-to-form quadruplex structures that are already verified therapeutic targets ready for further investigation. Other sequences should be first filtered by their score. We found that a score of 40 or over is a good threshold that represents over 97% of all known-to-form G4s examined (Supplementary Information S3: G-Quadruplex and G4-iM Grinder), and therefore represent the most plausible to form sequences. Genomic areas of interest (such as genes) or specific nucleotide arrangements within the quadruplex structure can be further used as filters. Alternatively, a frequency threshold can be employed that redirects the focus to the most recurrent high scoring structures. This way with few biophysical assays, many quadruplex targets with potentially greater biological repercussion can be confirmed. Known-NOT-to-form sequences can then be matched to filter out sequences that coincide. The resulting sequences are therefore the most interesting results to start an quadruplex in vitro evaluation.
i-Motifs and G4-iM Grinder
C-rich regions of DNA or RNA have the ability to fold into tetrameric structures known as i-Motif (31,32). These DNA assemblies consist of stranded duplexes sustained by hydrogen interactions between the intercalated nucleotide base pairs C·C+, which are stronger than the canonical G-C base pair when under acidic physiological conditions of temperature and ionic strength (33). Several consecutive Cs/C+s constitutes a run, and four C-runs arrange spatially as the final tetramer. The rest of the nucleotides in between C-runs form the minor or mayor grooves (loops). As an example, the telomeric sequence (CCCTAA)3CCC (34), which can be organized as loops and runs, is able to form an i-Motif, yet the rich in C sequence CTCCCTTCTCCTCTC cannot (35).
G4-iM Grinder was designed to allow the search and evaluation of Putative i-Motifs Sequences (PiMS). Its search engine can locate all sequences that can form these quadruplexes using the same flexible folding rule used for G4s, as both are comprised of runs and loops. This follows previous uses of G4 search engines to detect PiMS for in vitro evaluation (36).
If the user wishes, G4-iM Grinder can use its qualification functions to evaluate the PiMS’s in vitro formation potential. These include the application of the updated PQSfinder, cGcC, G4Hunter and Final.Score scoring algorithms, which operate (for i-Motifs) in an equal but contrary scale to G4s (where bigger negative values mean higher i-Motif probability; bigger positive values mean higher G4 probability).
Potentially, these scoring methods are useful for i-Motif punctuation as the algorithm’s evaluation characteristics (designed for G4s) are also expected to influence C-based structure stability. For example, G4Hunter and cGcC analyze the sequence G and C relationships whilst PQSfinder examines the tetrad size, bulges between tetrads and loop size to assess the potential of the sequences. Ninety-five known-to-form i-Motifs published in literature were located, listed and analyzed with G4-iM Grinder to test the use of these scoring systems in i-Motifs (Supplementary Information S4: i-Motifs and G4-iM Grinder). The mean score ± standard deviation (SD) of all these i-Motifs were: G4Hunter = −49.5 ± 17.0; PQSfinder = −62.1 ± 10.7; score(mean) = −55.8 ± 13.6, which indicates a strong relationship between the absolute high probability formation scores and the actual in vitro formation of the i-Motif (Supplementary Figure S4; Top: boxplots). These score relationships are very similar to that of the known-to-form G4s structures (Supplementary Information S3: Score (mean) = 52.9 ± 13.1) that validate the direct link between G4 in vitro formation and PQS score. In a similar fashion, these i-Motif results validate the link between i-Motif in vitro formation and the PiMS score. Furthermore, 87% of i-Motifs scored ≤ -40, and given the similar (absolute) mean and SD to the G4s examined, we propose to use the same absolute threshold of 40 (score ≤ -40) to filter off these results. The pH dependent melting temperatures (Tm) of all available i-Motifs were also listed and used to find potential relationships with their scores and length. An evident direct correlation linking Tm and scores was measured (especially for pH 5, R2 = 0.6), despite the deviating experimental conditions of the source’s Tm determination (Supplementary Figure S4; bottom: graphs).
RESULTS
Full genomic analysis with G4-iM Grinder
An initial performance run was executed on human chromosome 22 to test the performance of the algorithm (Supplementary Information S7: G4-iM Grinder performance). Then, a full analysis of all the human chromosomes was carried out with predefined variable configuration for the identification of G-based PQS and C-based PiMS. Due to the small difference in results between using different BulgeSize values (as it is constrained by the variable MaxIL, maximum number of bulges in a sequence), it was decided to accept only 1 different nucleotide per run, for a maximum total of 3 per sequence. It is well established that tetrad bulges are a factor for overall structure instability (37) and hence allowing too many of them would result in an increase in the detection rate of low probability in vitro-forming structures plus an increase in the computation processing time.
The complete human genomic analysis (M1, M2 and M3, A and B) took 12.6 h for G-based PQS and 10.4 h for C-based PiMS. In both cases, over 7.2 million potential sequences were detected with M2A (overlapping size-restricted method), and 2.9 million with M3A (non-overlapping size-unrestricted method; Figure 1B). These results include thousands of confirmed G-quadruplexes and i-Motifs. Overall, the non-unique sequences (percentage of sequences with a frequency of occurrence of over 1) represent 26 and 22% of all results (M2A and M3A, respectively). Some of these sequences are repeated over 30 000 times, although the average is six repetitions per non-unique sequence.
Following the workflow described previously, results were filtered by their scores to focus on the most probable to form in vitro sequences (Figure 2A). Doing so for PQS and PiMS reduced the number of results found with M2 3-fold whilst maintaining the ratio of unique to non-unique sequences. Of these (over 2 million results), 375 also had an absolute frequency higher than 100, including the confirmed G-Quadruplexes T30695_or_T30923 (38), 20 h (39), G4CT-pallidum (40), 93del (41) and 22Ag (42). None of these presented within known-NOT-to-form sequences. Similarly, applying the score filter decreased 4-fold (to 0.6 million) the original results of M3. Of these 0.6 million, only 42 sequences also had an absolute frequency of at least 100. The most frequent sequences after applying these filters revealed interesting relationships, including the combination of entries 2 and 4 to give the bigger sequences of entries 6 and 7 of Figure 2B. These combinations identify several widely spread high scoring sequences throughout the human genome, which can potentially form a changeable higher order structure or a fluctuating quadruplex.
Figure 2.

(A) Total found PQS (left) and found PQS after filtering by score (right) within the entire human genome per method of analysis. The results are divided into unique structures (found with a frequency of 1, in gray), and non-unique structures (found with a frequency of more than 1, in black). The percentage regarding the total is also shown in between parenthesis. (B) Top four most frequent PQS found within the filtered by score results per method of analysis. Sequence portions in blue are the detected G-runs, in green the run bulges and in red the loops. Score is the average between G4Hunter and PQSfinder. Abbreviations: Freq is frequency, IL is total run bulges and %G is the percentage of G in the sequence.,
The search for PiMS gave very similar results to that of PQS. Most noticeably, KRC6 (43), hTel (44), cJun (45) and C3T333 (46) were located within the 362 sequences found with M2 that score -40 or less and have a frequency of at least 100.
Potential higher order quadruplex sequences (PHOQS) and their analysis
Using M3A results, the longest of all possible higher order quadruplex sequences (PHOQS) was identified in chromosome 6. This structure potentially involves more than 2700 nucleotides and can be formed by over 300 possible PQS options, yet it was graded poorly because of its many bulges in between G-runs. Hence, the focus was set on the longest structures with the highest probability of formation (score ≥ 50, Table 2). PHOQS found this way include a 2343 long sequence in chromosome 11 (entry 1) and a 1005 segment in the end of chromosome X, rich in the telomeric and other known-to-form G4 sequences (entry 10).
Table 2.
Some of the longest potential higher order quadruplex sequence (PHOQS) found in the human genome, which scores at least 50 located with method 3A (M3A)
| Entry | Chrom. | Start | Length | Runs | IL | Strand | Score | %G | %C | %A | %T | Conf.Quad.Seqs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 11 | 400747 | 2343 | 305 | 13 | − | 58 | 66 | 17 | 7 | 10 | |
| 2 | X | 1587610 | 1666 | 293 | 37 | − | 61 | 70 | 1 | 29 | 1 | |
| 3 | 20 | 64093767 | 1646 | 151 | 13 | + | 53 | 60 | 8 | 13 | 19 | |
| 4 | 2 | 240923448 | 1351 | 156 | 0 | − | 54 | 59 | 9 | 7 | 25 | |
| 5 | X | 328170 | 1240 | 182 | 16 | − | 68 | 74 | 5 | 21 | 1 | |
| 6 | 10 | 131041965 | 1170 | 148 | 2 | + | 56 | 55 | 2 | 22 | 21 | |
| 7 | 7 | 470172 | 1125 | 136 | 0 | − | 54 | 58 | 8 | 17 | 16 | |
| 8 | 8 | 141812709 | 1102 | 142 | 8 | − | 54 | 60 | 9 | 13 | 18 | |
| 9 | 9 | 133668264 | 1089 | 189 | 11 | − | 51 | 66 | 1 | 27 | 6 | |
| 10 | X | 156029890 | 1005 | 164 | 6 | + | 52 | 56 | 0 | 13 | 31 | 26gtel4 (4), 22Ag (68), Tet22 (7), Gia18 (2), Scer21 (1) ,45Ag (51), 26gsc (1) |
| 11 | 2 | 239737366 | 990 | 163 | 4 | − | 60 | 63 | 19 | 4 | 14 | d(G4C2)4 (3) |
| 12 | 3 | 10005 | 978 | 159 | 1 | − | 52 | 55 | 2 | 15 | 29 | TSG24 (44), X3ACT (3), Tet22 (2), G4CT-pallidum (6) |
Conf.Quad.Seqs are identified G4 sequences within the found structure that are known to form in vitro, followed by the times detected in between parenthesis. Score is the mean of G4hunter and PQSfinder.
Abbreviations: Chrom.: chromosome; IL: run bulges.
%G, %C, %A and %T are percentage of that nucleotide in the sequence.
Attention was set on HoEBR1, a relatively small sized (<200 nucleotides to avoid excessive complexity), high scoring and frequent PHOQS. This 118 nucleotide-long PHOQS is repeated four times in the human complementary strand of chromosome 16. Here, it forms part of a nuclear pore complex interacting proteins (NPIPA1 and 2 genes), a polycystin 1 transient receptor potential channel and several other unidentified genes. HoEBR1 can be formed by a combination of its 32 potential PQS subunits (identified by extracting the results from M2A within the location of HoEBR1, Table 3). The known-to-form G4 sequence IV-1242540 was also located within these potential subunits (47).
Table 3.
HoEBR1 analysis and dissection into its core possible PQS subunits. In black and in the first row HoEBR1, and beneath are all the possible PQS units that can potentially form the PHOQS. Sequence portions in blue are the detected G-runs, in green the run bulges and in red the loops. Score is the average between G4Hunter and PQSfinder. C. Q. S. (confirmed quadruplex sequences) are identified G4 structures within the sequence that are known to form in vitro, followed by the times detected in between parenthesis, IL is total run bulges, %G, %C, %A and %T are percentage of that nucleotide in the sequence.
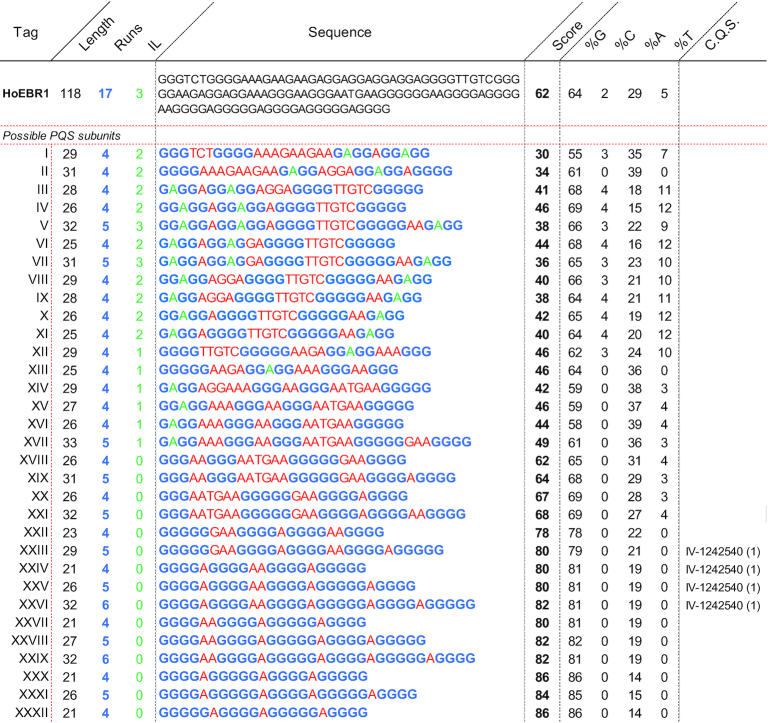
|
All these subunits overlap and will potentially compete to form the most stable structures. An algorithm was developed to predict the most interesting combinations of PQS subunits to form HoEBR1. Such a tool is included in the G4-iM Grinder package under the function GiG.M3Structure. The idea behind the code is to consider the PHOQS as several ‘seats’ for which all the subunits are candidates. When a candidate claims a ‘seat’, it will annul any other candidate with which it shares nucleotides. In our case, HoEBR1 can be potentially be formed by up to four ‘seats’ (Figure 3A).
Figure 3.

(A) The PHOQS HoEBR1 can be arranged into up to four ‘seats’. Greater number of ‘seats’ means smaller loops between units and potentially a gain in structure stability. (B). 1. High-scoring Sequential Allocation (HSA) conformation is based on assigning ‘seats’ sequentially to the highest scoring candidate and known-to-form G4s. Graph. After 10000 iterations of random seat allocation, all 307 candidate conformations of HoEBR1 were found and studied by the mean PQS score of the candidates forming the conformation. In red, blue and green, conformations with 2, 3 and 4 ‘seats’, respectively. 2. Random Allocation High-scoring (RAH) conformation is the highest mean PQS scoring arrangement. (C) Graph. The 307 conformations were normalized by the percentage of the PHOQS that is involved as a PQS to favor shorter loops between subunits and greater ‘seat’ density. 3. The focus was set on the best scoring conformations that present four ‘seats’. These can be occupied by a combination of 10 candidates. 4. Random Allocation normalized High-scoring (RAnH) conformation is the mean normalized highest scoring arrangement. Topologies are not accurate.
At first, ‘seat’ allocation was decided to be sequential, assigning a ‘seat’ first to the best scoring PQS with known-to-form G4 in their sequence (method HSA, Highest-score Sequential Assignment). This process yielded a unique organizational candidate that presented three seats and a poor overall score due to election of subunit XXVI as first ‘seat’. This election ultimately hinders the formation of two other interesting subunits in the tail of HoEBR1 that lowers its overall score (Figure 3B: 1. HSA Conformation).
An alternative method based on randomly assigning ‘seats’ to candidates was also developed and used. After 10 000 iterations, the process identified all possible 307 subunit conformations that can give rise to HoEBR1. This was repeated ten times to make sure no conformations had been excluded. The 307 arrangements were then analyzed by their mean ‘seat’ PQS scores (Figure 3B: Graph), as highest PQS scores are more probable to form G4 in vitro and therefore more probable to be the actual PHOQS subunits. Under such pretences, the highest mean score conformation is a three-‘seat’ structure composed by the PQSs: IV, XXII and XXXII (Figure 3B: 2. RAH Conformation).
The RAH conformation is based solely on PQS scores and therefore does not consider the loop size between subunits in its study. It can be argued that (as happens within G4s) longer loops are likely to decrease overall stability of the greater structure. Hence, the scores of the conformations were also normalized by the percentage of the PHOQS that is involved as PQS for that given conformation (Figure 3C: Graph). This way the method discriminates bigger loops between G4 in favor of higher PQS-density conformations. When applied to HoEBR1, 6 four-‘seat’ configurations scored highly (Scoren > 50, 2% of total conformations; Figure 3C), which are the results of electing 10 possible subunits (Figure 3C: 3). The highest scoring (normalized) arrangement found this way was the combination of the PQS Candidates: I, XII, XXI and XXXII PQS (Figure 3C: 4. RAnH Conformation). Here, over 96% of its nucleotides are involved as PQSs and <3% are loops between ‘seats’.
Potential quadruplex relevance in the genome of humans and other organisms
We used G4-iM Grinder’s results to analyze macroscopically the human genome. PQS and PiMS densities (per 100 000 nucleotides) were analyzed, taking into account both the overall and the most probable to form sequences (those that score at least 40; Figure 4). In addition, the genomic quadruplex uniqueness and the number of already confirmed G4s and i-Motif structures detected were calculated. The complete average genomic human density was then used as a reference to compare the human chromosomes. The search was then extended to other species that cause mortal and/or morbid pathologies in humans, including: viruses, fungi, bacteria and parasites. In doing so, we wanted to identify the diseases that could potentially be most effectively treated by targeting these therapeutic structures. We decided to use the genomic sequence density as the means to compare chromosomes and genomes because of the huge length differences between them all. Doing so enabled the most practical and simple method to efficiently and clearly compare the prevalence of these potential structures independently of the genomic length.
Figure 4.

G4-iM Grinder's Method 2A (M2A) results in humans and in several other disease-causing organisms. PQS and PiMS densities (per 100 000 nucleotides: [(M2A results)/(Genome length)] × 105), percentage of unique sequences (sequence percentage that have an occurrence frequency of 1: [(M2B results which frequency is 1)/(M2A results)] × 100) and number of results with Confirmed to form Quadruplex Sequences (CQS) are shown. Grayish bars are the unfiltered genomic densities whilst the colored bars are the filtered-by-score genomic densities (blue for PQS that score at least 40, yellow for PiMS that score -40 or less). Black dotted line is the human average density, and the red dotted line is the human average density that scores at least 40 for PQS and -40 for PiMS.
The combination of applying different scoring criteria and analytical methods to this -first of its class- quadruplex study allowed a wider context of result interpretation.
Depending on the G4-iM Grinder method employed and the scoring criteria used, the human genome potential structure density (both PQS and PiMS) oscillates between 10 and 300 per 100 000 nucleotides. Chromosome 19 showed the highest density (with over 3-fold the human average) followed by chromosomes 17, 22 and 16. Chromosome Y, by the contrary, revealed the smallest genomic density and the lowest percentage of unique sequences. In general, sequences found in the human genome present high frequency of repetition (with just 74% being unique) and high chance of in vitro formation, being a third of the total results over 40 in score. In all chromosomes, hundreds to thousands of already confirmed G4 and i-Motifs were detected. These values surpass most other species examined. However, some exceptions exist (Figure 4).
On the one hand, Leishmania (and to a less extent the Trypanosoma and Toxoplasma genus) have very rich quadruplex genomes with many known-to-form G4 and i-Motif sequences within. In Leishmania major for example, over 8000 PQS were detected containing the sequence 22Ag (42) with the motif GGGTTA. Also, more than 300 PQS containing T30695 and with less frequency T30177 (38), VEGF (42), Scer21 (48), 26gsc (49), Nef8528 (50), IV-1242540 (47), CEB1 (51), A, CC, C and Bc (52), B-raf (53), A3T (49), 96del (41), 27rap (49) and (TG5T)4 (42) were detected. Regarding i-Motifs, cMyb.S (54), cMyc.PY16 (55), KRC6 (43) and cJun (45) (besides the telomeric i-Motif (44)) were also found. In T. gondii, over 3000 sequences containing the known-to-form Ara24–1 (48) with the motif GGGTTTA, in addition to C, Bc (52), Chla27 (48) and 93del (41), together with the i-Motifs cMyb.S (54), cJun (45), cMyc.C20T (56) and RAD17.2 (57) were also localized. On the other hand, Plasmodium falciparum and Entamoeba histolytica (causers of malaria and amoebiasis, respectively) displayed very low quadruplex densities because of their high genomic AT content (80.6 and 75.2%, respectively). Still, these sequences identified within P. falciparum are the least unique of all analyzed as most are different variants of its telomeric sequence, PfTel -with the motif GGGTTXA (where X can be any nucleotide). The other helminthic parasites and fungi examined present lower densities that those found in the human genome although all had many known to form G4s and i-Motifs within their genomes.
Gram-positive bacteria display very low genomic quadruplex densities all together. The exceptions are the Mycobacterium genus—etiological cause of leprosy and tuberculosis—and the Corynebacterium bacteria, which causes diphtheria. These can surpass and even duplicate the human average. In opposition, Gram-negative bacteria have higher densities for those studied here. Pseudomonas aeruginosa is the most outstanding genome in this group with a genome 3-fold denser than its human counterpart. Brucella melitensis and Neisseria meningitides (causers of brucellosis and meningitis, respectively) follow next in density. Several confirmed known-to-form sequences were also found in Treponema pallidum, (40) indicating that G4s are already interesting targets against syphilis.
The viruses analyzed display a wide range of unique quadruplex densities. Most of them have different PQS and PiMS densities due to being single stranded genomes. For PQS, The Epstein–Barr virus and HIV present higher densities than the human average whilst Zika, Rubella, Rabies and Hepatitis C viruses have similar or slightly lower densities. Within HIV, the known-to-form sequence PRO1 (58) was located. Other viruses including Ebola, Influenza, Measles and Polio viruses were found to be totally void of PQS. When analyzing PiMS, Rubella genome was found to be extremely dense, followed by the Epstein–Barr virus that also presented the known cMyb.S (54) i-Motif. Both viruses have greater densities than the human average whilst Measles, Hepatitis C and HIV viruses have similar or slightly lower densities. In other genomes (including the Polio, Influenza and Zika viruses), no PiMS were found.
DISCUSSION
G4-iM Grinder is a fast, robust and highly adaptable algorithm capable of locating, identifying, qualifying and quantifying quadruplex DNA and RNA structures. These sequences include potential G-quadruplex, i-Motifs and their higher order forms. The adaptation of three scoring systems through machine learning, the structure frequency analysis and the ability to locate already known-to-form G4s and i-Motifs sequences makes G4-iM Grinder’s workflow a practical and easy way to find, filter and select the most interesting quadruplex therapeutic targets in a genome. Furthermore, the modular design and the extensive freedom of variable configuration of G4-iM Grinder gives the user full control of what and how these quadruplex are located and analyzed.
Using G4-iM Grinder, we examined the human genome to find new high scoring and frequent quadruplex G and C-based structures. These potential new targets may be so recurrent because they possibly form part of repetitive segments of conserved transposons. We also identified the longest and most probable higher order sequences to form, some of which have already several known-to-form G4 sequences within. The longest of these structures can involve thousands of nucleotides and hundreds of possible PQS combinations. For example, we analyzed HoEBR1 (a recurrent potential higher order quadruplex sequence with good score) and developed the methodology to calculate the best combinations of PQS subunits to form the structure.
A more macroscopic view of the human genome revealed chromosome 19 as the quadruplex densest chromosome and 13, 18 and Y as the least dense ones (with a fall of nearly 66%). The human genome is still denser and with less unique sequences than most other species examined. However, some parasites and bacteria, such as those in the Leishmania and mycobacterium genus, present very high densities surpassing by several fold the human average. Other bacteria, like Pseudomonas aeruginosa, Neisseria meningitides and Brucella melitensis are also very rich in potential quadruplex targets, as are the Trypanosoma and Toxoplasma parasites. In many of these organisms, we identified several sequences that form G4 and i-Motifs in vitro. The bacteria and viruses inspected barely presented these known-to-form sequences because these differ from those listed in the sources used to build the known-to-form database. Still the pathological causers of AIDS, hepatitis C, rubella, zika, measles and dengue fever showed very high densities of unique sequences that also exceed the human average. The sum of all these results reflects the great potential quadruplex have as therapeutic targets against these diseases that currently kill millions worldwide. Other bacteria, parasites and viruses are poorer in or void of quadruplexes and therefore may require less stringent search criteria to find potential targets (for example accepting G or C-runs of length 2).
Future work includes incorporating G4NN and Quadron (when fully developed) as scoring systems. A Shiny application will be developed for G4-iM Grinder and its subsequent result analysis. The quadruplex database will be maintained online to allow external contributions regarding new in vitro quadruplex sequences for their identification within G4-iM Grinder results.
DATA AVAILABILITY
The package and all the results can be found through GitHub (‘EfresBR/G4iMGrinder’). Instructions on how to install and use the package can be located in the Supplementary Information S8: G4-iM Grinder package. Results and related information can be located in the Supplementary Information S9: Genomes used and results with all methods.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank E. Belmonte-Garcia, B. Belmonte, M. Soto, M. Arévalo, P. Peñalver, S. Heselden, J. L. Mergny and L. Lacroix for their useful insights regarding this topic.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Spanish Ministerio de Economia y Competitividad [CTQ2015- 64275-P].
Conflict of interest statement. None declared.
REFERENCES
- 1. Biffi G., Tannahill D., McCafferty J., Balasubramanian S.. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013; 5:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eddy J., Vallur A.C., Varma S., Liu H., Reinhold W.C., Pommier Y., Maizels N.. G4 motifs correlate with promoter-proximal transcriptional pausing in human genes. Nucleic Acids Res. 2011; 39:4975–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kikin O., D’Antonio L., Bagga P.S.. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006; 34:W676–W682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hon J., Martínek T., Zendulka J., Lexa M.. pqsfinder: an exhaustive and imperfection-tolerant search tool for potential quadruplex-forming sequences in R. Bioinformatics. 2017; 33:3373–3379. [DOI] [PubMed] [Google Scholar]
- 5. Sahakyan A.B., Chambers V.S., Marsico G., Santner T., Di Antonio M., Balasubramanian S.. Machine learning model for sequence-driven DNA G-quadruplex formation. Sci. Rep. 2017; 7:14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guédin A., Gros J., Alberti P., Mergny J.-L.. How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res. 2010; 38:7858–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mukundan V.T., Phan A.T.. Bulges in G-quadruplexes: broadening the definition of G-quadruplex-forming sequences. J. Am. Chem. Soc. 2013; 135:5017–5028. [DOI] [PubMed] [Google Scholar]
- 8. Arora A., Nair D.R., Maiti S.. Effect of flanking bases on quadruplex stability and Watson-Crick duplex competition. FEBS J. 2009; 276:3628–3640. [DOI] [PubMed] [Google Scholar]
- 9. Huppert J.L. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005; 33:2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scaria V., Hariharan M., Arora A., Maiti S.. Quadfinder: server for identification and analysis of quadruplex-forming motifs in nucleotide sequences. Nucleic Acids Res. 2006; 34:W683–W685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bagga P., D’Antonio L., Kikin O., Zappala Z.. QGRS Mapper 2 | G-quadruplex analysis web tool. July 2017, date last accessedhttp://bioinformatics.ramapo.edu/QGRS2/index.php.
- 12. Dhapola P., Chowdhury S.. QuadBase2: web server for multiplexed guanine quadruplex mining and visualization. Nucleic Acids Res. 2016; 44:W277–W283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Varizhuk A., Ischenko D., Tsvetkov V., Novikov R., Kulemin N., Kaluzhny D., Vlasenok M., Naumov V., Smirnov I., Pozmogova G.. The expanding repertoire of G4 DNA structures. Biochimie. 2017; 135:54–62. [DOI] [PubMed] [Google Scholar]
- 14. Agrawal P., Lin C., Mathad R.I., Carver M., Yang D.. The major G-quadruplex formed in the human BCL-2 proximal promoter adopts a parallel structure with a 13-nt loop in K + solution. J. Am. Chem. Soc. 2014; 136:1750–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eddy J., Maizels N.. Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res. 2006; 34:3887–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bedrat A., Lacroix L., Mergny J.-L.. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016; 44:1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beaudoin J.-D., Jodoin R., Perreault J.-P.. New scoring system to identify RNA G-quadruplex folding. Nucleic Acids Res. 2014; 42:1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garant J.-M., Perreault J.-P., Scott M.S.. Motif independent identification of potential RNA G-quadruplexes by G4RNA screener. Bioinformatics. 2017; 33:3532–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Phan A.T., Kuryavyi V., Gaw H.Y., Patel D.J.. Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nat. Chem. Biol. 2005; 1:167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Omaga C.A., Fleming A.M., Burrows C.J.. The fifth domain in the G-quadruplex-forming sequence of the human NEIL3 promoter locks DNA folding in response to oxidative damage. Biochemistry. 2018; 57:2958–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marquevielle J., Kumar M.V.V., Mergny J.-L., Salgado G.F.. 1H, 13C, and 15N chemical shift assignments of a G-quadruplex forming sequence within the KRAS proto-oncogene promoter region. Biomol. NMR Assign. 2018; 12:123–127. [DOI] [PubMed] [Google Scholar]
- 22. Arévalo-Ruiz M., Doria F., Belmonte-Reche E., De Rache A., Campos-Salinas J., Lucas R., Falomir E., Carda M., Pérez-Victoria J.M., Mergny J.-L. et al.. Synthesis, binding properties, and differences in cell uptake of G-quadruplex ligands based on carbohydrate naphthalene diimide conjugates. Chem. Eur. J. 2017; 23:2157–2164. [DOI] [PubMed] [Google Scholar]
- 23. Guillon J., Cohen A., Gueddouda N.M., Das R.N., Moreau S., Ronga L., Savrimoutou S., Basmaciyan L., Monnier A., Monget M. et al.. Design, synthesis and antimalarial activity of novel bis{N -[(pyrrolo[1,2- a]quinoxalin-4-yl)benzyl]-3-aminopropyl}amine derivatives. J. Enzyme Inhib. Med. Chem. 2017; 32:547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ohnmacht S.A., Neidle S.. Small-molecule quadruplex-targeted drug discovery. Bioorg. Med. Chem. Lett. 2014; 24:2602–2612. [DOI] [PubMed] [Google Scholar]
- 25. Belmonte-Reche E., Martínez-García M., Guédin A., Zuffo M., Arévalo-Ruiz M., Doria F., Campos-Salinas J., Maynadier M., López-Rubio J.J., Freccero M. et al.. G-Quadruplex Identification in the Genome of Protozoan Parasites Points to Naphthalene Diimide Ligands as New Antiparasitic Agents. J. Med. Chem. 2018; 61:1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sahakyan A.B., Murat P., Mayer C., Balasubramanian S.. G-quadruplex structures within the 3′ UTR of LINE-1 elements stimulate retrotransposition. Nat. Struct. Mol. Biol. 2017; 24:243–247. [DOI] [PubMed] [Google Scholar]
- 27. Petraccone L., Trent J.O., Chaires J.B.. The tail of the telomere. J. Am. Chem. Soc. 2008; 130:16530–16532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petraccone L., Spink C., Trent J.O., Garbett N.C., Mekmaysy C.S., Giancola C., Chaires J.B.. Structure and stability of higher-order human telomeric quadruplexes. J. Am. Chem. Soc. 2011; 133:20951–20961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vorlícková M., Chládková J., Kejnovská I., Fialová M., Kypr J.. Guanine tetraplex topology of human telomere DNA is governed by the number of (TTAGGG) repeats. Nucleic Acids Res. 2005; 33:5851–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bauer L., Tlučková K., Tóhová P., Viglaský V.. G-quadruplex motifs arranged in tandem occurring in telomeric repeats and the insulin-linked polymorphic region. Biochemistry. 2011; 50:7484–7492. [DOI] [PubMed] [Google Scholar]
- 31. Gehring K., Leroy J.-L., Guéron M.. A tetrameric DNA structure with protonated cytosine-cytosine base pairs. Nature. 1993; 363:561–565. [DOI] [PubMed] [Google Scholar]
- 32. Kang C.H., Berger I., Lockshin C., Ratliff R., Moyzis R., Rich A.. Crystal structure of intercalated four-stranded d(C3T) at 1.4 A resolution. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:11636–11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang B., Rodgers M.T.. Base-pairing energies of proton-bound heterodimers of cytosine and modified cytosines: implications for the stability of DNA i -Motif conformations. J. Am. Chem. Soc. 2014; 136:282–290. [DOI] [PubMed] [Google Scholar]
- 34. Leroy J.-L., Guéron M., Mergny J.-L., Hélène C.. Intramolecular folding of a fragment of the cytosine-rich strand of telomeric DNA into an i-motif. Nucleic Acids Res. 1994; 22:1600–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Geinguenaud F., Liquier J., Brevnov M.G., Petrauskene O.V., Alexeev Y.I., Gromova E.S., Taillandier E.. Parallel self-associated structures formed by T,C-rich sequences at acidic pH. Biochemistry. 2000; 39:12650–12658. [DOI] [PubMed] [Google Scholar]
- 36. Bhavsar-Jog Y.P., Van Dornshuld E., Brooks T.A., Tschumper G.S., Wadkins R.M.. Epigenetic modification, dehydration, and molecular crowding effects on the thermodynamics of i-motif structure formation from C-rich DNA. Biochemistry. 2014; 53:1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Varizhuk A., Ischenko D., Tsvetkov V., Novikov R., Kulemin N., Kaluzhny D., Vlasenok M., Naumov V., Smirnov I., Pozmogova G.. The expanding repertoire of G4 DNA structures. Biochimie. 2017; 135:54–62. [DOI] [PubMed] [Google Scholar]
- 38. Mukundan V.T., Do N.Q., Phan A.T.. HIV-1 integrase inhibitor T30177 forms a stacked dimeric G-quadruplex structure containing bulges. Nucleic Acids Res. 2011; 39:8984–8991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stegle O., Payet L., Mergny J.-L., MacKay D.J.C., Huppert J.L.. Predicting and understanding the stability of G-quadruplexes. Bioinformatics. 2009; 25:i374–i1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rehm C., Hartig J.S.. Ogawa A. In vivo screening for aptazyme-based bacterial riboswitches. Artificial Riboswitches. 2014; 1111:Totowa: Humana Press; 237–249. [DOI] [PubMed] [Google Scholar]
- 41. Zhang S., Wu Y., Zhang W.. G-quadruplex structures and their interaction diversity with ligands. ChemMedChem. 2014; 9:899–911. [DOI] [PubMed] [Google Scholar]
- 42. Tran P.L.T., Largy E., Hamon F., Teulade-Fichou M.-P., Mergny J.-L.. Fluorescence intercalator displacement assay for screening G4 ligands towards a variety of G-quadruplex structures. Biochimie. 2011; 93:1288–1296. [DOI] [PubMed] [Google Scholar]
- 43. Manzini G., Yathindra N., Xodo L.E.. Evidence for intramolecularly folded i-DNA structures in biologically relevant CCC-repeat sequences. Nucleic Acids Res. 1994; 22:4634–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lannes L., Halder S., Krishnan Y., Schwalbe H.. Tuning the pH Response of i-Motif DNA Oligonucleotides. ChemBioChem. 2015; 16:1647–1656. [DOI] [PubMed] [Google Scholar]
- 45. Saxena S., Bansal A., Kukreti S.. Structural polymorphism exhibited by a homopurine·homopyrimidine sequence found at the right end of human c-jun protooncogene. Arch. Biochem. Biophys. 2008; 471:95–108. [DOI] [PubMed] [Google Scholar]
- 46. Gurung S.P., Schwarz C., Hall J.P., Cardin C.J., Brazier J.A.. The importance of loop length on the stability of i-motif structures. Chem. Commun. 2015; 51:5630–5632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Capra J.A., Paeschke K., Singh M., Zakian V.A.. G-quadruplex DNA Sequences Are Evolutionarily Conserved and Associated with Distinct Genomic Features in Saccharomyces cerevisiae. PLoS Comput. Biol. 2010; 6:e1000861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tran P.L.T., Mergny J.-L., Alberti P.. Stability of telomeric G-quadruplexes. Nucleic Acids Res. 2011; 39:3282–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stegle O., Payet L., Mergny J.-L., MacKay D.J.C., Huppert J.L.. Predicting and understanding the stability of G-quadruplexes. Bioinformatics. 2009; 25:i374–i1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Perrone R., Nadai M., Poe J.A., Frasson I., Palumbo M., Palù G., Smithgall T.E., Richter S.N.. Formation of a unique cluster of G-quadruplex structures in the HIV-1 nef coding region: implications for antiviral activity. PLoS ONE. 2013; 8:e73121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Adrian M., Ang D.J., Lech C.J., Heddi B., Nicolas A., Phan A.T.. Structure and conformational dynamics of a stacked dimeric G-quadruplex formed by the human CEB1 minisatellite. J. Am. Chem. Soc. 2014; 136:6297–6305. [DOI] [PubMed] [Google Scholar]
- 52. Dong D.W., Pereira F., Barrett S.P., Kolesar J.E., Cao K., Damas J., Yatsunyk L.A., Johnson F.B., Kaufman B.A.. Association of G-quadruplex forming sequences with human mtDNA deletion breakpoints. BMC Genomics. 2014; 15:677–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wei D., Todd A.K., Zloh M., Gunaratnam M., Parkinson G.N., Neidle S.. Crystal Structure of a Promoter Sequence in the B-raf Gene Reveals an Intertwined Dimer Quadruplex. J. Am. Chem. Soc. 2013; 135:19319–19329. [DOI] [PubMed] [Google Scholar]
- 54. Brazier J.A., Shah A., Brown G.D.. I-Motif formation in gene promoters: unusually stable formation in sequences complementary to known G-quadruplexes. Chem. Commun. 2012; 48:10739–10741. [DOI] [PubMed] [Google Scholar]
- 55. Simonsson T., Pribylova M., Vorlickova M.. A nuclease hypersensitive element in the human c-myc promoter adopts several distinct i-tetraplex structures. Biochem. Biophys. Res. Commun. 2000; 278:158–166. [DOI] [PubMed] [Google Scholar]
- 56. Dai J., Hatzakis E., Hurley L.H., Yang D.. I-motif structures formed in the human c-MYC promoter are highly dynamic–insights into sequence redundancy and I-motif stability. PLoS ONE. 2010; 5:e11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rogers R.A., Fleming A.M., Burrows C.J.. Rapid screen of potential i-motif forming sequences in DNA repair gene promoters. ACS Omega. 2018; 3:9630–9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Amrane S., Kerkour A., Bedrat A., Vialet B., Andreola M.-L., Mergny J.-L.. Topology of a DNA G-quadruplex structure formed in the HIV-1 promoter: a potential target for anti-HIV drug development. J. Am. Chem. Soc. 2014; 136:5249–5252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The package and all the results can be found through GitHub (‘EfresBR/G4iMGrinder’). Instructions on how to install and use the package can be located in the Supplementary Information S8: G4-iM Grinder package. Results and related information can be located in the Supplementary Information S9: Genomes used and results with all methods.