

Abstract
Long-read sequencing has substantial advantages for structural variant discovery and phasing of variants compared to short-read technologies, but the required and optimal read length has not been assessed. In this work, we used long reads simulated from human genomes and evaluated structural variant discovery and variant phasing using current best practice bioinformatics methods. We determined that optimal discovery of structural variants from human genomes can be obtained with reads of minimally 20 kb. Haplotyping variants across genes only reaches its optimum from reads of 100 kb. These findings are important for the design of future long-read sequencing projects.
INTRODUCTION
Long-read sequencing using Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) platforms has profound implications for genomics and genetics (1–4). In contrast to earlier generations of sequencing technologies, the read length routinely reaches tens to hundreds of kilobases and even up to megabases (5,6).
Long-read sequencing leads to more continuous de novo genome assemblies, but this does not necessarily make assembly a trivial task, especially for long segmental duplications and heterozygosity. Long reads also have advantages for genome resequencing in the context of structural variant (SV) discovery and variant phasing. It enables more comprehensive detection of genome-wide structural variation, owing to their higher mappability in repetitive regions and their ability to anchor alignments to both sides of the break point (7–9). SVs are defined as genomic variability of at least 50 bp with a change in copy number or location and include deletions, insertions, inversions and translocations (10). It has been shown that ∼29 000 SVs can be identified per human genome by combining multiple technologies (11), showing that current short-read sequencing approaches leave thousands of variants undiscovered. Phasing variants gains from long reads because of the higher chance of finding variants inherited from the same haplotype on a single read. Phasing has important implications in determining the pathogenicity of pairs of compound heterozygous variants and the effect of cis-regulation (12).
To our knowledge, the dependence of SV detection and variant phasing on the read length has not been formally assessed. In this work, we evaluated the influence of the read length on the accuracy and sensitivity of SV detection and on the length of contiguous stretches of phased nucleotides based on simulated long-read data from recent human genome assemblies.
MATERIALS AND METHODS
Commands for processing and analysis are included in the Supplementary Data. All scripts and commands are available at https://github.com/wdecoster/read_length_SV_discovery.
We include a high-quality phased genome assembly (2.9 Gb) of the Puerto Rican reference individual HG00733, obtained by combining 75× genome coverage of PacBio long reads with additional long-range information of conformational capture sequencing (Hi-C) assembled with FALCON-Phase (13,14) (NCBI Assembly identifier GCA_003634875.1, BioProject PRJNA483067), CHM13hTERT (draft v0.6), a complete hydatidiform mole (46,XX karyotype) (15) combining ONT and PacBio long-read sequencing, 10X Genomics linked-reads and Bionano Genomics optical maps assembled with Canu (16) and NA12878 (OCVW02, PRJEB23027), a well-characterized genome from European descent assembled using nanopore sequencing data assembled with Canu (6,16). The quality metrics and contiguity of the genome assemblies used in this work were evaluated using D-GENIES (17) and QUAST (18) (Supplementary Table S1).
We used SimLoRD (v1.0.2) (19) for simulation of 40× coverage of PacBio reads with a defined length between 100 bp and 700 kb. Reproducibility was assessed by simulating reads for HG00733 in triplicate. Simulated reads were aligned to the GRCh38 reference genome using minimap2 (v2.14) (20) followed by SV calling using Sniffles (v1.0.10) (21). Alignment files were sorted, indexed and downsampled using SAMtools (22). The obtained read depth was assessed with mosdepth (0.2.3) (23). The truth set of SVs was determined using paftools variant calling based on the alignment of the assembly to the GRCh38 reference alignment using minimap2 with the asm5 alignment presets and specific parameters for assembly-to-reference alignment, after splitting the diploid assembly by parental haplotypes when applicable (20,24). The performance metrics precision, recall and F-measure (harmonic mean of precision and recall) were evaluated for SVs with a minimal length of 50 nucleotides using surpyvor (7), which uses SURVIVOR (v1.0.5) for merging VCF (variant call format) files of SVs (25) and cyvcf2 (0.10.0) for parsing VCF files (26). VCF files of SVs were annotated with the read depth of the variants and their flanking sequences using duphold (v0.0.9) (27) and filtered based on the fold change for the read depth of copy number variants (CNVs) relative to its flanking regions using bcftools (v1.9) (28), to enrich for CNVs supported by deviation in read depth.
Single-nucleotide variants (SNVs) from HG00733 as identified in the 1000 Genomes Project (29,30) were phased with the simulated reads by WhatsHap (31), after which contiguous haplotyped segments (phase blocks) were compared to the Ensembl transcript annotation (GRCh38, v95) (32) using BEDTools (33). Data analysis and visualization was performed in Python and Jupyter Notebooks (34) using pandas (v0.23.4) (35), matplotlib (v3.0.0) (36) and joypy (L. Taccari; https://github.com/sbebo/joypy). Commands were parallelized using GNU Parallel (v20181022) (37).
RESULTS AND DISCUSSION
Long-read resequencing has promising applications for genomics, as it enables direct observation of SVs and inference of haplotypes (7,11,38). In this work, we formally assess the impact of increasing read length on the accuracy of SV identification and haplotyping of SNVs. While current long-read sequencing platforms allow sequencing of tens of kb to Mb reads, longer read lengths come with a number of disadvantages: They require more laborious manual DNA extraction from fresh tissue, which may not always be available. Avoiding fragmentation prior to and during library preparation is also essential. Furthermore, striving for ultra-long read lengths also seems to reduce the total yield (6). Due to these limitations and challenges, it is valuable to assess what the required and sufficient read length is to obtain an optimal balance between read length, accuracy and sensitivity. We approach this problem by simulating long reads based on recently assembled human genomes of HG00733 (14), CHM13 (15) and NA12878 (6).
The highest quality assembly in terms of contiguity is the one from HG00733 (Supplementary Table S1), which is also the only separated in maternal and paternal haplotypes. CHM13 is effectively a haploid genome, and NA12878 has both haplotypes superimposed, raising the possibility of incorrectly represented SVs. Notably, repetitive regions play a significant role in this analysis. These regions cannot always be resolved in assemblies, leading to separate contigs. Furthermore, these regions are known hot spots of structural variation, while spurious read alignment also leads to an inflated occurrence of false-positive and false-negative variants. As the HG00733 assembly is the most complete and thus presumably most correctly represents SVs, we mainly base our conclusions on this dataset. The truth set of SVs was based on a direct comparison of the assembly with the reference genome. In the case of HG00733, this alignment and variant calling was done separately per haplotype. This results in the identification of 25 139, 16 776 and 24 653 SVs larger than 50 bp for HG00733, CHM13 and NA12878, respectively, with a variant length distribution comparable to earlier reports (Supplementary Figure S1) (7,11,39).
For each genome, multiple datasets with 40× target coverage and a specific read length starting at 100 bp and up to 700 kb were simulated. A limitation of our analysis is that we use a fixed read length per dataset, while real long-read sequencing experiments typically produce a long-tailed log-normal distribution. Simulation of log-normal distributed reads requires additional complexity of specifying three parameters for the shape of the read length distribution. Testing of this variable read length distribution for HG00733 resulted in the same conclusion as using a single fixed read length (Supplementary Figure S2). We anticipate this simplification is therefore justified to provide approximate guidelines of optimal read length. After alignment of the simulated reads to the human reference genome GRCh38 with minimap2 (20), we obtained the expected read coverage of ∼40× (Supplementary Figure S3). SVs from simulated reads were called using Sniffles and compared to the truth set to calculate the precision, recall and F-measure (Figure 1). An SV is considered concordant (true positive) if it is of the same type and has maximally a pairwise distance of 500 bp between the beginning and end coordinates in the test set and the truth set.
Figure 1.

Precision (with and without filtering on duphold annotation), recall and F-measure (y-axis) for SV call sets of simulated reads from the HG00733 assembly with increasing read length (x-axis). Average of triplicate simulations.
For the HG00733 assembly, the SV precision reaches its maximum already at reads of 1000 bp, while recall no longer increases substantially after 20 kb (Figure 1). The F-measure indicates that optimal performance is reached approximately from reads of 15 kb and longer. Replicate simulations showed a high correlation of performance (Pearson's correlation coefficient >0.97). For the lesser contiguous assemblies, a shorter read length is sufficient to saturate the F-measure. For the haploid CHM13 genome, the performance metrics follow the same shape, but the F-measurement is already saturated at 4-kb reads (Supplementary Figure S4). For this dataset, the precision is higher than HG00733 at shorter read lengths and similar from 1-kb reads onward, while recall saturates at 20 kb, analogous to the findings for HG00733. CHM13 provides a valuable simplification as no heterozygous events are expected, while a diploid genome has been shown before to complicate SV discovery (40). The NA12878 assembly has both haplotypes compressed in a single haploid assembly, as such likely misrepresenting SVs. For this dataset, the F-measure already reaches its maximum from 1-kb reads (Supplementary Figure S5), while strikingly the precision is low, suggesting many false positives, and recall substantially higher. Interestingly, for all datasets recall decreases after 150 kb (discussed later). It is also worth considering that our results might be affected by differences in structural variability between individuals and populations relative to the reference genome (41).
For HG00733, two additional evaluations were performed using (i) lower coverage and (ii) variants filtered based on duphold annotation (27), which adds confidence to CNVs based on read depth information. The conclusion after downsampling the HG00733 alignments to 20× coverage is similar, although recall in general is lower (Supplementary Figure S6). Filtering false-positive CNVs on duphold annotation of read depth changes versus their flanking sequences substantially improved precision, especially for shorter read lengths (Figure 1), while only mildly penalizing recall (Supplementary Figure S7). As read depth changes are only applicable to CNVs, only the accuracy of deletions and duplications is improved, of which the latter is rarely identified by Sniffles in favor of more common insertion variants.
It is worth mentioning that in none of the variant sets from simulated data all variants from the assembly-based truth set are identified, highlighting the limitations of the variant caller or suggesting that some variants can only be identified using de novo assembly- or read depth-based methods. Alternatively, it cannot be fully excluded that the assembly-based SV identification contains false-positive events, is incomplete, or that coordinates of events that are inferred differently. Notably, as the size of the assemblies is 86.6–89.5% compared to the human reference genome GRCh38, some genomic content remained unassembled, presumably containing complex repetitive sequences for which longer reads are beneficial for both assembly and SV calling. As such, our estimate of minimal read length is probably an underestimation for these very long segmental duplications, but a sufficiently accurate guideline for the majority of the SVs in the nonrepetitive genome. With chromosome-scale, haplotype-resolved assemblies, a more accurate guideline could be calculated, a feature that is the promise of more recent ultra-long read libraries, highly accurate reads and tailored genome assembly methods (15,42–44).
Phasing 3.5 million SNVs called from short-read sequencing data shows a continuous increase in the length of phase blocks (contiguous haplotyped genomic fragments) with increasing read length, without reaching a point of saturation within the sizes we tested (Figure 2). Phasing variants across the length of genes is an important application to assess pathogenicity. With reads of 10 kb, ∼50% of the genes can be completely phased. This fraction of completely phased genes increases with read length up to a maximum of 90% with reads of 100 kb or longer (Figure 3). The longest phase blocks are megabases long but are limited by repetitive sequences, regions without identified small variants and structural variation leading to split read alignment. We anticipate that accurate SNV calling methods for long reads would further improve the length of phase blocks, as variants in repetitive sequences cannot be identified by short reads due to ambiguous alignments.
Figure 2.
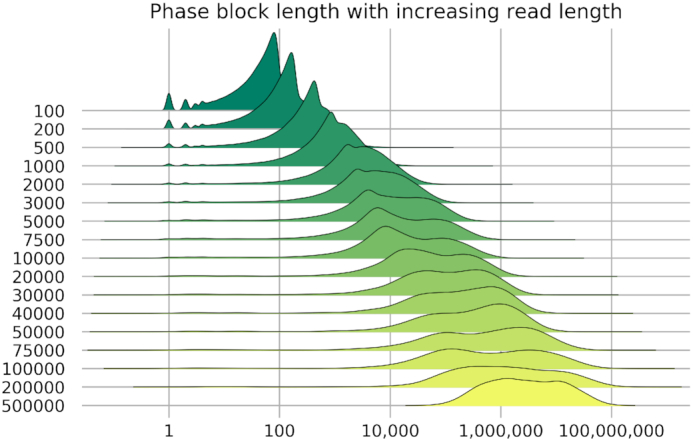
Ridge plot showing the distribution of the length of phase blocks with increasing read length simulated from HG00733. The x-axis is the genomic size of phase blocks, and the y-axis shows the length distribution. Datasets are stacked vertically on separate lines.
Figure 3.
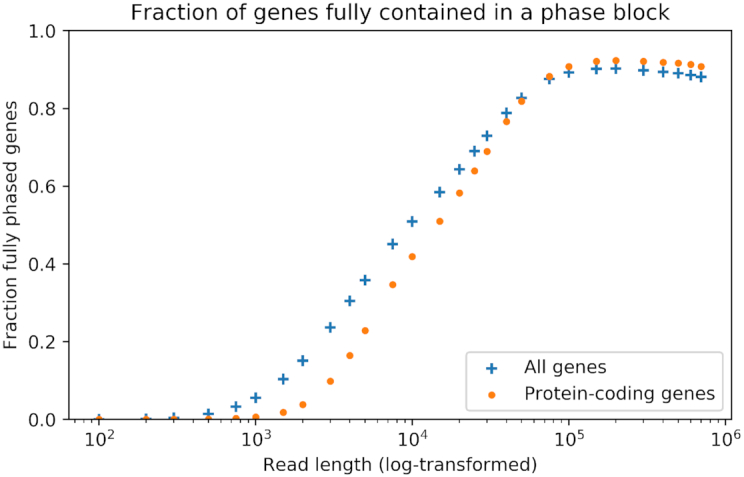
The fraction of genes entirely contained in a single phase block, reaching a plateau at 100 kb, enabling phasing of variants across the entire gene.
With very long reads (>150 kb), we surprisingly see that SV recall decreases while precision increases, and to a lesser extent, the proportion of phased genes decreases. As software, including aligners and SV callers, was not developed based on such extremely long read sizes, we hypothesize this reduction in performance is an analytic limitation and not due to the increased fragment length itself; e.g. highly complex combinations of SVs with multiple break points per read may be missed, leading to inaccurate alignment or break phase blocks. We can assume this can be mitigated by changing the assumptions of tools used for alignment, variant calling and phasing.
CONCLUSION
In the context of human long-read resequencing, our results show optimal performance for SV discovery for read lengths of 20 kb and longer and best phasing across genes from reads of 100 kb only, crucially guiding the experimental design of future long-read sequencing studies.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank employees from Pacific Biosciences and Phase Genomics for making the HG00733 assembly publicly available before publication.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
W.D.C is a recipient of a PhD scholarship from the Flemish Organisation for Innovation and Entrepreneurship (VLAIO) .
Conflict of interest statement. None declared.
REFERENCES
- 1. Loose M.W. The potential impact of nanopore sequencing on human genetics. Hum. Mol. Genet. 2017; 26:R202–R207. [DOI] [PubMed] [Google Scholar]
- 2. Ameur A., Kloosterman W.P., Hestand M.S.. Single-molecule sequencing: towards clinical applications. Trends Biotechnol. 2018; 37:72–85. [DOI] [PubMed] [Google Scholar]
- 3. van Dijk E.L., Jaszczyszyn Y., Naquin D., Thermes C.. The third revolution in sequencing technology. Trends Genet. 2018; 34:666–681. [DOI] [PubMed] [Google Scholar]
- 4. Pollard M.O., Gurdasani D., Mentzer A.J., Porter T., Sandhu M.S.. Long reads: their purpose and place. Hum. Mol. Genet. 2018; 27:R234–R241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Payne A., Holmes N., Rakyan V., Loose M.. BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics. 2018; 35:2193–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jain M., Koren S., Miga K.H., Quick J., Rand A.C., Sasani T.A., Tyson J.R., Beggs A.D., Dilthey A.T., Fiddes I.T. et al.. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018; 36:338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. De Coster W., De Roeck A., De Pooter T., D’Hert S., De Rijk P., Strazisar M., Sleegers K., Van Broeckhoven C.. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 2019; 29:1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chaisson M.J.P., Huddleston J., Dennis M.Y., Sudmant P.H., Malig M., Hormozdiari F., Antonacci F., Surti U., Sandstrom R., Boitano M. et al.. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2015; 517:608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Coster W., Van Broeckhoven C.. Newest methods for detecting structural variations. Trends Biotechnol. 2019; 37:973–982. [DOI] [PubMed] [Google Scholar]
- 10. Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H.-Y. et al.. An integrated map of structural variation in 2,504 human genomes. Nature. 2015; 526:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chaisson M.J.P., Sanders A.D., Zhao X., Malhotra A., Porubsky D., Rausch T., Gardner E.J., Rodriguez O.L., Guo L., Collins R.L. et al.. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019; 10:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castel S.E., Cervera A., Mohammadi P., Aguet F., Reverter F., Wolman A., Guigo R., Iossifov I., Vasileva A., Lappalainen T.. Modified penetrance of coding variants by cis-regulatory variation contributes to disease risk. Nat. Genet. 2018; 50:1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kronenberg Z.N., Hall R.J., Hiendleder S., Smith T.P.L., Sullivan S.T., Williams J.L., Kingan S.B.. FALCON-Phase: integrating PacBio and Hi-C data for phased diploid genomes. 2018; bioRxiv doi:21 May 2018, preprint: not peer reviewed 10.1101/327064. [DOI]
- 14. Porubsky D., Ebert P., Audano P.A., Vollger M.R., Harvey W.T., Munson K.M., Sorensen M., Sulovari A., Haukness M., Ghareghani M. et al.. A fully phased accurate assembly of an individual human genome. 2019; bioRxiv doi:26 November 2019, preprint: not peer reviewed 10.1101/855049. [DOI]
- 15. Miga K.H., Koren S., Rhie A., Vollger M.R., Gershman A., Bzikadze A., Brooks S., Howe E., Porubsky D., Logsdon G.A. et al.. Telomere-to-telomere assembly of a complete human X chromosome. 2019; bioRxiv doi:16 August 2019, preprint: not peer reviewed 10.1101/735928. [DOI] [PMC free article] [PubMed]
- 16. Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017; 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cabanettes F., Klopp C.. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ. 2018; 6:e4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gurevich A., Saveliev V., Vyahhi N., Tesler G.. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013; 29:1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stöcker B.K., Köster J., Rahmann S.. SimLoRD: simulation of long read data. Bioinformatics. 2016; 32:2704–2706. [DOI] [PubMed] [Google Scholar]
- 20. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sedlazeck F.J., Rescheneder P., Smolka M., Fang H., Nattestad M., von Haeseler A., Schatz M.C.. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods. 2018; 15:461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. 1000 Genome Project Data Processing Subgroup . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pedersen B.S., Quinlan A.R.. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics. 2018; 34:867–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H., Bloom J.M., Farjoun Y., Fleharty M., Gauthier L., Neale B., MacArthur D.. A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat. Methods. 2018; 15:595–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jeffares D.C., Jolly C., Hoti M., Speed D., Shaw L., Rallis C., Balloux F., Dessimoz C., Bähler J., Sedlazeck F.J.. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017; 8:14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pedersen B.S., Quinlan A.R.. cyvcf2: fast, flexible variant analysis with Python. Bioinformatics. 2017; 33:1867–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pedersen B.S., Quinlan A.R.. duphold: scalable, depth-based annotation and curation of high-confidence structural variant calls. GigaScience. 2019; 8:giz040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011; 27:2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zheng-Bradley X., Streeter I., Fairley S., Richardson D., Clarke L., Flicek P. 1000 Genomes Project Consortium . Alignment of 1000 Genomes Project reads to reference assembly GRCh38. GigaScience. 2017; 6:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. 1000 Genomes Project Consortium Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin M., Patterson M., Garg S., Fischer S.O., Pisanti N., Klau G.W., Schoenhuth A., Marschall T.. WhatsHap: fast and accurate read-based phasing. 2016; bioRxiv doi:14 November 2016, preprint: not peer reviewed 10.1101/085050. [DOI]
- 32. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G. et al.. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kluyver T., Ragan-Kelley B., Pérez F., Granger B., Bussonnier M., Frederic J., Kelley K., Hamrick J., Grout J., Corlay S. et al.. Loizides F, Schmidt B. Jupyter Notebooks—a publishing format for reproducible computational workflows. Positioning and Power in Academic Publishing: Players, Agents and Agendas. 2016; Amsterdam: IOS Press; 87–90. [Google Scholar]
- 35. McKinney W. pandas: a foundational Python library for data analysis and statistics. Python for High Performance and Scientific Computing. 2011; Seattle. [Google Scholar]
- 36. Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007; 9:90–95. [Google Scholar]
- 37. Tange O. GNU Parallel: the command-line power tool. USENIX Mag. 2011; 36:42–47. [Google Scholar]
- 38. De Coster W., Van Broeckhoven C.. Newest methods for detecting structural variations. Trends Biotechnol. 2019; 37:973–982. [DOI] [PubMed] [Google Scholar]
- 39. Cretu Stancu M., van Roosmalen M.J., Renkens I., Nieboer M.M., Middelkamp S., de Ligt J., Pregno G., Giachino D., Mandrile G., Espejo Valle-Inclan J. et al.. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017; 8:1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huddleston J., Chaisson M.J.P., Steinberg K.M., Warren W., Hoekzema K., Gordon D., Graves-Lindsay T.A., Munson K.M., Kronenberg Z.N., Vives L. et al.. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 2017; 27:677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Audano P.A., Sulovari A., Graves-Lindsay T.A., Cantsilieris S., Sorensen M., Welch A.E., Dougherty M.L., Nelson B.J., Shah A., Dutcher S.K. et al.. Characterizing the major structural variant alleles of the human genome. Cell. 2019; 176:663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wenger A.M., Peluso P., Rowell W.J., Chang P.-C., Hall R.J., Concepcion G.T., Ebler J., Fungtammasan A., Kolesnikov A., Olson N.D. et al.. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019; 37:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shafin K., Pesout T., Lorig-Roach R., Haukness M., Olsen H.E., Bosworth C., Armstrong J., Tigyi K., Maurer N., Koren S. et al.. Efficient de novo assembly of eleven human genomes using PromethION sequencing and a novel nanopore toolkit. 2019; bioRxiv doi:26 July 2019, preprint: not peer reviewed 10.1101/715722. [DOI] [PMC free article] [PubMed]
- 44. Garg S., Fungtammasan A., Carroll A., Chou M., Schmitt A., Zhou X., Mac S., Peluso P., Hatas E., Ghurye J. et al.. Efficient chromosome-scale haplotype-resolved assembly of human genomes. 2019; bioRxiv doi:18 October 2019, preprint: not peer reviewed 10.1101/810341. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.