

Abstract
Bioinformatics tools for fusion transcript detection from RNA-sequencing data are in general developed for identification of novel fusions, which demands a high number of supporting reads and strict filters to avoid false discoveries. As our knowledge of bona fide fusion genes becomes more saturated, there is a need to establish their prevalence with high sensitivity. We present ScaR, a tool that uses a supervised scaffold realignment approach for sensitive fusion detection in RNA-seq data. ScaR detects a set of 130 synthetic fusion transcripts from simulated data at a higher sensitivity compared to established fusion finders. Applied to fusion transcripts potentially involved in testicular germ cell tumors (TGCTs), ScaR detects the fusions RCC1-ABHD12B and CLEC6A-CLEC4D in 9% and 28% of 150 TGCTs, respectively. The fusions were not detected in any of 198 normal testis tissues. Thus, we demonstrate high prevalence of RCC1-ABHD12B and CLEC6A-CLEC4D in TGCTs, and their cancer specific features. Further, we find that RCC1-ABHD12B and CLEC6A-CLEC4D are predominantly expressed in the seminoma and embryonal carcinoma histological subtypes of TGCTs, respectively. In conclusion, ScaR is useful for establishing the frequency of known and validated fusion transcripts in larger data sets and detecting clinically relevant fusion transcripts with high sensitivity.
INTRODUCTION
Fusion genes and fusion transcripts are important in cancer biology and are often entirely cancer specific, making them attractive as biomarkers. Their attention started with the discovery of the Philadelphia chromosome and the resulting BCR-ABL1 fusion in patients with chronic myelogenous leukemia (CML) (1–4). In the 1980s and 90s, multiple recurrent fusions were discovered and characterized with chromosome banding and fluorescence in situ hybridization (FISH). These techniques were biased toward detection of fusion genes in hematological cancers and fusions arising from interchromosomal rearrangements (5). With the advent of high-throughput parallel RNA sequencing (RNA-seq) technology, the nomination rate of novel fusion transcripts in both hematological and solid tumor types has exploded. This is underlined with 20 731 fusion transcripts being detected in 9966 cancer samples (33 cancer types) from The Cancer Genome Atlas (TCGA) consortium alone (6). Importantly, 83% of these fusion transcripts are detected in single cancer samples and are thus not recurrent. This statistic underlines that fusion transcripts are commonly expressed in cancer, often as a result of increased genomic instability, and that only a minority of these are selected for and act as oncogenic drivers. Therefore, to minimize the detection of additional non-recurrent or nomination of even non-existing (false positive) fusion transcripts, most available fusion finder tools have focused on maximizing specificity.
Nevertheless, several recurrent fusion genes have been indicated as targetable molecular alterations in personalized cancer medicine. These include for example fusion genes involving the kinase-encoding genes ALK and ROS1 in non-small cell lung cancer, BCR-ABL1 in CML, and NTRK1, FGFR3 and BRAF in various cancer types (7). In fact, the FDA recently approved Vitrakvi (larotrectinib) as the second tumor-agnostic pan-cancer drug approved for patients harboring NTRK gene fusions without a known acquired resistance mutation, that are metastatic or where surgical resection is likely to result in severe morbidity and have no satisfactory alternative treatments (8). In addition, highly cancer specific fusion transcripts have potential as biomarkers for disease detection, monitoring and predicting treatment response.
The ability to detect these fusions at high sensitivity is therefore paramount. This will enable us to determine true prevalence of known, and previously validated, fusion transcripts in cohorts of cancer patients, where existing fusion finder tools would provide underestimates in efforts of avoiding the scoring of false positives. However, since we in those cases are not searching for novel fusions, we are not risking much by lowering the specificity demands. In more detail, much effort has been invested in developing approaches for fusion transcript detection without any prior knowledge from RNA-seq data, and >40 different tools have been developed for this task (Supplementary Table S1). The performance of fusion finder tools has been shown to vary according to the data set to which they are applied, and none achieve a perfect sensitivity (9,10). Most of the currently available tools use similar approaches to align reads, nominate fusion transcript breakpoints de novo based on supporting reads or read pairs and apply various filters to reduce noise from artifact fusion transcript sequences or the presence of chimeric transcripts in normal cells. A few of the tools available have an option to take a user provided list of known fusion genes and works to force the nominated fusion breakpoints through the list of strict filters (e.g. the –focus parameter in FusionCatcher). However, little effort has been made to develop tools that can validate the presence and prevalence of specific fusion transcripts, with the benefit of a priori knowledge (e.g. fusion breakpoint sequences or genomic junction coordinates) and thereby with increased sensitivity. This is underlined in a case where a simple search of a chimeric sequence in raw sequencing data, using the unix tool ‘grep’ , outperformed the sensitivity of several established fusion finder tools (11). As the knowledge of fusion transcripts and their clinical impact expands together with an increasing number of patients with RNA-seq data available, there is therefore a need for a tool that can establish the presence of already known and validated fusion transcripts in RNA-seq data with superior sensitivity.
A type of cancer for which no recurrent fusion genes have been established as biomarkers or drug targets is testicular germ cell tumors (TGCT), which is the most commonly diagnosed cancer among young men (12). In fact, not much effort has been done to introduce genomics based personalized medicine for this disease. Although TGCT patients have among the highest survival rates, the treatment choices are few and side effects are often profound. Further, since the patients are young, serious side effects may affect many decades of their life (13). Therefore, research on fusion genes as potential biomarkers or therapeutic targets in TGCT is of priority. We recently described the detection and characterization of recurrent fusion genes in TGCT (14). TGCT is a disease with distinct histological subtypes including seminomas and nonseminomas, where the latter can be subdivided into pluripotent embryonal carcinomas and more differentiated subtypes: teratomas, yolk sac tumors and choriocarcinomas. The pluripotent phenotype of malignant TGCTs has similarities to that of embryonic stem cells (15). Studying these cancers can therefore shed light on cancer biology in a context of pluripotency. We previously also showed that the expression of the fusion transcripts RCC1-ABHD12B and RCC1-HENMT1 is reduced upon in vitro differentiation of the EC cell line NTERA2 (14). It is therefore of interest to explore the frequency and distribution of the, sometimes weak, expression of these previously identified fusion transcripts in larger cohorts of TGCTs.
Based on the identified need for a sensitive approach to evaluate the recurrence of known fusion transcripts, we herein report the development of a new tool ScaR—Scaffold Realignment. We present benchmarking of ScaR on simulated data and apply it to investigate the prevalence of previously identified fusion transcripts in an extended cohort of TGCTs.
MATERIALS AND METHODS
RNA-sequencing data
We downloaded and processed paired-end RNA-seq raw fastq files of 150 TGCT samples from The Cancer Genome Atlas (TCGA) project (dbGAP accession: phs000178.v9.p8) (16). There was a median of 58.3 million pairs of reads per sample (min: 27.3 million and max: 107.3 million) with read length of 48 × 2 bp (see Supplementary Table S2 for detailed RNA-seq metrics and sample information). We further downloaded and processed paired-end RNA-seq raw fastq files of 198 normal testicular tissue samples of deceased individuals included in the Genotype-Tissue Expression (GTEx) project (dbGAP accession: phs000424.v6.p1) (17,18). All GTEx tissue samples were taken from healthy testis and the cause of death of individuals is not related to cancer, according to clinical data from GTEx (Supplementary Table S3). There was a median of 42.8 million pairs of reads (min: 27.8 million and max: 132.2 million) with read length of 76 × 2 bp (Supplementary Table S2). Paired-end RNA-seq data from the ES cell line Shef3, as described in Hoff et al. (14), were used together with simulated RNA-seq data from synthetic fusion transcripts for benchmarking (see Benchmarking and data simulation).
ScaR and the scaffold realignment approach
The main purpose of Scaffold realignment is to evaluate the presence of known fusion transcripts with breakpoint sites at exon boundaries or within exon regions (Figure 1). Scaffold realignment seeks two types of sequence reads to support fusion transcripts: split reads (a read mapping directly across the fusion transcript breakpoint sequence) and spanning reads (i.e. the paired reads map to one fusion partner gene each). Split reads are divided into two categories (Figure 1): discordant-split reads (i.e. the other read of the pair maps to the fusion gene partner A / B or across the fusion transcript breakpoint sequence) or singleton-split reads (i.e. the other read of the pair does not map to the transcriptome or genome). The pipeline is divided into four steps: (i) build reference sequences (scaffolds), (ii) read alignment to reference sequences, (iii) read re-alignment to genome sequences and (iv) summarize split read alignments across samples.
Figure 1.

Overview of the scaffold realignment approach — ScaR.
Build reference sequences
In the first step, a given breakpoint sequence supporting a fusion transcript is split in two fragments at the breakpoint site, which corresponds to Scaffold_partA and Scaffold_partB (Figure 1). If the sequences are longer than the read length used in the sequencing experiment, they are trimmed to match the length of the reads. Each sequence is then screened against all cDNA sequences of gene partners to match parental transcripts (3 transcriptome assembly annotations are packaged with the tool: Ensembl release 89 as default, and GENCODE release 27 or UCSC annotation based on GenBank release 225 and RefSeq release 86 as optional). ScaR also allows user-provided reference annotations if the breakpoint sequences are not previously annotated in the three transcriptome resources. If the sequence matches more than one transcript, the longest one (e.g. GeneA-T1 and GeneB-T1 marked as * in Figure 1) is selected to represent the gene. If the sequences are shorter than read length, the sequences are extended from the 5′-end of the matching transcript of Scaffold_partA and the 3′-end of the matching transcript of Scaffold_partB, respectively, to match the read length. The extended sequences are re-assembled to a new breakpoint sequence scaffold, which together with the sequences of the targeted transcripts from gene A / B serves as a reference sequence for read alignment. In addition, ScaR can accept a pair of fusion junction coordinates as input instead of a scaffold sequence. The scaffold sequences are then extracted from the longest matched annotated transcripts according to the coordinates.
Read alignment to reference sequences
To detect the presence of reads supporting the fusion breakpoint, paired-end reads are aligned to the custom scaffold reference using HISAT2 by default (see user’s manual for setting STAR as an optional aligner, although all of the results and benchmarking here have been performed with the HISAT2 aligner) (19). Briefly, an index of the custom scaffold reference sequence is built using hisat2-build with default parameters. Paired-end reads are then aligned to the reference sequences using –no-spliced-alignment model with –no-softclip setting. On the basis of the aligned SAM/BAM files, we retrieve three types of mapping reads: discordant-split reads, singleton-split reads and spanning reads. To increase mapping specificity, a minimum anchor length of 6 bp is required (by default) for split reads that map to the fusion breakpoint sequence (Figure 1). All supporting read-pairs of these three mapping types are extracted and saved as fastq files.
Read re-alignment to genome sequences
The supporting reads, both split and spanning reads, of a fusion breakpoint are further evaluated at a genomic level by aligning all extracted reads to the human reference genome (GRCH38) using HISAT2 –spliced-alignment model with –no-softclip setting (Figure 1). Supporting reads that are found to align to multiple locations are filtered out. This approach improves specificity and ensures that supporting reads that originate from repetitive sequences or gene homologs are not included in the support of a fusion breakpoint. In this step, singleton-split reads are also renamed as discordant-split reads if the unmapped read partner could be aligned uniquely to a gene partner at the genomic level.
Summary of fusion breakpoint support and cohort level statistics
A minimum support of two discordant-split reads is required to call a positive fusion breakpoint in a given sample. In addition, when the coverage of the fusion transcript is low, supported by only two or three split reads for each sample, the read coverage can show an uneven distribution between Scaffold_partA or Scaffold_partB regions. This uneven distribution can be attributed to either a sampling bias of a random distribution or an indication of artifact fusion sequences. For a better overview of the mapping distribution for a given scaffold, split reads across all samples in a cohort can be concatenated and aligned to the scaffold sequence. A Chi-squared test is then applied to test whether there is a significant bias in the distribution of the number of reads mapped to the upstream and downstream parts of the fusion scaffold sequence (Figure 1).
Benchmarking and data simulation
To compare the performance of our scaffold alignment approach on detecting known and previously validated fusion transcripts to that of established de novo fusion finders, we applied ScaR together with deFuse v.0.7.0 (20) and FusionCatcher v.1.00 (21) on the external TGCT and normal testis data sets from TCGA and GTEx. We used the Ensembl release 89 annotation database for all tools to avoid bias from different annotations. deFuse was run with ‘span_count_threshold = 1 & split_min_anchor = 6’, and FusionCatcher was run with ‘–paranoid-sensitive’, otherwise default parameters were used for both tools. We searched for the fusion transcripts RCC1-ABHD12B, RCC1-HENMT1, CLEC6A-CLEC4D and EPT1-GUCY1A3, as previously identified and characterized in TGCT by Hoff et al. (14). The Unix command line tool grep was also applied as a simple blunt tool for comparison to our scaffold approach. A string of 15 bp matching the gene on each side of the known breakpoints (30 bp total) was searched for in the fastq files and a minimum of two split reads were required for a positive call.
To further benchmark ScaR and the scaffold realignment approach on a controlled data set, we simulated RNA-seq reads from synthetic fusion transcripts using the MAQ v0.7.1 tool (22). Briefly, we simulated paired-end reads from in total 150 synthetic fusion transcripts, previously used for benchmarking in the SOAPfuse paper (23). After removing 20 fusion transcripts due to a high degree of sequence similarity between the gene partners and their paralogs and failure to lift over coordinates, a subset of 130 were finally used for benchmarking analysis in this study. Importantly, none of the fusion transcripts were between gene paralogs and also with an intergenic distance of >50 kb. Based on the breakpoint positions and genes listed in SOAPfuse (23), lifted over to GRCH38, we randomly selected one overlapping transcript isoform for each gene partner from Ensembl release 89 annotation and created the fused transcript from the paired transcript sequences. The minimum combined length of synthetic fusion transcripts was set to 500 bp, with a minimum upstream and downstream sequence length of 100 bp. Paired-end reads (76 bp each) with settings of background mutation rate, -r = 0.0001, fraction of indels, -R = 0.01 and a insert size of 170 bp (SD = 25 bp) were simulated. Further, different amounts of synthetic reads to match a gradient sequencing depth of the synthetic fusion transcripts (5X, 10X, 20X, 30X, 50X, 80X, 100X, 150X and 200X) were generated and then mixed with the RNA-seq reads from the embryonic stem cell line Shef3.
The fusion finder tools, deFuse, FusionCatcher and grep, were applied on the simulated data with identical settings to that previously described. To increase the robustness of our benchmarking, additional fusion finder tools (STAR-Fusion v.1.6.0 (24), FusionInspector v.2.0.0 (packaged with STAR-Fusion), SOAPfuse v.1.27 and JAFFA v.1.0.9 (25)) were applied on this synthetic data set. Genome indices were built with Ensembl release 89 annotation for all tools. STAR-Fusion was run with ‘–chimSegmentMin 6’, ‘–chimJunctionOverhangMin 6’, ‘–min_FFPM 0’, ‘–no_annotation_filter’ and otherwise default parameters. FusionInspector, which is packaged together with STAR-Fusion, was given a list of the 130 simulated fusion transcripts as input and run with ‘require_LDAS 0’, ‘–min_sum_frags 1’, ‘–min_junction_reads 0’, ‘–min_novel_junction_support 2’, ‘–min_spanning_frags_only 2’ and otherwise default parameters. SOAPfuse was run with ‘PA_s07_the_minimum_span_reads_for_junction_construction = 1’, ‘PA_s08_min_bases_covered_both_sides_around_fuse_point = 6’ and otherwise default parameters. JAFFA was run in hybrid mode with ‘overHang = 6’ and otherwise default parameters. A minimum of two supporting split reads was set as a cutoff for all applied fusion finder tools when calling the simulated fusion transcripts. Grep was also applied on the simulated data with identical search string patterns as previously described. For ScaR and the scaffold realignment approach, we generated scaffolds of the 130 synthetic transcripts and required a minimum of two discordant split reads as support. The sensitivity of these tools to detect the synthetic fusion transcripts in different mixtures was compared and reported.
Execution time and memory usage were compared for ScaR, FusionInspector and grep on both simulated data and real TCGA data. Benchmarking analyses were performed in the Abel high performance-computing cluster (16 CPU cores, 64 Gb size of physical memory per node and CentOS 6 operating system) at the University of Oslo. Four CPUs were allocated for the ScaR and FusionInspector jobs.
TGCT hierarchical clustering and differential expression analysis
To perform hierarchical clustering and differential expression analysis of the 150 TGCT samples from the TCGA cohort, we acquired raw gene count data produced by HTSeq-count from NCI’s Genomic Data Commons (http://xena.ucsc.edu) as well as clinical data including the International Classification of Diseases for Oncology (ICD-O) morphological codes (the latter being available for 134 of the 150 samples; Supplementary Table S3). Mutation data for the 150 samples were also acquired from cBioportal. The DESeq2 R package (26) was used to perform data normalization and differential expression analysis. Genes that were not expressed across the cohort were removed from further analyses. Prior to performing principal component analysis (PCA) and hierarchical clustering, variance stabilizing transformation was applied on the raw counts. PCA was then performed with the top 500 variable genes used for principal components. Hierarchical clustering was performed on the transformed raw counts using the top 50 most variable genes, clustering on both samples and genes. Clustered heat maps were produced with the pheatmap R package, plotted together with annotation tracks including ICD-O histological subtypes, fusion transcript status (determined by ScaR) and mutation data of known TGCT driver genes. Mutation status was plotted for genes previously implicated in TGCT and that were mutated in two or more samples in the TCGA cohort. Differential expression analysis was performed on RCC1-ABHD12B positive samples versus negative samples and CLEC6A-CLEC4D positive versus negative samples, both controlling for the effect of ICD-O histology subtypes.
RESULTS
Overview of the ScaR workflow
Here, we sought to establish the frequency of known and previously validated fusion transcripts in a larger cohort of TGCT patients and we report the development of ScaR—a tool for sensitive detection of known fusion transcripts, which is openly available at https://github.com/senzhaocode/ScaR. ScaR takes any fusion scaffold sequence, or genomic junction coordinates, as input together with raw RNA-seq data to return the number of spanning and discordant- / singleton- split reads supporting the scaffold sequence (Figure 1). Finally ScaR can summarize the number of supporting reads across a larger cohort. We applied ScaR to investigate the recurrence of four previously described fusion transcripts (RCC1-ABHD12B, CLEC6A-CLEC4D, RCC1-HENMT1 and EPT1-GUCY1A3) in 150 primary TGCT samples using RNA-seq data from TCGA. Overall, we find that ScaR has a sensitivity that is superior to tools such as deFuse and FusionCatcher and the basic grep method in detection of four known fusion transcripts in TGCT.
Optimization of ScaR parameters
To balance sensitivity and specificity for fusion transcript detection with ScaR, we investigated the sensitivity of detecting the TGCT fusion transcripts with a variable threshold. We also applied the fusion finder tools, deFuse and FusionCatcher, which calls fusion breakpoints in a de novo manner, as well as the basic grep method, to provide a reference for the performance of ScaR. As expected, the detection rate decreased with increasing the minimal threshold of required split reads for all four methods, but ScaR consistently achieved a higher sensitivity compared with the other three tools when setting the threshold below 5 required split reads (Figure 2). All of the four tools show a low sensitivity of detection and a high false negative rate when strict criteria (split read number > 5) are applied for fusion nomination. To evaluate the reads mapping to the different scaffold sequences, ScaR has the ability to concatenate all supporting split reads from a given cohort (in this case the 150 TGCT samples) and align them to the scaffold. For example from this cohort, 59 samples have detectable RCC1-HENMT1 with a threshold set to one split read, but 51 of them have only one discordant-split read support (Figure 2C and Supplementary Table S4). We found that 62 reads from 38 of the 51 samples aligned to a scaffold sequence that show a biased distribution around the scaffold breakpoint sequence with a shift toward the RCC1 part of the scaffold (P = 3 × 10−15; Chi-squared test; Supplementary Figure S1J), indicating that these are false positives. The same pattern was observed for the fusion scaffold RCC1-ABH12B_alt1 (Supplementary Figure S1B), but without a significant P-value, probably due to the small number of supporting reads. This coverage bias in the consensus of split read alignments indicates that the reads mapping to the breakpoint scaffold sequences of RCC1-HENMT1_alt1 and RCC1-ABH12B_alt1 are most likely mapping artifacts and that these fusion scaffolds represent false positives. These are therefore excluded from further analysis. Overall, from these results, we find that a minimal requirement of two discordant-split reads represents a good balance between sensitivity and specificity for fusion nomination by the ScaR approach, which is further used as a threshold for fusion detection in this study.
Figure 2.
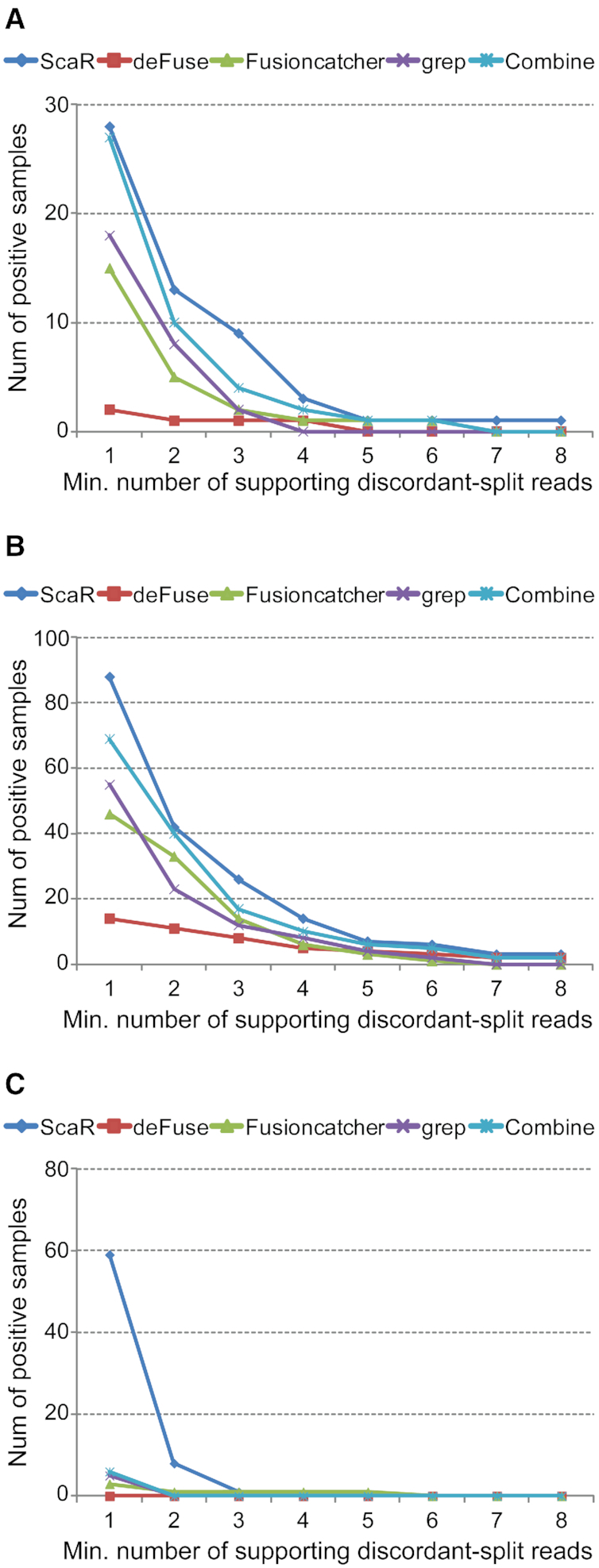
ScaR performance on TGCT data from TCGA. Comparison of sensitivity between ScaR and other tools (deFuse, FusionCatcher, grep and Combine; combination of the three other tools) for fusion transcripts RCC1-ABHD12B (A), CLEC6A-CLEC4D (B) and RCC1-HENMT1 (C) across 150 TCGA TGCT samples. The X-axes show an increasing threshold of minimum required supporting split reads. The Y-axes show the number of samples with positive detection.
ScaR—benchmarking using simulated fusion transcript read data
To evaluate the performance of ScaR on a controlled data set, we simulated RNA-seq data from 130 synthetic fusion transcripts (Supplementary Table S5). Various amounts of reads were simulated at 5X to 200X coverage of these synthetic fusion transcripts and mixed in silico with real RNA-seq data from the ES cell line Shef3. Briefly, 97.5% of the synthetic reads were found to map to the genome. The number of discordant split reads detected by ScaR for the synthetic fusion transcripts showed a perfect correlation with the number of simulated reads with the increase of simulated sequencing coverage (r = 0.99, P = 1 × 10−15, Pearson correlation). We further performed a comprehensive benchmark comparison of ScaR, deFuse, FusionCatcher, grep, SOAPfuse, STAR-Fusion, STAR-Fusion; FusionInspector and JAFFA to detect these fusion transcripts (Figure 3 and Supplementary Table S5). ScaR was able to detect 123 out of the 130 fusion transcripts (95%) at 5X coverage of simulated data, with median of four split reads and one spanning read. At all other sequencing depth levels, ScaR reached 100% detection rate of the synthetic fusion transcripts. In comparison, the best performing established fusion finder, in terms of sensitivity, at 5X coverage was deFuse with 105/130 synthetic fusion transcripts detected. However, it should be noted that deFuse was run with very non-stringent criteria and nominated on average 155917 fusion breakpoints per simulated sequencing depth level in the raw output file. The use of grep for a 30 bp search string (15 bp upstream and downstream of breakpoint) could identify 107/130 synthetic fusion transcripts at 5X coverage, while a combination of all the other fusion tools, together with grep, in total detected 122/130 at this level. Of note, none of the other fusion tools could achieve 100% detection rate at any level of coverage. However, a combination of the tools detected all 130 synthetic fusion transcripts at 10X coverage and above. Execution time and memory usage of ScaR, FusionInspector and grep were compared both for detecting the 130 synthetic fusion transcripts in simulated data (5X to 200X) and for detecting the RCC1-ABHD12B fusion transcript in real RNA-seq data from 150 TGCT samples. ScaR was the fastest tool for detecting the RCC1-ABHD12B fusion transcript per sample in TCGA data (Supplementary Table S6). For detecting the 130 synthetic fusion transcripts in simulated data, ScaR used considerably longer runtime compared to that of FusionInspector and grep (Supplementary Table S6). In terms of memory consumption, FusionInspector used the maximum amount with on average 41 Gb memory per sample, whereas ScaR used on average 6.3 Gb memory on simulated data (Supplementary Table S6).
Figure 3.
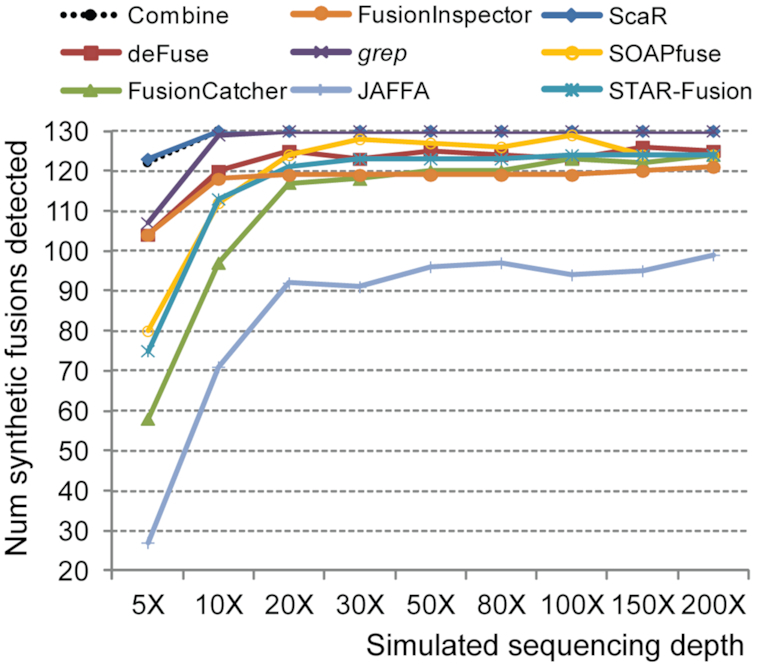
ScaR performance on simulated data. Benchmarking performance of ScaR, grep and all established fusion finder tools applied on simulated RNA-seq data showing the number of synthetic fusion transcripts detected at simulated coverage levels ranging from 5X to 200X. Combine indicates a union detection by all established fusion tools, including grep.
Known fusion transcripts in TGCT are frequently detected by applying ScaR to larger cohorts
To further evaluate the performance of ScaR on real biomedical data, we applied ScaR on the TGCT TCGA cohort to detect the previously described fusion transcripts. Specifically, for RCC1-ABHD12B (Figure 4A), ScaR detected the fusion transcript in 13 samples (8.7%) with at least two supporting discordant-split reads (Supplementary Table S4). In comparison, deFuse, FusionCatcher and grep detected the fusion in only one (0.6%), five (3.3%) and eight (5.3%) samples, respectively. By merging the results from these three tools, RCC1-ABHD12B was detected in 10 unique samples, where all except one sample (TCGA-XE-A8H4; Figure 4A) overlapped with the positive samples from ScaR. ScaR failed to report the fusion transcript in this sample because one of two supporting split reads is a singleton-split type (Supplementary Table S4). ScaR detected RCC1-ABHD12B in four additional unique samples compared to the other three tools. For CLEC6A-CLEC4D (Figure 4B), we evaluated six different fusion breakpoint scaffolds between the two neighboring genes, as have previously been reported ((14); Supplementary Table S4). Samples with reads supporting any of these scaffold sequences were regarded as positives. In total, ScaR detected the fusion transcript in 42 (28%) samples, which is higher compared to the frequency identified by deFuse (11; 7.3%), FusionCatcher (33; 22%) and grep (23; 14.7%). Importantly, five of 42 samples detected as positive by ScaR failed to be nominated by any of the three other tools. All positive samples except three cases detected by the deFuse, FusionCatcher or grep are also identified by ScaR. Two of these (TCGA-2X-A9D6 and TCGA-WZ-A8D5) are uniquely identified with grep and have two supporting split reads. For both samples, one of the reads show unspecific multiple alignment at genomic level and is therefore filtered out by ScaR. The third sample (TTCGA-VF-A8AA) is exclusively detected by deFuse. We found that the anchor length for supporting split read alignments for this sample is 4 bp, below the minimum requirement of ScaR. For RCC1-HENMT1, ScaR detected the fusion transcript in only one sample (TCGA-WZ-A7V3) when not regarding samples with support for the unreliable RCC1-HENMT1_alt1 scaffold. FusionCatcher detected RCC1-HENMT1 in a single sample (TCGA-XY-A8S3), while deFuse and grep failed to detect the fusion transcript in any of the 150 samples (Supplementary Table S4). The RCC1-HENMT1 breakpoint sequence nominated by FusionCatcher was found to span from the 3′UTR region of RCC1 to an intronic region of HENMT1, with the downstream part of the breakpoint sequence having a high number of homologues sequences sharing a high percentage of sequence identity. The fusion EPT1-GUCY1A3 could not be rediscovered by any of these four tools, with zero spanning and split reads identified. These findings indicate that EPT1-GUCY1A3 is most likely a private fusion event. We further investigated the scaffold alignments for the samples that were uniquely called by ScaR and not by any of the other tools. For RCC1-ABHD12B and CLEC6A-CLEC4D that were detected uniquely by ScaR in four and five samples, respectively, we found that the mapping qualities of the reads at the breakpoint sites were of high quality with an even distribution to upstream and downstream regions, suggesting that these samples are indeed positive (Figure 4A and B).
Figure 4.

Fusion transcript detection in TGCTs. Overview of TGCT samples from TCGA (n = 150) that are positive for the fusion transcripts RCC1-ABHD12B (A) and CLEC6A-CLEC4D (B) among the four tools: ScaR, deFuse, FusionCatcher and Grep. Split read alignments of positive samples uniquely identified by ScaR are visualized using IGV. The ICD-O histology codes are shown as annotated by TCGA.
TGCT fusion transcripts are malignancy specific and not detected in normal testis tissue samples from the GTEx consortium
We furthermore evaluated the prevalence of these fusion transcripts in 198 normal testicular samples from GTEx project using ScaR. In brief, none of the investigated fusion transcripts could be detected in any of the normal samples (Supplementary Table S7). For the fusion transcript CLEC6A-CLEC4D where the two genes are located only 30 kb apart on chromosome arm 12p, we detected only two split reads and one spanning read all in distinct samples across the 198 samples. Therefore, none of the samples pass the threshold for detection. Similarly, no split or spanning reads are identified for RCC1-ABHD12B and one spanning read is identified for RCC1-HENMT1 across all 198 GTEx samples. Importantly, reads from the GTEx data aligned to genome show a mapping percentage with a median value of 93.5% (only one sample < 85%) compared to 95.5% for the TCGA tumor samples. Additionally, the GTEx samples have a median sequencing output of 6.5 Gbp compared to the median sequencing output of the TCGA tumor samples of 5.6 Gbp. These results indicate that the failure to detect the investigated fusion transcripts in normal GTEx samples is not due to differences in sequencing power between cohorts, and that these fusion transcripts are specifically present in TGCT and not in normal tissue of the testis. This is in accordance with previously published experimental RT-PCR data (14), although then from relatively few samples.
CLEC6A-CLEC4D and RCC1-ABHD12B are more frequently detected in the undifferentiated seminoma and embryonal carcinoma like subgroups, respectively
To investigate the biological associations of the frequently identified fusion transcripts CLEC6A-CLEC4D and RCC1-ABHD12B in data from the TCGA cohort, we performed principal component analysis on gene expression data from the 150 TGCT samples. Not surprisingly, we found that the samples cluster roughly into three groups that correspond well to the annotated ICD-O histological subtypes by TCGA (Supplementary Figure S2A). The three groups comprise mostly of seminomas, embryonal carcinomas and a third subgroup with the more differentiated histological subtypes and a high frequency of mixed tumors. Further, we performed hierarchical clustering with the 50 most variable genes across the cohort and annotated the samples with somatic mutation calls in known TGCT driver genes, as well as the fusion transcript status, as determined by ScaR (Figure 5). Among the top 50 most variable genes, we found some of the commonly described stem cell associated genes, such as NANOG, POU5F1 and SOX2. Intriguingly, we saw a clear enrichment of CLEC6A-CLEC4D expressing samples within the seminoma-like subgroup (P < 0.0001, Fisher’s exact test; Figure 5 and Supplementary Figure S2C) together with frequent KIT and KRAS mutations. For RCC1-ABHD12B there was a clear association with the embryonal carcinoma-like subgroup, with 12/13 positive samples clustering within this group (P < 0.0001; Figure 5 and Supplementary Figure S2B). CLEC6A-CLEC4D and RCC1-ABHD12B were also largely mutually exclusive, except for two samples that had either a mixed germ cell tumor or unavailable histological subtype. Further, by differential expression analysis we also found that RCC1 and ABHD12B were significantly upregulated in the RCC1-ABHD12B positive subgroup (Supplementary Figure S3A). Also, both CLEC6A and CLEC4D were among the highest ranked upregulated genes in the CLEC6A-CLEC4D subgroup (Supplementary Figure S3B).
Figure 5.

Fusion transcripts in TGCT and associated molecular features. Heat map showing fusions, somatic mutations and RNA expression (normalized RNA-seq counts) of the 50 most variable genes across the TCGA cohort. Individual samples are clustered along the horizontal axis while genes are clustered on the vertical axis. Annotation tracks include ICD-O histology codes, and fusion transcript status for RCC1-ABHD12B, RCC1-HENMT1 and CLEC6A-CLEC4D. Somatic mutation status for genes known to be recurrently mutated in TGCT are also shown and colored according to mutation type.
DISCUSSION
We have developed, tested and applied the bioinformatics tool ScaR for sensitive assessment of the prevalence of known fusion transcripts in cohorts of cancer samples using RNA-seq data. ScaR efficiently implements a direct scaffold realignment approach, and we have benchmarked the tool on simulated data. Importantly, we have evaluated previously described fusion genes in TGCTs in larger cohorts from the TCGA and GTEx consortia, and demonstrated that ScaR achieves a high sensitivity for detecting known fusions compared to established fusion finder tools that are developed to call fusion transcripts de novo.
The improved sensitivity will be of value as an expanding array of fusion genes with clinical impact are uncovered. Already, multiple fusion genes occurring in cancer are predictive for response to kinase inhibitors, and establishing the presence of such fusion genes and their fusion transcript products in patients prior to treatment is of importance. Improved detection sensitivity for a fusion transcript biomarker can also be important in monitoring a patient’s response to treatment or in detecting minimal residual disease, e.g. detecting the presence of BCR-ABL1 in CML patients undergoing treatment with the kinase inhibitor imatinib. By looking specifically for the fusion transcripts of interest and thereby circumventing the need for strict filters and thresholds to avoid false positives, due to biological and technical noise in RNA-seq data, our approach with ScaR could be better suited for these purposes. Also, as RNA-seq data from more patients and cancer types are becoming available, establishing the prevalence of known and validated fusion transcripts in expanded and new cohorts is of importance. For instance, fusion genes involving the kinases ALK, RET, ROS1 and BRAF have been found in multiple cancer types, expanding the repertoire of cancers that kinase inhibitors could target (27).
We show that our tool has an improved sensitivity compared to established fusion gene detection tools, both by using simulated data and on real data from TCGA. The improvement in sensitivity is not unexpected, as ScaR uses an approach for the detection of known fusion transcripts with a prior knowledge compared to most other approaches that nominate fusion transcripts without any prior knowledge of the fusion events. Although there are >40 different fusion finder tools available (Supplementary Table S1), most of them build on similar de novo approaches by read-alignment, detection of reads or read-pairs that support a fusion breakpoint and applying different filtering criteria. We applied deFuse and FusionCatcher in our search for TGCT fusion transcripts in data from TCGA, on the basis that deFuse has been an established fusion finder tool for many years (and still maintained) and that FusionCatcher has repeatedly performed well in independent comparison studies on multiple data sets (9,10,20). In the benchmarking analysis of simulated synthetic fusion transcript data, we further conducted a comprehensive evaluation by adding more fusion finder tools (SOAPfuse, STAR-Fusion, FusionInspector and JAFFA). SOAPfuse, FusionCatcher and JAFFA were among the best performing tools in an independent comparison study including 15 fusion finder tools, and were suggested combined in a meta-caller (10). STAR-Fusion is a recent, maintained and widely adopted fusion finder that is built on the popular STAR RNA-seq read aligner (28), and it performed well in comparison with other established fusion finders (24). In addition, FusionInspector was included in our benchmarking because this tool is designed for detecting and validating fusion transcript predictions or known and validated fusions in a similar manner to ScaR. Our benchmarking results show that ScaR is able to detect the synthetic fusion transcripts with improved sensitivity compared to all tested tools individually and even combined at a low level (5X) coverage. Although FusionInspector performed better than STAR-fusion at low coverage, its sensitivity was lower than ScaR at all levels. Interestingly, FusionInspector detected fewer fusion transcripts at higher coverage compared to STAR-Fusion, which probably is a result of some additional filters applied in this pipeline. Similarly, none of the established tools were able to detect all the 130 synthetic fusion transcripts at any level of coverage, indicating that some of the synthetic fusion transcripts fail to pass the stringent filters applied. However, the combination of all tools showed a 100% detection rate from 10X coverage, underlining that there is not a systematic bias for some of the synthetic fusion transcripts calling and that different filters for the different tools may lead to the observed results. Comparison of execution time and memory usage showed that ScaR has a relatively small memory requirement compared to that of FusionInspector. Although the performance of FusionInspector in runtime was more efficient than ScaR for detecting hundreds of fusion transcripts in the nine levels of simulated data, ScaR was the fastest tool for detecting the RCC1-ABHD12B fusion transcript in the 150 TGCT samples. Nevertheless, the main purpose of ScaR is not to be the fastest tool, but to enable efficient establishment of the prevalence of one or a few fusion breakpoints in large patient cohorts.
Most fusion tools that work without prior knowledge of fusion events relies on spanning reads to nominate gene partners of fusion genes and split reads are consequently used to refine the exact breakpoint sequences. The amount of spanning read pairs for a given fusion breakpoint is highly dependent on the insert size of the read-pairs in each RNA-seq library. In fact, in sequencing libraries with very short or negative insert sizes (overlapping single-end reads) the number of supporting spanning reads may be very low or completely absent leading to a reduced sensitivity of detection. By providing ScaR with an already known fusion breakpoint, we can avoid this bias of insert size. In addition, for some fusions, the breakpoint in the upstream gene partner can be close to the transcript start site, with distance less than the read length. As a consequence, the sequence from the upstream gene partner can be too short to detect spanning reads, which can reduce the sensitivity of other fusion tools. ScaR not only uses split-reads as the main support for a given fusion breakpoint, but also provides the supporting spanning reads in the output, which may be used for downstream purposes. This is one of the major impacts on the improved sensitivity we see with ScaR compared to established fusion tools. The unix tool grep, which we also compared to, has been used as a direct approach to indicate the presence of fusion transcripts from RNA-seq data (11). However, this approach suffers from requiring a perfect match to the query sequence in RNA-sequencing reads, not allowing for single mismatches, indels or variable anchor lengths (In Supplementary Figure S4, a few samples are shown to have fusion supporting split reads with mismatched bases that grep fails to detect). The search string given to grep is in addition static, and in this study matching 15 bp on each side of the fusion breakpoints (30 bp total). Using grep to allow a junction overhang of 6 bp to resemble parameters of ScaR and other tools included would result in many unspecific hits if a 12 bp total length query sequence is applied for search, or requiring multiple grep commands with a dynamic sliding window string. The latter option is possible, but computationally demanding and time-consuming (e.g. requiring 19 search strings with a total length of 30 bp and 6 bp overhang to grep). Indeed, execution time for ScaR was shorter for detecting the RCC1-ABHD12B fusion in TCGA RNA-seq data compared to grep for the same fusion, even when using single static 30 bp sequences. Also, supporting reads from the grep approach are not confirmed to be unambiguously mapping to the breakpoint sequence, or if they potentially map ambiguously to multiple sequences in the genome. ScaR circumvents these drawbacks and improves the sensitivity while balancing specificity by using a dedicated aligner for aligning reads to a fusion specific scaffold sequence and further mapping supporting reads back to the genome to avoid ambiguous supporting reads. For example, in the 150 synthetic fusion transcripts previously used for benchmarking in the SOAPfuse paper, 16 fusions were identified with different degrees of multi-mapping split and spanning reads from 5X to 200X coverage (Supplementary Table S8), as one or two of their partner genes had paralogs with moderate to high level of sequence similarity. These 16 fusion transcripts were excluded from the final simulation data and benchmarking analyses to avoid biases to fusion calling.
ScaR requires the use of a transcriptome annotation and generates the fusion scaffold from exonic sequences of transcripts matched to the input breakpoint sequence. Currently, we include three options of major transcriptome annotation resources (Ensembl, GENCODE and UCSC) in ScaR. In addition, we allow a user-defined annotated reference sequence as input, which could involve non-coding sequences from intronic and intergenic regions. It extends the functionality of ScaR to evaluate fusion transcripts from alternative promoter or new splicing events that are not previously annotated in any of the three major transcriptome annotations.
Here, our aim was to validate the presence and explore on the prevalence of fusion transcripts we previously discovered to be recurrent in a small cohort of TGCTs (14), in a larger cohort from TCGA. Admittedly, we initially found that the frequency of samples positive for these fusion transcripts was much lower than what we previously established with quantitative real-time PCR in our cohort of TGCTs. We therefore explored if these fusion transcripts could be expressed at low levels in a larger number of samples, and that more sensitive approaches were needed to detect this signal in RNA-seq data. By developing and applying ScaR, we discovered that these fusion transcripts, especially the read-through CLEC6A-CLEC4D and the interchromosomal fusion RCC1-ABHD12B, are detectable in a higher frequency of TGCTs than what could be established with previously established fusion finder tools. Importantly, we also show that our sensitive detection approach with ScaR does not uncover these fusion transcripts in any samples from a large cohort of normal testis samples (GTEx), indicating a high specificity of ScaR and that these fusion transcripts, albeit being expressed at low levels, are cancer-specific. Further, by hierarchical clustering on gene expression data from the TCGA, we show that the TGCT samples cluster according to their histological subtypes (16), in line with previous publications on gene expression in TGCT (29). From the heat map in Figure 5, we see that CLEC6A-CLEC4D is significantly enriched in samples of the undifferentiated seminoma-like cluster, while RCC1-ABHD12B is significantly enriched in samples of the undifferentiated embryonal carcinoma-like cluster. These findings support our previous results that showed that RCC1-ABHD12B expression, but not CLEC6A-CLEC4D expression, was significantly reduced when a pluripotent embryonal carcinoma cell line (NTERA2) was differentiated in vitro (14). These observations support a biological significance of these fusion transcripts being markers of pluripotent TGCTs.
In conclusion, we have developed ScaR, a tool that uses a scaffold alignment approach for sensitive detection of known fusion transcripts in RNA-seq data. Such sensitive detection of known fusion transcripts will be of importance in personalized cancer medicine. Further, we have used ScaR to establish that the RCC1-ABHD12B and CLEC6A-CLEC4D fusion transcripts are frequently detected in TGCTs and associated with the undifferentiated embryonal carcinoma and seminoma histological subtypes.
DATA AVAILABILITY
ScaR is freely available via GitHub and its implementation is explained in the manual and tutorials: https://github.com/senzhaocode/ScaR.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Jonas M. Strømme for his valuable suggestions and input on the study.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Norwegian Cancer Society [PR-2007-0166 to S.Z., A.M.H., R.I.S.]; Research Council of Norway [FRIPRO project number, 262529]; NorStore [storage of computational data, NS9013K]; Notur [CPU hours from the Abel supercomputer, NN9313K].
Conflict of interest statement. None declared.
REFERENCES
- 1. Nowell P.C., Hungerford D.A.. A minute chromosome in human chronic granulocytic leukemia. Science. 1960; 142:1497. [DOI] [PubMed] [Google Scholar]
- 2. Shtivelman E., Lifshitz B., Gale R.P., Canaani E.. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature. 1985; 315:550–554. [DOI] [PubMed] [Google Scholar]
- 3. Heisterkamp N., Stephenson J.R., Groffen J., Hansen P.F., de Klein A., Bartram C.R., Grosveld G.. Localization of the c-abl oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature. 1983; 306:239. [DOI] [PubMed] [Google Scholar]
- 4. Groffen J., Stephenson J.R., Heisterkamp N., de Klein A., Bartram C.R., Grosveld G.. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984; 36:93–99. [DOI] [PubMed] [Google Scholar]
- 5. Mertens F., Johansson B., Fioretos T., Mitelman F.. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 2015; 15:371–381. [DOI] [PubMed] [Google Scholar]
- 6. Hu X., Wang Q., Tang M., Barthel F., Amin S., Yoshihara K., Lang F.M., Martinez-Ledesma E., Lee S.H., Zheng S. et al.. TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 2017; 46:D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoshihara K., Wang Q., Torres-Garcia W., Zheng S., Vegesna R., Kim H., Verhaak R.G.W.. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2014; 34:4845–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. U.S. Food & Drug Administration 2018; FDA approves an oncology drug that targets a key genetic driver of cancer, rather than a specific type of tumor http://www.fda.gov/news-events/press-announcements/fda-approves-oncology-drug-targets-key-genetic-driver-cancer-rather-specific-type-tumor.
- 9. Kumar S., Vo A.D., Qin F., Li H.. Comparative assessment of methods for the fusion transcripts detection from RNA-Seq data. Sci. Rep. 2016; 6:21597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu S., Tsai W.-H., Ding Y., Chen R., Fang Z., Huo Z., Kim S., Ma T., Chang T.-Y., Priedigkeit N.M. et al.. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2015; 44:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Panagopoulos I., Gorunova L., Bjerkehagen B., Heim S.. The ‘grep’ command but not FusionMap, FusionFinder or ChimeraScan captures the CIC-DUX4 fusion gene from whole transcriptome sequencing data on a small round cell tumor with t(4;19)(q35;q13). PloS One. 2014; 9:e99439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Znaor A., Lortet-Tieulent J., Jemal A., Bray F.. International variations and trends in testicular cancer incidence and mortality. Eur. Urol. 2014; 65:1095–1106. [DOI] [PubMed] [Google Scholar]
- 13. Haugnes H.S., Bosl G.J., Boer H., Gietema J.A., Brydøy M., Oldenburg J., Dahl A.A., Bremnes R.M., Fosså S.D.. Long-Term and late effects of germ cell testicular cancer treatment and implications for Follow-Up. J. Clin. Oncol. 2012; 30:3752–3763. [DOI] [PubMed] [Google Scholar]
- 14. Hoff A.M., Alagaratnam S., Zhao S., Bruun J., Andrews P.W., Lothe R.A., Skotheim R.I.. Identification of novel fusion genes in testicular germ cell tumors. Cancer Res. 2016; 76:108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andrews P.W., Matin M.M., Bahrami A.R., Damjanov I., Gokhale P., Draper J.S.. Embryonic stem (ES) cells and embryonal carcinoma (EC) cells: opposite sides of the same coin. Biochem. Soc. Trans. 2005; 33:1526–1530. [DOI] [PubMed] [Google Scholar]
- 16. Shen H., Shih J., Hollern D.P., Wang L., Bowlby R., Tickoo S.K., Thorsson V., Mungall A.J., Newton Y., Hegde A.M. et al.. Integrated molecular characterization of testicular germ cell tumors. Cell Rep. 2018; 23:3392–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carithers L.J., Ardlie K., Barcus M., Branton P.A., Britton A., Buia S.A., Compton C.C., DeLuca D.S., Peter-Demchok J., Gelfand E.T. et al.. A novel approach to high-quality postmortem tissue Procurement: The GTEx project. Biopreserv Biobank. 2015; 13:311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N. et al.. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McPherson A., Hormozdiari F., Zayed A., Giuliany R., Ha G., Sun M.G.F., Griffith M., Heravi Moussavi A., Senz J., Melnyk N. et al.. deFuse: An algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 2011; 7:e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nicorici D., Satalan M., Edgren H., Kangaspeska S., Murumagi A., Kallioniemi O., Virtanen S., Kilkku O.. FusionCatcher - a tool for finding somatic fusion genes in paired-end RNA-sequencing data. 2014; bioRxiv doi:19 November 2014, preprint: not peer reviewed 10.1101/011650. [DOI]
- 22. Li H., Ruan J., Durbin R.. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008; 18:1851–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jia W., Qiu K., He M., Song P., Zhou Q., Zhou F., Yu Y., Zhu D., Nickerson M.L., Wan S. et al.. SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 2013; 14:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haas B.J., Dobin A., Li B., Stransky N., Pochet N., Regev A.. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019; 20:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davidson N.M., Majewski I.J., Oshlack A.. JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015; 7:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zehir A., Benayed R., Shah R.H., Syed A., Middha S., Kim H.R., Srinivasan P., Gao J., Chakravarty D., Devlin S.M. et al.. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10, 000 patients. Nat. Med. 2017; 23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Skotheim R.I., Lind G.E., Monni O., Nesland J.M., Abeler V.M., Fosså S.D., Duale N., Brunborg G., Kallioniemi O., Andrews P.W. et al.. Differentiation of human embryonal carcinomas in vitro and in vivo reveals expression profiles relevant to normal development. Cancer Res. 2005; 65:5588–5598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ScaR is freely available via GitHub and its implementation is explained in the manual and tutorials: https://github.com/senzhaocode/ScaR.