

Abstract
Multiple sequence alignments (MSAs) play a pivotal role in studies of molecular sequence data, but nobody has developed a minimum reporting standard (MRS) to quantify the completeness of MSAs in terms of completely specified nucleotides or amino acids. We present an MRS that relies on four simple completeness metrics. The metrics are implemented in AliStat, a program developed to support the MRS. A survey of published MSAs illustrates the benefits and unprecedented transparency offered by the MRS.
INTRODUCTION
Multiple sequence alignments (MSAs) are widely used during annotation and comparison of molecular sequence data, allowing us to identify medically important substitutions (1), infer the evolution of species (2), detect lineage- and site-specific changes in the evolutionary processes (3) and engineer new enzymes (4). There is a wide range of computational tools for obtaining MSAs, and two of these (i.e. Clustal W (5) and Clustal X (6)) are now among the 100 most cited papers in science (7).
In addition to the completely specified nucleotides (i.e. A, C, G, T/U) or amino acids (i.e. A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y), MSAs may contain ambiguous characters (i.e. incompletely specified nucleotides or amino acids). Frequently, they also contain alignment gaps (i.e. ‘–’) inserted between the nucleotides or amino acids of some of the sequences. Alignment gaps are inserted to maximize the homology of residues from different sequences (alignment gaps should only be used to improve alignment whereas N and X should only be used to signal missing data). A correct MSA is necessary for accurate genome annotation, phylogenetic inference and ancestral sequence reconstruction. However, deciding where to put the alignment gaps may be more art than science. This is because homology is defined as similarity due to historical relationships by descent (8). Most of these relationships belong to the unobservable distant past, so it is impossible to measure the accuracy of most MSAs inferred from real sequence data.
Without this ability, reporting the completeness of MSAs may be the best that can be achieved. So far, the only metric sometimes used is the percent missing data for a sequence (9) or an alignment (10), but neither is sufficiently transparent and informative. Recently, a guideline for systematic reporting of sequence alignments has been suggested (11), but it did not include completeness of MSAs—instead, it focused on quality indicators of alignment, but it did not define any of these or point to relevant literature. To rectify this, we developed a minimum reporting standard (MRS) for MSAs.
MATERIALS AND METHODS
Metrics for measuring completeness of MSAs
The MRS uses four metrics to quantify the completeness of different attributes of MSAs. Given an MSA with 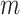 sequences and
sequences and 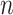 sites, we may compute four metrics:
sites, we may compute four metrics: 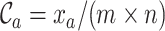 ,
, 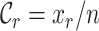 ,
, 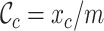 and
and 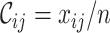 , where
, where 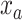 is the number of completely specified characters (12) in the MSA,
is the number of completely specified characters (12) in the MSA, 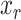 is the number of completely specified characters in the
is the number of completely specified characters in the 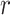 -th sequence of the MSA,
-th sequence of the MSA, 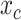 is the number of completely specified characters in the
is the number of completely specified characters in the 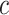 -th column of the MSA and
-th column of the MSA and 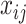 is the number of homologous sites with completely specified characters in both sequences (
is the number of homologous sites with completely specified characters in both sequences (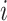 and
and 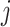 ). In summary,
). In summary, 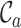 ,
, 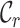 ,
, 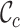 and
and 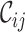 measure the completeness of the alignment, the
measure the completeness of the alignment, the 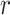 -th sequence, the
-th sequence, the 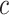 -th site, and the
-th site, and the 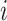 -th and
-th and 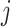 -th sequences, respectively.
-th sequences, respectively.
The first of these metrics (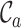 ) is related to the percent missing data used previously, but it is also, as shown in Figure 1A, the least useful completeness metric considered here: alignments A and B differ greatly, but they have the same
) is related to the percent missing data used previously, but it is also, as shown in Figure 1A, the least useful completeness metric considered here: alignments A and B differ greatly, but they have the same 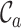 value (i.e. 0.7). The
value (i.e. 0.7). The 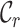 ,
, 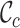 and
and 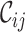 metrics, on the other hand, are able to detect these differences. For example, the
metrics, on the other hand, are able to detect these differences. For example, the 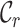 values range from 0.3 to 1.0 for alignment A and from 0.4 to 1.0 for alignment B, raising greater concern, from a sequence-centric perspective, about alignment A than about alignment B. If we were to omit any sequence from alignment A, then it would be sensible to omit the one with the smallest
values range from 0.3 to 1.0 for alignment A and from 0.4 to 1.0 for alignment B, raising greater concern, from a sequence-centric perspective, about alignment A than about alignment B. If we were to omit any sequence from alignment A, then it would be sensible to omit the one with the smallest 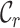 value. The
value. The 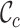 values range from 0.2 to 1.0 for alignment A and from 0.5 to 0.8 for alignment B. Again, there is greater concern about alignment A than about alignment B (due to the lower
values range from 0.2 to 1.0 for alignment A and from 0.5 to 0.8 for alignment B. Again, there is greater concern about alignment A than about alignment B (due to the lower 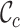 scores and the greater range of values). The
scores and the greater range of values). The 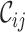 values range from 0.3 to 1.0 for alignment A and from 0.0 to 0.9 for alignment B. There is cause for great concern if
values range from 0.3 to 1.0 for alignment A and from 0.0 to 0.9 for alignment B. There is cause for great concern if  is detected because it means that sequences
is detected because it means that sequences 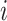 and
and 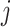 have no shared homologous sites with completely specified characters in both sequences. Evolutionary distances between such sequences cannot be estimated unless the MSA contains at least one other sequence that overlaps both
have no shared homologous sites with completely specified characters in both sequences. Evolutionary distances between such sequences cannot be estimated unless the MSA contains at least one other sequence that overlaps both 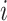 and
and 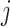 . When such a case occurs, the evolutionary distance between sequences
. When such a case occurs, the evolutionary distance between sequences 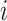 and
and 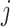 is inferred by proxy. Currently, the prevalence of this problem is unknown.
is inferred by proxy. Currently, the prevalence of this problem is unknown.
Figure 1.

Example, based on two multiple sequences alignments (A), illustrating the corresponding distributions of completeness scores for rows (B), columns (C) and pairs of sequences (D).
Figures 1B and 1C reveal the distributions of 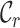 and
and 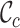 for alignments A and B, offering additional insight into the alignments’ completeness. Conveniently, the
for alignments A and B, offering additional insight into the alignments’ completeness. Conveniently, the 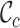 scores may be used to selectively omit the least complete sites. This masking of sites in MSAs is popular in phylogenetics and many methods (13–21) are now available. Additional information can be obtained by analyzing heat maps generated from the
scores may be used to selectively omit the least complete sites. This masking of sites in MSAs is popular in phylogenetics and many methods (13–21) are now available. Additional information can be obtained by analyzing heat maps generated from the 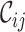 values. Figure 1D shows the heat maps obtained from alignments A and B. The most obvious things to note are that in alignment A Tagliatelle stands out as being the least complete sequence whereas Capellini and Spaghetti share no homologous sites with completely specified nucleotides in both sequences in alignment B. Although this was easy to detect in Figure 1A, it will be more difficult to do if
values. Figure 1D shows the heat maps obtained from alignments A and B. The most obvious things to note are that in alignment A Tagliatelle stands out as being the least complete sequence whereas Capellini and Spaghetti share no homologous sites with completely specified nucleotides in both sequences in alignment B. Although this was easy to detect in Figure 1A, it will be more difficult to do if 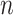 and/or
and/or 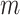 were larger, as is typically the case in phylogenomic data.
were larger, as is typically the case in phylogenomic data.
The benefits offered by the new completeness metrics are clear, but embedding figures like those in Figure 1 in publications may be impractical. Alternatively, the essential details may be reported in a table (Table 1), or in one line (e.g. alignment B: 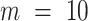 ,
, 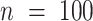 ,
, 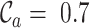 ,
, 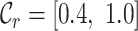 ,
, 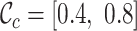 and
and 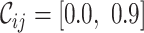 ). The closer to 1.0 the four
). The closer to 1.0 the four 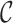 scores are, the more complete an alignment is. If, on the other hand, the values are closer to 0.0 than to 1.0, users may consider masking some of the sequences and/or sites before starting a phylogenetic analysis of the data.
scores are, the more complete an alignment is. If, on the other hand, the values are closer to 0.0 than to 1.0, users may consider masking some of the sequences and/or sites before starting a phylogenetic analysis of the data.
Table 1.
Example of the MRS for the alignments in Figure 1A
| Feature | Alignment A | Alignment B |
|---|---|---|
| Sequences | 10 | 10 |
| Sites | 100 | 100 |
| Alphabet | Nucleotides | Nucleotides |
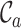
|
0.7 | 0.7 |
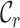 [min–max] [min–max] |
0.3–1.0 | 0.4–1.0 |
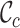 [min–max] [min–max] |
0.1–1.0 | 0.4–0.8 |
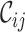 [min–max] [min–max] |
0.3–1.0 | 0.0–0.9 |
Given their potential to inform researchers across a wide range of scientific disciplines, we argue that 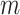 ,
, 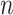 ,
, 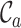 ,
, 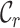 ,
, 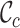 and
and 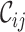 should be combined into what we henceforth call an MRS for MSAs, and that publications that report all of these values be labeled compliant with the MRS for MSAs. To our knowledge, this has never been done beforehand, leading to widespread ignorance about the MSAs that are relied upon in ground-breaking biomedical research.
should be combined into what we henceforth call an MRS for MSAs, and that publications that report all of these values be labeled compliant with the MRS for MSAs. To our knowledge, this has never been done beforehand, leading to widespread ignorance about the MSAs that are relied upon in ground-breaking biomedical research.
AliStat: a program supporting the MRS for MSAs
To enable compliance with the MRS for MSAs, we developed AliStat, which is written in C++. To our knowledge, it is the first program to compute the four completeness scores presented above.
AliStat reads a text file with sequences of single nucleotides (i.e. a 4-state alphabet), di-nucleotides (i.e. a 16-state alphabet), codons (a 64-state alphabet) and amino acids (a 20-state alphabet), which are aligned and saved in the FASTA format. If the sequences comprise single nucleotides, then the characters may be ‘lumped’ to form six 3-state alphabets (i.e. CRT, AGY, ACK, GMT, AST and CGW) and seven 2-state alphabets (i.e. RY, KM, SW, AB, CD, GH and TV)—here R = A or G, Y = C or T, K = A or C, M = G or T, B = C or G or T, D = A or G or T, H = A or C or T, and V = A or C or G. If the 3- and 2-state alphabets are used, the letters R, Y, K, M, S, W, B, D, H and V are considered completely specified characters, unlike normal practice (12).
AliStat can be run in two modes: Brief mode or Full mode. Execution in brief mode is done using the following command:
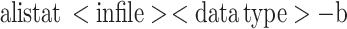 |
and results in the following output format being printed to the terminal:
File name, #seqs, #sites,
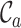 ,
, 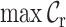 ,
, 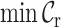 ,
, 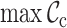 ,
, 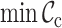 ,
, 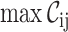 ,
, 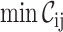
The brief-mode execution was included to allow users to quickly obtain the essential values from a great number of alignments (e.g. when comparing genomes phylogenetically).
The full-mode execution (default option) allows other options to be used and is intended when a more detailed examination of an MSA is required. For example, the –t option is used to indicate what types of 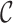 scores should be printed in output files, the –m option is used to set a threshold for masking sites and the –i option is used to indicate that a heat map is needed. Other options and how all of the options may be used are described in the AliStat manual. The same information can be obtained by typing
scores should be printed in output files, the –m option is used to set a threshold for masking sites and the –i option is used to indicate that a heat map is needed. Other options and how all of the options may be used are described in the AliStat manual. The same information can be obtained by typing
 |
in the command-line.
The output files appear in the .txt, .csv, .R, .dis, .svg and .fst formats, which can be processed by other software packages. The .txt file summarizes the results. The .csv files present the 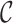 scores and may be examined using R. For example, if a user wishes to generate a histogram of the
scores and may be examined using R. For example, if a user wishes to generate a histogram of the 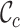 scores, the Table_2.csv file may be analyzed using the Histogram_Cr.R file. In some cases, users may want to infer a tree or network based on the
scores, the Table_2.csv file may be analyzed using the Histogram_Cr.R file. In some cases, users may want to infer a tree or network based on the 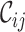 score (or the
score (or the 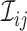 score, where
score, where 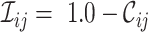 ). In such cases, .dis files may be analyzed by, for example, SplitsTree (22). The heat map, which may be triangular or square, is stored in the .svg file and may be opened using Adobe Illustrator™. If the –m option is used, the original MSA is split into two, with all sites having a
). In such cases, .dis files may be analyzed by, for example, SplitsTree (22). The heat map, which may be triangular or square, is stored in the .svg file and may be opened using Adobe Illustrator™. If the –m option is used, the original MSA is split into two, with all sites having a 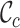 score larger than a user-specified threshold saved in a file called Mask.fst and the other sites saved in a file called Disc.fst. The two .fst files may be analyzed separately by other means (e.g. phylogenetic programs).
score larger than a user-specified threshold saved in a file called Mask.fst and the other sites saved in a file called Disc.fst. The two .fst files may be analyzed separately by other means (e.g. phylogenetic programs).
RESULTS AND DISCUSSON
The MRS may help to identify dubious MSAs. These alignments occur regularly in biomedical research and may also be present in large phylogenomic research, due to problems that might have arisen during the assembly, orthology assignment and alignment procedures.
Typically, MSAs comprise more sequences and sites than those in Figure 1A, so to facilitate using the MRS, we implemented AliStat, a fast, flexible and user-friendly program for surveying MSAs. AliStat computes the 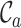 ,
, 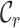 ,
, 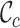 and
and 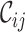 values from MSAs of nucleotides, di-nucleotides, codons and amino acids., AliStat lists the results on the command-line or in files that can be accessed by other programs.
values from MSAs of nucleotides, di-nucleotides, codons and amino acids., AliStat lists the results on the command-line or in files that can be accessed by other programs.
The benefit of the MRS for MSAs is underlined in two surveys of large MSAs (Table 2). In the first case, surveying an MSA of the enzyme carboxyl/cholinesterase (23) revealed that some of the 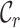 and
and 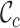 scores are closer to 0.0 than 1.0, and that at least two sequences have no homologous sites in common with completely specified characters in both sequences. Further inspection of the output files revealed large proportions of low
scores are closer to 0.0 than 1.0, and that at least two sequences have no homologous sites in common with completely specified characters in both sequences. Further inspection of the output files revealed large proportions of low 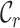 ,
, 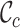 and
and 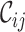 scores (Supplementary Figures, S1–3), so it might be wise to mask some of the sequences or sites before phylogenetic analysis of these data. Given the main objective of the original analysis of these data (to annotate the genes in two major crop pests), masking sites with completeness scores below
scores (Supplementary Figures, S1–3), so it might be wise to mask some of the sequences or sites before phylogenetic analysis of these data. Given the main objective of the original analysis of these data (to annotate the genes in two major crop pests), masking sites with completeness scores below 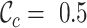 had a big impact on the
had a big impact on the 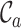 score (it increased from 0.2262 to 0.9562) and, hence, also on the maximum scores of
score (it increased from 0.2262 to 0.9562) and, hence, also on the maximum scores of 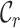 and
and 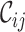 (Supplementary Table S1).
(Supplementary Table S1).
Table 2.
Example of the MRS for two published MSAs
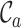
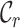
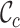
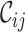
In the second case, surveying a massive concatenation of MSAs of nuclear genes (24) revealed a more complete alignment but also low 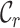 ,
, 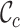 and
and 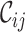 values. The presence of these values shows that additional masking of this MSA might have been wise (Supplementary Figures S4–6). For example, omitting the two most incomplete sequences (i.e. the genera Leucoptera and Pseudopostega) could have been considered (Supplementary Figures S4 and 6).
values. The presence of these values shows that additional masking of this MSA might have been wise (Supplementary Figures S4–6). For example, omitting the two most incomplete sequences (i.e. the genera Leucoptera and Pseudopostega) could have been considered (Supplementary Figures S4 and 6).
The MRS for MSAs is a robust and sensible solution to a large and so-far-neglected problem: how do we report, as transparently and informatively as possible, the completeness of the MSAs used in biomedical research? Better transparency about the completeness of MSAs is clearly needed, because MSAs represent a foundational cornerstone in many biomedical research projects and, as revealed by the example in Figure 1, MSAs may look different but have the same percentage of missing data. So far, information on the completeness of MSAs used in biomedical research has been largely absent, leaving readers unable to critically evaluate the merits of scientific discoveries made on the basis of MSAs. It is critical to recognize, and acknowledge, that many MSAs are the result of scientific procedures. Therefore, it is necessary to present the results of these procedures more transparently and comprehensively. Many scientific papers now include links to the MSAs used, but the MSAs are often so large that it is impossible to form a comprehensive picture about the completeness of these MSAs.
Our MRS enables a radical change in scientific behavior, allowing authors to report their results more transparently and readers the ability to critically assess discoveries made from analyses of sequence data stored in MSAs.
DATA AVAILABILITY
AliStat is available from http://github.com/thomaskf/AliStat/ under an CSIRO Open Source Software License Agreement (variation of the BSD / MIT License).
Supplementary Material
ACKNOWLEDGEMENTS
We thank staff at the Australian National University and University College Dublin for feedback on the color scheme used in the heat map; many of the respondents are color-blind. At last, we wish to thank three reviewers for their constructive comments.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
CSIRO.
Conflict of interest statement. None declared.
REFERENCES
- 1. Higgs D.R., Wood W.G.. Genetic complexity in sickle cell disease. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:11595–11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Misof B., Liu S.L., Meusemann K., Peters R.S., Donath A., Mayer C., Frandsen P.B., Ware J., Flouri T., Beutel R.G. et al.. Phylogenomics resolves the timing and pattern of insect evolution. Science. 2014; 346:763–767. [DOI] [PubMed] [Google Scholar]
- 3. Jayaswal V., Wong T.K.F., Robinson J., Poladian L., Jermiin L.S.. Mixture models of nucleotide sequence evolution that account for heterogeneity in the substitution process across sites and across lineages. Syst. Biol. 2014; 63:726–742. [DOI] [PubMed] [Google Scholar]
- 4. Wilding M., Peat T.S., Kalyaanamoorthy S., Newman J., Scott C., Jermiin L.S.. Reverse engineering: transaminase biocatalyst development using ancestral sequence reconstruction. Green Chem. 2017; 19:5375–5380. [Google Scholar]
- 5. Thompson J.D., Higgins D.G., Gibson T.J.. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994; 22:4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F., Higgins D.G.. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997; 25:4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Noorden R., Maher B., Nuzzo R.. The top 100 papers. Nature. 2014; 514:550–553. [DOI] [PubMed] [Google Scholar]
- 8. Morrison D.A. Is sequence alignment an art or a science. Syst. Bot. 2015; 40:14–26. [Google Scholar]
- 9. Wiens J.J. Missing data, incomplete taxa, and phylogenetic accuracy. Syst. Biol. 2003; 52:528–538. [DOI] [PubMed] [Google Scholar]
- 10. Driskell A.C., Ane C., Burleigh J.G., McMahon M.M., O’Meara B.C., Sanderson M.J.. Prospects for building the tree of life from large sequence databases. Science. 2004; 306:1172–1174. [DOI] [PubMed] [Google Scholar]
- 11. Vihinen M. Guidelines for systematic reporting of sequence alignments. Biol. Methods Protoc. 2020; 5:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cornish-Bowden A. Nomenclature for incompletely specified bases in nucleic acid sequences: recommendations 1984. Nucleic Acids Res. 1985; 13:3021–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Castresana J. Selection of conservative blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000; 17:540–552. [DOI] [PubMed] [Google Scholar]
- 14. Talavera G., Castresana J.. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007; 56:564–577. [DOI] [PubMed] [Google Scholar]
- 15. Dress A.W.M., Flamm C., Fritzsch G., Grunewald S., Kruspe M., Prohaska S.J., Stadler P.F.. Noisy: identification of problematic columns in multiple sequence alignments. Algorith. Mol. Biol. 2008; 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hartmann S., Vision T.J.. Using ESTs for phylogenomics: can one accurately infer a phylogenetic tree from a gappy alignment. BMC Evol. Biol. 2008; 8:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Capella-Gutierrez S., Silla-Martinez J.M., Gabaldon T.. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009; 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Misof B., Misof K.. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst. Biol. 2009; 58:21–34. [DOI] [PubMed] [Google Scholar]
- 19. Kück P., Meusemann K., Dambach J., Thormann B., von Reumont B.M., Wägele J.W., Misof B.. Parametric and non-parametric masking of randomness in sequence alignments can be improved and leads to better resolved trees. Front. Zool. 2010; 7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Criscuolo A., Gribaldo S.. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010; 10:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wu M.T., Chatterji S., Eisen J.A.. Accounting for alignment uncertainty in phylogenomics. PLoS One. 2012; 7:e30288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huson D.H., Bryant D.. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006; 23:254–267. [DOI] [PubMed] [Google Scholar]
- 23. Pearce S.L., Clarke D.F., East P.D., Elfekih S., Gordon K.H.J., Jermiin L.S., McGaughran A., Oakeshott J.G., Papanikolaou A., Perera O.P. et al.. Genomic innovations, transcriptional plasticity and gene loss underlying the evolution and divergence of two highly polyphagous and invasive Helicoverpa pest species. BMC Biol. 2017; 15:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kawahara A.Y., Plotkin D., Espeland M., Meusemann K., Toussaint E.F.A., Donath A., Gimnich F., Frandsen P.B., Zwick A., dos Reis M. et al.. Phylogenomics reveals the evolutionary timing and pattern of butterflies and moths. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:22657–22663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
AliStat is available from http://github.com/thomaskf/AliStat/ under an CSIRO Open Source Software License Agreement (variation of the BSD / MIT License).