

Abstract
Understanding transcription has been a central goal of the scientific community for decades. However, much is still unknown, especially concerning how it is regulated. In bacteria, a single DNA-directed RNA-polymerase performs the whole of transcription. It contains multiple subunits, among which the σ factor that confers promoter specificity. Besides the housekeeping σ factor, bacteria encode several alternative σ factors. The most abundant and diverse family of alternative σ factors, the extracytoplasmic function (ECF) family, regulates transcription of genes associated with stressful scenarios, making them key elements of adaptation to specific environmental changes. Despite this, the evolutionary history of ECF σ factors has never been investigated. Here, we report on our analysis of thousands of members of this family. We show that single events are in the origin of alternative modes of regulation of ECF σ factor activity that require partner proteins, but that multiple events resulted in acquisition of regulatory extensions. Moreover, in Bacteroidetes there is a recent duplication of an ecologically relevant gene cluster that includes an ECF σ factor, whereas in Planctomycetes duplication generates distinct C-terminal extensions after fortuitous insertion of the duplicated σ factor. At last, we also demonstrate horizontal transfer of ECF σ factors between soil bacteria.
INTRODUCTION
Transcription is an essential process for all DNA-based organisms. Eukaryotes use three distinct RNA polymerases that are responsible for transcription of different genes. In contrast, bacteria contain a single DNA-directed RNA polymerase, a multi-subunit enzyme whose catalytic subunits (β and β’) are similar to their eukaryotic counterparts. The bacterial specific σ subunit is essential for transcription initiation and is the one determining promoter specificity (1). σ factors can be assigned to one of four groups according to their overall function and domain architecture (1–5). All bacterial cells contain an essential group 1 σ factor, which initiates transcription of housekeeping genes and hence ensures growth. Group 1 σ factors contain four conserved domains: σ1 acts to prevent promiscuous binding of the σ factor to DNA when not associated with the RNA polymerase; σ2 is required for binding to the RNA polymerase, promoter recognition and DNA melting; σ3 interacts with the promoter; and σ4 is necessary for promoter recognition. The large majority of bacteria additionally contain alternative σ factors belonging to the three other groups. Group 2 contains alternative (non-essential) σ factors with the same domain architecture as those of group 1. Group 3 is composed of σ factors that lack the σ1 domain and regulate expression of heat-shock, flagellar and sporulation genes. Group 4 gathers the extracytoplasmic function (ECF) σ factors that contain only σ2 and σ4 domains and regulate transcription of subsets of genes whose expression is only necessary in very specific, usually stressful, situations.
Over the last decade many studies about σ factors reported the analysis of a few hundred genomes of Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria and Planctomycetes (6–10). Several ECF groups were proposed based on sequence similarity, regulatory mechanism, genomic context conservation and target promoter sequence. These studies brought to light the high number of ECF σ factors encoded in each genome, their widespread distribution among bacteria, the diversity of their target promoter sequence and regulatory mechanisms.
The classical ECF σ factor regulatory mechanism involves a membrane-bound anti-σ factor that sequesters the ECF σ factor and prevents its interaction with the RNA polymerase in the absence of an input signal. Upon stimulus perception, three proteolytic cleavage steps lead to the degradation of the anti-σ factor and subsequent release of the ECF σ factor (11–13). Alternative regulatory mechanisms rely on (i) conformational changes of soluble anti-σ factors (14–17), (ii) transcriptional activation by two-component systems, (18,19), (iii) conserved C-terminal extensions of the minimal σ2 and σ4 architecture (9,20,21) or (iv) N-terminal extensions (10).
Since regulating ECF σ factor activity is crucial for cell homeostasis and due to the complexity associated with that regulation, often requiring additional proteins or domains, we investigated the evolutionary origin of such mechanisms. Here we characterize the evolutionary steps that led to the high diversity and abundance observed in ECF σ factors by doing a comparative genomics analysis on 16,606 ECF sequences. Specifically, we determined the phylogenetic relationships between ECF σ factors, investigated recent ECF gene duplication events and tested for horizontal gene transfer, to understand the abundance and diversity of ECF σ factors encoded in single bacterial genomes.
We show that single events are in the origin of alternative modes of regulation of ECF σ factor activity that require partner proteins but that, in contrast, multiple events resulted in the acquisition of regulatory extensions. We also identified recent duplications in Bacteroidetes and Planctomycetes and show evidences that in the first they originate from the duplication of an ecologically relevant gene cluster. In the former, the duplication and fortuitous insertion of the duplicated σ factor generated unrelated C-terminal extensions. At last, we show that an event of horizontal gene transfer is responsible for the presence of a group of ECF σ factors in Streptomyces.
MATERIALS AND METHODS
ECF σ factor phylogeny inference
All ECF σ factors sequences used for phylogenetic inference were downloaded from the National Center for Biotechnology Information (NCBI) protein database (Supplementary File1) and were previously published (6–10). Conserved protein domains were identified using HMMER version 3.1b2 (22) to search PfamA version 31.0 profiles (23) against our ECF sequences with an E-value cut-off of 10. Sequences that were assigned to either Sigma70_r2 (PF04542; σ2 region) and Sigma70_r4 or Sigma70_r4_2 (PF04545 or PF08281; σ4 region) domains and were at least 40 amino acids long, were extracted using a customized Python (24) script (Supplementary File 2). A multiple sequence alignment was performed for each domain using a local installation of MAFFT (25) with default settings. Pairwise sequence identities and distances calculated with the BLOSUM45 matrix are shown in Supplementary Figure S1. ECF sequences were then clustered according to their percent identity using customized Python (24) script. Briefly, all pairwise identities were calculated from the multiple sequence alignment and sequence pairs with a percent identity > 50% were put in the same cluster (Supplementary Files 3–6). One sequence of each cluster was randomly selected and a maximum-likelihood tree was generated with IQ-TREE (26) using the LG+F+R5 model of evolution (amino acid exchange rate matrices: general matrix (27); amino acid frequency: empirical amino acid frequencies from the data; rate heterogeneity across sites: free rate model (28,29)), chosen using the Bayesian Information Criterion from the available 542 protein evolution models available in IQ-TREE (30) and including 1000 fast bootstraps (31) (Supplementary File 7). Additionally, the sequences of the most divergent pair in each cluster were selected and maximum-likelihood tree was generated with IQ-TREE (26) using the LG+F+R6 (amino acid exchange rate matrices: general matrix (27); amino acid frequency: empirical amino acid frequencies from the data; rate heterogeneity across sites: free rate model (28,29)) chosen using the Bayesian Information Criterion from the available 542 protein evolution models available in IQ-TREE (30) and including 1000 fast bootstraps (31) (Supplementary File 35).
ECF σ factor duplication in Bacteroidetes
We selected 99 organisms belonging to this phylum whose genomes were classified as representative in the NCBI genome database (Supplementary File 8). The complete proteomes were downloaded and HMMER version 3.1b2 (22) was used to search PfamA version 31.0 profiles Sigma70_r1_2 (PF00140), Sigma70_r1_1 (PF03979), Sigma70_ner (PF04546), Sigma70_r2 (PF04542), Sigma70_r3 (PF04539), Sigma70_r4 (PF04545) and Sigma70_r4_2 (PF08281) (23) against the complete proteomes, with an E-value cut-off of 10. ECF σ factors were identified as those proteins with hits of Sigma70_r2 and Sigma70_r4 or Sigma70_r4_2 but no hits of other profiles (Supplementary Files 8 and 9). A multiple sequence alignment was done for each separate domain using MAFFT (25) with default settings. The concatenated alignment was used to generate a maximum-likelihood tree with IQ-TREE (26,30,31) (model of evolution: LG+R9, chosen using the Bayesian Information Criterion; amino acid exchange rate matrix: general matrix; rate heterogeneity across sites: free rate model (32–34)) (Supplementary File 10). To generate the species tree for comparison, we used the protein sequences of housekeeping genes gyrB, metG, trxB, nth, lon, mutS2, secA and argS for each organism (Supplementary File 11). Sequences were aligned with MAFFT (25) with default settings and then concatenated to generate a maximum-likelihood tree with IQ-TREE (26,30,31) (model of evolution: LG+F+R8, chosen using the Bayesian Information Criterion; amino acid exchange rate matrix: general matrix; amino acid frequency: empirical amino acid frequencies from the data; rate heterogeneity across sites: free rate model (32–34) (Supplementary File 12)). Genomic context conservation was analyzed with Microbial Genomic context Viewer (35), centered on the ECF σ factors with a context range of 15 000 nt and an identical gene orientation; genes are represented by their locus tag and custom color coded.
ECF σ factor duplication in Planctomycetes
We selected 23 organisms belonging to the Planctomycetes whose genomes were classified as representative in the NCBI genome database (Supplementary File 13). The complete proteomes were downloaded and HMMER version 3.1b2 (22) was used to search PfamA version 31.0 profiles Sigma70_r1_2 (PF00140), Sigma70_r1_1 (PF03979), Sigma70_ner (PF04546), Sigma70_r2 (PF04542), Sigma70_r3 (PF04539), Sigma70_r4 (PF04545) and Sigma70_r4_2 (PF08281) (23) against the complete proteomes with an E-value cut-off of 10. ECF σ factors were identified as those proteins containing hits of Sigma70_r2 and Sigma70_r4 or Sigma70_r4_2 but no hits of the other profiles (Supplementary Files 13 and 14). Their Sigma70_r2 and Sigma70_r4 or Sigma70_r4_2 sequences were extracted, aligned with MAFFT (25) and concatenated to generate a maximum-likelihood tree with IQ-TREE (26,30,31) (model of evolution: LG+F+R5, chosen using the Bayesian Information Criterion; amino acid exchange rate matrix: general matrix; amino acid frequency: empirical amino acid frequencies from the data; rate heterogeneity across sites: free rate model (32–34) (Supplementary File 15)). Trees were also inferred for σ2 and σ4 domains and C-terminal extensions (Supplementary Files 16 and 17) of the ECF σ factors from the monophyletic group containing all duplication (models of evolution: LG+F+R7 and VT+F+R6, respectively, chosen using the Bayesian Information Criterion; amino acid exchange rate matrices: general and general ‘variable time’ matrices, respectively; amino acid frequency: empirical amino acid frequencies from the data; rate heterogeneity across sites: free rate model (26,30–34,36)). Concordance factors between the two trees were calculated with IQ-TREE (37). HMMER version 3.1b2 (22) was used to search PfamA version 31.0 profiles against the C-terminal extension sequences of the ECF σ factors of the monophyletic group containing all duplicated clusters. The E-value of the best domain hit was considered as a measure of the confidence (Supplementary File 18). BLASTx (38) was used to search the nucleotide sequence of each C-terminal extension against the non-redundant database of NCBI (Supplementary File 19). Results were filtered to include solely hits whose percent identity was above 30% and whose E-value was below 0.001. The assignment of NCBI taxon IDs to the NCBI accession numbers was done using the taxonomizr R package (39,40). Lineage information was automatically retrieved from NCBI taxonomy database using a custom Python (24) script (Supplementary File 20).
Horizontal gene transfer of ECF σ factors
We selected 238 organisms belonging to genus Streptomyces whose genomes were classified as representative genomes in the NCBI genome database (Supplementary File 21). The complete proteomes were downloaded and HMMER version 3.1b2 (22) was used to search PfamA version 31.0 profiles Sigma70_r1_2 (PF00140), Sigma70_r1_1 (PF03979), Sigma70_ner (PF04546), Sigma70_r2 (PF04542), Sigma70_r3 (PF04539), Sigma70_r4 (PF04545) and Sigma70_r4_2 (PF08281) (23) against the complete proteomes with an E-value cut-off of 10. ECF σ factors were identified as those proteins containing hits of Sigma70_r2 and Sigma70_r4 or Sigma70_r4_2 but no hits of the other profiles (Supplementary Files 21 and 22). Their Sigma70_r2 and Sigma70_r4 or Sigma70_r4_2 sequences were extracted, aligned with MAFFT (25) and concatenated to generate a maximum-likelihood tree with IQ-TREE (26,30,31) (model of evolution: LG+F+R8, chosen using the Bayesian Information Criterion; amino acid exchange rate matrix: general matrix; amino acid frequency: empirical amino acid frequencies from the data; rate heterogeneity across sites: free rate model (32–34) (Supplementary File 23). Horizontal gene transfer of ECF σ factors was first investigated using HGT-Finder (41). BLASTp (38) was used to search the NCBI non-redundant protein database using each ECF σ factor sequence as query and also to perform a search of each ECF σ factor sequence against itself. The taxon ID was retrieved for each organism from the NCBI Taxonomy database. HGT-Finder was run locally with R-values of 0.2, 0.2, 0.4, 0.5, 0.6, 0.7, 0.8 and 0.9 (Supplementary File 24). ECF groups were classified using ECFfinder (6). HMMER version 3.1b2 (22) was used to search the hidden Markov models of the relevant ECF groups (6) against the non-redundant protein database of NCBI. The assignment of NCBI taxon IDs to the NCBI accession numbers was done using the taxonomizr R package (39,40). Lineage information was automatically retrieved from NCBI using a custom Python (24) script (Supplementary File 20). Phylogenetic reconciliation was performed with Notung 2.9 (42). The species tree was generated with the protein sequences of housekeeping genes atpD, recA, rpoB and trpB (Supplementary Files 21 and 25). The sequences were extracted aligned with MAFFT (25) and used to generate a maximum-likelihood tree with IQ-TREE (26,30) (model of evolution: LG+F+R9, chosen using the Bayesian Information Criterion; amino-acid exchange rate matrix: general matrix; amino-acid frequency: empirical amino-acid frequencies from the data; rate heterogeneity across sites: FreeRate model (32–34) (Supplementary File 26)). Tree of the ECF20 σ factors was generated the same way as the tree with all the ECF σ factors (model of evolution LG+F+G4; Supplementary Files 21 and 27). Dotplots for the pairwise comparison of the 10 kbp region surrounding the ECF20 coding genes (Supplementary File 21) were generated with the seqinr R package (43) and a window size of 100 bps, a window step of 100 bps and a minimum number of matches needed to produce a dot of 50 bps.
Graphical representation of the data
Unless stated otherwise, the graphics presented here were generated using R (39) and packages ape (44), gplots (45), seqinr (43) and ggplot2 (46). Supplementary Files 28 to 31 containing the R scripts used to generate the main figures and Supplementary Files 32 to 34 those used to generate the Supplementary Figures.
RESULTS
Mapping the alternative regulatory mechanisms of ECF σ factor activity onto a gene tree
Previously, over 5000 ECF σ factors were identified in almost 700 bacterial genomes (6–10), but despite having been classified in groups based on a number of functional criteria (6), their evolutionary relationships are broadly uncharacterized. We started our analysis by clustering all 5000 ECF σ factors according to the percent identity of their conserved σ2 and σ4 regions and then used one sequence of each cluster to infer a reference ECF σ factor phylogeny (Figure 1). Since it has been shown before that ECF σ factors belonging to the same group have the same regulatory mechanism (6,20,47,48), we assigned a regulatory mechanism to each cluster according to what has been previously proposed for the ECF group(s) represented in each cluster (49). We considered six types of mechanisms that depend on: (i) membrane-bound anti-σ factors; (ii) soluble anti-σ factors; (iii) transcriptional control; (iv) C-terminal extensions; (v) N-terminal extensions; or (vi) serine/threonine protein kinases. This grouping clusters the regulatory mechanisms based on their extensive major similarities despite the small differences sometimes observed between members of the same group.
Figure 1.

Phylogenetic distribution of ECF σ factor activity regulatory mechanisms. Maximum-likelihood tree of ECF σ factors built with one representative sequence of each cluster. The tree tips are labeled with the cluster number (001–427), the ECF group represented in the cluster and color coded according to the regulatory mechanism proposed for the cluster members. *Refer to Supplementary File 6 for more details on the distribution of ECF groups in the cluster and Supplementary Figure S5 for a tree of the most divergent pair of each cluster.
The distribution of the regulation types on the ECF σ factor phylogeny confirms the assumption that most ECF σ factors (74% of the clusters with assigned mechanisms) are regulated by membrane bound anti-σ factors (dark blue in Figure 1). The data also suggest multiple independent origins of soluble anti-σ factor-dependent regulation (light blue in Figure 1) and of regulatory C-terminal extensions (red in Figure 1). Although speculative at the moment, we hypothesize that soluble anti-σ factors have evolved from membrane-bound anti-σ factors either by accumulation of mutations that compromised the hydrophobicity of the transmembrane helix and consequent membrane association or by domain-splitting of the cytoplasmic and extracytoplasmic domains of the anti-σ factor. The remaining regulatory mechanisms—transcriptional control (green in Figure 1), serine/threonine protein kinases (yellow in Figure 1) and N-terminal extensions (orange in Figure 1)—seem to have been acquired only once, with the N-terminal extension appearing in a serine/threonine protein kinase regulated ancestor.
The genomic context of ECF σ factor duplications
The characterization of recent ECF σ factor duplications was done first on Bacteroidetes, as bacteria of this phylum contain several members of group ECF10 (6), suggestive of an increased level of duplication of these ECF σ factors. We selected 99 organisms belonging to this phylum, whose genomes were classified as representative in the NCBI genome database. We identified 2257 ECF σ factors and five recent duplications clusters, i.e. monophyletic groups of ECF σ factors all found in the same genome and hence originating after the last speciation event (Figure 2A and Supplementary Table S1). Figure 2B shows how the duplications are not shared between the Bacteroidetes, but are specific to only some organisms. In order to assess whether the duplications included the ECF σ factor alone or a larger portion of the genome, we analyzed the genomic context conservation around the duplicated ECF σ factor genes (Figure 2C). For duplicated clusters 1 and 5, the duplications included not only the ECF σ factor, but also adjacent genes (a total of five and three genes, respectively). Instead, for duplicated clusters 2, 3 and 4, a total of two additional genes were also duplicated. The duplicated genes code for a Sus-like system, used by Bacteroidetes in the gut to import and degrade glycans (50).
Figure 2.

ECF σ factor duplication in Bacteroidetes. (A) Maximum-likelihood tree of Bacteroidetes ECF σ factors. The tree tips are labeled with the NCBI accession number and color coded according to the duplicated cluster. (B) Bacteroidetes species and ECF σ factor trees. The species tree is a maximum-likelihood tree build from sequences of GyrB, MetG, TrxB, Nth, Lon, MutS2, SecA and ArgS. Species tree tips are labeled with the organism name. The ECF σ factor tree is the same represented in panel A. Connecting lines represent the association between the ECF σ factors belonging to the identified duplicated clusters and their species, and are color coded accordingly: duplicated cluster 1, dark blue; duplicated cluster 2, light blue; duplicated cluster 3, green; duplicated cluster 4, red; duplicated cluster 5, orange. (C) Genome context conservation inside each cluster. Locus and organisms are indicated on the left side. Conserved genes are color coded according to the legend in the bottom.
We then proceeded to investigate the duplications in Planctomycetes using 23 genomes classified as representative in the NCBI genome database. We identified 1134 ECF σ factors and detected four recently duplicated clusters (Figure 3A and Supplementary Table S2). These ECF σ factors have C-terminal extensions which, unlike the highly conserved σ2 and σ4 regions, show low percent identity levels (Figure 3B). This is influencing the low similarity found for the full-length protein sequences, as the C-terminal regions can be quite large (Figure 3B and F). To determine whether the different domains had similar origins, we inferred maximum-likelihood phylogenetic trees of the C-terminal extensions and the σ2 and σ4 regions for the group of proteins included in the monophyletic group that contains all recent duplicated clusters (Figure 3C). This approach allowed us to see that recent duplicated clusters that can be found in the σ2 and σ4 tree are not seen in the C-terminal extension tree and that in most cases (concordance factors shown in Supplementary Figure S2), there are likely multiple origins for the C-terminal extensions present in ECF σ factors of a single cluster.
Figure 3.

ECF σ factor duplication in Planctomycetes. (A) Maximum-likelihood tree of Planctomycetes ECF σ factors. The tree tips are labeled with the NCBI accession number and color coded according to the duplicated cluster. (B) Boxplot showing the distribution of pairwise percent identity on each cluster. Dc1, duplicated cluster 1; Dc2, duplicated cluster 2; Dc3, duplicated cluster 3; Dc4, duplicated cluster 4; FL, full length sequence; R2, region σ2; R4, region σ4; CTE, C-terminal extension. Boxes are colored according to the duplicated cluster: duplicated cluster 1, dark blue; duplicated cluster 2, light blue; duplicated cluster 3, green; duplicated cluster 4, red. (C) Maximum-likelihood trees of ECF σ factors on the monophyletic group that includes all Planctomycetes duplicated clusters. On the left is the tree inferred exclusively from regions σ2 and σ4; on the right the tree inferred exclusively from the C-terminal extensions. Connecting lines represent the association between the ECF σ factors and those belonging to the recent duplicated clusters: duplicated cluster 1, dark blue; duplicated cluster 2, light blue; duplicated cluster 3, green; duplicated cluster 4, red. (D) Heat map of the PfamA profiles found in the C-terminal extensions of the ECF σ factors on the monophyletic group that includes all Planctomycetes duplicated clusters. Each row corresponds to a C-terminal extension in the same order as the CTE tree in panel C. Each column corresponds to a PfamA profile with at least one hit in the analyzed sequences. The heat map is color coded according to the E-value of the best hit found in the sequence for that PfamA profile; the color scale is depicted in the bottom. (E) Each row corresponds to a C-terminal extension in the same order as the CTE tree in panel C. The first bar refers to those cases in which an intergenic origin of the C-terminal extension is hypothesized (marked in blue). The second bar refers to the percent identity between the C-terminal extension and the first hit of the BLASTx search that is no longer an ECF σ factor. The third bar represents a measure of the taxonomical proximity between the organisms to which the ECF σ factor belongs to and the organism to which the best first hit that is no longer an ECF σ factor belongs to. 1, organisms belong to different superkingdoms; 2, organisms belong to different phyla; 3, organisms belong to different classes; 4, organisms belong to different orders; 5, organisms belong to different families; 6, organisms belong to different genus; 7, organisms belong to different species. The color scales for the second and third bars are shown in the bottom of the panel. Supplementary Table S3 provided detailed BLASTx data for the four recent duplicated clusters. (F) Histogram showing the distribution of C-terminal extension sizes among the ECF σ factors on the monophyletic group that includes all Planctomycetes duplicated clusters.
We expect that the evolutionary constraints on the C-terminal regions are more relaxed than on regions σ2 and σ4, which need to interact with both the DNA and the RNA polymerase, but the large diversity of PfamA profile hits in these C-terminal extensions (Figure 3D) and an extensive size variation (Figure 3F), cannot be explained solely by the unconstrained accumulation of mutations. Alternatively, what could have happened is the duplication of only the σ2 and σ4 regions and subsequent incorporation of the region adjacent to the chromosomal region of insertion as a C-terminal extension, thereby creating the pattern in Figure 3C. This is supported by the σ2 and σ4 regions and the C-terminal extensions: (i) having different levels of conservation (Figure 3B and F), (ii) not sharing the same evolutionary history (Figure 3C), (iii) having different putative functions (Figure 3D) and at last (iv) by the lack of genomic conservation inside the recent duplicated clusters (data not shown). In this scenario, the duplicated σ2 and σ4 regions could (Supplementary Figure S3): (a) insert in an intergenic region, thereby turning a previous non-coding region into a coding region until an in frame stop codon is reached; (b) insert in-frame in a pre-existing coding region, interrupting it and turning it's C-terminal portion in a C-terminal extension; (c) insert out-of-frame in a pre-existing coding region, interrupting it and turning it's C-terminal portion in a C-terminal extension with a different sequence until an in frame stop codon is found; (d) insert in an intergenic region, turning a previous non coding region into a coding region and sequestering the in-frame downstream gene; (e) insert in an intergenic region, turning a previous non-coding region into a coding region and sequestering the downstream gene, out-of-frame and until an in-frame stop codon is found.
To check whether the C-terminal extensions where part of pre-existing coding regions, we used BLASTx to map the nucleotide sequence of the C-terminal extension in all its six frames against the NCBI non-redundant protein database. For 46 C-terminal extensions, no similar proteins were found or those found represent similar ECF σ factors in other organisms, suggesting that those C-terminal extensions were previously non-coding sequences, supporting the scenario in which the inserted duplicated ECF σ factor containing the σ2 and σ4 regions often sequesters an intergenic region as a C-terminal extension (Figure 3E and Supplementary Figure S3, scenario A). For the remaining 159 C-terminal extensions, similar non-ECF σ factor proteins were found in the database with percent identities varying between 30 and 100% (Figure 3E). We argue that if these represent in fact the originally disrupted genes by the insertion of the ECF σ factor gene, similar proteins should be found encoded in genomes of closely related organisms. To investigate this, we determined the taxon ID corresponding to each protein, retrieved the lineage information stored at NCBI and compare the ECF σ factor and the putative disrupted gene's lineages. In 35% of the cases, the ECF σ factor and the putatively disrupted gene were found in organisms of the same genus; in 19% of the cases, they were found in organisms of the same family but different genus; in 24% they were found in organisms of the same order but different families; in 1% in organisms of the same class but different orders; in 3% in organisms of the same phylum but different classes; in 16% in organisms of different phyla; and only in 2% from organisms outside the Bacteria (Figure 3E). The fact that only a small number of Planctomycetes genomes has been sequenced to date, that most of them are still not completely classified and that 82% of the putatively disrupted genes are present in the genome of organisms of the same phylum agree with our hypothesis that some C-terminal extensions where previously part of coding regions disrupted by the insertion of the σ2 and σ4 coding sequences (Supplementary Figure S3, scenarios B, C, D and E).
Horizontal gene transfer of ECF σ factors
We next investigated horizontal gene transfer in the Streptomyces genus, an ECF σ factor rich group (6) where the occurrence of horizontal gene transfer has been suggested for an individual ECF σ factor of group ECF01 in Streptomyces coelicolor (51). We analyzed 238 Streptomyces spp. genomes using HGT-Finder (41) which does not require a priori assumptions about the nature or origin of the genes being transferred. Unlike other approaches that rely on synteny or species and gene trees (52,53), this one allowed us to focus the analysis on ECF σ factors alone, which makes the search more specific to this protein family. The HGT-Finder approach is, however, dependent on and hence biased by the composition of the NCBI non-redundant protein database, as many other approaches.
The HGT-Finder identified monophyletic groups of ECF σ factors whose probability of being acquired by the Streptomyces spp. by horizontal gene transfer was increased (Figure 4A). Those ECF σ factors preferentially belonged to four ECF groups (Figure 4B) (6): ECF20, ECF118, ECF121 and ECF123. The search for ECF σ factors of these groups on the NCBI non-redundant database revealed that ECF118, ECF121 and ECF123 are predominantly found in Actinobacteria (the bacterial phylum to which Streptomyces spp. belong), while those of ECF20 are predominantly found in Proteobacteria (Figure 4C). In fact, for the case of ECF20, HGT-Finder suggests a transfer from Sinorhizobium spp. (Proteobacteria) to Streptomyces spp.. We then proceeded to use gene tree reconciliation to investigate the origin of the horizontal gene transfer. We used 238 Streptomyces spp. and 19 Sinorhizobium spp. species together with their ECF σ factors of group ECF20 and performed phylogenetic tree reconciliation. This analysis identified four losses (all in the Sinorhizobium lineage) and 13 transfers (2 from a Sinorhizobium sp. to a Streptomyces sp.; 1 from a Streptomyces sp. to a Sinorhizobium sp.; 6 between Streptomyces spp. and 4 between Sinorhizobium spp.) (Figure 4D). Hence, the reconciliation supported the HGT-Finder predictions, confirming that the origin of the ECF20 σ factors in Streptomyces was via horizontal gene transfer from a Sinorhizobium sp.
Figure 4.
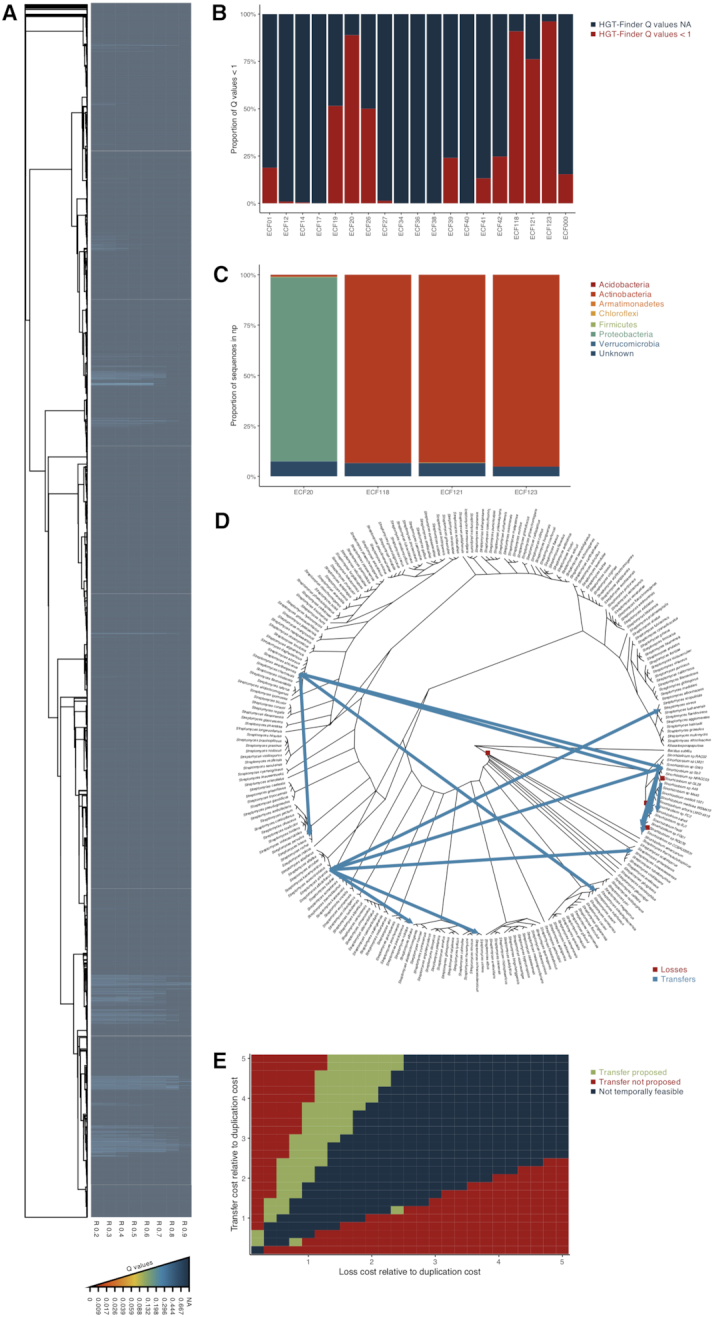
Horizontal gene transfer of ECF σ factors. (A) Heat map of the Q-values of HGT-Finder horizontal gene transfer predictions with R-values varying from 0.2 to 0.9 (R 0.2, R 0.3, R 0.4, R 0.5, R 0.6, R 0.7, R 0.8, R 0.9). The tree on the left side is a maximum-likelihood tree of Streptomyces spp. ECF σ factors. Q-values are color coded as shown in the bottom of the panel. (B) Bar graph showing the proportion of ECF σ factors that passed the initial HGT-Finder cut-off for putative horizontal transferred ECF σ factors (Q-values < 1) and those that did not (NA) and the ECF group they belong to, as determined using ECFfinder. (C) Taxonomical distribution of ECF σ factors of groups ECF20, ECF118, ECF121 and ECF123 (those with higher proportion of Q-values < 1 in panel B) on the NCBI non-redundant protein database. Phyla are color coded as shown on the right. (D) Species tree of the analyzed Streptomyces spp. and Sinorhizobium spp. with the evolutionary events predicted through phylogenetic reconciliation highlighted. Each type of event is color coded as shown on the bottom right side of the tree. Supplementary Table S4 provides a list of the events shown. (E) Graphical representation of the outcome of the phylogenetic reconciliation on the parameter space. The parameter space was screened on 0.2 intervals with the loss and transfer costs varying between 0 and 5 relative to the duplication cost. In green are represented all parameter combinations whose phylogenetic reconciliation predicted a transfer from Sinorhizobium sp. to Streptomyces sp.; in red are represent all parameter combinations whose phylogenetic reconciliation did not predict a transfer from Sinorhizobium sp. to Streptomyces sp.; in blue is represented the parameter space for which no temporally feasible solutions could be found.
The influence of the choice of parameters in the final outcome of a phylogenetical reconciliation analysis is well documented (54). Therefore we have further explored the parameter space (Figure 4E) to exclude bias in the selection of the event costs. The transfer from a Sinorhizobium sp. to a Streptomyces sp. is suggested as the most likely scenario to explain the origin of ECF20 σ factors in the Streptomyces spp., in all temporally feasible solutions in which the cost of transfer is above that of the loss, and only not when the cost of transfer is over four times higher than the cost of the loss. In these cases, the most parsimonious solutions suggest very high numbers of losses to explain the patchy distribution if these ECF σ factors in these organisms.
The analysis of similarity between the 10 kbp region surrounding the ECF20 coding gene in the Sinorhizobium spp. (Supplementary File 21) identified extensive homology, consistent with the vertical inheritance of the ECF coding gene. In contrast, no significant homology was identified among the Streptomyces spp. or between the Streptomyces spp. and the Sinorhizobium spp. (Supplementary Figure S4) suggesting that either the ECF gene is the only one being transfer or that extensive genomic instability in that region breaks synteny.
DISCUSSION
The ECF σ factor family is frequently referred to as the most abundant and diverse family of σ factors. This statement is indeed supported by census of a few hundred genomes (6–8,10) but the question remains as to how has this family of alternative σ factors become this important.
One of the features leading to the diversity of ECF σ factors is their variety of regulatory mechanisms (55), which can be roughly divided into six categories: (i) sequestration via membrane-bound (13) or (ii) soluble (56) anti-σ factors, (iii) transcriptional control (19), (iv) C- or (v) N-terminal extensions (10,21), or (vi) phosphorylation by serine/threonine protein kinases (57). When we mapped the regulatory mechanisms on the phylogeny of the ECF σ factor family (Figure 1), we found that a widespread distribution of membrane-bound anti-σ factors (74% of the tree tips with an assigned mechanism), which is in line with this being the ancestral regulatory mechanism. The use of soluble anti-σ factors seems to have been acquired multiple times, possibly through domain-splitting events (as proposed already for other protein families (58)). Independent events leading to convergent evolution in the type of regulation also seems to be the case for C-terminal extensions, which could simply arise from multiple events of gene fusion [Figure 3, (58)]. In contrast, the more complex mechanisms that require additional partner proteins (two-component systems and regulation via serine/threonine protein kinases) are only observed in four lineages of ECF σ factors (and represent 6% of the tree tips in with an assigned regulatory mechanism (Figure 1)).
We therefore propose that a small number of events gave rise to the acquisition of complex regulatory mechanisms that depend on additional partner proteins, while numerous and recurring events gave rise to the remaining mechanisms. We note that the assignments of regulatory mechanisms to each ECF group relied mostly on the presence of conserved neighbouring genes whose functions are deduced via automatic annotation or hidden-Markov model hits (6,8,10), which are known to be prone to error (59). Nonetheless, a few of those assignments have already been successfully experimentally confirmed (20,47,57), which strongly strengthens the validity of the predictions. Hence, although we advocate caution regarding the absolute counts of events for the acquisition of complex and simple regulatory approaches, we believe that there is an extensive body of evidence supporting this general trend.
Another driver of diversification in the ECF σ factor family is duplication, but it is unknown how these duplications occurred. To investigate this we analyzed recent duplications in two phyla for which a high number of ECF σ factors of the same group has been noted (6,7): Bacteroidetes, a group of important human gastrointestinal commensals (60), and Planctomycetes, a group of environmentally relevant bacteria due to their role in the nitrogen cycle (61). We observed two very distinct scenarios.
In Bacteroidetes, the duplication involved a complete gene cluster that includes the genes necessary for the ECF-mediated signal transduction (the ECF σ factor and anti-σ factor of the FecI/R type) and those necessary for the uptake and hydrolysis of glycans (TonB-dependent receptor plug, glycoside hydrolase, and Sus-like system). Complex glycans are an abundant carbohydrate source in the gut environment (62), and the duplication of this gene cluster might increase the fitness of the organism, making this a potential case of adaptive duplication (63). Since Bacteroidetes are commensal in the human gut, the fact that the genes responsible for glycan utilization are connected with ECF σ factors whose homologs have been implicated in ferric citrate sensing, could be reminiscent of an anticipatory behaviour (64): the high concentration of iron in the colon (65) could drive bacteria to express the machinery required for processing the also abundant glycans (66). Alternatively, this might represent simply a shared signal transduction mechanism for the utilization of two different substrates—i.e. iron and glycans.
In Planctomycetes, ECF σ factor duplication was already proposed in Gemmata obscuriglobus (7) based on the detection of increased number of ECF σ factors of one particular group. We showed that these duplications involved a ‘minimal’ ECF σ factor, i.e. only regions σ2 and σ4 were duplicated and then inserted into either a non-coding region (converting it in a coding sequence) or in a coding region (sequestering the downstream gene product as a C-terminal extension). Such a process of duplication and fortuitous insertion can generate a lot of variability upon which natural selection can act and hence be part of an effective adaptive strategy. However, this unusual scenario raises questions concerning: (i) the generation of other (nowadays) very conserved C-terminal extensions (20,21,67); (ii) the functionality of an ECF σ factor that would still need to productively interact with an RNA polymerase carrying a C-terminal extension that can be up to 10 times bigger that the ECF σ factor itself (Figure 3F); and (iii) if such random extensions could actually provide any regulatory function. Experimentally addressing these questions would certainly bring light into the success of such strategy.
Horizontal gene transfer has an important role shaping bacterial evolution, as adaptation can be accelerated by the acquisition of foreign DNA. It has already been suggested for an individual ECF σ factor that is part of a genomic island in Streptomyces coelicolor (51), but only demonstrated once between closely related bacteria already containing similar ECF σ factors (68). Acquisition of a foreign transcription factor might have dramatic consequences to the recipient, given that the binding sites of the new transcription factors might change the recipient's transcription profile in deleterious ways. In the specific case of ECF σ factors, unregulated expression might have deleterious effects on the recipient through competition for the pool of RNA polymerases (69,70). However, the implementation of ECF σ factors from distantly phylogenetically related organisms, when successful, seems to have minimal effect on the host's gene expression (71).
Hence, to strengthen the confidence on possible claims of horizontal ECF σ factor transfer, we followed an unbiased and stringent approach to look for horizontally transferred ECF σ factors. The hypothesis of a transfer of an ECF σ factor of group ECF20 from Sinorhizobium sp. to Streptomyces sp. was suggested by HGT-Finder and supported by the taxonomical distribution of the ECF σ factors (Figure 4). We then tested this hypothesis using the gold standard method to demonstrate horizontal gene transfer, i.e. phylogenetic tree reconciliation. The hypothesis was confirmed and is predicted in a biologically reasonable parameter space, given even more support to this prediction.
The work presented here aimed at providing insights into the mechanisms involved in making the ECF σ factor family the most abundant and diverse family of alternative σ factors. We have provided proof of the multiple origins of regulatory mechanisms dependent on gene fusions or domain-splitting (C-terminal extensions and soluble anti-σ factors), and single origins of regulation mechanisms that depend on additional partner proteins (transcription control by two-component systems, serine/threonine protein kinases). We also characterized events of adaptive duplication in the Bacteroidetes and C-terminal extension generation by gene sequestration in the Planctomycetes. At last, we have demonstrated horizontal transfer of ECF σ factors between soil bacteria of different phyla. Altogether, our analysis revealed a complex evolutionally history of the ECF σ factor family and opened new directions for further research.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Mark Buttner, Thorsten Mascher, Christian Jogler and Anna Nagy-Staroń for critically reading an earlier version of this manuscript.
Notes
Present address: Daniela Pinto, Biosystems and Integrative Sciences Institute (BioISI), Edificio TecLabs, Campus da FCUL, Campo Grande, 1749-016, Lisbon, Portugal.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
European Molecular Biology Organization Short-term Fellowship [7462 to D.P.]; Danish National Research Foundation [DNRF96 to R.R.F.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Helmann J.D., Chamberlin M.J.. Structure and function of bacterial sigma factors. Annu. Rev. Biochem. 1988; 57:839–872. [DOI] [PubMed] [Google Scholar]
- 2. Gruber T.M., Gross C.A.. Multiple sigma subunits and the partitioning of bacterial transcription space. Annu. Rev. Microbiol. 2003; 57:441–466. [DOI] [PubMed] [Google Scholar]
- 3. Helmann J.D. The extracytoplasmic function (ECF) sigma factors. Adv. Microb. Physiol. 2002; 46:47–110. [DOI] [PubMed] [Google Scholar]
- 4. Lonetto M., Gribskov M., Gross C.A.. The sigma 70 family: sequence conservation and evolutionary relationships. J. Bacteriol. 1992; 174:3843–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lonetto M.A., Brown K.L., Rudd K.E., Buttner M.J.. Analysis of the Streptomyces coelicolor sigE gene reveals the existence of a subfamily of eubacterial RNA polymerase sigma factors involved in the regulation of extracytoplasmic functions. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:7573–7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Staroń A., Sofia H.J., Dietrich S., Ulrich L.E., Liesegang H., Mascher T.. The third pillar of bacterial signal transduction: classification of the extracytoplasmic function (ECF) sigma factor protein family. Mol. Microbiol. 2009; 74:557–581. [DOI] [PubMed] [Google Scholar]
- 7. Jogler C., Waldmann J., Huang X., Jogler M., Glöckner F.O., Mascher T., Kolter R.. Identification of proteins likely to be involved in morphogenesis, cell division, and signal transduction in Planctomycetes by comparative genomics. J. Bacteriol. 2012; 194:6419–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang X., Pinto D., Fritz G., Mascher T.. Environmental sensing in Actinobacteria: a comprehensive survey on the signaling capacity of this phylum. J. Bacteriol. 2015; 197:2517–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gómez-Santos N., Pérez J., Sánchez-Sutil M.C., Moraleda-Muñoz A., Muñoz-Dorado J.. CorE from Myxococcus xanthus is a copper-dependent RNA polymerase sigma factor. PLoS Genet. 2011; 7:e1002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wiegand S., Jogler M., Boedeker C., Pinto D., Vollmers J., Rivas-Marín E., Kohn T., Peeters S., Heuer A., Rast P. et al.. Deep-cultivation and phenomics of the phylum Planctomycetes unveil novel, unsuspected bacterial biology. Nat. Microbiol. 2019; 5:126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chaba R., Grigorova I.L., Flynn J.M., Baker T.A., Gross C.A.. Design principles of the proteolytic cascade governing the sigmaE-mediated envelope stress response in Escherichia coli: keys to graded, buffered, and rapid signal transduction. Genes Dev. 2007; 21:124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Heinrich J., Wiegert T.. Regulated intramembrane proteolysis in the control of extracytoplasmic function sigma factors. Res. Microbiol. 2009; 160:696–703. [DOI] [PubMed] [Google Scholar]
- 13. Ho T.D., Ellermeier C.D.. Extra cytoplasmic function σ factor activation. Curr. Opin. Microbiol. 2012; 15:182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li W., Stevenson C.E.M., Burton N., Jakimowicz P., Paget M.S.B., Buttner M.J., Lawson D.M., Kleanthous C.. Identification and structure of the anti-sigma factor-binding domain of the disulphide-stress regulated sigma factor σR from Streptomyces coelicolor. J. Mol. Biol. 2002; 323:225–236. [DOI] [PubMed] [Google Scholar]
- 15. Zdanowski K., Doughty P., Jakimowicz P., O’Hara L., Buttner M.J., Paget M.S.B., Kleanthous C.. Assignment of the zinc ligands in RsrA, a redox-sensing ZAS protein from Streptomyces coelicolor. Biochemistry. 2006; 45:8294–8300. [DOI] [PubMed] [Google Scholar]
- 16. Dufour Y.S., Landick R., Donohue T.J.. Organization and evolution of the biological response to singlet oxygen stress. J. Mol. Biol. 2008; 383:713–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paget M.S., Molle V., Cohen G., Aharonowitz Y., Buttner M.J.. Defining the disulphide stress response in Streptomyces coelicolor A3(2): identification of the sigmaR regulon. Mol. Microbiol. 2001; 42:1007–1020. [DOI] [PubMed] [Google Scholar]
- 18. Merighi M., Majerczak D.R., Stover E.H., Coplin D.L.. The HrpX/HrpY two-component system activates hrpS expression, the first step in the regulatory cascade controlling the Hrp regulon in Pantoea stewartii subsp. stewartii. Mol. Plant. Microbe. Interact. 2003; 16:238–248. [DOI] [PubMed] [Google Scholar]
- 19. Paget M.S., Leibovitz E., Buttner M.J.. A putative two-component signal transduction system regulates sigmaE, a sigma factor required for normal cell wall integrity in Streptomyces coelicolor A3(2). Mol. Microbiol. 1999; 33:97–107. [DOI] [PubMed] [Google Scholar]
- 20. Wecke T., Halang P., Staroń A., Dufour Y.S., Donohue T.J., Mascher T.. Extracytoplasmic function σ factors of the widely distributed group ECF41 contain a fused regulatory domain. Microbiologyopen. 2012; 1:194–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu Q., Pinto D., Mascher T.. Characterization of the widely distributed novel ECF42 group of extracytoplasmic function σ factors in Streptomyces venezuelae. J. Bacteriol. 2018; 200:e00437-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mistry J., Finn R.D., Eddy S.R., Bateman A., Punta M.. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013; 41:e121–e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. et al.. The Pfam protein families database in 2019. Nucleic Acids Res. 2019; 47:D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Python Software Foundation 2010; Python Software Foundation Python Language Reference, version 2.7https://www.python.org.
- 25. Yamada K.D., Tomii K., Katoh K.. Application of the MAFFT sequence alignment program to large data—reexamination of the usefulness of chained guide trees. Bioinformatics. 2016; 32:3246–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q.. IQ-TREE: A fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 2015; 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le S.Q., Gascuel O.. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008; 25:1307–1320. [DOI] [PubMed] [Google Scholar]
- 28. Yang Z. A space-time process model for the evolution of DNA sequences. Genetics. 1995; 139:993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Soubrier J., Steel M., Lee M.S.Y., Der Sarkissian C., Guindon S., Ho S.Y.W., Cooper A.. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 2012; 29:3345–3358. [DOI] [PubMed] [Google Scholar]
- 30. Kalyaanamoorthy S., Minh B.Q., Wong T.K.F., von Haeseler A., Jermiin L.S.. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017; 14:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoang D.T., Chernomor O., von Haeseler A., Minh B.Q., Vinh L.S.. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018; 35:518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Le S.Q., Gascuel O.. An improved general amino acid replacement matrix. Mol. Biol. Evol. 2008; 25:1307–1320. [DOI] [PubMed] [Google Scholar]
- 33. Yang Z. A space-time process model for the evolution of DNA sequences. Genetics. 1995; 139:993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Soubrier J., Steel M., Lee M.S.Y., Der Sarkissian C., Guindon S., Ho S.Y.W., Cooper A.. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 2012; 29:3345–3358. [DOI] [PubMed] [Google Scholar]
- 35. Overmars L., Kerkhoven R., Siezen R.J., Francke C.. MGcV: the microbial genomic context viewer for comparative genome analysis. BMC Genomics. 2013; 14:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Müller T., Vingron M.. Modeling amino acid replacement. J. Comput. Biol. 2000; 7:761–776. [DOI] [PubMed] [Google Scholar]
- 37. Minh B.Q., Hahn M.W., Lanfear R.. New methods to calculate concordance factors for phylogenomic datasets. 2018; bioRxiv doi:05 December 2018, preprint: not peer reviewed 10.1101/487801. [DOI] [PMC free article] [PubMed]
- 38. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 39. R Core Team R: A language and environment for statistical computing. R Found. Stat. Comput. 2018; Vienna. [Google Scholar]
- 40. Sherrill-Mix S. taxonomizr: Functions to Work with NCBI Accessions and Taxonomy. R Packag. version 0.5.1. 2018; [Google Scholar]
- 41. Nguyen M., Ekstrom A., Li X., Yin Y.. HGT-Finder: a new tool for horizontal gene transfer finding and application to Aspergillus genomes. Toxins (Basel). 2015; 7:4035–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stolzer M., Lai H., Xu M., Sathaye D., Vernot B., Durand D.. Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics. 2012; 28:i409–i415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Charif D., Lobry J.R.. Bastolla U, Porto M, Roman HE, Vendruscolo M. Seqin{R} 1.0-2: a contributed package to the {R} project for statistical computing devoted to biological sequences retrieval and analysis. Structural Approaches to Sequence Evolution: Molecules, Networks, Populations. 2007; NY: Springer Verlag; 207–232. [Google Scholar]
- 44. Paradis E., Schliep K.. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 2018; 35:526–528. [DOI] [PubMed] [Google Scholar]
- 45. Warnes G.R., Bolker B., Bonebakker L., Gentleman R., Liaw W.H.A., Lumley T., Maechler M., Magnusson A., Moeller S., Schwartz M. et al.. gplots: Various R Programming Tools for Plotting Data. R Packag. version 3.0.1. 2016; [Google Scholar]
- 46. Wickham H. ggplot2. 2009; NY: Springer. [Google Scholar]
- 47. Liu Q., Pinto D., Mascher T.. Characterization of the widely distributed novel ECF42 group of extracytoplasmic function σ factors in Streptomyces venezuelae. J. Bacteriol. 2018; 200:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu H., Liu Q., Casas-Pastor D., Dürr F., Mascher T., Fritz G.. The role of C-terminal extensions in controlling ECF σ factor activity in the widely conserved groups ECF41 and ECF42. Mol. Microbiol. 2019; 12:498–514. [DOI] [PubMed] [Google Scholar]
- 49. Pinto D., Mascher T.. de Bruijn FJ. The ECF classification: a phylogenetic reflection of the regulatory diversity in the extracytoplasmic function σ factor protein family. Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria. 2016; Hoboken: John Wiley & Sons, Inc; 64–96. [Google Scholar]
- 50. Bolam D.N., Koropatkin N.M.. Glycan recognition by the Bacteroidetes Sus-like systems. Curr. Opin. Struct. Biol. 2012; 22:563–569. [DOI] [PubMed] [Google Scholar]
- 51. Kao C.M., Karoonuthaisiri N., Weaver D., Vroom J.A., Gai S.A., Ho M.-L., Patel K.G.. A genomic island of Streptomyces coelicolor with the self-contained regulon of an ECF sigma factor. 2018; bioRxiv doi:14 February, 2018, preprint: not peer reviewed 10.1101/247056. [DOI]
- 52. Adato O., Ninyo N., Gophna U., Snir S.. Detecting horizontal gene transfer between closely related taxa. PLoS Comput. Biol. 2015; 11:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakhleh L. Computational approaches to species phylogeny inference and gene tree reconciliation. Trends Ecol. Evol. 2013; 28:719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Libeskind-Hadas R., Wu Y.-C., Bansal M.S., Kellis M.. Pareto-optimal phylogenetic tree reconciliation. Bioinformatics. 2014; 30:i87–i95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mascher T. Signaling diversity and evolution of extracytoplasmic function (ECF) σ factors. Curr. Opin. Microbiol. 2013; 16:148–155. [DOI] [PubMed] [Google Scholar]
- 56. Rajasekar K.V., Zdanowski K., Yan J., Hopper J.T.S., Francis M.-L.R., Seepersad C., Sharp C., Pecqueur L., Werner J.M., Robinson C.V. et al.. The anti-sigma factor RsrA responds to oxidative stress by reburying its hydrophobic core. Nat. Commun. 2016; 7:12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bayer-Santos E., Lima L.D.P., Ceseti L., de M., Ratagami C.Y., de Santana E.S., da Silva A.M., Farah C.S., Alvarez-Martinez C.E.. Xanthomonas citri T6SS mediates resistance to Dictyostelium predation and is regulated by an ECF σ factor and cognate Ser/Thr kinase. Environ. Microbiol. 2018; 20:1562–1575. [DOI] [PubMed] [Google Scholar]
- 58. Wu Y.-C., Rasmussen M.D., Kellis M.. Evolution at the subgene level: domain rearrangements in the drosophila phylogeny. Mol. Biol. Evol. 2012; 29:689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nikel P.I. Unexpected functions of automatically annotated genes: a lesson learnt from Bacillus subtilis. Environ. Microbiol. 2017; 19:5–6. [DOI] [PubMed] [Google Scholar]
- 60. Gibiino G., Lopetuso L.R., Scaldaferri F., Rizzatti G., Binda C., Gasbarrini A.. Exploring bacteroidetes: metabolic key points and immunological tricks of our gut commensals. Dig. Liver Dis. 2018; 50:635–639. [DOI] [PubMed] [Google Scholar]
- 61. Fuerst J.A., Sagulenko E.. Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat. Rev. Microbiol. 2011; 9:403–413. [DOI] [PubMed] [Google Scholar]
- 62. Koropatkin N.M., Cameron E.A., Martens E.C.. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 2012; 10:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hastings P., Rosenberg S.M.. In pursuit of a molecular mechanism for adaptive gene amplification. DNA Repair (Amst). 2002; 1:111–123. [DOI] [PubMed] [Google Scholar]
- 64. Tagkopoulos I., Liu Y.-C.Y.-C., Tavazoie S.. Predictive behavior within microbial genetic networks. Science (80-.). 2008; 320:1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kortman G.A.M., Raffatellu M., Swinkels D.W., Tjalsma H.. Nutritional iron turned inside out: intestinal stress from a gut microbial perspective. FEMS Microbiol. Rev. 2014; 38:1202–1234. [DOI] [PubMed] [Google Scholar]
- 66. Koropatkin N.M., Cameron E.A., Martens E.C.. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 2012; 10:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Marcos-Torres F.J., Pérez J., Gómez-Santos N., Moraleda-Muñoz A., Muñoz-Dorado J.. In depth analysis of the mechanism of action of metal-dependent sigma factors: characterization of CorE2 from Myxococcus xanthus. Nucleic Acids Res. 2016; 44:5571–5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. López-Leal G., Cevallos M.A., Castillo-Ramírez S.. Evolution of a sigma Factor: An All-In-One of gene duplication, horizontal gene transfer, purifying selection, and promoter differentiation. Front. Microbiol. 2016; 7:581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mauri M., Klumpp S.. A model for sigma factor competition in bacterial cells. PLoS Comput. Biol. 2014; 10:e1003845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kurata H., El-Samad H., Iwasaki R., Ohtake H., Doyle J.C., Grigorova I., Gross C.A., Khammash M.. Module-based analysis of robustness tradeoffs in the heat shock response system. PLoS Comput. Biol. 2006; 2:0663–0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rhodius V.A., Segall-Shapiro T.H., Sharon B.D., Ghodasara A., Orlova E., Tabakh H., Burkhardt D.H., Clancy K., Peterson T.C., Gross C.A. et al.. Design of orthogonal genetic switches based on a crosstalk map of σs, anti-σs, and promoters. Mol. Syst. Biol. 2013; 9:702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.