

Abstract
Misidentification and contamination of biobank samples (e.g. cell lines) have plagued biomedical research. Short tandem repeat (STR) and single-nucleotide polymorphism assays are widely used to authenticate biosamples and detect contamination, but with insufficient sensitivity at 5–10% and 3–5%, respectively. Here, we describe a deep NGS-based method with significantly higher sensitivity (≤1%). It can be used to authenticate human and mouse cell lines, xenografts and organoids. It can also reliably identify and quantify contamination of human cell line samples, contaminated with only small amount of other cell samples; detect and quantify species-specific components in human–mouse mixed samples (e.g. xenografts) with 0.1% sensitivity; detect mycoplasma contamination; and infer population structure and gender of human samples. By adopting DNA barcoding technology, we are able to profile 100–200 samples in a single run at per-sample cost comparable to conventional STR assays, providing a truly high-throughput and low-cost assay for building and maintaining high-quality biobanks.
INTRODUCTION
Cell lines, organoids, and xenograft and homograft models are useful model systems in oncology and other biomedical research. Model authentication and characterization helps their proper utilization and alleviates a series of problems such as misidentification and misuse, cross-contamination, erroneous cancer classification, undetected genomic change due to longtime culture and genetic drift, which are all well noted especially in cell lines due to their popular use (1–8). For example, various studies have reported ∼10–40% misidentification/contamination rates for cell line banks (9–17).
A number of methods have been reported for authenticating cell lines, including examining cell morphology, isoenzymology, cytogenetic analysis (karyotyping and fluorescence in situ hybridization), human lymphocyte antigen typing, short tandem repeat (STR) profiling, single-nucleotide polymorphism (SNP) typing, and DNA and RNA sequencing (RNA-seq) (18,19). Among these technologies, STR profiling has been most widely used and there is an STR standard (ASN-0002) for guiding human cell line authentication (20). A panel of 19 STR markers for mouse cell lines was also developed (21). The sensitivity of STR assays for detecting contaminants is ∼5–10% (8). In recent years, SNP typing has been increasingly used for cell line and biosample authentication owing to its improved accuracy, sensitivity and reduced cost (8,22–29). Current SNP assays have detection sensitivity at ∼3–5% (8,30,31). There are also databases with STR, SNP and other information for cell lines to facilitate their authentication and characterization (8,32–34).
However, STR and SNP assays have several limitations: (i) they are low-throughput and labor-intensive methods, and therefore are costly and cumbersome to use for authenticating large batches of samples; (ii) their ability in detecting contamination can be much lower than the commonly claimed sensitivity; for example, a >20% contamination was not detected in a mixture of two unrelated cell lines by a 96-SNP assay (27); and (iii) they are monofunctional assays, and we need to use other assays for, say, checking mycoplasma contamination. All these shortcomings call for better approaches, especially for large biobanks with many different types of biosamples that bring in additional complications, as elaborated below.
Besides cell lines, organoids and mouse tumor models are widely used in oncology research and drug development. Organoids are in vitro three-dimensional cultures deriving from stem cells, primary and engineered tumor samples, and xenografted human tumors that maintain many organ structures and functions (35). Mouse tumor models are in vivo systems including patient-derived xenografts (PDXs), cell line-derived xenografts, syngeneic or mouse cell line-derived models, mouse homograft models, etc. Some of these models, such as PDXs, more faithfully capture primary tumor histopathology and genomics than cell lines (36–39). Like cell lines, these tumor models have similar quality control issues, but there are also additional problems. In xenograft models, tumors contain human tumor cells and mouse stromal cells, the latter gradually replacing human counterparts during the passaging of models (40), which, when compounded with genomic heterogeneity, implantation site differences (subcutaneous and orthotopic), growth variation and dissection randomness, makes the human–mouse genetic composition of tumors from even the same PDX differ considerably, to the extent that some samples are nearly pure human or mouse content. Such tumor–host mixing and interference occurs with all implanted tumor models, causing fluctuation of allele frequencies for STR markers and SNPs, therefore adversely impacting traditional STR- and SNP-based authentication methods. Large-scale sample authentication is also a logistic burden and error-prone, especially for biobanks where many kinds of in vitro and in vivo models are simultaneously maintained and used.
In this article, we report a deep NGS-based multifunctional assay that simultaneously solves all these problems. This assay can (i) authenticate and characterize hundreds of samples in a single run, (ii) authenticate all kinds of human and mouse samples, including cell lines, xenografts, organoids, mouse tumor models and clinical samples, (iii) detect mycoplasma contamination, (iv) estimate mouse percentage in mouse–human mixed samples such as xenograft tumors, (v) analyze genetic admixture and population structure of human samples and (vi) infer gender for human samples. It should be noted that this assay is more suitable for large-scale sample authentication, as commonly performed in biobanks or centered facilities, while conventional STR/SNP assays are still a convenient tool for sporadic sample check.
MATERIALS AND METHODS
Nucleic acid extraction
Genomic DNA from cells, PDXs and PDX-derived organoids (PDXOs) was purified using DNeasy Blood & Tissue Kit (QIAGEN, Valencia, CA, Cat. 69506) according to the manufacturer’s instructions. DNA integrity was determined by 2100 Bioanalyzer (Agilent) and quantified using NanoDrop (Thermo Scientific). One aliquot of high-quality DNA sample (OD260/280 = 1.8–2.0, OD260/230 ≥ 2.0, >1 μg) was used for deep NGS sequencing and WES sequencing. Total RNA from cells, PDXs and PDXOs was purified using RNeasy Mini Kit (QIAGEN, Cat. 74106) according to the manufacturer’s instructions. Integrity of the total RNA was determined by 2100 Bioanalyzer (Agilent) and quantified using NanoDrop (Thermo Scientific). One aliquot of high-quality RNA sample (OD260/280 = 1.8–2.2, OD260/230 ≥ 2.0, RIN ≥ 8.0, >1 μg) was used for deep NGS sequencing and RNA-seq.
Cell line mixture preparation
A cell line mixture was prepared by mixing cells from two cell lines with given ratios. Based on cell growth rate, cells were seeded in 15 ml medium in T75 that allowed cell confluence to reach 60–80%, followed by overnight incubation in a CO2 water-jacketed incubator (SANYO). Cells were harvested during the logarithmic growth period, and counted with a hemocytometer (Chongguang) for the calculation of concentration. Cells from two cell lines were then mixed according to predefined ratios to create a cell line mixture that was subsequently centrifuged at 3000 rpm for 5 min. Supernatant was aspirated and cell pellets were stored at −20°C for DNA extraction.
Human–mouse DNA mixture preparation
Serial dilutions of mouse–human DNA mixture benchmark samples were prepared by mixing mouse spleen DNA and human genomic DNA (Thermo Scientific, Cat. 4312660). Mouse spleen DNA was purified using DNeasy Blood & Tissue Kit (QIAGEN, Cat. 69506) according to the manufacturer’s instructions and quantified using the NanoDrop (Thermo Scientific). Mouse spleen DNA and human genomic DNA were diluted to 200 ng/μl, and then mixed by predefined mouse ratios, including 90%, 80%, 70%, 50%, 30%, 20%, 10%, 7%, 5% and 0%. The DNA mixture was used for the deep NGS sequencing later. To further assess the NGS assay’s sensitivity in detecting mouse content in human samples, a second series of mouse–human DNA mixture benchmark samples was prepared by mixing mouse genomic DNA (Promega, G3091) and human lymphoma cell line K562 genomic DNA (Promega, E4931). Mouse and human DNA were diluted to 40 ng/μl, and then mixed by predefined mouse ratios, including 80%, 40%, 20%, 10%, 5%, 2.50%, 1.25%, 0.63%, 0.31% and 0.16%.
Barcode deep NGS sequencing
Multiplex PCR was used to prepare target sequencing libraries for Illumina sequencers with a paired-end read length of 150 bp (pE150). The NGS deep sequencing covered 630 amplicons, sizes of which ranged from 160 to 260 bp. Genomic DNA was amplified using IGT-EM808 polymerase mixture (iGeneTech Bioscience Co., Ltd; 95°C for 3 min 30 s, 18 cycles of incubation at 98°C for 20 s and 60°C for 8 min, hold at 72°C for 5 min) and then purified by AMPure XP beads (Beckman, Cat. A63881).
Barcoding was executed by a second round of amplification. Briefly, purified target amplicons were taken as templates and added with upstream IGT-I5 index (10 μM), downstream IGT-I7 index (10 μM) and polymerase mixture for the PCR reaction. The mixture was then placed in a thermal cycler for amplification with the following settings: 95°C for 3 min 30 s, nine cycles of incubation at 98°C for 20 s, 58°C for 1 min and 72°C for 30 s, hold at 72°C for 5 min. The barcoded library was then purified by using AMPure XP beads (Beckman, Cat. A63881).
After library construction, Qubit 3.0 Fluorometer dsDNA HS Assay (Thermo Fisher Scientific) was used to quantify concentrations of the resulting sequencing libraries. 2100 Bioanalyzer (Agilent) was used to analyze size distribution ranging from 280 to 420 bp. Paired-end sequencing was performed using an Illumina system following Illumina-provided protocols for 2 × 150 bp paired-end sequencing.
RNA-seq and WES sequencing
In RNA-seq, the mRNA-focused sequencing libraries were constructed from total RNA. PolyA mRNA was purified from total RNA using oligo-dT-attached magnetic beads and then fragmented by fragmentation buffer. Using the short fragments as templates, first-strand cDNA was synthesized using reverse transcriptase and random primers, followed by second-strand cDNA synthesis. The synthesized cDNA was then subjected to end repair, phosphorylation and ‘A’ base addition according to the library construction protocol. Following this, sequencing adapters were added to both ends of the cDNA fragments. After PCR amplification for cDNA fragments, the targeted 250–350 bp fragments were cleaned up. After library construction, Qubit 3.0 Fluorometer dsDNA HS Assay (Thermo Fisher Scientific) was used to quantify concentrations of the resulting sequencing libraries, while the size distribution was analyzed using 2100 Bioanalyzer (Agilent). After library validation, Illumina cBOT cluster generation system with HiSeq PE Cluster Kits (Illumina) was used to generate clusters. Paired-end sequencing was performed using an Illumina system following Illumina-provided protocols for 2 × 150 paired-end sequencing.
WES was performed by Wuxi Nextcode Co. Ltd. (Shanghai, China). Briefly, genomic DNA was extracted and fragmented to an average size of 180–280 bp. DNA libraries were generated by Illumina’s manufacturer paired-end protocols. Exons were captured by Agilent SureSelect Human All Exon V6, and subsequently sequenced by the Illumina NovaSeq platform (Illumina Inc., San Diego, CA, USA) to generate 150 bp paired-end reads.
SNP selection and profiling
We selected 200 SNPs for human sample authentication by several criteria: (i) SNPs are in exons; (ii) SNPs are located on all 22 autosomes and are sufficiently away from each other since chromosome abnormality, including deletions and duplications of large chromosome segments, is common in tumors; and (iii) SNPs are in highly expressed genes. Of the 200 SNPs, 132 were categorized in the International HapMap Project (41). We added another 13 SNPs also in the HapMap catalog (release 3), so a total of 145 SNPs were used for population structure analysis based on three reference populations, namely Han Chinese (CHB), Nigeria Yoruba (YRI) and Utah residents with Northern and Western European ancestry from the CEPH collection (CEU).
Benchmark samples and data
Two cell line benchmark sample sets were prepared. The first set had 78 samples for three pairs of cell lines, including PANC-1 and RT4, MV-4-11 and ‘LNCaP clone FGC’, and CAL27 and Raji. Each pair has 26 samples including the two pure cell lines and three replicates for eight mix ratios by cell count (Supplementary Table S1). The second set had 22 cell lines each contaminated by a known second cell line by a mostly small but unspecified ratio (Supplementary Table S2).
Estimating heterogeneity ratios
There are six informative genotype combinations that can be used to estimate heterogeneity ratios from the deep NGS sequencing data (Table 1). They exhibit four distinct nucleotide frequency patterns. Combinations 1 and 2 generate the same pattern, and we used an average formula to calculate the percentage of the minor component S2, or the heterogeneity ratio. The formula produced an exact estimate of the ratio when the two combinations occur with equal frequency, a scenario that should be closely approximated when the number of SNPs is large. A similar averaging approach is used for combinations 4 and 5. When the heterogeneity ratio is low, sequencing error may interfere the inference of heterogeneity ratio. To alleviate this, we used a two-step statistical procedure. Assuming sequencing error is e = 0.001 and the sequencing depth is n (n≥ 500, any SNP with n< 500 is discarded) at a given SNP site, the probability of observing k erroneous nucleotides follows a binomial distribution with parameters n and e:
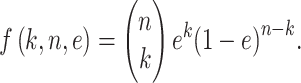 |
Table 1.
Six informative genotype combinations to estimate heterogeneity/contamination ratioa
| Combination | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| S1 genotype | AA | AA | AA | AT | AT | AT |
| S2 genotype | TT | AT | TG | GG | AG | GC |
| S2 ratio (SNP heterogeneity ratio) | T/(A + T)b | 2T/(A + T)b | (T + G)/(A + T + G) | G/(A + T + G)c | 2G/(A + T + G) | (G + C)/(A + T + G + C) |
| Nucleotide frequency pattern | Large A, small T | Large A, small T | Large A, small T and G | Large A and T, small G | Large A and T, small G | Large A and T, small G and C |
aS1 is the major component and S2 is the minor/contaminating component in the mixed sample. Each combination uses specific nucleotides to represent a class of combinations; for example, the first combination denotes that both are homozygous genotypes with different nucleotides. In the formulas for calculating S2 ratio, a nucleotide denotes its count (total number of reads) in NGS sequencing data.
bCombinations 1 and 2 cannot be distinguished from observed NGS data, so 1.5T/(A + T) is used for both.
cCombinations 4 and 5 cannot be distinguished from observed NGS data, so 1.5G/(A + T + G) is used for both.
For each n, we calculated the cumulative density function and obtained a threshold h so that the probability of observing more than h erroneous nucleotides out of the n nucleotides was <0.01. In the sequencing data, any low-frequency nucleotide with number of reads smaller than a corresponding threshold h was discarded. We then used an expectation–maximization algorithm [package mclust in R, version 3.5.3 (42)] to estimate parameters of a Gaussian mixture (with one to three components) that models the distribution of nucleotide frequencies smaller than a maximal heterogeneity (0.2 used for all samples in this study). If there was only a single Gaussian component or the Gaussian component with smallest mean accounted for >60% of all data points, median of all data points was taken as the sample heterogeneity ratio; otherwise, median of data points in the other Gaussian component(s) was taken as the sample heterogeneity ratio.
Determining major component of a sample
The genotype at an SNP site was determined using only nucleotides with allele frequencies larger than a threshold, 10% for reference samples and 25% for test samples that may be contaminated. The genotype similarity between a reference sample and a test sample was the percentage of SNPs with identical genotypes, excluding SNPs with sequencing depth <500 in the test sample. The major component of the test sample was the reference sample with the highest genotype similarity, which must be >90% (or 80%) if the heterogeneity ratio of the test sample was <10% (or >10%). Otherwise, no major component was called.
Determining minor component of a sample
After the estimation of heterogeneity ratio and determination of major component, we determined the minor component of a test sample. For a mixture of the major component and one of the other reference samples (e.g. all cell lines with genomic data), we obtained a chimeric genotype, with possibly one to four nucleotides, at every SNP site. Frequencies of nucleotides were calculated using the heterogeneity ratio. Similarly, we defined the chimeric genotype of the test sample. The two chimeric genotypes were considered identical if they harbored the same nucleotides and frequencies of each nucleotide were within 3-fold. We then calculated the genotype similarity between the test sample and each reference sample combined with the major component. The set of all pairwise genotype similarities was then fitted by a beta distribution with parameters (α, β):
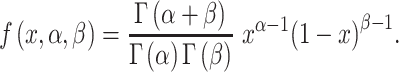 |
In this equation, Γ(α) is the gamma function and x is genotype similarity. Its parameters were estimated by package fitdistrplus in R (version 3.5.3). From the fitted beta distribution, we then calculated the probability of observing any genotype similarity larger than a specific value. A quantile–quantile graph with 99% confidence band was plotted for all observed genotype similarities for visualization. A reference sample was considered the minor component if (i) it had the highest genotype similarities, (ii) its genotype similarity was above the 99% confidence upper bound in the quantile–quantile graph and (3) its P-value was <1.0E−6 in the fitted beta distribution.
Estimating mixture ratio of two cell lines
We used cell lines to explain the estimation of mix ratio for two reference samples. Assume that two cell lines S1 and S2 were mixed with ratio θ for S1 and (1 − θ) for S2, where 0 ≤ θ≤ 1. From deep NGS sequencing data, we accurately estimated nucleotide frequencies of all n SNPs in both cell lines. For an SNP, we denoted its four nucleotide frequencies, which sum to 1, as {A1, T1, G1, C1} for cell line S1 and {A2, T2, G2, C2} for cell line S2. In principle, one of the frequencies is close to 1 if the SNP is homozygous, and two frequencies are both close to 0.5 if the SNP is heterozygous. Actual data may have some deviations due to sequencing errors and randomness, as well as multiclonality of cell lines.
From sequencing data of the mix sample, we denoted the actual occurrences of the four nucleotides as 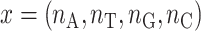 . The likelihood of such observation is
. The likelihood of such observation is
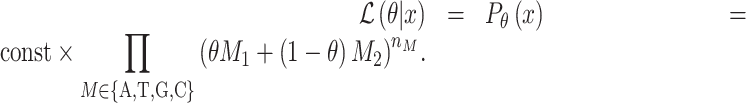 |
The likelihood 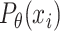 can be calculated for any SNP
can be calculated for any SNP 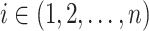 with observed data
with observed data 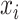 ; the likelihood of observing data
; the likelihood of observing data 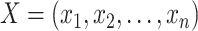 for all SNPs is
for all SNPs is
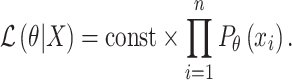 |
The log likelihood is therefore
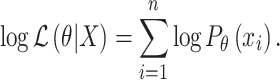 |
We then solved for θ that maximized the likelihood by stepwise increment of θ. The above procedure can also be used for a mixture of any two human samples.
Simulation of cell line mixture for contaminant detection
A simulation was performed for three cell line pairs including PANC-1 and RT4, MV-4-11 and ‘LNCaP clone FGC’, and CAL27 and Raji. All six cell lines were profiled by deep NGS sequencing to obtain their SNP fingerprints. Two cell lines in a pair were mixed in silico where the ratio of the first cell line was r, and r takes the following values: 0.15%, 0.30%, 0.625%, 1.25%, 2.5%, 5%, 10%, 15% and 20%. For each SNP site, we obtained 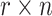 nucleotides from the first cell line where n was a random integer from 500 to 5000; we further distributed
nucleotides from the first cell line where n was a random integer from 500 to 5000; we further distributed 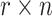 into four nucleotides (A, T, G, C) according to their frequencies in the first cell line. Similarly, we obtained
into four nucleotides (A, T, G, C) according to their frequencies in the first cell line. Similarly, we obtained 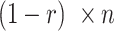 nucleotides from the second cell line. We then reversed the ratio, so a symmetric sampling was done with ratio r for the second cell line.
nucleotides from the second cell line. We then reversed the ratio, so a symmetric sampling was done with ratio r for the second cell line.
Estimating mouse ratio from RNA-seq and WES datasets
Sequencing reads were mapped to human reference (hg19) and mouse reference (mm10) genomes using mapping tools STAR (43) for RNA-seq data and BWA (44) for WES data with default parameters. If a read was only mapped to a human genome, or had fewer mismatches to a human genome than to a mouse genome, it was classified as a human read. Mouse reads were similarly assigned. If a read was mapped to both genomes with a close number of mismatches, off by at most 2, the read was unclassifiable and discarded. The mouse ratio was the proportion of mouse reads out of all kept reads.
RESULTS
Human sample authentication and contamination detection
SNP profiling and fingerprint
A panel of 200 SNPs was selected for authenticating human samples, including cell lines, xenografts and organoids (Supplementary Figure S1 and Supplementary Table S3). SNPs were profiled by deep NGS sequencing with an average depth of 3000×. Each sample has a unique SNP fingerprint consisting of both nucleotide identities and frequencies for each of the SNPs. It should be emphasized that a cell line can have fluctuating SNP fingerprints between passages and among biobanks due to genetic drift and heterogeneity (45), so a current SNP fingerprint can be profiled for better curation. The SNP fingerprints can be generated, with reduced precision, by relatively low-depth NGS data. In this study, we generated SNP fingerprints for 1055 cell lines from RNA-seq data profiled by us and CCLE (46), which serve as references (Supplementary Data S1).
We illustrated the authentication, characterization, and intraspecies and interspecies contamination detection using SNP profiling data from deep NGS sequencing for 217 cell line samples, 220 PDX samples and 31 PDXO samples. For the cell line samples, there are 72 mixtures of two cell lines with known mix ratios from serial dilutions and six corresponding pure cell lines (Table 2, Supplementary Table S1), 22 mixtures of two cell lines with unknown mix ratios (Table 3, Supplementary Table S2) and 117 unmixed cell lines (Supplementary Table S4).
Table 2.
Authentication and contaminant detection of three cell line pairs with serial dilutionsa
| Cell line mixture | Major component | Minor component (contaminant) | Minor component ratio (percentage)b | No. of informative SNPs | Heterogeneity ratio (percentage) | Major component inferredc | Minor component (contaminant) inferredd | Contamination ratio (percentage)c | P-valuee |
|---|---|---|---|---|---|---|---|---|---|
| PANC-1:RT4 | PANC-1 | 118 | 1.4 | PANC-1 (99.46%) | |||||
| RT4 | 122 | 1.65 | RT4 (98.54%) | ||||||
| RT4 | PANC-1 | 5 | 86 | 3.3 | RT4 (97.78%) | PANC-1 (96.73%) | 2.88 | 2.98E−16 | |
| RT4 | PANC-1 | 2.5 | 80 | 1.85 | RT4 (97.93%) | PANC-1 (94.79%) | 1.08 | 6.65E−12 | |
| RT4 | PANC-1 | 1.25 | 81 | 1.1 | RT4 (97.76%) | PANC-1 (88.14%) | 0.41 | 3.97E−09 | |
| RT4 | PANC-1 | 0.625 | 80 | 1.06 | RT4 (97.76%) | ||||
| PANC-1 | RT4 | 5 | 131 | 8.09 | PANC-1 (99.50%) | RT4 (98.33%) | 7.21 | 5.01E−17 | |
| PANC-1 | RT4 | 2.5 | 128 | 4.06 | PANC-1 (99.35%) | RT4 (95.50%) | 2.81 | 5.01E−17 | |
| PANC-1 | RT4 | 1.25 | 132 | 2.75 | PANC-1 (99.49%) | RT4 (90.73%) | 1.48 | 5.01E−17 | |
| PANC-1 | RT4 | 0.625 | 139 | 2.62 | PANC-1 (99.47%) | RT4 (81.61%) | 1.08 | 1.67E−08 | |
| LNCAPCLONEFGC:MV-4-11 | LNCAPCLONEFGC | 97 | 1.005 | LNCAPCLONEFGC (99.03%) | |||||
| MV-4-11 | 93 | 0.965 | MV-4-11 (99.03%) | ||||||
| MV-4-11 | LNCAPCLONEFGC | 5 | 99 | 9 | MV-4-11 (99.45%) | LNCAPCLONEFGC (96.83%) | 9.08 | 5.01E−17 | |
| MV-4-11 | LNCAPCLONEFGC | 2.5 | 111 | 4.51 | MV-4-11 (99.50%) | LNCAPCLONEFGC (98.34%) | 4.14 | 5.01E−17 | |
| MV-4-11 | LNCAPCLONEFGC | 1.25 | 117 | 2.18 | MV-4-11 (99.18%) | LNCAPCLONEFGC (90.32%) | 2.01 | 1.67E−09 | |
| MV-4-11 | LNCAPCLONEFGC | 0.625 | 112 | 1.58 | MV-4-11 (99.00%) | LNCAPCLONEFGC (89.44%) | 1.48 | 1.67E−09 | |
| LNCAPCLONEFGC | MV-4-11 | 5 | 102 | 2.35 | LNCAPCLONEFGC (98.99%) | MV-4-11 (94.57%) | 2.14 | 5.01E−17 | |
| LNCAPCLONEFGC | MV-4-11 | 2.5 | 101 | 1.67 | LNCAPCLONEFGC (99.04%) | MV-4-11 (91.58%) | 0.88 | 2.37E−11 | |
| LNCAPCLONEFGC | MV-4-11 | 1.25 | 98 | 1.49 | LNCAPCLONEFGC (99.03%) | MV-4-11 (87.77%) | 0.71 | 2.76E−11 | |
| LNCAPCLONEFGC | MV-4-11 | 0.625 | 105 | 1.36 | LNCAPCLONEFGC (99.03%) | ||||
| CAL28:RAJI | CAL27 | 39 | 1.39 | CAL27 (97.39%) | |||||
| RAJI | 114 | 1.18 | RAJI (98.56%) | ||||||
| RAJI | CAL27 | 5 | 116 | 5.36 | RAJI (98.66%) | CAL27 (99.12%) | 5.54 | 5.01E−17 | |
| RAJI | CAL27 | 2.5 | 127 | 4.17 | RAJI (98.32%) | CAL27 (94.70%) | 4.01 | 8.37E−11 | |
| RAJI | CAL27 | 1.25 | 121 | 1.84 | RAJI (98.51%) | CAL27 (90.83%) | 2.21 | 8.37E−06 | |
| RAJI | CAL27 | 0.625 | 116 | 2.49 | RAJI (98.50%) | CAL27 (90.43%) | 2.28 | 1.67E−07 | |
| CAL27 | RAJI | 5 | 121 | 7.11 | CAL27 (99.17%) | RAJI (99.51%) | 5.41 | 5.01E−17 | |
| CAL27 | RAJI | 2.5 | 113 | 3.79 | CAL27 (98.94%) | RAJI (92.06%) | 2.41 | 4.30E−13 | |
| CAL27 | RAJI | 1.25 | 112 | 2.14 | CAL27 (98.61%) | RAJI (83.42%) | 1.21 | 4.20E−07 | |
| CAL27 | RAJI | 0.625 | 112 | 1.4 | CAL27 (98.49%) | RAJI (83.75%) | 0.61 | 1.59E−07 |
aAverage values for each cell line mixture with three technical replicates, except the unmixed ones (see Supplementary Table S2 for full data).
bPercentage of the minor cell line based on cell counts.
cGenotype similarity shown in parentheses.
dChimeric genotype similarity shown in parentheses.
eProbability that the inferred minor component is incorrect.
Table 3.
Authentication and contaminant detection of 22 cell line mixtures
| Cell line mixturea | No. of informative SNPs | Heterogeneity ratio (percentage) | Major component inferredb | Minor component (contaminant) inferredc | Contamination ratio (percentage) | P-valued |
|---|---|---|---|---|---|---|
| ME180:143B | 119 | 6.54 | ME180 (98.06%) | 143B (97.09%) | 7.01 | 5.01E−17 |
| 143B:ME180 | 135 | 3.24 | 143B (98.55%) | ME180 (94.17%) | 3.21 | 5.01E−17 |
| JEG-3:A-875 | 104 | 1.63 | JEG-3 (98.49%) | A-875 (87.94%) | 1.21 | 5.01E−13 |
| A-875:JEG-3 | 93 | 15.50 | A-875 (91.71%) | JEG-3 (99.00%) | 16.71 | 5.01E−17 |
| HT3:C33A | 115 | 3.54 | HT3 (100%) | C33A (97.06%) | 3.41 | 5.01E−17 |
| C33A:HT3 | 90 | 4.34 | C33A (99.01%) | HT3 (100%) | 4.61 | 0 |
| DOTC24510:CASKI | 136 | 5.47 | DOTC24510 (98.99%) | CASKI (93.97%) | 5.21 | 5.01E−17 |
| CASKI:DOTC24510 | 129 | 4.26 | CASKI (98.98%) | DOTC24510 (91.84%) | 4.11 | 5.01E−17 |
| HLE:HCC94 | 163 | 2.62 | HLE (99.0%), HLF (96.08%) | HCC94 (91.46%) | 2.91 | 5.01E−17 |
| HCC94:HLE | 133 | 10.65 | HCC94 (97.6%) | HLE (96.63%), HLF (96.63%) | 10.11 | 5.01E−17 |
| NCIH1993:LS174T | 141 | 3.97 | NCIH1993 (98.05%) | LS174T (95.12%), LS180 (95.12%), HM7 (94.63%) | 4.21 | 5.01E−17 |
| LS174T:NCIH1993 | 114 | 4.88 | LS174T (99.02%), LS180 (99.03%), HM7 (98.54%) | NCIH1993 (97.06%) | 4.71 | 5.01E−17 |
| OSC19:SF763 | 152 | 7.03 | OSC19 (98.08%) | SF763 (96.15%) | 5.71 | 5.01E−17 |
| SF763:OSC19 | 133 | 3.35 | SF763 (99.02%) | OSC19 (90.15%) | 2.91 | 2.51E−16 |
| SW626:SJCRH30 | 155 | 11.67 | SW626 (95.63%) | SJCRH30 (98.54%) | 13.21 | 5.01E−17 |
| SJCRH30:SW626 | 88 | 1.79 | SJCRH30 (98.55%) | SW626 (94.2%) | 2.01 | 1.58E−16 |
| A-875:ME180 | 115 | 2.68 | A-875 (98.56%) | ME180 (95.67%) | 2.31 | 5.01E−17 |
| DOTC24510:CASKI | 144 | 1.75 | DOTC24510 (98.5%) | CASKI (86%) | 1.71 | 1.00E−15 |
| OSC19:SF763 | 130 | 2.68 | OSC19 (98.56%) | SF763 (93.27%) | 2.11 | 5.01E−17 |
| NOZ:SW626 | 127 | 0.82 | NOZ (97.56%) | SW626 (82.93%) | 0.71 | 3.98E−11 |
| SNU739:MM1R | 121 | 2.29 | SNU739 (99.01%) | MM1R (94.03%), MM1S (94.03%) | 1.71 | 5.01E−17 |
| U251:SR | 127 | 1.09 | U251 (98.54%) | SR (89.76%) | 1.11 | 5.01E−17 |
aIn the format of major cell line:minor/contaminating cell line.
bGenotype similarity shown in parentheses.
cChimeric genotype similarity shown in parentheses.
dProbability that the inferred minor component is wrong.
Authentication of human samples
The identity of a sample, or the major component of a contaminated sample, is determined by its genotype similarity to a library of reference samples. From 217 tested cell line samples, genotype similarities between the same cell lines are always >90% with an average of 98.6%; the lowest genotype similarity is 91.7% for an A-875 cell culture with 16.7% contamination of JEG-3 (Figure 1A and Table 3). In contrast, genotype similarities between unrelated cell lines are almost always below 50%. Still there are cell lines that are closely related or in the same synonymous group for various reasons, including mislabeling, contamination, deriving from the same patient, one cell line being parental to another, etc. (8) For example, HCT-15 and HCT-8 were likely derived from the same patient (47); QGY-7701 is contaminated and a HeLa derivative (48). Genotype similarities for 16 such cell line pairs in our dataset range from 84% to 96% (Supplementary Table S5). These cell line pairs can be distinguished, except for almost identical ones such as HLE and HLF. Genotype similarities between the same models on average are 98.0% (87.2–100%) for 220 PDX and 31 PDXO samples, and nearly all are below 50% between different models.
Figure 1.
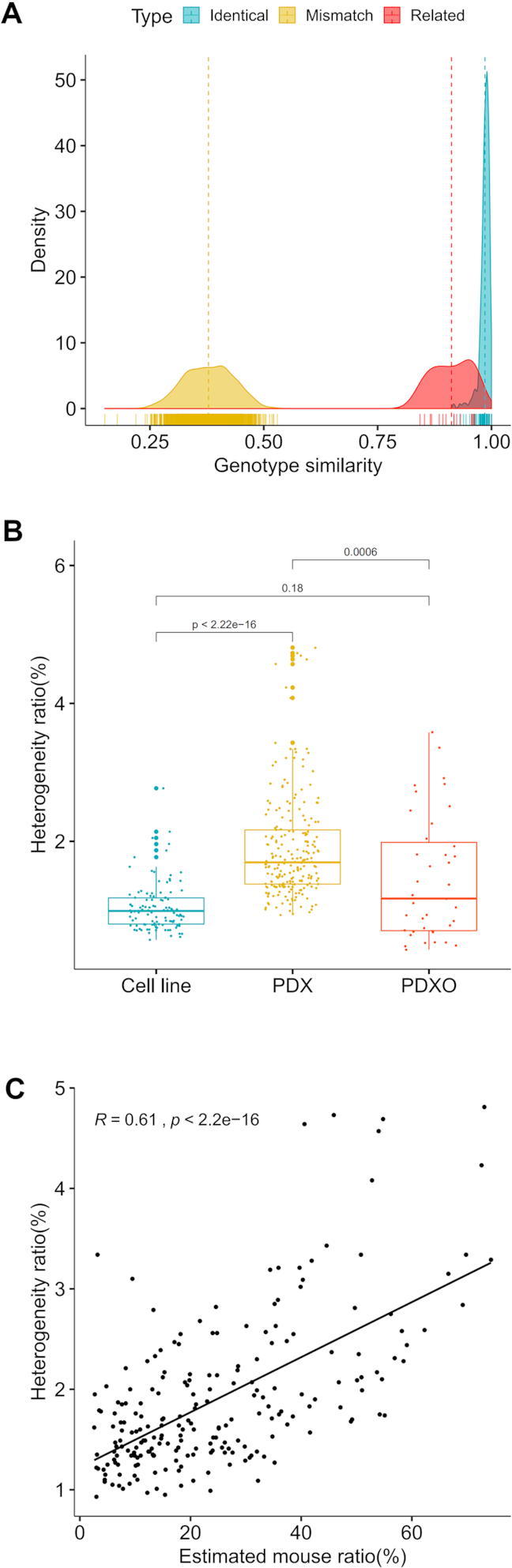
Cell line authentication and sample genetic heterogeneity. (A) Genotype similarities for unrelated/mismatch, identical and closely related cell line pairs. Genotype similarities are calculated for both uncontaminated and contaminated cell lines. Therefore, contaminated cell lines have reduced genotype similarities to their uncontaminated counterparts. The lowest genotype similarity is 91.7% between identical pairs, for an uncontaminated A-875 to a contaminated A-875 with 16.7% of JEG-3. (B) Heterogeneity ratios in 118 uncontaminated cell lines, 220 PDX models and 31 PDXO models. (C) Heterogeneity ratio is positively correlated with mouse ratio in PDX models.
Estimation of genetic heterogeneity using ‘informative SNPs’
If a sample is uncontaminated and is purely monoclonal diploid, then an SNP site is either homozygous or heterozygous, and the observed nucleotide frequency is close to 1 or 0.5 in deep NGS sequencing data; differences only arise from errors and randomness in sequencing. In reality, cell lines may have minor clones, are aneuploid or are contaminated (contaminants), so we observe not only frequencies far from 0.5 and 1, but also three or four nucleotides at an SNP site (46,49). We can use such information to estimate the genetic heterogeneity of a sample.
The dominant clone is the major component of a sample; minor clones and contaminants are the minor component. There are six informative genotype combinations of the major and minor components that can be used to estimate SNP heterogeneity ratios, based on the four observed nucleotide frequency patterns (Table 1). An SNP site is informative if it emits one of the four patterns. Subsequently, the sample heterogeneity ratio is estimated from individual SNP heterogeneity ratios by a statistical modeling approach (see the ‘Materials and Methods’ section). Using the test samples, we found that uncontaminated cell lines on average have 107 informative SNP sites, while contaminated cell lines have a slightly higher value of 112. On average, PDX and PDXO models have 156 and 111 informative SNP sites, respectively, which reflects higher genetic heterogeneity and/or mouse contamination in PDX models.
Detection and quantification of contamination by ‘heterogeneity ratio’
We detect sample contamination by combining three analyses. First, contaminated samples can have high heterogeneity ratios, while uncontaminated samples do not. In our test samples, 115 of 118 (97.5%) presumably uncontaminated cell lines have heterogeneity ratios <2% and all test samples have ratios <3% (Figure 1B). In contrast, we observed high heterogeneity ratios for contaminated cell lines; for example, an A-875 cell culture mixed with JEG-3 cells had a heterogeneity ratio of 15.5% (Table 3). We will later demonstrate that the heterogeneity ratio is proportional to the contamination ratio (percentage of contaminants), and therefore is a good indicator of contamination. Human tumors dissected from PDX models contain mouse stroma, and indeed we see higher heterogeneity ratios in PDX tumors (Figure 1B), caused by mouse contamination (Figure 1C). PDXOs, as in vitro culture of PDXs, have significantly smaller heterogeneity ratios due to much smaller, and often only trace, amounts of mouse cells generally due to the loss of mouse components in culture (Figure 1B).
Contamination is also indicated by a distinct right peak in the probability density of SNP heterogeneity ratios for a sample (Figure 2 and Supplementary Figures S2–S4). The peak shifts right as contamination and heterogeneity ratios increase, and sometimes splits into two peaks. The bi-/trimodal distribution vanishes, or only marginally appears, for uncontaminated cell lines or cell lines with very low contamination ratios (<1%) and heterogeneity ratios (<2%).
Figure 2.

The heterogeneity ratio can be used to detect and quantify contamination. (A–D) Serial mixes of cell lines MV-4-11 (MV411) and LNCaP clone FGC (LNCAPCLONEFGC) with cell ratios of 5%, 2.5%, 1.25% and 0.625% for the latter; (E) pure LNCaP clone FGC cell line; and (F) pure MV-4-11 cell line. Each tick above the horizontal axis represents an informative SNP site with corresponding SNP heterogeneity ratio. Probability density was estimated by assuming a two/three-component Gaussian mixture. Sample serial number is labeled in the top-right box with the major component cell line in parentheses. Sample heterogeneity ratio is shown underneath.
Finally, we can directly detect contaminants by statistical modeling that provides intuitive visualization and rigorous probabilistic measurement (see the ‘Materials and Methods’ section, Figure 3A and Supplementary Figures S5–S7). In 94 cell line samples each mixed with another cell line, we always correctly inferred the minor contaminant cell line when the heterogeneity ratio is ≥2% (Figure 3B). Accuracy is reduced to ∼80% and ∼50% when the heterogeneity ratio is 1–2% and <1%, respectively. For the eight missed samples, seven samples were characterized as clean and only one was marked by the incorrect contaminating cell line (Supplementary Table S1). It should be noted that such inference is only feasible when the contaminating cell line also has a known SNP fingerprint. We detected several contaminated cell lines in our biobank; one example is cell line ‘G-292 clone A141B1’ that had a high heterogeneity ratio of 7.62% (Figure 3C), and was contaminated by 6.21% OCI-AML-2 (Figure 3D).
Figure 3.

Contamination detection, contaminant inference and contamination ratio estimation. (A) Sample 19R58129 is MV-4-11 mixed with minor contaminating cell line LNCaP clone FGC (LNCAPCLONEFGC). LNCAPCLONEFGC was correctly identified as the contaminant (P-value = 5.01E−17) with a contamination ratio of 1.41%. LNCaP-C4-2 (C42) and LNCAPCLONEFGC were both derived from LNCaP and share high genetic identity (32). In the quantile–quantile plot, each dot is a reference cell line; theoretical and sample quantiles were calculated from a beta distribution fitted to genotype similarities between MV-4-11 and 1055 reference cell lines. The 99% confidence band is shaded. (B) Accuracy of inferring the contaminating second cell line in a cell line under different heterogeneity ratios. A total of 94 cell line samples with known contaminating second cell line were tested; samples were binned by heterogeneity ratio. (C) Cell line ‘G-292 clone A141B1’ had a sample heterogeneity ratio of 7.62% with a distinct right peak in the probability density of SNP heterogeneity ratios, indicating it was contaminated. (D) OCI-AML-2 was inferred as the contaminant (P-value = 1.58E−07) in cell line ‘G-292 clone A141B1’ with a contamination ratio of 6.21%. (E) Near-perfect correlation between estimated and known contamination ratios in simulated cell line mixtures. (F) High correlation between heterogeneity ratios and contamination ratios for cell line samples with known contamination.
After identifying the contaminating cell line, we can estimate the contamination ratio (i.e. percentage of the second cell line) using a maximum-likelihood approach (see the ‘Materials and Methods’ section). Simulation studies show that the estimated contamination ratios are extremely close to known ratios (Figure 3E). We observed a tight linear correlation between heterogeneity ratios and contamination ratios (Figure 3F). Therefore, as stated above, the heterogeneity ratio is a good estimator of contamination, and is particularly useful when contaminants are not standard cell lines. Within contaminated samples, contaminants contribute only a part (although this is sometimes the majority) of the genetic heterogeneity; consequently, contamination ratios are generally smaller than corresponding heterogeneity ratios (Table 3, Supplementary Tables S1 and S2), and the few outliers we observed were caused by data processing methods.
In summary, the heterogeneity ratio, by its value and distribution, is a reliable contamination measure for human samples. Cell line samples with a heterogeneity ratio of ≥2% are highly likely to be contaminated, and when the contaminant is another cell line also with a known SNP fingerprint, we can infer its identity and estimate the contamination ratio with an unprecedented sensitivity of ≤1%, measured by cell or DNA mix ratios (Tables 2 and 3).
Mouse tumor model authentication by a special set of SNPs
We used a total of 199 mouse SNPs for authenticating 32 syngeneic mouse tumor models commonly used in preclinical immunomodulatory drug development, including 4T1, A20, B16-BL6, B16-F0, B16-F1, B16-F10, C1498, Colon26, CT26WT, E.G7-Ova, EL4, EMT6, H22, Hepa1-6, J558, J774A1, JC, KLN205, L1210, L5178-R, LLC, MBT2, MC38, MPC-11, Neuro-2a, P388D1, P815, Pan02, Renca, RM1, S91 and WEHI164 (Supplementary Table S6). Authentication of these models achieved 100% accuracy (data not shown). Below, we explain the authentication protocol.
Most syngeneic models have a unique six-SNP signature. For example, 4T1 has the signature ‘TGGTGA’ at its six characteristic SNP sites across five chromosomes (namely 5_136026554, 1_91387260, 19_47898131, 11_100695233, 12_110649884 and 11_69740416 in the format of chromosome_location in the mouse reference genome mm10). All other models have the signature ‘CTCGAG’. Therefore, a syngeneic model can be unequivocally identified as 4T1 if we observe ‘TGGTGA’ at the six SNP sites. It should be noted that all the six SNP sites are heterozygous in 4T1, so we also observe the alternative nucleotides, namely ‘CTCGAG’. However, for non-4T1 models, the six SNP sites are all homozygous as ‘CTCGAG’.
There are two sets of models that are exceptions. The first set is Colon26 and CT26WT, both of which are mouse colon adenocarcinoma models originating from the BALB/c mouse strain. We use the first signature ‘AAATAA’ to identify a model as one from this set, and then assign the model as Colon26 if we observe ‘AGAACC’ for the second signature, and as CT26WT if we observe ‘GTTGGC’ for the third signature.
The second set is B16-BL6, B16-F0, B16-F1 and B16-F10, all of which are mouse melanoma cell lines in the C57BL/6 mouse strain, and which were all derived from the B16 cell line. Specifically, B16 is the parental line of B16-F0, which in turn is the parental line of B16-F1. B16-F10 is the 10th serial passage of B16-F0 and is the parental line of B16-BL6 (32). The four lines have high genetic similarity. We first use a seven-SNP signature ‘GGAGACC’ to assign a test cell line into this group, and then assign the cell line to B16-F0 by a second signature ‘GTGGTA’, or to B16-F10 by a third signature ‘CACTCT’ or to B16-BL6 by a fourth signature ‘TGAAAG’; if none of the three signatures is observed, then the cell line is identified as B16-F1.
Human–mouse interspecies contamination detection based on divergent segments between two species
We compared human hg19 and mouse mm10 genomes, and identified 108 100–300 bp segments such that each segment significantly diverged (by insertion, deletion and point mutation) between human and mouse (31–97% sequence similarities), yet has identical flanking sequences ensuring that a common pair of primers can be readily designed (Supplementary Table S7). After NGS sequencing, we can separate human and mouse reads, calculate mouse ratios for all segments and take the median of these ratios as the mouse ratio in a human–mouse mixed sample. This method demonstrated extremely high accuracy in a set of benchmark samples in which mouse and human DNA was mixed by serial dilution (Figure 4A). We further prepared a second set of serial dilution samples with low mouse ratios, and demonstrated that the method can reliably detect mouse contamination at ∼0.1% (Supplementary Table S8).
Figure 4.

Estimation of mouse ratio in human–mouse mixtures. (A) Accurate estimation of mouse ratio by the deep NGS sequencing in a serial dilution of human–mouse DNA mixtures with mouse ratios of 90%, 80%, 70%, 50%, 30%, 20%, 10%, 7%, 5% and 0%. (B, C) Mouse ratios estimated in 220 PDX and 31 PDXO models by three approaches, assayed on the same sample for each model. (D) A quadratic relationship between mouse ratios estimated by the deep NGS sequencing and WES in 220 PDX models.
We also developed methods of estimating mouse content from RNA-seq and WES data (see the ‘Materials and Methods’ section). We compared three methods in estimating mouse ratios in 220 PDX and 31 PDXO models (Figure 4B and C). DNA (for WES and the deep NGS sequencing) and RNA (for RNA-seq) were extracted and sequenced from the same sample of a model to remove sample variance. PDXO models generally have low mouse content. In PDX models, mouse ratios accurately estimated from deep NGS sequencing data are the highest, followed by RNA-seq and then WES. This observed lower ratio for WES is likely due to the exon-capture kit used in WES, which was designed to enrich human exons and had low hybridization affinity to homologous mouse exons. RNA-seq used a polyA-enrichment protocol with no species preference; however, gene expression has great temporospatial variability in human tumor and mouse stroma of PDX. Indeed, we observed a very strong quadratic relationship for mouse ratios between the deep NGS sequencing data and WES data (R = 0.96, Figure 4D), but a much weaker linear correlation between the deep sequencing data and RNA-seq data (R = 0.62).
Mycoplasma detection using a specially designed SNP set
Mycoplasma contamination is a major concern in laboratory cell and tissue culture, impacting experiment conduction and causing false positive/negative errors. There are several ways to detect mycoplasma contamination, including PCR, enzymatic, indirect DNA DAPI staining and microbial culture methods. The PCR method uses oligonucleotide primers to amplify conserved 16S rRNA regions, and was shown to be most sensitive (50). In our NGS assay, we used one such pair of universal primers to detect all mycoplasma species (51), which produce a 425-bp amplicon easily identified from the high-depth sequencing data.
In addition, we used 11 pairs of species-specific primers, with proven effectiveness, to detect 11 common mycoplasma species, including Acholeplasma laidlawii, Mycoplasma arginini, M. fermentans, M. genitalium, M. hominis, M. hyorhinis, M. orale, M. pirum, M. pneumoniae, M. salivarium and Ureaplasma urealyticum (51). These primers generate amplicons ranging from 300 to 335 bp, which are also the perfect size for NGS detection. It is possible that a sample can be contaminated by more than one mycoplasma species, which can be discerned by our NGS assay. Mycoplasma can be eradicated by antibiotics, including BM-Cyclin, ciprofloxacin and other removal agents (51). Treated samples can be routinely checked by the NGS assay to ensure that they are mycoplasma free. We identified one mycoplasma-contaminated cell line in our biobank by the NGS assay and subsequently validated the result by a mycoplasma detection kit (Supplementary Table S9).
Population structure analysis and gender determination
Individuals carry genetic variants, including SNPs. Related individuals share some of these variants and the overlapping percentage is a measurement of genetic similarity. A collection of population-specific SNPs has been identified by international collaborations such as the International HapMap Project (41) and the 1000 Genomes Project Consortium (52). We can perform population structure analysis using full or part of these SNPs, so that an individual with unknown ancestry can be inferred and the proportion of each reference population is deduced.
Of the 200 SNPs used for human sample authentication, 132 were characterized by the International HapMap Project. We added another 13 SNPs, making a total of 145 SNPs in HapMap (release 3). These SNPs, by design, are all located on 22 autosomes and are sufficiently far enough away from each other to ensure very limited linkage disequilibrium. Furthermore, their MAFs are close to 0.5 in three reference populations (CHB, YRI and CEU; see the ‘Materials and Methods’ section). Therefore, they are ideal for inferring population structure. To evaluate the performance, we used fastSTRUCTURE (53) to analyze the three reference populations. Indeed, all 406 individuals were unambiguously assigned to the correct reference populations with high probabilities (Supplementary Table S10).
We profiled 423 PDX models derived from East Asian patients and 634 PDX models derived from Western patients in the United States. The analysis showed that all the East Asian PDX models have dominant CHB composition with only one exception. The majority of the Western PDX models have predominantly CEU composition; the rest have major CHB or YRI compositions, or are mixtures of two or three of the reference populations (Figure 5).
Figure 5.
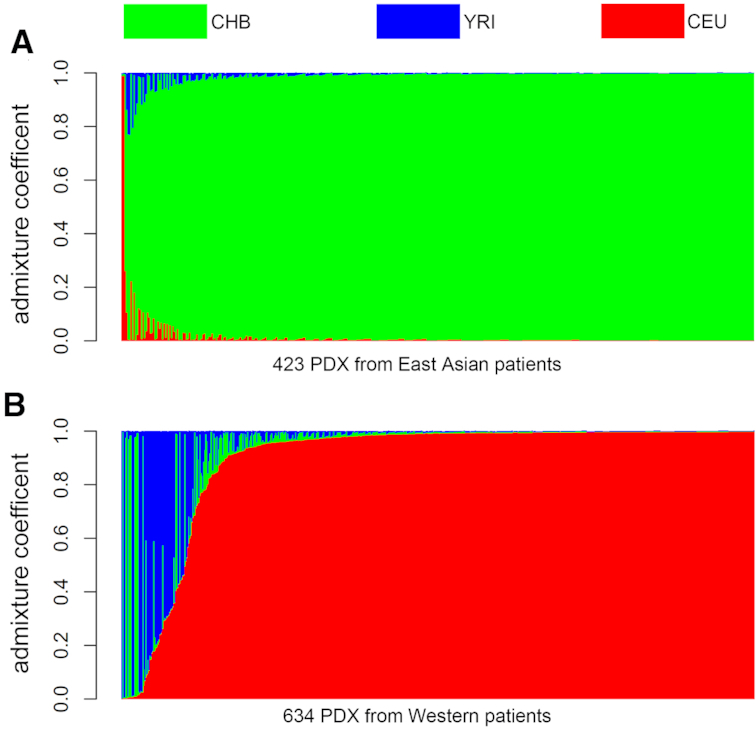
Inferred population structures in PDX models. (A) Four hundred twenty-three PDXs derived from East Asian patients. (B) Six hundred thirty-four PDXs derived from Western patients. The three reference populations from the International HapMap Project are CHB, YRI and CEU (41).
To infer gender of human samples, we amplified three segments (109, 137 and 189 bp) on the Y chromosome (Supplementary Table S11). The three short segments have no homologous sequences in autosomes or X chromosome. Therefore, a non-trivial amplification of them, defined as the total number of reads >300, indicated a male sample. When tested on 541 PDX samples, the NGS assay correctly inferred genders for 528 samples, while the other 13 male samples were erroneously labeled as female due to segment deletion or total loss of the Y chromosome (Supplementary Table S12).
DISCUSSION
There are three levels of biosample authentication. Level 1 authentication matches a sample to a reference (e.g. standard cancer cell lines). Conventional STR and SNP assays largely used genotype-based Tanabe–Masters algorithm and its variations (8,54,55). STR assays generate analog signals for a dozen markers, while SNP assays often genotype many more SNPs. Therefore, higher similarity thresholds are often used by SNP assays to identify two samples as a match (3,8). However, the matching power of conventional assays can be severely compromised for contaminated samples even with ∼100 SNPs (27). Our method performed high-depth (3000×) sequencing of 200 SNP sites for human samples, and showed 100% accuracy in identifying a sample or the major component of contaminated samples, which is a significant improvement over the conventional STR/SNP assays.
Level 2 authentication detects contamination in biosamples. The sensitivity for detecting contamination in cell lines is ∼5–10% for STR assays and 3–5% for SNP assays. However, as previously stated, performance of these assays can be rather unstable. Our method consistently reaches 2% sensitivity when using only the heterogeneity ratio, by both its value and distinct bi-/trimodal distribution. The sensitivity can reach ≤1% if the contaminant is in a library of reference samples with an SNP fingerprint. Such sensitivity virtually reaches the theoretic detection limit, because uncontaminated cell lines, due to multiclonality and sequencing errors, exhibit a comparable level of genetic heterogeneity to cell line samples with ∼1% contamination.
Level 3 authentication identifies the contaminant in a contaminated sample, which is difficult to achieve, but is made practically possible by our reported method. For example, PANC-1 is the contaminant in a cell mix of 97% RT4 and 3% PANC-1 cells. Level 3 capability is available within our method, but not in conventional STR and SNP assays. Cross-contamination of cell lines is common in biobanks. The composition of a contaminated culture changes over time due to different growth rates of cell lines. Cell lines also differ in genomics such as gene mutations and may respond differently to drug treatment, causing erroneous results in drug screening. We constructed an SNP fingerprint library for over 1000 cancer cell lines, enabling a contaminating cell line to be unambiguously identified. Furthermore, we can accurately estimate the contamination ratio. Alongside checking cell line quality, this capacity can have other uses such as monitoring the dynamic composition of two cell lines under biological or chemical interference.
For cell line authentication, it is normally sufficient to identify the cell line and determine whether it is contaminated, which can be achieved by conventional STR assays due to them being readily available. As detailed above, such assays suffer from their limited ability to discern contamination. Their sensitivity also depends greatly on the threshold for calling matches, which cell lines are within a mix, subjective determination of STR bands and the number of loci. When there is interspecies contamination, such as PDX tumors in which human tumor cells are mixed with murine stromal cells, STR assays are problematic in detecting mouse cell contamination in human cells. All of these problems are satisfactorily solved by our NGS method. Due to its high-throughput nature, our method is more suitable for use by large biobanks with multiple types of biospecimens, including cell lines, organoids, PDX models, syngeneic mouse models, human samples, etc.
Besides intraspecies contamination, our method is able to accurately detect and quantify interspecies contamination between human and mouse. Here, we do not use SNPs, instead employing 108 homologous DNA segments that are diverged between the two species but have identical flanking nucleotide sequences. This allows common primers to be designed for unbiased amplification of human and mouse DNA segments. This approach showed perfect performance in a serial dilution of mouse–human DNA mixture benchmark samples. The homology-based principle can be used for detecting other interspecies contaminations. We used the amplification of three Y-chromosome segments to infer gender of human sample, which exhibited complete accuracy except for samples with partial or total loss of Y chromosome. The problem can be largely alleviated or removed for samples with only partial loss of Y chromosome by amplifying more segments that spread across the Y chromosome. For example, amplicons can be designed around these Y-chromosome sites: 2822023, 7235632, 21765821 and 28479069.
The power of our method comes from several novel features. First, deep NGS sequencing was used to obtain both genotype and nucleotide frequency of SNPs. Conventional STR and SNP assays only profile SNP genotypes. Second, our method performs additional targeted sequencing for detecting mycoplasma contamination and estimating mouse–human mix ratios. Third, a suite of statistical models and algorithms was devised to exploit deep NGS sequencing data, making the authentication process automatic, robust and objective. Finally, DNA barcode technology is used to enable parallel sequencing of 100–200 samples simultaneously that drastically reduces cost.
In conclusion, we have developed a high-throughput low-cost method that can be routinely used by biobanks to maintain authentic and high-quality samples. The method can be broadly adapted for samples from other species and even the microbiome, and can be implemented on any NGS sequencing platform. With NGS technology becoming a standard platform for different applications, our method could potentially become the future assay of choice for sample authentication and quality controls.
DATA AVAILABILITY
Raw NGS data in FASTQ format were deposited to the Sequence Read Archive with accession number PRJNA647262 (https://www.ncbi.nlm.nih.gov/sra/PRJNA647262). Perl/R scripts implementing the algorithms, along with processed NGS data, are available at https://github.com/guosheng437/NGSQC.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to express their gratitude to the Biobank and Genomics team members at Crown Bioscience, Inc. for data production, to Dr Xiao Cong for critical discussion, to Dr Jody Barbeau for manuscript proofreading and editing, and to iGeneTech for NGS technical assistance.
Contributor Information
Xiaobo Chen, Crown Bioscience, Inc., 218 Xinghu Road, Suzhou, Jiangsu 215400, China.
Wubin Qian, Crown Bioscience, Inc., 218 Xinghu Road, Suzhou, Jiangsu 215400, China.
Zhenzhen Song, Crown Bioscience, Inc., 218 Xinghu Road, Suzhou, Jiangsu 215400, China.
Qi-Xiang Li, Crown Bioscience, Inc., 16550 W Bernardo Dr, Building 5, San Diego, CA 92127, USA.
Sheng Guo, Crown Bioscience, Inc., 218 Xinghu Road, Suzhou, Jiangsu 215400, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Crown Bioscience, Inc.
Conflict of interest statement. This research was funded by Crown Bioscience, Inc. and all authors were employees thereof at the time the study was performed. The authors declare no other competing financial interests.
REFERENCES
- 1. Editorial. Identity crisis. Nature. 2009; 457:935–936. [DOI] [PubMed] [Google Scholar]
- 2. American Type Culture Collection Standards Development Organization Workgroup ASN-0002. Cell line misidentification: the beginning of the end. Nat. Rev. Cancer. 2010; 10:441–448. [DOI] [PubMed] [Google Scholar]
- 3. Capes-Davis A., Reid Y.A., Kline M.C., Storts D.R., Strauss E., Dirks W.G., Drexler H.G., MacLeod R.A., Sykes G., Kohara A. et al.. Match criteria for human cell line authentication: where do we draw the line. Int. J. Cancer. 2013; 132:2510–2519. [DOI] [PubMed] [Google Scholar]
- 4. Gartler S.M. Apparent HeLa cell contamination of human heteroploid cell lines. Nature. 1968; 217:750–751. [DOI] [PubMed] [Google Scholar]
- 5. Lacroix M. Persistent use of ‘false’ cell lines. Int. J. Cancer. 2008; 122:1–4. [DOI] [PubMed] [Google Scholar]
- 6. Lorsch J.R., Collins F.S., Lippincott-Schwartz J.. Cell biology. Fixing problems with cell lines. Science. 2014; 346:1452–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fusenig N.E., Capes-Davis A., Bianchini F., Sundell S., Lichter P.. The need for a worldwide consensus for cell line authentication: experience implementing a mandatory requirement at the International Journal of Cancer. PLoS Biol. 2017; 15:e2001438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu M., Selvaraj S.K., Liang-Chu M.M., Aghajani S., Busse M., Yuan J., Lee G., Peale F., Klijn C., Bourgon R. et al.. A resource for cell line authentication, annotation and quality control. Nature. 2015; 520:307–311. [DOI] [PubMed] [Google Scholar]
- 9. Bian X., Yang Z., Feng H., Sun H., Liu Y.. A combination of species identification and STR profiling identifies cross-contaminated cells from 482 human tumor cell lines. Sci. Rep. 2017; 7:9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horbach S., Halffman W.. The ghosts of HeLa: how cell line misidentification contaminates the scientific literature. PLoS One. 2017; 12:e0186281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Maagd R.A., Bakker P., Staykov N., Dukiandjiev S., Stiekema W., Bosch D.. Identification of Bacillus thuringiensis delta-endotoxin Cry1C domain III amino acid residues involved in insect specificity. Appl. Environ. Microbiol. 1999; 65:4369–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Azari S., Ahmadi N., Tehrani M.J., Shokri F.. Profiling and authentication of human cell lines using short tandem repeat (STR) loci: report from the National Cell Bank of Iran. Biologicals. 2007; 35:195–202. [DOI] [PubMed] [Google Scholar]
- 13. Wu M.L., Liao L.C., Chen C.Y., Lee S.Y., Yuan G.F., Hwang S.M.. A 2-yr service report of cell line authentication. In Vitro Cell. Dev. Biol. Anim. 2013; 49:743–745. [DOI] [PubMed] [Google Scholar]
- 14. Masters J.R. HeLa cells 50 years on: the good, the bad and the ugly. Nat. Rev. Cancer. 2002; 2:315–319. [DOI] [PubMed] [Google Scholar]
- 15. MacLeod R.A., Dirks W.G., Matsuo Y., Kaufmann M., Milch H., Drexler H.G.. Widespread intraspecies cross-contamination of human tumor cell lines arising at source. Int. J. Cancer. 1999; 83:555–563. [DOI] [PubMed] [Google Scholar]
- 16. Cosme B., Falagan-Lotsch P., Ribeiro M., Napoleao K., Granjeiro J.M., Moura-Neto R.. Are your results valid? Cellular authentication a need from the past, an emergency on the present. In Vitro Cell. Dev. Biol. Anim. 2017; 53:430–434. [DOI] [PubMed] [Google Scholar]
- 17. Ye F., Chen C., Qin J., Liu J., Zheng C.. Genetic profiling reveals an alarming rate of cross-contamination among human cell lines used in China. FASEB J. 2015; 29:4268–4272. [DOI] [PubMed] [Google Scholar]
- 18. Freedman L.P., Gibson M.C., Wisman R., Ethier S.P., Soule H.R., Reid Y.A., Neve R.M.. The culture of cell culture practices and authentication: results from a 2015 survey. BioTechniques. 2015; 59:189–190. [DOI] [PubMed] [Google Scholar]
- 19. Nims R.W., Reid Y.. Best practices for authenticating cell lines. In Vitro Cell. Dev. Biol. Anim. 2017; 53:880–887. [DOI] [PubMed] [Google Scholar]
- 20. Almeida J.L., Cole K.D., Plant A.L.. Standards for cell line authentication and beyond. PLoS Biol. 2016; 14:e1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Almeida J.L., Dakic A., Kindig K., Kone M., Letham D.L.D., Langdon S., Peat R., Holding-Pillai J., Hall E.M., Ladd M et al.. Interlaboratory study to validate a STR profiling method for intraspecies identification of mouse cell lines. PLoS One. 2019; 14:e0218412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zaaijer S., Gordon A., Speyer D., Piccone R., Groen S.C., Erlich Y.. Rapid re-identification of human samples using portable DNA sequencing. eLife. 2017; 6:e27798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yousefi S., Abbassi-Daloii T., Kraaijenbrink T., Vermaat M., Mei H., van ’t Hof P., van Iterson M., Zhernakova D.V., Claringbould A., Franke L. et al.. A SNP panel for identification of DNA and RNA specimens. BMC Genomics. 2018; 19:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jobling M.A., Gill P.. Encoded evidence: DNA in forensic analysis. Nat. Rev. Genet. 2004; 5:739–751. [DOI] [PubMed] [Google Scholar]
- 25. Sanchez J.J., Phillips C., Borsting C., Balogh K., Bogus M., Fondevila M., Harrison C.D., Musgrave-Brown E., Salas A., Syndercombe-Court D. et al.. A multiplex assay with 52 single nucleotide polymorphisms for human identification. Electrophoresis. 2006; 27:1713–1724. [DOI] [PubMed] [Google Scholar]
- 26. Didion J.P., Buus R.J., Naghashfar Z., Threadgill D.W., Morse H.C. 3rd, de Villena F.P.. SNP array profiling of mouse cell lines identifies their strains of origin and reveals cross-contamination and widespread aneuploidy. BMC Genomics. 2014; 15:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liang-Chu M.M., Yu M., Haverty P.M., Koeman J., Ziegle J., Lee M., Bourgon R., Neve R.M.. Human biosample authentication using the high-throughput, cost-effective SNPtrace™ system. PLoS One. 2015; 10:e0116218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pengelly R.J., Gibson J., Andreoletti G., Collins A., Mattocks C.J., Ennis S.. A SNP profiling panel for sample tracking in whole-exome sequencing studies. Genome Med. 2013; 5:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morgan A.P., Fu C.P., Kao C.Y., Welsh C.E., Didion J.P., Yadgary L., Hyacinth L., Ferris M.T., Bell T.A., Miller D.R. et al.. The mouse universal genotyping array: from substrains to subspecies. G3 (Bethesda). 2015; 6:263–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Castro F., Dirks W.G., Fahnrich S., Hotz-Wagenblatt A., Pawlita M., Schmitt M.. High-throughput SNP-based authentication of human cell lines. Int. J. Cancer. 2013; 132:308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El-Hoss J., Jing D., Evans K., Toscan C., Xie J., Lee H., Taylor R.A., Lawrence M.G., Risbridger G.P., MacKenzie K.L. et al.. A single nucleotide polymorphism genotyping platform for the authentication of patient derived xenografts. Oncotarget. 2016; 7:60475–60490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bairoch A. The Cellosaurus, a cell-line knowledge resource. J. Biomol. Tech. 2018; 29:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ruitberg C.M., Reeder D.J., Butler J.M.. STRBase: a short tandem repeat DNA database for the human identity testing community. Nucleic Acids Res. 2001; 29:320–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van der Meer D., Barthorpe S., Yang W., Lightfoot H., Hall C., Gilbert J., Francies H.E., Garnett M.J.. Cell Model Passports: a hub for clinical, genetic and functional datasets of preclinical cancer models. Nucleic Acids Res. 2019; 47:D923–D929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tuveson D., Clevers H.. Cancer modeling meets human organoid technology. Science. 2019; 364:952–955. [DOI] [PubMed] [Google Scholar]
- 36. Day C.P., Merlino G., Van Dyke T.. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. 2015; 163:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo S., Qian W., Cai J., Zhang L., Wery J.P., Li Q.X.. Molecular pathology of patient tumors, patient-derived xenografts, and cancer cell lines. Cancer Res. 2016; 76:4619–4626. [DOI] [PubMed] [Google Scholar]
- 38. Khaled W.T., Liu P.. Cancer mouse models: past, present and future. Semin. Cell Dev. Biol. 2014; 27:54–60. [DOI] [PubMed] [Google Scholar]
- 39. Li Q.X., Feuer G., Ouyang X., An X.. Experimental animal modeling for immuno-oncology. Pharmacol. Ther. 2017; 173:34–46. [DOI] [PubMed] [Google Scholar]
- 40. Chao C., Widen S.G., Wood T.G., Zatarain J.R., Johnson P., Gajjar A., Gomez G., Qiu S., Thompson J., Spratt H. et al.. Patient-derived xenografts from colorectal carcinoma: a temporal and hierarchical study of murine stromal cell replacement. Anticancer Res. 2017; 37:3405–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. International HapMap, C. The International HapMap Project. Nature. 2003; 426:789–796. [DOI] [PubMed] [Google Scholar]
- 42. R, Core Team. R: A Language and Environment for Statistical Computing, 3.5.3 ed. R Foundation for Statistical Computing. 2018; Vienna. [Google Scholar]
- 43. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H., Durbin R.. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fasterius E., Al-Khalili Szigyarto C.. Analysis of public RNA-sequencing data reveals biological consequences of genetic heterogeneity in cell line populations. Sci. Rep. 2018; 8:11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ghandi M., Huang F.W., Jane-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R. 3rd, Barretina J., Gelfand E.T., Bielski C.M., Li H. et al.. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019; 569:503–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vermeulen S.J., Chen T.R., Speleman F., Nollet F., Van Roy F.M., Mareel M.M.. Did the four human cancer cell lines DLD-1, HCT-15, HCT-8, and HRT-18 originate from one and the same patient?. Cancer Genet. Cytogenet. 1998; 107:76–79. [DOI] [PubMed] [Google Scholar]
- 48. Rebouissou S., Zucman-Rossi J., Moreau R., Qiu Z., Hui L.. Note of caution: contaminations of hepatocellular cell lines. J. Hepatol. 2017; 67:896–897. [DOI] [PubMed] [Google Scholar]
- 49. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehar J., Kryukov G.V., Sonkin D. et al.. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Molla Kazemiha V., Bonakdar S., Amanzadeh A., Azari S., Memarnejadian A., Shahbazi S., Shokrgozar M.A., Mahdian R.. Real-time PCR assay is superior to other methods for the detection of mycoplasma contamination in the cell lines of the National Cell Bank of Iran. Cytotechnology. 2016; 68:1063–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Molla Kazemiha V., Shokrgozar M.A., Arabestani M.R., Shojaei Moghadam M., Azari S., Maleki S., Amanzadeh A., Jeddi Tehrani M., Shokri F.. PCR-based detection and eradication of mycoplasmal infections from various mammalian cell lines: a local experience. Cytotechnology. 2009; 61:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Genomes Project, C. Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Raj A., Stephens M., Pritchard J.K.. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics. 2014; 197:573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Masters J.R., Thomson J.A., Daly-Burns B., Reid Y.A., Dirks W.G., Packer P., Toji L.H., Ohno T., Tanabe H., Arlett C.F. et al.. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc. Natl Acad. Sci. U.S.A. 2001; 98:8012–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tanabe H., Takada Y., Minegishi D., Kurematsu M., Masui T., Mizusawa H.. Cell line individualization by STR multiplex system in the cell bank found cross-contamination between ECV304 and EJ-1/T24. Tissue Culture Res. Commun. 1999; 18:329–338. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw NGS data in FASTQ format were deposited to the Sequence Read Archive with accession number PRJNA647262 (https://www.ncbi.nlm.nih.gov/sra/PRJNA647262). Perl/R scripts implementing the algorithms, along with processed NGS data, are available at https://github.com/guosheng437/NGSQC.