

Abstract
The study of bacterial symbioses has grown exponentially in the recent past. However, existing bioinformatic workflows of microbiome data analysis do commonly not integrate multiple meta-omics levels and are mainly geared toward human microbiomes. Microbiota are better understood when analyzed in their biological context; that is together with their host or environment. Nevertheless, this is a limitation when studying non-model organisms mainly due to the lack of well-annotated sequence references. Here, we present gNOMO, a bioinformatic pipeline that is specifically designed to process and analyze non-model organism samples of up to three meta-omics levels: metagenomics, metatranscriptomics and metaproteomics in an integrative manner. The pipeline has been developed using the workflow management framework Snakemake in order to obtain an automated and reproducible pipeline. Using experimental datasets of the German cockroach Blattella germanica, a non-model organism with very complex gut microbiome, we show the capabilities of gNOMO with regard to meta-omics data integration, expression ratio comparison, taxonomic and functional analysis as well as intuitive output visualization. In conclusion, gNOMO is a bioinformatic pipeline that can easily be configured, for integrating and analyzing multiple meta-omics data types and for producing output visualizations, specifically designed for integrating paired-end sequencing data with mass spectrometry from non-model organisms.
INTRODUCTION
Symbiosis is a widespread relationship present in all groups of organisms but intensely developed between animals and bacteria that benefit from each other in order to survive. Consequently, both acquire an evolutionary advantage in comparison to individuals lacking this relationship. Two different types of symbiosis can be distinguished: ectosymbiosis, in which bacteria are attached to the surface of the host, and endosymbiosis, which usually is a mutualistic relationship, where bacteria live intracellularly in the host and are transmitted vertically (1,2). To understand these evolutionary relationships host and symbionts are best studied together. In mutualistic symbiosis, the eukaryotes provide a safe environment for endosymbiotic bacteria that live in close interaction with the host. In return, the endosymbionts provide nutrients and metabolites (such as essential amino acids or vitamins) to the host that cannot be obtained in any other way. For example, it has been estimated that around 15% of insect species maintain endosymbiotic associations with bacteria that supply the host with the nutrients that are lacking in their diets (3) On the other hand, most insects possess a gut microbiome that affects the physiology of the host by, for example, contributing to metabolic and nutritional needs, and the immune system development (4). Recently, many studies have been performed in humans to study the gut microbiota (5), but non-model organisms require further investigations to better understand this specific type of symbiosis. In this context, cockroaches are a suitable model, because they have two symbiotic systems, i.e. an endosymbiont (Blattabacterium cuenoti) in the fat body and a rich and complex gut microbiota (6,7). The German cockroach Blattella germanica is a hemimetabolous insect (it has an incomplete metamorphosis) with three developmental stages. Regarding its symbionts, genome analysis demonstrated that the endosymbiont Blattabacterium contributes to the nitrogen (N) recycling and the synthesis of essential amino acids (8), but the function of the gut microbiota in cockroaches still has to be elucidated. It has been shown that the gut microbiome of cockroaches shows much overlap with the one in humans probably reflecting a similar omnivorous diet (6,9–10).
Recently, research interests in microbial communities have been strongly increased due to findings on the impact of the microbiome on human health (11,12). Microbiome studies often employ meta-omics techniques such as metagenomics (13) that aims to analyze the genetic material from all members in a microbial community sample. Despite many advantages, metagenomics still presents a static gene-centric approach that cannot assess temporal dynamics and functional activities of complex microbial populations (14). To gain insights into the dynamic functional repertoire of microbial communities, further techniques such as metatranscriptomics and metaproteomics have been established in recent years (15,16). Beyond the genome level, these meta-omics analysis approaches allow studying complex microbial systems and their host interactions at the gene expression level (transcripts and proteins, respectively). Used separately, metagenomics, metatranscriptomics and metaproteomics are already powerful because they complement and mutually support each other. However, the bioinformatics analysis still faces various specific challenges that concern, for example, the identification of genes and proteins, the construction of multi-organism databases, the database selection process influencing the taxonomic and functional assignment (17), and the use of different sample extraction or data analysis protocols making the results comparison difficult (18). Finally, the lack of properly annotated reference genomes and proteomes is also a typical overseen issue in this context (19). These challenges must be overcome to design optimized and standardized meta-omics pipelines for analysing microbiome data.
In the past, powerful tailored bioinformatic solutions have been developed for the individual meta-omics analysis levels (13,15–16). However, the true strength unfolds when these analysis techniques are integrated (20,21). As a holistic approach, a complete meta-omics integration can extend the capabilities of microbiome and host-related studies in various ways. Most importantly, integrating multiple meta-omics levels allows to expand the possibilities of biological interpretation and to investigate biological pathways from a more comprehensive perspective. Compared to single-omics strategies, an integrative approach provides a deeper and more thorough understanding of how the key players of microbial communities regulate underlying pathway mechanisms (22).
While the integration of meta-omics has been described in previous studies (23), its potential has not been fully exploited so far. In particular, the data analysis is challenging, because studies often present customized in-house workflows that cannot be fully automated or are not reproducible. In general, automated multi-omics analysis pipelines are rare and limited to few meta-omics levels (24) and are not tailored for host and microbiome analyses of non-model organisms.
Here, we present gNOMO, a meta-omics software pipeline that allows integrating three different levels of omics analyses, derived from metagenomics, metatranscriptomics and metaproteomics experiments. It provides two different, optionally iterative operating modes: (i) each of the three omics levels can be analyzed separately and independently of each other and subsequently, (ii) up to three omics layers can be analyzed in a fully integrated fashion. The workflow of gNOMO starts from raw data to essential processing steps and finally provides output visualizations for taxonomic classification, functional metabolic pathway profiling and differential sample analysis. The integration of metagenomics, metatranscriptomics and metaproteomics data is possible due to the production of a tailored proteogenomic database, which optimizes the identification and quantification of peptides in metaproteomics data (25,26). As microbiota needs to be analyzed in its context, the host is also studied together with the microbiome. Host data can be analyzed without a reference database, which allows to study non-model organisms, and proteins of the host are also identified with a tailored host database obtained from genomics and transcriptomic sequences. The pipeline has been implemented using the Python-based Snakemake (27) framework to perform fully automated and reproducible multi-omics analyses of host and microbiome samples. So far, gNOMO has been developed and optimized for data from non-model organism samples, but it is fully executable on generic sample types, for example, from human or mouse microbiomes. With gNOMO, we aim to fill the gap of barely existing multi-omics pipelines for microbial community samples being able to compare and integrate data at the genome, transcriptome and proteome level.
MATERIALS AND METHODS
gNOMO is a pipeline that integrates multiple bioinformatic methods and software tools to analyze metagenomics, metatranscriptomics and metaproteomics data and to provide the results with an easily readable final output. One of the main purposes of integrating such different kinds of multi-omics data is to directly improve the analysis of microbial populations and to investigate their function in poorly characterized environments, such as non-model organisms. At the genome and transcriptome level, our pipeline includes both quality control and data preparation steps, of which parameters can be adjusted depending on the quality of the input data. In addition, gNOMO allows to directly create a proteogenomic database from metagenomics and metatranscriptomics data. This important processing step makes it possible to connect the metagenomics and metatranscriptomics analysis to the protein identification at the metaproteomics level. In particular, the proteogenomic database generation step leads to the full integration of all three omics levels.
The complete gNOMO pipeline is built in Snakemake (27), a management system for bioinformatic workflows, that allows obtaining standardized and reproducible output data. The input data and parameters of programs that are used in Snakemake are defined by editing a single configuration file. Further, the gNOMO pipeline including all dependencies is available at the BioConda channel (28). Tools added to BioConda provide a user-friendly installation because the required tools and libraries are easily incorporated and automatically installed with the use of Snakemake environments. Due to the high computational needs of some parts of the workflow, we recommend a system with at least 16 available cores and at least 200 GB RAM. The storage requirements are data-dependent and were in our case about 1 TB of free storage. The runtime highly depends on the number of available cores because Snakemake is able to parallelize non-dependent tasks and decreases the runtime this way substantially. On a cluster node with 16 cores and 200 GB RAM the analysis of the B. germanica microbiome took about 72 h. The runtime of gNOMO can vary from run to run as it not only depends on CPU power but network speeds used, for example, for database updates as well. In addition, it should be stated that the Snakemake workflow engine is compatible and scalable in cluster environments (e.g. using the SLURM Workload Manager). The gNOMO pipeline typically consists of five main steps (Figure 1): (i) pre-processing, (ii) metagenomics and metatranscriptomics data analysis, (iii) proteogenomic database creation, (iv) metaproteomics data analysis and (iv) data integration. In the following paragraphs, these individual steps are described in more detail.
Figure 1.

Workflow overview of the gNOMO pipeline. Each box represents a processing step in the pipeline. Box colors indicate the types of steps: input (orange), processing step (blue), optional step (red) and output (green). A legend with the colors is also incorporated. In each step, the process is indicated as well as the program used (in blue, red or green boxes), or which kind of input is required (in yellow boxes). Each blue, green and red box is marked with a number in parentheses indicating to which pipeline step it belongs: (1) pre-processing, (2) metagenomic and metatranscriptomic data analysis, (3) proteogenomics database construction, (4) metaproteomics data analysis, (5) final output visualizations based on the meta-omics integration.
Pre-processing
The first step includes various pre-processing mechanisms improving metagenomics and metatranscriptomics read quality, including: (i) FastQC (29) for reviewing the quality of the reads, (ii) PrinSeq (30) for cleaning and for trimming the sequences, (iii) a second quality control with FastQC and Fastq-join (31) for binning the pair-end reads. This binning step is included because our workflow is designed for paired-end reads.
Metagenomic and metatranscriptomic analysis
In the metagenomic and metatranscriptomic analysis step, the pre-processed paired-end sequences are analyzed using pre-configured tools. These tools include (i) a genome mapping against the NCBI non-redundant (nr) database (accessed 5 July 2019) using Kaiju (32), (ii) an assembly using Ray, (iii) and protein prediction using both Prodigal (33) for bacterial proteins and (iv) Augustus (34) for host proteins. The contigs obtained through the genome assembly are used to increase the accuracy of the protein predictions. Bacterial proteins are predicted using Prodigal, a program specifically designed to predict bacterial open reading frames. Host proteins are predicted, with an engine (Augustus, (34)), from the same samples as bacterial proteins, because our pipeline is designed to analyze mixtures of host hindgut cells and bacterial cells. In this experiment, the vivisection process has been performed to ensure the only acquisition of hindgut tissue, essential to properly integrate bacterial data in its context, which is the hindgut of the host. Functional annotation of these predicted proteins is performed using EggNOG (version 1.0 accessed 5 June 2019) (35) to obtain KEGG Orthology (KO) identifiers. An optional step is included that requires the installation of InterProScan (36). This software is not implemented in BioConda but will be automatically installed locally with the snakemake script and allows a TIGRFAM (37) functional annotation. Details regarding the quality of the annotation in metagenomics and metatranscriptomics are available in the Supplementary Table S1.
Proteogenomic database generation
The output of the previous bacterial prediction from the metagenomics and metatranscriptomics data is used to create a proteogenomic database. This database includes bacterial and host proteins from metagenomics, metatranscriptomics or both kinds of data. A database with both kinds of information provides a comprehensive reference for peptide and protein identification (see next paragraph). The proteogenomic database obtained from the validation data has been built with the sequences resulting from the bacterial protein prediction performed with Prodigal. This database (data of creation: 19 November 2019) contains 1 014 200 sequences, of which 850 455 are unique (i.e. occur only once in the database).
Metaproteomic data analysis
For peptide and protein identification, MS-GF+ (38) is used as database search engine, employing the custom proteogenomic database as reference for peptide-to-spectrum matching. Both taxonomic and functional annotations of the peptides are performed with Unipept version 4.0 (39). The output obtained from this step is a taxonomic annotation at three different levels (phylum, family and genus) and the Enzyme Commission (EC) number associated with each peptide. To assess the performance of our tailored database, we compared the peptide identification yield with a very complete human gut microbial protein database: NIH Human Microbiome Project Gastrointestinal database (accessed 25 November 2019) (Supplementary Table S2). With our tailored database we obtained four times more peptides identified than using the NIH Gastrointestinal database. The search parameters are available in the modifications file for msgf plus (mods) and the config file. These results are consistent with previous studies on the use of metagenomic sequences for constructing proteogenomics databases (40).
Meta-omics data integration and visualization
The final step concerns the integration and visualization of all three-level meta-omics data and results. The integration of all three meta-omics data levels is performed in the following stages: (i) parallelized meta-omics analysis, (ii) proteogenomic database construction and (iii) pathway visualization.
First, both metagenomics and metatranscriptomics data are analyzed in parallel, which allows a reliable integration of them. The taxonomic annotation of the microbiome is visualized with KronaPlots (41). These plots show the taxonomic distribution in each sample reads for metagenomics and metatranscriptomics data. To analyze this information further, linear discriminant analysis (LDA) effect size (LEfSe) (42) is used that performs a statistical analysis on the microbiome data. LEfSe identifies features most likely to explain differences between conditions by coupling standard statistical tests with additional tests encoding biological consistency and effect relevance. The statistics performed are Kruskal-Wallis rank-sum test on classes, Wilcoxon rank-sum test among subclasses and LDA score on relevant features. Taking account of the effect size is essential to properly analyze microbiomes. The outcome of the statistical analysis is depicted in a graph with up to two levels of classification, and only the features with an LDA score over 2 are shown. This allows visualizing different conditions and different data within the same graph.
For the functional annotation, the representation of the metabolic pathways is included using Pathview (43), which allows pathway integration. The Pathview plots represent the log2 ratio of the means of the different conditions and data compared (i.e. 10d and 20d, metagenomic, metatranscriptomic and metaproteomic data, see below), after a fold change normalization. These log2 ratios are calculated for the proteins predicted from the contigs assembled from each sample. The database used to identify the peptides in the metaproteomics data is based on the protein prediction from the metagenomics and metatranscriptomics data. This proteogenomics approach creates a sample-specific protein database and therefore optimizes the peptide and protein identification at the metaproteome level, and provides a full integration of three datasets: metagenomics, metatranscriptomics and metaproteomics. The log2 ratio of the means of the peptides identified are then included in the Pathview visualization. When integrating all three datasets (metagenomics, metatranscriptomics and metaproteomics), the log2 ratios are compared between pairs of datasets (transcripts/gene, protein/gene, protein/transcript). Pathview shows these ratios as a color gradient, indicating which dataset is over-represented in the comparison. We can interpret if the transcriptional activity is high (transcripts over-represented among genes), or if the protein production is low (genes over-represented among proteins). This R-based tool shows the differential expression of the enzymes on graphs visualizing the selected metabolic pathways. Pathview itself uses functional pathway information from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (44).
Validation data
Blattella germanica population originated from a stable laboratory population housed by Dr X. Bellés’ group at the Institute of Evolutionary Biology (CSIC-UPF, Barcelona). It was reared in chambers at the Institute for Integrative Systems Biology (University of Valencia) at 25°C, 60% humidity and a photoperiod of 12L:12D. Cockroaches were fed dog-food pellets (Teklad global 21% protein dog diet 2021C, Envigo, Madison, WI, USA) and water ad libitum. Samples were taken at 10 days and 20 days after becoming adults, conditions names 10d and 20d, respectively. Vivisections of CO2-anesthetized females were performed to obtain the hindgut of each individual. DNA and RNA samples were obtained from the same hindgut, with a total of 12 samples (six replicates per condition). Protein samples were obtained from individuals of the same age and population, with a total of eight samples (with four replicates per condition). Hindgut was ground with a sterile plastic pestle. DNA and RNA extraction of each hindgut was performed using Nucleospin RNA XS and Nucleospin DNA/RNA Buffer Set (Macherey-Nagel, France). Protein extraction of each hindgut was performed solubilizing the ground hindgut with lysis buffer (7 M urea, 2 M thiourea, 4% (w/v) CHAPS). Metagenomic sequencing using the Illumina MiSeq (2 × 300 bp) technology was done at the FISABIO (Valencia, Spain). Metaproteomics shotgun sequencing was performed by the Proteomics Unit of the Servei Central de Suport a la Investigació Experimental (SCSIE) at the University of Valencia.
A small subset of human data has been also analyzed in order to show the plasticity of the pipeline. The dataset consisted of two samples of metagenomics and metaproteomics data from the study of Tanca et al. (45). Both samples correspond to faecal samples from healthy Sardinian individuals: a female and a male.
RESULTS
To illustrate the outputs and analysis that can be obtained from this pipeline, we used a complex gut microbiota dataset from the non-model organism B. germanica, which genome has been sequenced (without being fully annotated) (46). This dataset consists of metagenomics, metatranscriptomics and metaproteomics data of two different adult conditions: 10d and 20d.
Comparison of metagenomics and metatranscriptomics/ metaproteomics datasets for one-condition sample (multi-meta-omic approach)
Assessing bacterial composition from metagenomics and metatranscriptomics data
The analysis of microbial community samples often raises the question of which bacteria form a given population. To answer this question, we performed two different types of analysis using gNOMO. First, we processed and analyzed metagenomics data to investigate the taxonomic composition of a given sample. Second, we analyzed and compared samples of two different conditions: 10d and 20d.
For the first analysis, the output was visualized using a Krona plot that is produced for each metagenomics and metatranscriptomics sample automatically within the gNOMO pipeline. For the first-condition (10d) sample, we observed that the main phyla present in this population were Bacteroidetes, Firmicutes and Proteobacteria (Figure 2). After analyzing the taxonomic distribution differences between the 10d and 20d samples, we observed no significant abundance differences in a preliminary analysis (Supplementary Tables S3 and 4). In this analysis, the relative abundance of the main phyla and families was calculated in relation to the mean abundance of the two conditions. We observed that the four most abundant phyla distributions match our previous published studies based on 16S gene sequencing, while others (e.g. Planctomycetes, Deferribacteres and Actinobacteria) do not match exactly previous studies on this topic (10) (Supplementary Table S3). We made similar observations regarding taxonomic abundances at the family level (Supplementary Table S4). In general, this can be explained by the difference concerning the method and annotation between 16S rRNA gene sequencing analysis and metagenomics. 16S rRNA gene sequencing focuses on bacterial data and can be useful in environmental studies due to the lack of fully sequenced bacterial genomes in these kinds of scenarios. In contrast, metagenomics offers higher resolution, enabling a more specific taxonomic classification of sequences as well as the detection of new bacterial genes and genomes (47).
Figure 2.

KronaPlot of the taxonomic annotation of a metagenomics sample (condition 10d). Bacterial taxa distribution of metagenomics data, corresponding to condition 10d. The bacterial taxa are classified by taxonomic hierarchy levels, from higher levels in the center of the chart (Kingdom Bacteria) progressing outward until genus level.
As described previously, our first analysis provided no clearly visible abundance differences between the two conditions, as we were expecting when studying such a stable situation (both are adult individuals differing in 10 days of development). However, we decided to validate this finding by a more sensitive statistical approach. To investigate this issue further, we used LEfSe (42) as a well-established statistical method for comparing the taxonomic distribution at genus level between 10d and 20d conditions. LEfSe has the advantage of recognizing the hierarchy of the taxonomic classification and accurately calculate statistically significant differences (represented as LDA scores) between different conditions.
Using LEfSe, we found, for example, that Fusobacterium (Fusobacteriaceae family), was more abundant at 10 days (LDA score > 3) in both metagenomics and metatranscriptomics data (Figure 3). The role of Fusobacterium on cockroaches’ gut microbiome deserve a detailed study due to these results and some interesting findings about this groups’ role in other organisms: Fusobacterium has been related to disease and stress situations in the human gut microbiota (48), but is has also been related to the infants gut microbiota (49). Conversely, an unidentified genus belonging to the family Ruminococcaceae, has been found more abundant in 20d than 10d condition (LDA score > 3) in metagenomics data (Figure 3A), but no differences between conditions have been found in metatranscriptomics data (Figure 3B). Various genera belonging to the family Ruminococcaceae have been related to a healthy gut microbiota, like Ruminococcus and Faecalibacterium. These have been linked to degradation of starch in the human colon making it available for other bacteria in the gut (50), and degradation of cellulose in herbivorous mammals (51). These differences between 10d and 20d conditions could suggest that, even if the population is very stable along adult stages, it is being rearranged to its final composition. This rearrangement would imply a reduction in Fusobacterium and an increase of Ruminococcaceae along time (10d against 20d, Figure 3A). On the other hand, Pseudomonas genus and an unclassified genus belonging to the family Pelagibacteraceae are more abundant only in metatranscriptomics analysis at 20d against 10d (Figure 3B). Pelagibacteraceae has been described as a bacterial family localized in marine and freshwater environments (52), but has also been detected in the mouse gut microbiome (53) Pseudomonas genus has been related to pathogenicity in animals and plants, and is a commonly detected taxa in the gut of cockroaches (54). These results suggest that these taxa increase their transcriptional activity but not their abundance in the population along time. By the same reason the unidentified genus of Ruminococcaceae reduce its transcriptional activity (is over-represented at metagenomics level but not at metatranscriptomics level in 20d sample). More importantly, for the present work is the integration of this level of comparison that allows detection of particular taxa that differ significantly in their abundance in different conditions.
Figure 3.
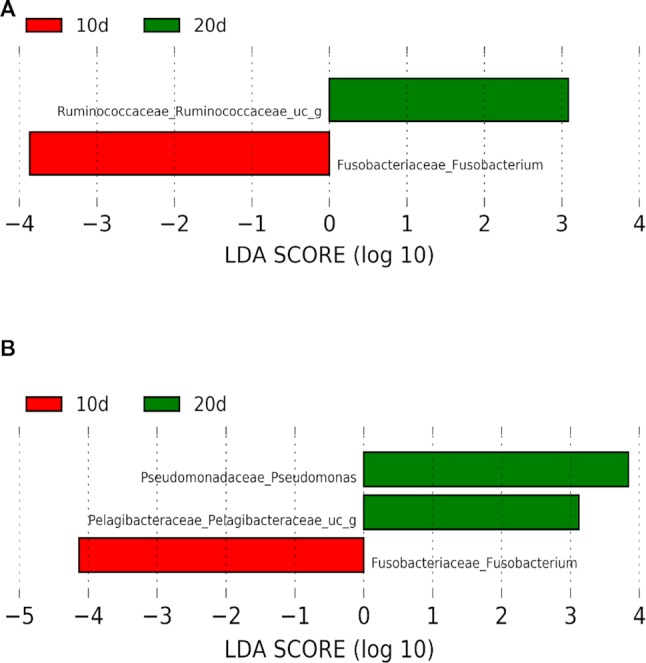
LEfSe graph of taxonomic annotation of metagenomics (top) and metatranscriptomics (bottom) data comparing the two conditions: 10d and 20d. Taxa with significant different distribution among the two conditions are identified. Only taxa with LDA scores over 2 are shown. Positive LDA scores are assigned to the taxa over-represented in the condition 20d (green), and negative LDA scores to the taxa over-represented in the condition 10d (red). Metagenomics data (A) and metatranscriptomics data (B) are represented.
Functional analysis from integrated metagenomics and metatranscriptomics data for one-condition sample
Next steps concern the functional analysis of each microbiome dataset and the qualitative and quantitative differences of assigned functional annotations. To assess the level of transcriptional activity of the population, we compare the metagenomics data (gene pool) and the metatranscriptomics data (transcripts) corresponding to the microbiota of the 10d condition. Integrating metagenomics and metatranscriptomics allows calculating transcript/gene ratios that indicate gene transcriptional activation or repression. For this purpose, we applied LEfSe based on the functional role (or subrole) assignment using TIGRFAM (Figure 4 and Supplementary Table S5). We observed that energy metabolism (both anaerobic and aerobic metabolisms) and protein production are the most active metabolic pathways (Figure 4), which indicates that the bacterial population is active.
Figure 4.

LEfSe graph comparing metagenomics and metatranscriptomics data of TIGRFAM annotation (role and subrole levels) of condition 10d. Taxa with significant different distribution among metagenomics and metatranscriptomics data are identified. Only taxa with LDA scores above 2 are shown. Positive LDA scores are assigned to the functional categories over-represented in the metatranscriptomics data (RNA, green), and negative LDA scores to the functional categories over-represented in metagenomics data (DNA, red).
Alternatively, a pathway analysis enables discovering differences between states by using the Pathview R package. An analysis with Pathview shows which specific metabolic pathways (KEGG pathways) have statistically significant correlations between sample types and/or conditions and thereby complements the information provided by LEfSe. In a Pathview graph, an increase of the gene activity involved in a certain pathway can be observed. Our exemplary analysis using Pathview here focuses on the tricarboxylic acid cycle (TCA cycle) of the gut microbiota, comparing again gene pool (metagenomics data) against transcripts (metatranscriptomics data) (Figure 5). The TCA cycle consists of a series of oxidative reactions to finally obtain energy (adenosine triphosphate) from oxidative degradation of the acetyl group, in the form of acetyl-CoA, to carbon dioxide. The full cycle can be performed by bacteria in aerobic conditions, but some autotrophic bacteria are also able to perform the reverse TCA cycle (rTCA), and even some anaerobic bacteria are able to carry out an incomplete TCA cycle, defining the pan-metabolic capabilities for this pathway of the gut microbiota.
Figure 5.

KEGG Pathview graph of the TCA cycle metabolism route comparing metagenomics versus metatranscriptomics data of the microbiota of 10d and 20d conditions. Some nodes are split between two colors, indicating 10d (left) and 20d (right) conditions. Light blue (−1) depicts genes under-represented in metagenomics (but over-represented in metatranscriptomics), while those marked in pink (1) depicts over-represented genes in metagenomics (but under-represented in metatranscriptomics). In purple, values close to 0 in the ratio metatranscriptomics/metagenomics, indicating no differences in frequency.
We have found that most enzymes that take part in the TCA cycle are over-represented at the transcript level. This confirms our previous observations related to energy metabolism (Figure 4). With both analysis methods and their visualizations, we were able to study different levels of complexity of the pan-metabolism of all bacterial populations. We observed that the microbiome actively produces energy and proteins to grow and maintain a very complex population. Beyond the use case shown above, depending on the particular study, other pathways could be analyzed.
Meta-omics integration: comparing metagenomics, metatranscriptomics and metaproteomics data at the functional pathway level
Each meta-omics level data provides unique information in various ways, but their integration is crucial to gain a complete overview of the metabolic capabilities of the studied bacterial populations. Metaproteomics data incorporation to the integrated analysis of microbiomes is essential to have a realistic overview of the functional capabilities of the bacterial populations. For this purpose, we analyzed these meta-omics data together, as an example, focusing on the N metabolism pathway, corresponding to the N cycle, the set of reactions by which different inorganic N compounds are transformed into ammonia, a biologically reduced form of N that can be mainly introduced into synthesis of amino acids (glutamine and glutamate). We were interested in this pathway due to previous findings related to N metabolism of the host (B. germanica) and the endosymbiont Blattabacterium. As explained previously, Blattabacterium participates in the N recycling from stored urates to ammonia that can be used to synthesize glutamine and glutamate, connecting with the amino acid biosynthesis pathway (6). Here, the aim was to study N metabolism in the host gut microbiome and then to assess if the bacterial population has the metabolic capability to produce a form of usable N.
In this analysis, we investigated how variable or stable the overall N metabolism is at the gene, transcript and protein level along time (10d against 20d) in the investigated pathway (Figure 6). While metagenomics and metatranscriptomics show almost complete coverage of the N metabolism pathways and very variable along time, only a few enzymes were observed in the metaproteomics data and very stable along time. These results suggest that while the gene pool (the population) can be variable, the final transcripts and at least the four detected proteins remain stable, which could point in the direction of a functional redundancy at the protein level, as has been previously described for human gut microbiota (55). However, deeper coverage of the metaproteomics data would be necessary to confirm these findings.
Figure 6.

KEGG Pathview graph of the N metabolism route comparing metagenomics/metatranscriptomics/metaproteomics data of the microbiome at 10d and 20d. Some nodes are split between different colors, indicating metagenomics (left), metatranscriptomics (middle) and metaproteomics (right) data. Light blue (−1) depicts genes/transcripts/proteins over-represented in 10d (but under-represented in 20d), while those marked in pink (1) depicts genes/transcripts/proteins over-represented in 20d (but under-represented in 10d). In purple, values close to 0 in the ratio 10d/20d, indicating no differences in frequency.
Comparison of host and microbiome data
Microbiota metabolism and functions are better understood when studied together with its host. gNOMO includes the analysis of the host data in parallel with its microbiome, so we can integrate and compare the metabolic pathways of host and microbiome. In the case of B. germanica, we have studied the N metabolism pathway that we had analyzed before with the focus on the microbiota data (Figure 6) integrating the host data (Figure 7). We have observed which enzymes can be found in the bacterial population data and which ones can be explained by the host data (Figures 6 and 7).
Figure 7.

KEGG Pathview graph of the N metabolism pathways comparing metagenomics/metatranscriptomics/metaproteomics data of the host between 10d and 20d conditions. Some nodes are split between different colors, indicating metagenomics (left), metatranscriptomics (middle) and metaproteomics (right) data. Light blue (−1) depicts genes/transcripts/proteins over-represented in 10d (but under-represented in 20d), while those marked in pink (1) depicts genes/transcripts/proteins over-represented in 20d (but under-represented in 10d). In pruple, values close to 0 in the ratio 10d/20d, indicating no differences in frequency.
We expected to find a maximum of four enzymes in the host data, as in most eukaryotes only four enzymes of this pathway are present, and we could detect those in the host pathway. While these four enzymes were the only ones detected in the host, its gut microbiome possesses most of the enzymes present in the N metabolism pathway.
If we study these four enzymes present in the host data in detail, it can be observed that all of them are over-represented at 10d against 20d condition in metaproteomics data, and in metagenomics and metatranscriptomics data, they are almost undetectable (Figure 7). When looking at the microbiome metatranscriptomics data, these proteins have a stable abundance over the whole time (Figure 6). These findings could indicate that the production of these proteins in the hindgut of the host is reduced along time, but its production by the microbiome remains stable.
After analyzing the bacterial and the host capabilities together regarding this metabolic pathway, we find that the N metabolism corresponding to the N cycle is mostly performed by the microbiome. These data show the importance of the meta-omics integration, as different levels of cell function are represented, each of them with different implications. DNA (in metagenomics) is more stable and can represent the gene pool of a population, but it can be misunderstood as also dead bacteria and genes which are not active are being represented with this methodology. RNA (in metatranscriptomics) shows the levels of active transcription, essential to understand the activity of a microbiome, which can differ substantially from the gene pool, both in bacterial and eukaryotic cells (Figures 6 and 7). The identified proteins for both microbial and host data, have been decisive to conclude that the N cycle is active in the German cockroaches’ hindgut due to its microbiome (Figure 6). This conclusion is reinforced by the host data, as it has been proven that the host is not actively taking part of the N cycle (Figure 7). The importance of these findings should be analyzed in the future, including other pathways and improving the metaproteomics coverage of the microbiota.
Human microbiome dataset
In order to evaluate the applicability of gNOMO to other microbiome data, we performed an analysis on human microbiome data. In this analysis, we processed metagenomics and metaproteomics data of two healthy Sardinian individuals gut microbiota (45). The results of this exemplary analysis are included as two tables and two figures in the Supplementary File.
The basic statistics of the metagenomics data used are available in Supplementary Table S6. The output of the human dataset analysis includes the average taxonomic distribution of the metagenomics data of these samples in Supplementary Table S7. Our taxonomic identification at levels of phylum and family corresponds with the ranges obtained in the original study.
To exemplify the functional annotation output in the human dataset, we have included two Pathview graphs of the glycolysis/gluconeogenesis KEGG pathway. In Supplementary Figure S1, the two chosen conditions (male/female) are compared in both metagenomics and metaproteomics data. In this figure, the metatranscriptomics data possible spot in blank, which implies that the pipeline works even with the lack of one of the meta-omics data, and in general, the pipeline also works with all three meta-omics levels (as shown in the previous text). It should be noted that these exemplary data cannot be directly compared to the results of the original study, as their authors had not compared the microbiota between sexes. The results indicate an overall similar behavior of both bacterial populations, but with punctual strong divergences between individuals, which is in line with the results from the original study.
Finally, the ratio between metagenomics and metaproteomics data are studied in both conditions. The results show very different abundances between metagenomics and metaproteomics data, which indicates high or low translational activity, depending on the positive or negative value of the ratio (Supplementary Figure S2). These findings also confirm the results obtained from the Sardinian cohort study.
DISCUSSION
The aim of our software design and implementation was to provide a complete pipeline to analyze omics data from a non-model host and its microbiome. Based on these requirements, we developed the gNOMO software that presents an end-to-end workflow covering all the required data analysis steps starting from the processing of raw omics data to the final output visualization of the results. gNOMO performs the analysis of up to three different meta-omics data: metagenomics, metatranscriptomics and metaproteomics, and their integration.
gNOMO is designed for paired-end sequencing of metagenomics and metatranscriptomics data, the pipeline includes a preprocessing and binning step designed for this type of datasets. A tailored proteogenomic database is generated to perform a highly efficient database search for protein identification in the metaproteomics data analysis without a reference microbiome. To obtain this database metagenomics and metatranscriptomics data are assembled into contigs, which are then used to predict the proteins present in the samples. Together with the microbiome data, host data is obtained from the same samples and analyzed de novo in order to be able to analyze microbiota of non-model organisms integrated with the host information. Host databases can also be provided to analyze human or other model organisms data.
The pipeline is developed using the modular Snakemake framework that allows to incorporate software tools and libraries with different requirements. These tools are available at the BioConda channel and their installation is incorporated in the workflow. Snakemake makes use of programming languages Python and Bash, which are commonly used in bioinformatics. Parameters can be specified in the configuration file provided to Snakemake, so it can be adapted to any kind of host or microbiome analyzed. The use of Snakemake makes gNOMO fully automated, efficient and reproducible.
Previously published meta-omics workflows such as IMP (24) incorporate two layers of meta-omics information by integrating metagenomics and metatranscriptomics data. Such workflows focus on the analysis of the microbiome and often consider host information as contaminant reads: thus, instead of providing a host data analysis, the host genome is only used to remove the host information from the microbiome data. To overcome this issue, gNOMO offers the possibility to analyze host data in parallel to microbiome data and both datasets can be studied simultaneously. gNOMO includes the analysis of metaproteomics data and creates a tailored proteogenomic database to achieve better and more efficient protein identification. The incorporation of the metaproteomics data to the study of the microbiome gives another dimension to the analysis of the microbiome because the proteome provides the functional profile and thereby gives insights on the actual interaction between microbial populations and their host.
The visualization output provided by gNOMO pipeline includes krona charts for taxonomic distribution, and KO categories are plotted using Pathview graphs. The functional distribution represented with Pathview permits to investigate two different aspects: first, the completeness of the metabolic pathways by visualizing each enzyme in the route, and second, the differences in abundance of each enzyme by comparing datasets (metagenomics, metatranscriptomics and metaproteomics) or conditions. This integration in gNOMO is highly useful, for example, when information regarding the presence and abundance of specific enzymes is needed. The integration is developed at three different stages: the parallelization of the meta-omics datasets, the integration of the functional annotation in Pathview pathways, and the construction of a proteogenomics database with metagenomics and metatranscriptomics information to identify peptides and proteins from the metaproteomics dataset.
With the study of a small human dataset, we can show the plasticity and adaptation capability of the pipeline to any type of dataset. The results obtained from this study validate the results from the paper the exemplary dataset was obtained from (45), which also proves that gNOMO is a robust and reproducible workflow to work with.
In conclusion, gNOMO is a standardized and reproducible bioinformatic pipeline designed to integrate and analyze metagenomics, metatranscriptomics and metaproteomics microbiota data of non-model organisms. It incorporates preprocessing, binning, assembly steps, taxonomic and functional annotations, and the production of a proteogenomic database to improve the metaproteomics analysis. gNOMO also includes the analysis of both microbiota and host data in parallel, which makes it a useful tool to analyze the microbiome of non-model organisms, as it was demonstrated using experimental data of the German cockroach B. germanica. In general, gNOMO can also be applied to data from human or other model organism sample types. Finally, gNOMO generates output and visualization of multiple meta-omics results in a single automated pipeline.
DATA AVAILABILITY
gNOMO is an open source software available in the GitHub repository: https://gitlab.com/rki_bioinformatics/gnomo and https://gitlab.com/gaspilleura/gnomo.
The validation data have been deposited with Zenodo under the accession number 3569690 (https://doi.org/10.5281/zenodo.3569690), metagenomics and metatranscriptomics data have been deposited with ENA under the accession number PRJEB37860 (http://www.ebi.ac.uk/ena/data/view/PRJEB37860) and metaproteomics data have been submitted to PRIDE under the accession number (PXD018642).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Nuria Jiménez and FISABIO for the help in the processing and sequencing of the metagenomics and metatranscriptomics samples and the Proteomics Service of the SCSIE for the processing and sequencing of the metaproteomics samples.
Notes
Present address: Felix Hartkopf, Section eScience (S.3), Federal Institute for Materials Research and Testing, Berlin 12205, Germany.
Present address: Vior C. Piro, Hasso-Plattner-Institute, Faculty of Digital Engineering, University of Potsdam, Potsdam 14482, Germany.
Present address: Bernhard Y. Renard, Hasso-Plattner-Institute, Faculty of Digital Engineering, University of Potsdam, Potsdam 14482, Germany.
Present address: Thilo Muth, Section eScience (S.3), Federal Institute for Materials Research and Testing, Berlin 12205, Germany.
Contributor Information
Maria Muñoz-Benavent, Institute for Integrative Systems Biology (I2SysBio), Universitat de València/CSIC, Paterna (València) 46980, Spain.
Felix Hartkopf, Bioinformatics Unit (MF 1), Department for Methods Development and Research Infrastructure, Robert Koch Institute, Berlin 13353, Germany.
Tim Van Den Bossche, VIB - UGent Center for Medical Biotechnology, VIB, Ghent 9000, Belgium; Department of Biomolecular Medicine, Faculty of Medicine and Health Sciences, Ghent University, Ghent 9000, Belgium.
Vitor C Piro, Bioinformatics Unit (MF 1), Department for Methods Development and Research Infrastructure, Robert Koch Institute, Berlin 13353, Germany.
Carlos García-Ferris, Institute for Integrative Systems Biology (I2SysBio), Universitat de València/CSIC, Paterna (València) 46980, Spain; Departament de Bioquímica i Biologia Molecular, Universitat de València. Burjassot (València) 46100, Spain.
Amparo Latorre, Institute for Integrative Systems Biology (I2SysBio), Universitat de València/CSIC, Paterna (València) 46980, Spain; Área de Genómica y Salud, Fundación para el Fomento de la Investigación Sanitaria y Biomédica de la Comunidad Valenciana (FISABIO), València 46020, Spain.
Bernhard Y Renard, Bioinformatics Unit (MF 1), Department for Methods Development and Research Infrastructure, Robert Koch Institute, Berlin 13353, Germany.
Thilo Muth, Bioinformatics Unit (MF 1), Department for Methods Development and Research Infrastructure, Robert Koch Institute, Berlin 13353, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
European Regional Development Fund (ERDF), Ministerio de Ciencia, Innovación y Universidades [PGC2018-099344-B-I0 to A.L.]; Generalitat Valenciana [PROMETEO/2018/133 to A.L.], Research Foundation Flanders [1S90918N to T.V.D.B.]; Deutsche Forschungsgemeinschaft [RE 3474/2-2 to B.Y.R.]; Ministerio de Ciencia, Innovación y Universidades, FPU Fellowship [FPU15/01203 to M.M.B.]; Federation of European Biochemical Societies [FEBS Summer Fellowship to TVDB].
Conflict of interest statement. None declared.
REFERENCES
- 1. Gil R., Latorre A.. Unity makes strength: a review on mutualistic symbiosis in representative insect clades. Life. 2019; 9:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moya A., Peretó J., Gil R., Latorre A.. Learning how to live together: genomic insights into prokaryote–animal symbioses. Nat. Rev. Genet. 2008; 9:218–229. [DOI] [PubMed] [Google Scholar]
- 3. Douglas A.E. Lessons from studying insect symbioses. Cell Host Microbe. 2011; 10:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moran N.A., Ochman H., Hammer T.J.. Evolutionary and ecological consequences of gut microbial communities. Annu. Rev. Ecol. Evol. Syst. 2019; 50:451–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heintz-Buschart A., May P., Laczny C.C., Lebrun L.A., Bellora C., Krishna A., Wampach L., Schneider J.G., Hogan A., de Beaufort C. et al.. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat. Microbiol. 2017; 2:16180. [DOI] [PubMed] [Google Scholar]
- 6. Carrasco P., Pérez-Cobas A.E., van de Pol C., Baixeras J., Moya A., Latorre A.. Succession of the gut microbiota in the cockroach Blattella germanica. Int. Microbiol. 2014; 17:99–109. [DOI] [PubMed] [Google Scholar]
- 7. López-Sánchez M.J., Neef A., Peretó J., Patiño-Navarrete R., Pignatelli M., Latorre A., Moya A.. Evolutionary convergence and Nitrogen metabolism in Blattabacterium strain Bge, Primary endosymbiont of the cockroach Blattella germanica. PLoS Genet. 2009; 5:e1000721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Patiño-Navarrete R., Piulachs M.D., Belles X., Moya A., Latorre A., Peretó J.. The cockroach Blattella germanica obtains nitrogen from uric acid through a metabolic pathway shared with its bacterial endosymbiont. Biol. Lett. 2014; 10:20140407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pérez-Cobas A.E., Gosalbes M.J., Friedrichs A., Knecht H., Artacho A., Eismann K., Otto W., Rojo D., Bargiela R., Von Bergen M. et al.. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut. 2013; 62:1591–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rosas T., García-Ferris C., Domínguez-Santos R., Llop P., Latorre A., Moya A.. Rifampicin treatment of Blattella germanica evidences a fecal transmission route of their gut microbiota. FEMS Microbiol. Ecol. 2018; 94:fiy002. [DOI] [PubMed] [Google Scholar]
- 11. Cani P.D. Human gut microbiome: hopes, threats and promises. Gut. 2018; 67:1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mohajeri M.H., Brummer R.J.M., Rastall R.A., Weersma R.K., Harmsen H.J.M., Faas M., Eggersdorfer M.. The role of the microbiome for human health: from basic science to clinical applications. Eur. J. Nutr. 2018; 57:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Piro V.C., Matschkowski M., Renard B.Y.. MetaMeta: integrating metagenome analysis tools to improve taxonomic profiling. Microbiome. 2017; 5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Knight R., Vrbanac A., Taylor B.C., Aksenov A., Callewaert C., Debelius J., Gonzalez A., Kosciolek T., McCall L.-I., McDonald D. et al.. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018; 16:410–422. [DOI] [PubMed] [Google Scholar]
- 15. Martinez X., Pozuelo M., Pascal V., Campos D., Gut I., Gut M., Azpiroz F., Guarner F., Manichanh C.. MetaTrans: an open-source pipeline for metatranscriptomics. Sci. Rep. 2016; 6:25447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muth T., Behne A., Heyer R., Kohrs F., Benndorf D., Hoffmann M., Lehtevä M., Reichl U., Martens L., Rapp E.. The MetaProteomeAnalyzer: a powerful Open-Source software suite for metaproteomics data analysis and interpretation. J. Proteome Res. 2015; 14:1557–1565. [DOI] [PubMed] [Google Scholar]
- 17. Heyer R., Schallert K., Zoun R., Becher B., Saake G., Benndorf D.. Challenges and perspectives of metaproteomic data analysis. J. Biotechnol. 2017; 261:24–36. [DOI] [PubMed] [Google Scholar]
- 18. Wang W.-L., Xu S.-Y., Ren Z.-G., Tao L., Jiang J.-W., Zheng S.-S.. Application of metagenomics in the human gut microbiome. World J. Gastroenterol. 2015; 21:803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shakya M., Lo C.-C., Chain P.S.G.. Advances and challenges in metatranscriptomic analysis. Front. Genet. 2019; 10:904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Manzoni C., Kia D.A., Vandrovcova J., Hardy J., Wood N.W., Lewis P.A., Ferrari R.. Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018; 19:286–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hernández-de-Diego R., Tarazona S., Martínez-Mira C., Balzano-Nogueira L., Furió-Tarí P., Pappas G.J., Conesa A.. PaintOmics 3: a web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018; 46:W503–W509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moya A., Ferrer M.. Functional redundancy-induced stability of gut microbiota subjected to disturbance. Trends Microbiol. 2016; 24:402–413. [DOI] [PubMed] [Google Scholar]
- 23. Franzosa E.A., Morgan X.C., Segata N., Waldron L., Reyes J., Earl A.M., Giannoukos G., Boylan M.R., Ciulla D., Gevers D. et al.. Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E2329–E2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Narayanasamy S., Jarosz Y., Muller E.E.L., Heintz-Buschart A., Herold M., Kaysen A., Laczny C.C., Pinel N., May P., Wilmes P.. IMP: a pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol. 2016; 17:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruggles K. V., Krug K., Wang X., Clauser K.R., Wang J., Payne S.H., Fenyö D., Zhang B., Mani D.R.. Methods, tools and current perspectives in proteogenomics. Mol. Cell. Proteomics. 2017; 16:959–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schiebenhoefer H., Van Den Bossche T., Fuchs S., Renard B.Y., Muth T., Martens L.. Challenges and promise at the interface of metaproteomics and genomics: an overview of recent progress in metaproteogenomic data analysis. Expert Rev. Proteomics. 2019; 16:375–390. [DOI] [PubMed] [Google Scholar]
- 27. Köster J., Rahmann S.. Snakemake-a scalable bioinformatics workflow engine. Bioinformatics. 2012; 28:2520–2522. [DOI] [PubMed] [Google Scholar]
- 28. Grüning B., Dale R., Sjödin A., Chapman B.A., Rowe J., Tomkins-Tinch C.H., Valieris R., Köster J.. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods. 2018; 15:475–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andrews Si. FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformatics. 2010; http://www.bioinformatics.babraham.ac.uk/projects/. [Google Scholar]
- 30. Schmieder R., Edwards R.. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011; 27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aronesty E. Comparison of sequencing utility programs. Open Bioinform. J. 2013; 7:1–8. [Google Scholar]
- 32. Menzel P., Ng K.L., Krogh A.. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016; 7:11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hyatt D., Chen G.-L., LoCascio P.F., Land M.L., Larimer F.W., Hauser L.J.. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stanke M., Morgenstern B.. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005; 33:W465–W467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jensen L.J., Julien P., Kuhn M., von Mering C., Muller J., Doerks T., Bork P.. eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2007; 36:D250–D254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G. et al.. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014; 30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haft D.H., Selengut J.D., White O.. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003; 31:371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim S., Pevzner P.A.. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014; 5:5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gurdeep Singh R., Tanca A., Palomba A., Van der Jeugt F., Verschaffelt P., Uzzau S., Martens L., Dawyndt P., Mesuere B.. Unipept 4.0: Functional analysis of metaproteome data. J. Proteome Res. 2019; 18:606–615. [DOI] [PubMed] [Google Scholar]
- 40. Tanca A., Palomba A., Fraumene C., Pagnozzi D., Manghina V., Deligios M., Muth T., Rapp E., Martens L., Addis M.F. et al.. The impact of sequence database choice on metaproteomic results in gut microbiota studies. Microbiome. 2016; 4: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ondov B.D., Bergman N.H., Phillippy A.M.. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. 2011; 12:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C.. Metagenomic biomarker discovery and explanation. Genome Biol. 2011; 12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luo W., Pant G., Bhavnasi Y.K., Blanchard S.G., Brouwer C.. Pathview Web: user friendly pathway visualization and data integration. Nucleic Acids Res. 2017; 45:W501–W508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tanca A., Abbondio M., Palomba A., Fraumene C., Manghina V., Cucca F., Fiorillo E., Uzzau S.. Potential and active functions in the gut microbiota of a healthy human cohort. Microbiome. 2017; 5:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harrison M.C., Jongepier E., Robertson H.M., Arning N., Bitard-Feildel T., Chao H., Childers C.P., Dinh H., Doddapaneni H., Dugan S. et al.. Hemimetabolous genomes reveal molecular basis of termite eusociality. Nat. Ecol. Evol. 2018; 2:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jovel J., Patterson J., Wang W., Hotte N., O’Keefe S., Mitchel T., Perry T., Kao D., Mason A.L., Madsen K.L. et al.. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 2016; 7:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saito K., Koido S., Odamaki T., Kajihara M., Kato K., Horiuchi S., Adachi S., Arakawa H., Yoshida S., Akasu T. et al.. Metagenomic analyses of the gut microbiota associated with colorectal adenoma. PLoS One. 2019; 14:e0212406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rinninella E., Raoul P., Cintoni M., Franceschi F., Miggiano G., Gasbarrini A., Mele M.. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms. 2019; 7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Flint H.J., Scott K.P., Louis P., Duncan S.H.. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012; 9:577–589. [DOI] [PubMed] [Google Scholar]
- 51. Douglas A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 2009; 23:38–47. [Google Scholar]
- 52. Ortmann A.C., Santos T.T.L.. Spatial and temporal patterns in the Pelagibacteraceae across an estuarine gradient. FEMS Microbiol. Ecol. 2016; 92:fiw133. [DOI] [PubMed] [Google Scholar]
- 53. Dranse H.J., Zheng A., Comeau A.M., Langille M.G.I., Zabel B.A., Sinal C.J.. The impact of chemerin or chemokine-like receptor 1 loss on the mouse gut microbiome. PeerJ. 2018; 6:e5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moges F., Eshetie S., Endris M., Huruy K., Muluye D., Feleke T., G/Silassie F., Ayalew G., Nagappan R.. Cockroaches as a source of high bacterial pathogens with multidrug resistant strains in Gondar Town, Ethiopia. Biomed. Res. Int. 2016; 2016:2825056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lozupone C.A., Stombaugh J.I., Gordon J.I., Jansson J.K., Knight R.. Diversity, stability and resilience of the human gut microbiota. Nature. 2012; 489:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
gNOMO is an open source software available in the GitHub repository: https://gitlab.com/rki_bioinformatics/gnomo and https://gitlab.com/gaspilleura/gnomo.
The validation data have been deposited with Zenodo under the accession number 3569690 (https://doi.org/10.5281/zenodo.3569690), metagenomics and metatranscriptomics data have been deposited with ENA under the accession number PRJEB37860 (http://www.ebi.ac.uk/ena/data/view/PRJEB37860) and metaproteomics data have been submitted to PRIDE under the accession number (PXD018642).