

Abstract
Large-scale metagenomic assemblies have uncovered thousands of new species greatly expanding the known diversity of microbiomes in specific habitats. To investigate the roles of these uncultured species in human health or the environment, researchers need to incorporate their genome assemblies into a reference database for taxonomic classification. However, this procedure is hindered by the lack of a well-curated taxonomic tree for newly discovered species, which is required by current metagenomics tools. Here we report DeepMicrobes, a deep learning-based computational framework for taxonomic classification that allows researchers to bypass this limitation. We show the advantage of DeepMicrobes over state-of-the-art tools in species and genus identification and comparable accuracy in abundance estimation. We trained DeepMicrobes on genomes reconstructed from gut microbiomes and discovered potential novel signatures in inflammatory bowel diseases. DeepMicrobes facilitates effective investigations into the uncharacterized roles of metagenomic species.
INTRODUCTION
Shotgun metagenomic sequencing provides unprecedented insight into the critical functional roles of microorganisms in human health and the environment (1). One of the fundamental analysis steps in metagenomic data interpretation is to assign individual reads to their taxon-of-origin, which is termed taxonomic classification. Many of the large-scale metagenomic assembly efforts have reconstructed thousands of uncultivated novel species from metagenome samples (2–4), which hugely expands the known diversity of microbiomes in specific habitats like the human gut. Developing methods for investigating the role of these novel uncultured organisms in the health state of their hosts remains an important and unsolved challenge in microbiome research.
Incorporating these metagenomic species (MGS) into reference databases for use with current metagenomics tools for taxonomic classification proves difficult and time consuming. Metagenome-assembled genomes (MAGs) are typically highly fragmented compared to genomes obtained using whole genome sequencing from cultures. This fragmentation degrades the effectiveness of traditional alignment tools. Tools using rare or unique short sequences (k-mers) for classification also suffer performance losses with the presence of unknown microbes. Kraken (5), for example, builds a lowest common ancestor (LCA) database to store k-mer information of each organism. Unfortunately, this process relies on a well-curated taxonomic tree retrieved from the taxonomy database maintained by National Center for Biotechnology Information (NCBI). Many of the newly discovered MGS do not have representative taxon nodes in the database.
Machine learning techniques provide a possible solution to bypass the curation of a taxonomic tree. Previous machine learning algorithms for taxonomic classification mainly utilize handcrafted sequence composition features such as oligonucleotide frequency (6,7). These approaches either underperform alignment methods in terms of precision and recall or require prohibitive running times when processing large datasets (8). Deep learning is a class of machine learning algorithms capable of modeling complex dependencies between input data (e.g. genomic fragments) and target output variables (e.g. species-of-origin) in an end-to-end fashion (9). In addition, the fragmentation of reference genomes become a negligible problem since genomes are cut to the length of sequencing reads for training.
Here we describe DeepMicrobes, a deep learning-based computational framework for taxonomic classification of short metagenomics sequencing reads. To illustrate its application in MGS investigation, we trained DeepMicrobes on the previously defined complete bacterial repertoire of the human gut microbiota (2). The repertoire is composed of 2505 species, most of which are identified by metagenome assembly of human gut microbiomes. The general usage outline of DeepMicrobes is presented in Figure 1A. We show that DeepMicrobes surpasses state-of-the-art taxonomic classification tools in genus or species identification and performs at least comparably in abundance estimation on the gut-derived data. We reanalyzed a gut microbiome dataset from the Integrative Human Microbiome Project (iHMP) (10) using DeepMicrobes and discovered potential uncultured species signatures in inflammatory bowel diseases.
Figure 1.

Overview of DeepMicrobes. (A) DeepMicrobes facilitates taxonomic classification for cohorts of interest using newly discovered species in large-scale metagenomic assembly studies. (B) The deep neural network architecture of DeepMicrobes. (C–E) The algorithm details of k-mer embedding, bidirectional LSTM and the self-attention mechanism, respectively. LSTM, long short-term memory.
MATERIALS AND METHODS
Data for model training
We downloaded 2505 representative genomes of human gut species identified previously by a large-scaled assembling study of human gut microbiomes, as well as the taxonomy assigned to them above the species level, from ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/umgs_analyses. This species collection is composed of 1952 unclassified metagenomic species (UMGS) and 553 gut species from the human-specific reference (HR) database, hereafter referred to as HGR (Supplementary Table S1).
We trained separate models for species and genus classification. The genomes were excluded from training the genus model if they were not assigned at the genus level. For each classification category, namely species or genus, we simulated equal proportion of 150 bp reads with the ART Illumina read simulator (11) using HiSeq 2500 error model (HS25), paired-end reads with insert size of 400 and standard deviation of 50 bp. The ART simulator automatically sampled reads from forward and reverse complement genome strands. A pair of reads were treated as two single-end reads during training. Reads of all the categories were shuffled before training. The number of reads to simulate depended on how many training steps were required for models to converge. After simulation, we randomly trimmed the reads from 3′ end to 75–150 bp in equal probability. Each read was given a numerical label according to the species or genus that it was simulated from. Reads along with their labels were converted to the TensorFlow format TFRecord, a binary format that facilitates reading input instances into the learning model.
We created evaluation sets for the species and genus models using the methods described above, except that we ran ART and trimming using a random seed different from the one used to generate the training sets and ran the models using paired-end mode. The evaluation sets were used to search for optimal hyperparameters and decide when to stop training. They were not seen during training to protect against overfitting the models.
Benchmark datasets
Simulation of reads from gut-derived MAGs
We downloaded 3269 high-quality MAGs reconstructed from human gut microbiomes using the ENA study accession ERP108418 (2) (Supplementary Table S2). We used the following criteria to select high-quality MAGs for benchmark: >90% completeness, 0% contamination and 0% strain heterogeneity, which were determined with CheckM (12). The genomes used to generate the training set had been excluded. We used the MAGs assignment method described previously (2) to assign species label to the MAGs using the scripts available at https://github.com/Finn-Lab/MGS-gut. Briefly, the MAGs and training genomes were first converted into a MinHash sketch with default k-mer and sketch sizes respectively (13). The closest relative of each MAG in the training set was then determined based on the lowest Mash distance. Subsequently, each pair of genomes were aligned with MUMmer 3.23 (14) to obtain the fraction of the MAG aligned (aligned query, AQ) and average nucleotide identity (ANI) between them. According to previously established standards for species delineation (15,16), only MAGs with AQ >60% and ANI >95% were labeled as the same species as their closest relatives. The pipeline was also used to compute the similarity between each training genome and its closest training genome in other species categories.
These gut-derived MAGs were used to generate the benchmark datasets used to compare the performance of different model architectures and the performance of the best model on reads derived from different sequencing platforms, respectively. To select the best model, we simulated 10 000 paired-end reads per MAG with ART simulator using HiSeq 2500 error model with an insert size of 400 and standard deviation of 50 bp. We trimmed the 150 bp reads from 3′ end to 75–150 bp as described above. To simulate reads with different lengths and error-profiles depending on sequencing platforms, we simulated five fixed-length datasets comprised 10 000 paired-end reads per MAG using different ART error models (-m 400, -s 50): 75 bp, GenomeAnalyzer II; 100 bp, HiSeq 2000; 125 bp and 150 bp, HiSeq 2500; 250 bp, MiSeq v3.
Generation of mock communities from gastrointestinal bacterial isolates
We downloaded 258 whole genome-sequenced bacterial isolate sequencing data from the Human Gastrointestinal Bacteria Culture Collection (HBC) (17) using ENA accession ERP105624 and ERP012217 (Supplementary Table S3). Any isolates ambiguously assigned at the genus level were excluded. For each of the ten mock communities, we simulated relative abundances for each isolate using a different random seed from a lognormal distribution, as this method is widely used to model microbial abundance distribution. We used the rlnorm function in R for random generation with the mean set to 1 and the standard deviation to 2 (18). We normalized the sum of the random numbers to 1 by dividing each number by their sum and randomly sampled 10 million paired-end reads in total for each mock community. This dataset was used to compare the performance of DeepMicrobes with the other taxonomic classification tools with regard to precision, recall, abundance estimation, classification rate and speed. The ground truth abundance profiles were generated by summing the relative abundances, which is the read count proportion, of the isolates according to their genus or species assignment. The ground truth profiles for genus and species are available in Supplementary Table S4 and 5, respectively.
Simulation of reads from species absent from reference databases
We downloaded the 7903 genomes previously reconstructed from the metagenomes of a wide range of habitats using NCBI BioProject accession PRJNA348753 (19). These genomes were then aligned to the reference databases of different taxonomic classification tools using the pipeline described above to determine their distance (AQ and ANI) to the species included in each database. For CLARK (20) and CLARK-S (21) the genomes automatically downloaded via set_targets.sh were taken as the reference for genome alignment. To avoid disadvantaging Kraken and Kraken 2 (which provide pre-built database indexes), we excluded the genomes released after the update dates of their pre-built databases from the RefSeq complete prokaryotic genome database downloaded on 20 September 2019. For DeepMicrobes the 2505 genomes used to create the training set were taken as the reference for genome alignment. We defined the absence of the species from the reference databases as genome alignments with both AQ <60% and ANI <95% to their closest relatives in the databases. We further retained the genomes whose AQ >10%, yielding a total of 121 genomes whose species were absent from the databases and prone to false positive classifications (Supplementary Table S6). We simulated 1× coverage of paired-end reads with length 150 bp using ART simulator (-ss HS 25, -f 1, -m 400, -s 50) for each of the 121 genomes and randomly trimmed the reads to 75–150 bp.
Performance metrics
Species and genus level performances of DeepMicrobes were benchmarked using the species and genus classification models, respectively.
Read-level precision and recall
We use read-level precision and recall to determine the threshold for the confidence score. For each model architecture we select the threshold making read-level precision of species classification >0.95 measured on the benchmark dataset simulated from gastrointestinal-derived MAGs. For threshold selection, precision and recall are calculated considering the 32 690 000 paired-end reads as a whole dataset. The number of total reads is 10 000 when measuring the metrics for each MAG. The read-level precision and recall of genus classification are computed for each of the mock communities. Formally, read-level precision and recall are defined as follows:
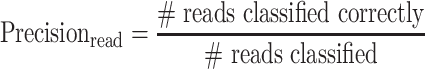 |
 |
Community-level precision and recall
Community-level precision and recall describe whether the presence or absence of taxa (i.e. species or genus) in a microbial community is correctly identified by a taxonomic classifier, where
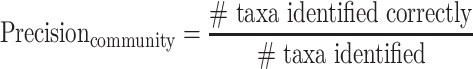 |
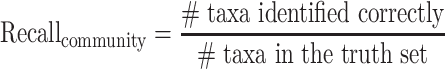 |
To assess the community-level precision and recall given an abundance cutoff, we normalize the read count at the genus level of each taxonomic classifier to sum of 1. We applied two abundance cutoffs (0.01% and 0.0001%) on the profiles and only consider predicted genus above the cutoffs when calculating community-level precision and recall. Community-level precision and recall are not computed at the species level, because a large fraction of the isolates represent unclassified novel species and only a small fraction of the species is shared between our training set and the databases of the other taxonomic classification tools. When comparing the number of identified species for each simulated dataset, we require at least one supporting reads for the identification of a species.
Classification rate
We do not perform filtering by abundance or read count for the classification rate and abundance estimation benchmarks. We define the classification of reads in this paper as confidence score >0.50. In our testing, this confidence threshold results in both the species and genus classification models achieving read-level precision >0.95 on both the gastrointestinal-derived MAGs and the mock communities simulated benchmarks. DeepMicrobes provides the confidence score as an adjustable parameter for users to modify depending on their different requirements.
Tools like Kraken output hierarchical read counts that are the sum of assignments at least made at a specific taxon level. For tools that do not output these values, we generate them by summing up the number of assignments at and below the level. For example, we summed up the number of hits at the level of genus, species, subspecies, and leaf to calculate the number of reads classified by Centrifuge (22) at the genus level.
Abundance estimation
To generate the abundance profiles for each taxonomic classifier, we divided each taxon sum by the total number of reads classified at the species or genus level yielding an abundance vector summing to one. A previous abundance estimation benchmark study adopted the L2 (Euclidean) distance for use with taxonomic classification tools (23). The L2 distance between predicted and ground truth abundance vectors was calculated using the norm function implemented in R package PET (https://CRAN.R-project.org/package = PET). For species quantification based on the 14 species shared by the database/training set of all classifiers, we generated species abundance profiles for each classifier as described above, but only the abundance of these species was considered when calculating L2 distance.
Model architectures
In this section, we provide the technical details of the deep learning algorithms used and give the mathematical description of the network layers and computational modules. This includes descriptions of the sequence encoding schemes and the network models tested. The architecture of DeepMicrobes is described below and a schematic representation is presented in Figure 1B. The technical details of the other tested architectures are available in Supplementary Material.
One-hot encoding and k-mer embedding
We tried two strategies to encode DNA sequences into numeric matrices, namely one-hot encoding and k-mer embedding. For one-hot encoding we convert DNA into 4 × L matrix, where A = [1, 0, 0, 0], C = [0, 1, 0, 0], G = [0, 0, 1, 0] and T = [0, 0, 0, 1]. Specifically, the convolutional model, hybrid convolutional and recurrent model, and seq2species (24) model take as input one-hot encoded DNA, whereas embedding-based models utilize k-mer embedding as the first layer of deep neural networks (DNNs). For k-mer embedding, we split a DNA sequence of length L into a list of substrings of length K with a stride of one, yielding L – K + 1 substrings. The length of K is chosen to reach balance between the model's fitting capacity and computational resources since the vocabulary size grows exponentially in K by 4K (Supplementary Table S7). We use 12-mers unless otherwise stated. Notably, we confirmed that the final best architecture using 12-mers performs much better than the variants using 8-mers to 11-mers (Supplementary Figures S7 and 11).
The k-mer vocabulary is constructed using Jellyfish (25). We only retain canonical k-mers as representatives (-C parameter of Jellyfish), which downsizes the vocabulary. We include a word symbol <unk> in the vocabulary to represent k-mers with Ns. Each k-mer is indexed with a positive integer V according to its lexical order in the vocabulary (V = [1, 2, …, i]). The position of 0 is reserved to denote zero-padding for variable-length sequences, because the TensorFlow input pipeline require that all sequences in a mini-batch should be in the same length. The padding does not affect the performance of the final best model, because its dynamic long short-term memory (LSTM) layer automatically dismisses the padding regions and outputs fixed-length feature maps.
The embedding layer of the DNN utilizes these indexes (for fast look-ups in the implementation) to map each k-mer to a dembed dimensional dense vector (hereafter referred to as k-mer embedding vector). See below for details.
Embedding-based recurrent self-attention model (Embed + LSTM + Attention)
Suppose we have a DNA sequence, which is composed of 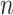 k-mers, represented in a sequence of k-mer embedding vectors (Figure 1C).
k-mers, represented in a sequence of k-mer embedding vectors (Figure 1C).
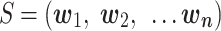 |
where 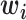 is a vector standing for a dembed dimensional k-mer embedding for the
is a vector standing for a dembed dimensional k-mer embedding for the 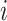 -th k-mer (
-th k-mer (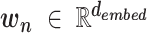 ).
). 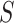 is a 2-D matrix concatenating all the k-mer embedding vectors together.
is a 2-D matrix concatenating all the k-mer embedding vectors together. 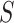 has the shape dembed-by-
has the shape dembed-by-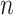 .
.
We use a bidirectional LSTM to process the 2-D embedding matrix 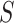 generated with the embedding layer (Figure 1D). Let the hidden unit number of each unidirectional LSTM be dlstm. Formally,
generated with the embedding layer (Figure 1D). Let the hidden unit number of each unidirectional LSTM be dlstm. Formally,
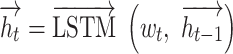 |
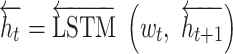 |
We concatenate each 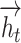 and
and 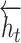 to obtain the hidden state as
to obtain the hidden state as 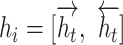 . For simplicity, we denote all the
. For simplicity, we denote all the 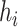 output from the bidirectional LSTM as
output from the bidirectional LSTM as 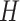 who has a shape of 2dlstm-by-
who has a shape of 2dlstm-by-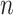 .
.
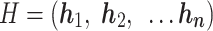 |
We apply a self-attention mechanism (26) between the bidirectional LSTM and fully connected layers (Figure 1E). The self-attention mechanism computes a linear combination of the 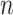 LSTM hidden vectors in
LSTM hidden vectors in 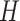 and outputs a vector of attention weights
and outputs a vector of attention weights 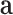 . Formally,
. Formally,
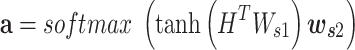 |
where 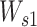 is a weight matrix sized 2dlstm-by-da (da is a hyperparameter that can be set arbitrarily) and
is a weight matrix sized 2dlstm-by-da (da is a hyperparameter that can be set arbitrarily) and 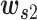 is a weight vector sized da.
is a weight vector sized da.
To allow the model to focus on multiple components in a DNA sequence, we perform 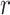 rows of attention (i.e. generate
rows of attention (i.e. generate 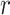 different attention weightings over length of the DNA sequence) and form the multi-head attention matrix
different attention weightings over length of the DNA sequence) and form the multi-head attention matrix 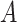 whose shape is
whose shape is 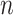 -by-
-by-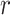 . Thus,
. Thus,
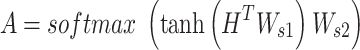 |
where 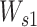 and
and 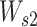 are both weight matrices of the linear transformations and
are both weight matrices of the linear transformations and 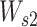 is extended into a da-by-
is extended into a da-by-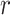 matrix. Here softmax function (see ‘Model Training’ section for details) is performed along the
matrix. Here softmax function (see ‘Model Training’ section for details) is performed along the 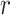 dimension of the input to ensure each column of attention scores sum up to 1. The attention scores indicate the relative importance of each k-mer.
dimension of the input to ensure each column of attention scores sum up to 1. The attention scores indicate the relative importance of each k-mer.
These importance scores are then used to weight the LSTM hidden vectors generated from each k-mer. This allows the model to pay attention to some specific parts of a DNA sequence which might contribute most to classification. To this end, we multiply the LSTM hidden states 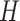 and the attention matrix
and the attention matrix 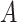 . The resulting matrix
. The resulting matrix 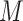 has a shape of 2dlstm-by-
has a shape of 2dlstm-by-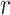 that is irrelevant to the input sequence length
that is irrelevant to the input sequence length 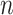 .
.
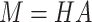 |
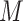 is passed through a three layer fully connected classifier and softmax function that converts output activations to class probabilities (see ‘Model Training’ section below for details of softmax function; a fully connected layer is also known as a dense layer). The fully connected classifier, also termed multilayer perceptron (MLP), consists of three linear transformations with ReLU activations in between. Formally,
is passed through a three layer fully connected classifier and softmax function that converts output activations to class probabilities (see ‘Model Training’ section below for details of softmax function; a fully connected layer is also known as a dense layer). The fully connected classifier, also termed multilayer perceptron (MLP), consists of three linear transformations with ReLU activations in between. Formally,
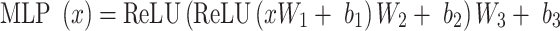 |
where the dimensions of 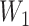 and
and 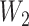 are tunable hyperparameters and the dimension of
are tunable hyperparameters and the dimension of 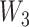 depends on the number of output classification categories.
depends on the number of output classification categories.
Model training
The DNNs were implemented using the TensorFlow framework. We used NVIDIA Tesla P40 24GB GPU to accelerate computation. We trained the models until they converged on the evaluation set. For each architecture of DNN, we performed random search to pick the optimal combination of hyperparameters. In detail, we randomly sampled 30 candidate hyperparameters setting from the search space and picked the models which performed best on the evaluation set. The optimal hyperparameters for each model are listed in Supplementary Table S8.
For the final best architecture, namely the embedding-based recurrent self-attention model, we used a batch size of 2048 and initialized training using a learning rate of 0.001 with a decay rate of 0.05. We did not use regularization methods like dropout or L2 normalization.
We used Adam as the optimizer and minimized the objective function, which is the cross-entropy loss computed between softmax activated prediction output and one-hot encoded ground truth label. The softmax function takes as input a C-dimensional vector x and outputs a C-dimensional vector y of values between 0 and 1. More formally, the softmax function computes
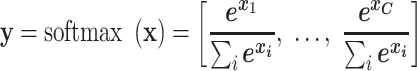 |
where C is the number of classification categories (i.e., species or genus). The denominator 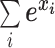 makes sure that
makes sure that 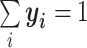 . Thus, y can be seen as the probability distribution of prediction over all the categories. The cross-entropy loss objective is defined as
. Thus, y can be seen as the probability distribution of prediction over all the categories. The cross-entropy loss objective is defined as
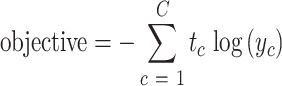 |
where 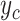 is the probability that the input DNA sequence is of taxon c and
is the probability that the input DNA sequence is of taxon c and 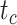 is the binary value (0 or 1) indicates whether taxon c is the correct assignment.
is the binary value (0 or 1) indicates whether taxon c is the correct assignment.
Software versions and databases of other taxonomic classifiers
We compared the performance of DeepMicrobes with Kraken, Kraken 2, Centrifuge, CLARK, CLARK-S, Kaiju (27), DIAMOND-MEGAN and BLAST-MEGAN (28). These tools were run with default options. The tools were run in paired-end mode, except for DIAMOND-MEGAN and BLAST-MEGAN. For paired-end data we averaged the softmax probability distributions generated by DeepMicrobes for two ends of reads. We ran Kraken (v1.0) using the pre-built MiniKraken 8GB database included complete bacterial, archaeal and viral genomes in RefSeq (as of 18 October 2017). We ran Kraken 2 (v2.0.6) using pre-built MiniKraken2 v1 8GB database including RefSeq bacteria, archaea and viral libraries (available on 23 April 2019). Centrifuge (v1.0.3) was run using pre-built NCBI nucleotide non-redundant sequence database (updated on 3 March 2018). The bacteria (and archaea) database for CLARK and CLARK-S (v1.2.5) was downloaded via the set_targets.sh script (on 25 August 2018). Kaiju (v1.5.0) was run using pre-built microbial subset of the NCBI nr database (as of 16 May 2017). To run DIAMOND-MEGAN, we queried unpaired reads using DIAMOND (v0.9.22.123) against nr database downloaded from NCBI (on 27 August 2018). To run BLAST-MEGAN, we queried unpaired reads using BLAST executable (v2.6.0+) against nt index downloaded from NCBI (on 25 August 2018) with Megablast mode and an E-value of 1e-20. After database query, we ran MEGAN (v5.3.11) on the tabular files generated with DIAMOND and BLAST to summarize the LCA taxon for each read.
We created a custom database for Kaiju with the 2505 genomes of human gut species used to train DeepMicrobes. Protein sequences were predicted with Prodigal (29) (v2.6.3) using the default single mode as previously described (2). We assigned a different pseudo species-level taxonomic identifier to each species. These taxonomic identifiers did not duplicate any existing NCBI taxonomic identifiers. The parent nodes of each species were retrieved from taxonomy_hgr.tab and taxonomy_umgs.tab available at ftp://ftp.ebi.ac.uk/pub/databases/metagenomics/umgs_analyses. The custom Kaiju index was created using kaiju-mkbwt and kaiju-mkfmi.
Computational environment
DeepMicrobes and other taxonomic classifiers were benchmarked on a compute node having 256 Gb of memory and two Intel E5–2650 v4 processors, each of which with 12 cores (24 threads). DeepMicrobes was further accelerated utilizing a NVIDIA Tesla P40 24GB GPU during both training and testing. Specifically, CPUs extract and transform the training and testing data and then feed it to a model running on a GPU. We parallelized the data preparation across 8 CPU cores using the num_parallel_calls argument of the TensorFlow input pipeline. The run-time of DeepMicrobes includes the time used for TFRecord conversion. The computational time benchmark for other classifiers was measured by running a single instance of each classifier provided all 48 threads and memory. We also tried running other classifiers provided 8 threads and all memory and compared the time with 48 threads.
Uncultured species signatures of inflammatory bowel diseases
We downloaded the gut metagenome samples from 106 subjects with or without inflammatory bowel diseases (Crohn's disease or ulcerative colitis) using SRA BioProject accession PRJNA398089 (10), which is part of the Integrative Human Microbiome Project (iHMP). We randomly chose one sample as representative if multiple samples for a subject were available. The dataset is composed of 26 healthy subjects, 50 subjects with Crohn's disease and 30 subjects with ulcerative colitis. The samples were quality controlled using Trimmomatic (v0.36) (30) with minimum read length 75 bp. Host reads were further removed using KneadData (v0.6.1). We then analyzed the samples using the species model of DeepMicrobes with confidence score 0.50 and generated species abundance profiles using the method described above. We used LEfSe (31) to determine the species most likely to explain differences between the three subject groups. Briefly, we used the non-parametric factorial Kruskal–Wallis sum-rank test to detect species with significant abundance (P < 0.05) with respect to each group. The resulting subset of species was used to build a linear discriminant analysis (LDA) model to estimate the effect size of each differentially abundant species. The species whose LDA effect size >2.0 were retained and ranked according to the effect size.
RESULTS
A deep learning architecture for taxonomic classification
Deep learning has been applied for the classification of 16S rRNA reads (24) and representation learning from metagenomic reads longer than 1 kb (32). However, taxonomic classification of short shotgun sequencing reads is more challenging. The model should learn genome-wide patterns during training, whereas only information from a short genomic fragment is available during application.
To determine what kind of deep neural network (DNN) is suitable for modeling the taxonomic signatures of shotgun metagenomic sequencing reads, we presented a systematic exploration of DNN architectures with different combinations of network architectural building blocks, DNA encoding schemes and other hyperparameters. To train the models for species classification, we simulated equal proportion of variable-length reads between 75 and 150 bp for each of the 2505 gut species (2) (‘Materials and Methods’ section). To test the models, we simulated variable-length reads from a held-out set of 3269 MAGs reconstructed from human gut microbiomes (‘Materials and Methods’ section), which represent phylogenetic diversity within the gut ecosystem spanning multiple populations. The distribution of read-level precision and recall across these MAGs is used as the metric for model selection. The confidence threshold to decide whether reads are classified or not is determined by benchmarking a gradient of confidence score with a stride of 0.05 on the whole test set (i.e. reads simulated from all the MAGs are considered as a single set). The minimum confidence scores ensuring >0.95 read-level precision are chosen for each architecture (Figure 2; Supplementary Figures S6 and 7).
Figure 2.

Performance of different DNN methods. (A) The read-level precision and recall of different models on simulated reads from the gut-derived MAGs. Each point represents the metric measured on a MAG. (B) The read-level precision and recall of the three of the best models across a series of confidence score. The minimum confidence threshold where precision >0.95 is selected for each model.
We first tried three DNNs that take as input one-hot encoded DNA matrices, including a ResNet-like convolutional neural network (CNN), a hybrid DNN of CNN and bidirectional long short-term memory (LSTM), and the seq2species model proposed for short 16S rRNA read classification (‘Materials and Methods’ section). The ResNet-like CNN (Supplementary Figures S1) and the hybrid DNN (Supplementary Figures S2) are representative of architectures that achieved state-of-the-art performance in predicting the impact of mutations (33) and transcription factor binding (34), respectively. However, the accuracy and overall prediction confidence of the three DNNs are low, with seq2species performs best relatively, followed by the hybrid DNN (Figure 2A; Supplementary Tables S9 and 10). This implies that taxonomic classification for short metagenomic reads requires a distinct deep learning scheme.
One likely reason for the low performance of the DNNs above may be one-hot encoding. Apart from being information-sparse, such encoding scheme represents complementary strands of a DNA sequence as two unrelated matrices. To overcome these limitations, we make an analogy between k-mers and words and used k-mer embedding to represent DNA sequences (Figure 1C), which is a common practice in natural language processing (NLP). Reverse complement k-mers are treated as the same word. To assess the contribution of this encoding scheme to model performance, we trained a baseline model whose only trainable parameters are the weights in the embedding layer (Supplementary Figure S3). In addition, we trained two variants of the baseline model by applying CNN or bidirectional LSTM after the embedding layer, respectively (Materials and Methods; Supplementary Figures S4 and S5). We found that the baseline model outperforms previous DNNs that use one-hot encoding, which indicates that the k-mer embedding layer is capable of embedding taxonomic attributes in each k-mer vector. Interestingly, the CNN variant performs worse than the baseline, though it contains more trainable parameters. In contrast, the LSTM variant further improves the baseline (Figure 2A; Supplementary Tables S9 and 10).
We also proposed a third variant (Figure 1B), where a self-attention mechanism (26) is applied to the hidden states generated by the LSTM variant (Materials and Methods). Self-attention enables the model to focus on specific regions of an input DNA sequence and generate sequence-level representation. The self-attention variant achieves higher read-level precision (mean = 0.942) and recall (mean = 0.428) than the second-best LSTM variant (read-level precision mean = 0.893, read-level recall mean = 0.155). Although we chose different confidence thresholds for different models, we observed that both the read-level precision and recall of the self-attention variant are better than the LSTM variant and the baseline model across a series of confidence scores (Figure 2B), surpassing other DNNs utilizing convolution (Supplementary Figure S6). Therefore, this self-attention augmented embedding-based recurrent model is selected for DeepMicrobes.
The diversity of the MAGs used to create the test set further provides us with the opportunity to explore what factors affect the performance of DeepMicrobes (Supplementary Table S2). As expected, the degree of similarity between tested MAGs and the representative genomes in training set is the major factor that affects the read-level recall (Supplementary Figure S8). In addition, species with relatively small genomes tend to be high in recall, as their genomic features could be easier to grasp than species with large genomes, given a theoretical upper bound on the model's total capacity. What affects the read-level precision most is the similarity between different categories (i.e. species). In general, DeepMicrobes achieves near-perfect precision when the aligned proportion <50% between a pair of most similar categories (Supplementary Figure S9). We did not observe a clear relationship between performance and specific taxonomic groups (Supplementary Figure S10).
The test set we mentioned above is comprised of variable-length reads. To investigate the impact of sequencing platform on performance, we simulated five additional test sets, each of which represents read length and error profile of a specific next-generation sequencing platform (‘Materials and Methods’ section). Generally, the read-level precision of DeepMicrobes is high for reads ≥ 100 bp and from the commonly-used HiSeq and MiSeq platforms (Supplementary Figure S11 and Tables S11-12). The results also show that both the read-level precision and recall are higher for longer reads, even when the read length is not seen during training. This implies that DeepMicrobes generalizes well and performs even better on MiSeq reads with length, for example, 300 or 400 bp.
Comparison of DeepMicrobes with other taxonomic classification tools
We next evaluate whether the DeepMicrobes, which is trained on a bacterial repertoire of the human gut microbiota, has an advantage over state-of-the-art metagenomics tools for taxonomic classification of gut metagenome sequences. Although it is the most ideal choice to benchmark on genuine metagenomic reads, such data would not provide us with read-level and community-level ground truth for taxon identification and abundance estimation. One common alternative is to create mock communities by combining real reads obtained from whole genome sequencing for microbial isolates (5,22). Thus, we created 10 such microbial communities by random sampling reads from the isolates cultured from human fecal samples (Supplementary Table S3), many of which represent candidate novel species yet to be named (‘Materials and Methods’ section).
The lack of overlap in reference databases of different tools at the species level lead us to focus our comparisons on genus-level performance. We classified each mock sample using DeepMicrobes and other taxonomic classification tools, including Kraken, Kraken 2, Centrifuge, CLARK, CLARK-S, Kaiju, DIAMOND-MEGAN and BLAST-MEGAN. The confidence threshold for the genus model is determined according to read-level classification accuracy measured on these real reads (Supplementary Table S13). We observed that the genus model achieves a read-level precision of 0.969 and a recall of 0.866 on average using threshold 0.50, which is the default setting for DeepMicrobes.
We benchmarked the performance of genus identification using two abundance cutoffs, 0.01% and 0.0001%, representing two analysis scenarios. The first scenario is useful for detecting high-abundance taxa (e.g. studies in metabolic disorders), and the second favors high sensitivity for detecting low-abundance taxa (e.g. pathogen detection). In general, a low abundance cutoff increases the community-level recall at the cost of precision. We found that only DeepMicrobes succeeded in identifying all the genera using abundance cutoff 0.01% (Figure 3A). The genomes of microorganisms living in a specific habitat might be divergent from their representatives in standard databases like RefSeq and NCBI non-redundant databases. This may partially explain the poor performance of other tools. Although Kaiju, DIAMOND-MEGAN and BLAST-MEGAN identified all the genera using cutoff 0.0001%, their community-level precision decreased dramatically (Figure 3A). DeepMicrobes also surpasses the other tools in community-level precision under the low-abundance cutoff and ranks second after BLAST-MEGAN under the high cutoff. In addition, the classification speed of DeepMicrobes is acceptable (Supplementary Figure S12).
Figure 3.

Benchmark results of DeepMicrobes and other taxonomic classification tools on the ten mock communities. (A) Genus-level precision and recall measured at the community level using abundance cutoff 0.01% and 0.0001%. Each point represents a mock community. A random jitter of 0.005 is added on the recall to reduce overplotting. (B) Distance between the genus abundance profile for each tool compared with the true composition. (C) Genus-level classification rate for each tool. (D) Distance between the species abundance profile for each tool compared with the true composition. These results consider the 14 species included in the reference databases of all the tools in abundance estimation. (E) Species-level classification rate for each tool. The error bars represent standard error.
Next, we compared DeepMicrobes with other taxonomic classification tools with respect to the accuracy of abundance estimation. Our results show that DeepMicrobes outperforms other tools in genus quantification (Figure 3B and Supplementary Figure S13). Moreover, the genus model of DeepMicrobes classified on average 89.40% of the reads in the mock communities, much higher than the second most sensitive tools, Kaiju, which classified on average 68.27% of the reads (Figure 3C). In addition, we sought to compare the performance of species quantification using the 14 species shared by all the reference datasets, though they only represent a limited fraction of the whole communities. We found that DeepMicrobes is at least comparable to other tools in species-level abundance estimation (Figure 3D). The proportion of reads classified by the species model of DeepMicrobes is slightly lower than Kraken and CLARK (Figure 3E). However, the proportion of false positive classifications might vary among different tools.
To separate the source of performance increase with the habitat specific database that contains MAGs and the deep learning algorithm, we compared DeepMicrobes with Kaiju, which is one of the best competitor tools especially in genus-level quantification and classification rate, using the same reference database. We created a custom Kaiju database composed of the genomes used to train DeepMicrobes (‘Materials and Methods’ section). We observed that the custom Kaiju classified on average 83.25% of the reads at the genus level and correctly recalled more genera than the original Kaiju that uses the microbial subset of the NCBI nr database (Supplementary Figure S14). This demonstrates that both the deep learning algorithm and the MAG-containing database contribute to the improvement in sensitivity. Surprisingly, the performance of genus quantification of the custom Kaiju is worse than the original Kaiju, which implies that the improvement in abundance estimation should be mostly attributed to the deep learning algorithm. In addition, the community-level precision of Kaiju greatly improved using the gut specific reference database. Notably, the community-level precision of custom Kaiju is comparable to DeepMicrobes using abundance cutoff 0.0001%. This suggests that the habitat specific database is the major contributor to precise taxon identification from the metagenomes of the corresponding habitat.
The absence of a species from the reference database could be a major source of false positives. We assessed how such species affect the community-level precision of DeepMicrobes and other tools in terms of the proportion of misclassified reads and the number of misidentified species from that species. Here we benchmarked with the four tools (Kraken, Kraken 2, CLARK and CLARK-S) that performed best apart from DeepMicrobes by taking taxa identification, abundance estimation, and also classification speed into consideration. We used 121 genomes (19), whose species is absence from all the databases, spanning different degrees of relationship to the closest genome in the databases. We simulated 1× coverage variable-length reads for each genome (‘Materials and Methods’ section). Generally, other tools misclassify more reads and misidentify more species as the relationship gets closer, except that less misidentifications are produced when ANI is higher due to more concentrated distribution of misclassified reads (Figure 4 and Supplementary Table S6). In contrast, the species model of DeepMicrobes produces far fewer false positives than other tools regardless of different degrees of similarity that were tested. This indicates that DeepMicrobes is higher in species-level precision than other tools, especially when the microbial communities harbor many unknown species.
Figure 4.

Species-level false positive classification and identification measured on the species absent from the databases. Each point represents the proportion of misclassified reads at the species level or the number of misidentified species measured for a simulated dataset of one species-absent genome. The ANI between each genome and its closest genome in database and the proportion of each genome aligned to its closest genome in database (i.e. the proportion of genome used for ANI calculation) are calculated with MUMmer.
Taken together, DeepMicrobes outperforms state-of-the-art taxonomic classification tools in genus and species identification and achieves better or at least comparable accuracy in abundance estimation. Therefore, DeepMicrobes is ready to serve as a relatively reliable tool to help us explore the important but yet to be discovered roles of novel MGS, which complements results generated with other taxonomic classification tools using standard and universal databases.
Discovery of uncultured species related to inflammatory bowel diseases
We used the species model of DeepMicrobes to classify reads from 106 gut metagenomes sequenced as part of the iHMP (10) (‘Materials and Methods’ section). The fecal samples were collected from healthy subjects and patients with inflammatory bowel diseases (IBD) including Crohn's disease (CD) and ulcerative colitis (UC). The previous study used MetaPhlAn2 for taxonomic analysis and identified a series of species that are differentially abundant in CD or UC (10). However, the species included in MetaPhlAn2 database are mainly well-defined ones (35). To determine whether the uncultured species, which are newly discovered by genome reconstruction from gut microbiomes, possess unexplored associations with IBD, we analyzed the species abundance profiles generated by DeepMicrobes with LEfSe (‘Materials and Methods’ section). The result show that most of the identified candidate biomarkers are unclassified MGS (UMGS) defined previously (2) (Figure 5A and Supplementary Table S14).
Figure 5.

Potential uncultured species biomarkers of IBD identified with LEfSe. The group names (Crohn's disease, Ulcerative colitis and Non-IBD) for each species are assigned by LEfSe. The P-values (Kruskal–Wallis test) calculated by LEfSe are shown. The assigned taxa for each species in the highest resolution are indicated in brackets. (A) The LDA scores for 10 of the most differentially abundant species. The species are ranked using LDA scores. (B) Relative abundance distribution for UMGS368, as a ratio of the median relative abundance in non-IBD individuals. (C) Relative abundance distributions for UMGS37 and UMGS41, as a ratio of the median relative abundance in non-IBD individuals.
We observed that some of the identified species are new members of the taxa whose correlation with CD or UC has been reported. For example, the previous study found that Alistipes species, such as Alistipes shahii, Alistipes finegoldii and Alistipes putredinis are depleted in IBD (10). Here we identified another Alistipes species, UMGS368, whose abundance also decreases in CD and UC (Figure 5B). In addition, we identified some uncultured species whose genera have not been found to increase in UC, such as UMGS37 and UMGS41, which are Lachnospira and Anaeromassilibacillus species, respectively (Figure 5C). These results complement previous findings and might potentially provide new insight into the diagnosis and treatment of IBD.
DISCUSSION
Metagenomic assembly efforts so far have greatly expanded the known diversity of uncultured microorganisms living in specific habitats. For example, the human microbiome harbors a large fraction of species without any representatives in standard reference databases which mainly include genomes obtained using culture-dependent approaches. These species might encode a number of newly identified protein families which possess distinctive metabolic functional capacities (2). Furthermore, the pan-genomes of species in specific environments might diverge from their representatives in universal databases (4).
In this study, we present DeepMicrobes, a deep learning-based computational framework that aims to facilitate effective utilization of the new taxonomic knowledge acquired in large-scale metagenomic assemblies into tools for microbiome research. We can train a model to classify metagenomic reads at any taxonomic rank provided with any collection of training genomes representing different categories. Specifically, we are allowed to bypass the laborious and time-consuming curation of a taxonomic tree, which is required by other taxonomic classification tools like Kraken for database creation.
One limitation of our current framework is that adding new species requires retraining the entire deep neural network. Future efforts to address this issue may include incremental learning. The goal of incremental learning is to retain the knowledge acquired from the old classes and meanwhile learn the new classes (36). This could allow continuous learning as new classes (e.g. new species) of data arrive (37).
Accurate taxonomic classification of short shotgun metagenomic reads requires a distinct DNA encoding approach and DNN architecture. We found that k-mer embedding significantly boosts model performance. Interestingly, k-mer embedding has recently been showed to surpass one-hot encoding in predicting transcription factor binding (38). This suggests the general applicability of k-mer embedding in other biological fields. Notably, the k-mer length we used in this study is optimized for typical data volume of thousands of genomes generated in large-scale metagenomic assembly projects. We suggest that researches who may want to use k-mer embedding in other scenarios should try different k-mer lengths (e.g. 6–12 bp) to finally find a balance between underfitting and overfitting, especially when training on only a few categories.
Our finding that LSTM surpasses CNN highlights the importance of order and context of oligonucleotides in taxonomic classification. Given the evidence from image classification, CNNs might not take into account the spatial ordering of local motifs (39). This can have little impact on tasks where only the occurrence of a few nucleotides is the key to classification (e.g. transcription factor binding site detection). However, it is more complex to model taxonomic signatures, such as single-nucleotide variants and insertions and deletions especially for short microbial sequencing reads. In contrast, LSTM understands a k-mer better with the help of knowledge from the previous and next k-mer. Hence, ordering and contextual information are retained and passed to the next layer.
To our knowledge, DeepMicrobes is the first deep learning architecture that incorporates self-attention mechanisms for DNA sequence analysis. The better performance of DeepMicrobes than the other embedding-based models implies that the model should focus on some specific parts of a DNA sequence rather than treat the whole sequence equally. In addition to boosting performance, attention scores potentially provide simple and straightforward method for identifying the regions of the DNA sequences that contribute most to prediction making the algorithm more interpretable than black-box approaches. Other attention architectures, such as hierarchical attention networks (40) and the Transformer (41), take up too much memory to be feasible in our task. Nonetheless, their applications on genomic sequences are promising for investigation. Another bonus of the self-attention mechanism is that it enables the model to encode variable-length DNA sequences into a fixed-size representation. As a result, DeepMicrobes can be directly applied to longer DNA sequences such as those generated using long-read sequencing platforms without any modification in the model architecture. However, here we focus on next-generation sequencing reads and retraining might be required to adapt to reads whose lengths and error profiles are strongly different.
We trained DeepMicrobes on the complete bacterial repertoire of human gut microbiota defined previously. The benchmark results on real sequencing reads show that DeepMicrobes outperforms state-of-the-art taxonomic classification tools in species and genus identification. Specifically, the algorithm of DeepMicrobes produces far fewer false positives than other tools. As for abundance estimation, DeepMicrobes surpasses other tools in genus quantification and performs comparably to them in species quantification.
We reanalyzed the IBD gut metagenome dataset and discovered potential signatures related to CD, UC or healthy state within these uncultured species, some of which corroborate previous findings at the genus level while others constitute novel findings. This suggests that the uncultured members in gut microbiome might have underappreciated roles in human health and disease. We believe that DeepMicrobes, together with other taxonomic classification tools, will provide a comprehensive picture of microbiome structure and pave the way for the discovery of the functional roles of uncharacterized MGS.
DATA AVAILABILITY
The DeepMicrobes program, trained model parameters, hyperparameters and the implementation of the other DNN architectures are provided at GitHub (https://github.com/MicrobeLab/DeepMicrobes). The sequences of benchmark datasets, the abundance profiles of different taxonomic classification tools on the ten mock communities, the species profiles of the IBD dataset generated with DeepMicrobes, the predicted protein sequences and the custom database for Kaiju are available at GitHub (https://github.com/MicrobeLab/DeepMicrobes-data). The command lines used to run the taxonomic classification tools and the R scripts used to generate figures for benchmarking are available at https://github.com/MicrobeLab/DeepMicrobes-data/tree/master/scripts.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all members of the Wei Laboratory for their support and discussion.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
National Basic Research Program of China [2015CB964601]; National Natural Science Foundation of China [81570828]. Funding for open access charge: National Basic Research Program of China [2015CB964601].
Conflict of interest statement. None declared.
REFERENCES
- 1. Quince C., Walker A.W., Simpson J.T., Loman N.J., Segata N.. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017; 35:833–844. [DOI] [PubMed] [Google Scholar]
- 2. Almeida A., Mitchell A.L., Boland M., Forster S.C., Gloor G.B., Tarkowska A., Lawley T.D., Finn R.D.. A new genomic blueprint of the human gut microbiota. Nature. 2019; 568:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stewart R.D., Auffret M.D., Warr A., Walker A.W., Roehe R., Watson M.. Compendium of 4, 941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 2019; 37:953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pasolli E., Asnicar F., Manara S., Zolfo M., Karcher N., Armanini F., Beghini F., Manghi P., Tett A., Ghensi P. et al.. Extensive unexplored human microbiome diversity revealed by over 150, 000 genomes from metagenomes spanning age, Geography, and Lifestyle. Cell. 2019; 176:649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wood D.E., Salzberg S.L.. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014; 15:R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosen G.L., Reichenberger E.R., Rosenfeld A.M.. NBC: the naïve Bayes classification tool webserver for taxonomic classification of metagenomic reads. Bioinformatics. 2011; 27:127–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vervier K., Mahé P., Tournoud M., Veyrieras J.B., Vert J.P.. Large-scale machine learning for metagenomics sequence classification. Bioinformatics. 2016; 32:1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McIntyre A.B.R., Ounit R., Afshinnekoo E., Prill R.J., Hénaff E., Alexander N., Minot S.S., Danko D., Foox J., Ahsanuddin S. et al.. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol. 2017; 18:182–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eraslan G., Avsec Ž., Gagneur J., Theis F.J.. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 2019; 20:389–403. [DOI] [PubMed] [Google Scholar]
- 10. Lloyd-Price J., Arze C., Ananthakrishnan A.N., Schirmer M., Avila-Pacheco J., Poon T.W., Andrews E., Ajami N.J., Bonham K.S., Brislawn C.J. et al.. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019; 569:655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang W., Li L., Myers J.R., Marth G.T.. ART: a next-generation sequencing read simulator. Bioinformatics. 2012; 28:593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W.. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015; 25:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ondov B.D., Treangen T.J., Melsted P., Mallonee A.B., Bergman N.H., Koren S., Phillippy A.M.. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016; 17:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kurtz S., Phillippy A., Delcher A.L., Smoot M., Shumway M., Antonescu C., Salzberg S.L.. Versatile and open software for comparing large genomes. Genome Biol. 2004; 5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Varghese N.J., Mukherjee S., Ivanova N., Konstantinidis K.T., Mavrommatis K., Kyrpides N.C., Pati A.. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015; 43:6761–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jain C., Rodriguez-R L.M., Phillippy A.M., Konstantinidis K.T., Aluru S.. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018; 9:5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forster S.C., Kumar N., Anonye B.O., Almeida A., Viciani E., Stares M.D., Dunn M., Mkandawire T.T., Zhu A., Shao Y. et al.. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol. 2019; 37:186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fritz A., Hofmann P., Majda S., Dahms E., Dröge J., Fiedler J., Lesker T.R., Belmann P., Demaere M.Z., Darling A.E. et al.. CAMISIM: Simulating metagenomes and microbial communities. Microbiome. 2019; 7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parks D.H., Rinke C., Chuvochina M., Chaumeil P.A., Woodcroft B.J., Evans P.N., Hugenholtz P., Tyson G.W.. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017; 2:1533–1542. [DOI] [PubMed] [Google Scholar]
- 20. Ounit R., Wanamaker S., Close T.J., Lonardi S.. CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics. 2015; 16:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ounit R., Lonardi S.. Higher classification sensitivity of short metagenomic reads with CLARK-S. Bioinformatics. 2016; 32:3823–3825. [DOI] [PubMed] [Google Scholar]
- 22. Kim D., Song L., Breitwieser F.P., Salzberg S.L.. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016; 26:1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ye S.H., Siddle K.J., Park D.J., Sabeti P.C.. Benchmarking metagenomics tools for taxonomic classification. Cell. 2019; 178:779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Busia A., Dahl G.E., Fannjiang C., Alexander D.H., Dorfman E., Poplin R., McLean C.Y., Chang P.-C., DePristo M.. A deep learning approach to pattern recognition for short DNA sequences. 2019; bioRxiv doi:10 August 2019, preprint: not peer reviewed 10.1101/353474. [DOI]
- 25. Marçais G., Kingsford C.. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011; 27:764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin Z., Feng M., Santos C.N. dos, Yu M., Xiang B., Zhou B., Bengio Y.. A structured self-attentive sentence embedding. 2017; arXiv doi:09 March 2017, preprint: not peer reviewedhttps://arxiv.org/abs/1703.03130.
- 27. Menzel P., Ng K.L., Krogh A.. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016; 7:11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huson D.H., Beier S., Flade I., Górska A., El-Hadidi M., Mitra S., Ruscheweyh H.J., Tappu R.. MEGAN Community Edition - Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016; 12:e1004957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hyatt D., Chen G.-L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J.. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C.. Metagenomic biomarker discovery and explanation. Genome Biol. 2011; 12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rojas-Carulla M., Tolstikhin I., Luque G., Youngblut N., Ley R., Schölkopf B.. GeNet: Deep Representations for Metagenomics. 2019; arXiv doi:30 January 2019, preprint: not peer reviewedhttps://arxiv.org/abs/1901.11015.
- 33. Sundaram L., Gao H., Padigepati S.R., McRae J.F., Li Y., Kosmicki J.A., Fritzilas N., Hakenberg J., Dutta A., Shon J. et al.. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 2018; 50:1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quang D., Xie X.. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016; 44:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Segata N., Waldron L., Ballarini A., Narasimhan V., Jousson O., Huttenhower C.. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods. 2012; 9:811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castro F.M., Marín-Jiménez M.J., Guil N., Schmid C., Alahari K.. Ferrari V, Hebert M, Sminchisescu C, Weiss Y. End-to-End Incremental Learning. Proceedings of the European Conference on Computer Vision (ECCV). 2018; Germany: Springer International Publishing; 241–257. [Google Scholar]
- 37. Xiao T., Zhang J., Yang K., Peng Y., Zhang Z.. Error-Driven Incremental Learning in Deep Convolutional Neural Network for Large-Scale Image Classification. Proceedings of the 22nd ACM international conference on Multimedia. 2014; Orlando: ASSOC COMPUTING MACHINERY; 177–186. [Google Scholar]
- 38. Shen Z., Bao W., Huang D.-S.. Recurrent neural network for predicting transcription factor binding sites. Sci. Rep. 2018; 8:15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brendel W., Bethge M.. Approximating CNNs with bag-of-local-features models works surprisingly well on ImageNet. 2019; arXiv doi:20 March 2019, preprint: not peer reviewedhttps://arxiv.org/abs/1904.00760.
- 40. Sinha K., Dong Y., Cheung J.C.K., Ruths D.. Riloff E, Chiang D, Hockenmaier J, Tsujii J. A hierarchical neural attention-based text classifier. Proceedings of the 2018 Conference on Empirical Methods in Natural Language Processing. 2018; Brussels: Association for Computational Linguistics; 817–823. [Google Scholar]
- 41. Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser L., Polosukhin I.. Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S, Garnett R. Attention is all you need. Advances in Neural Information Processing Systems. 2017; Long Beach: Neural information processing systems (NIPS)5998–6008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The DeepMicrobes program, trained model parameters, hyperparameters and the implementation of the other DNN architectures are provided at GitHub (https://github.com/MicrobeLab/DeepMicrobes). The sequences of benchmark datasets, the abundance profiles of different taxonomic classification tools on the ten mock communities, the species profiles of the IBD dataset generated with DeepMicrobes, the predicted protein sequences and the custom database for Kaiju are available at GitHub (https://github.com/MicrobeLab/DeepMicrobes-data). The command lines used to run the taxonomic classification tools and the R scripts used to generate figures for benchmarking are available at https://github.com/MicrobeLab/DeepMicrobes-data/tree/master/scripts.