

Abstract
N6-adenosine methylation (m6A) is the most abundant internal RNA modification in eukaryotes, and affects RNA metabolism and non-coding RNA function. Previous studies suggest that m6A modifications in mammals occur on the consensus sequence DRACH (D = A/G/U, R = A/G, H = A/C/U). However, only about 10% of such adenosines can be m6A-methylated, and the underlying sequence determinants are still unclear. Notably, the regulation of m6A modifications can be cell-type-specific. In this study, we have developed a deep learning model, called TDm6A, to predict RNA m6A modifications in human cells. For cell types with limited availability of m6A data, transfer learning may be used to enhance TDm6A model performance. We show that TDm6A can learn common and cell-type-specific motifs, some of which are associated with RNA-binding proteins previously reported to be m6A readers or anti-readers. In addition, we have used TDm6A to predict m6A sites on human long non-coding RNAs (lncRNAs) for selection of candidates with high levels of m6A modifications. The results provide new insights into m6A modifications on human protein-coding and non-coding transcripts.
INTRODUCTION
Epigenetically, RNAs are elaborated with some chemical modifications affecting cellular activities. In addition to the well-known 5′ cap and 3′ poly(A) modifications, internal RNA modifications are also found in eukaryotes, such as N6-methyladenosine (m6A), N1-methyladenosine (m1A), 2′-O-dimethyladenosine (m6Am), 5-methylcytosine (m5C) and 5-hydroxymethylcytosine (hm5C) (1). N6-adenosine methylation (m6A) is the most abundant internal mRNA modification with the prevalence of one per 700–800 nucleotides (nt) for poly(A)+ nuclear RNAs and one per 800–900 nt for cytoplasmic RNAs (1,2). High-throughput single-nucleotide-resolution mapping of m6A has revealed that m6A modifications are present in thousands of transcripts (3–5). These modifications are mainly clustered in the 3′ UTR near the stop codon and long internal exons, but may also be found in the 5′ UTR and coding regions of mRNAs. Moreover, m6A modifications have been shown to be different in various brain regions and neural cells (6), indicating that this process is under tissue- or cell-type-specific regulation.
RNA m6A modifications are controlled by a methyltransferase complex formed by METT3, METT14, WTAP and KIAA1429, and by two potential demethylases, FTO and ALKBH5 (1). Many cellular activities are modulated by m6A modifications. For instance, m6A is selectively recognized by the human YTH domain family 2 (YTHDF2) reader protein to regulate mRNA degradation and localization from the translatable pool to decay sites (7), whereas YTHDF1 interacts with translation initiation factors to increase the translational efficiency of m6A-marked transcripts (8). Interestingly, m6A modifications are important for the functions of some long non-coding RNAs (lncRNAs), such as XIST (X-inactive specific transcript), which has at least 78 m6A sites required for its function in transcriptional gene silencing on the X chromosome (9). Moreover, m6A modifications have been shown to impact many other cellular processes, including mRNA alternative splicing (10), microRNAs biogenesis (11), stem cell differentiation (12–14), circadian clock control (15), heat shock response (16), DNA damage response (17) and cancer development (18).
However, not all RNA adenosines are methylated. Previous studies suggest that m6A modifications in mammals preferably occur in the consensus sequence of DRACH (D = A/G/U, R = A/G, H = A/C/U) (3,19,20). Furthermore, only a fraction of DRACH-conformed adenosines may actually be methylated (21). The underlying sequence determinants are still unclear. To date, machine learning models have been developed to predict m6A sites in different species and to learn features that may be important for m6A modifications. m6Apred (22) and iRNA-Methyl (23) are support vector machine (SVM) models for yeast m6A site prediction, and the model performance may be limited by the small amount of available training data. With the availability of single-nucleotide-resolution mapping of m6A modifications, machine learning models with improved performance have been developed. SRAMP (24), a random forest (RF) model with sequence-derived features, was the first machine learning model for mammalian m6A site prediction. Recently, by integrating sequence and genomic features, WHISTLE, an SVM model, has been developed for accurate prediction of human m6A sites (25). Although good model performance may be achieved using conventional machine learning algorithms, SVM or RF model construction requires careful and considerable hand-crafted work to transform raw sequences into suitable feature vectors (26). In contrast, deep learning methods can automatically learn high-level features from transcript sequences, and have thus been widely applied to biological problems (27). By combining bidirectional gated recurrent unit (BGRU) and RF, BERMP was developed for multi-species m6A site prediction (28). Convolutional neural networks (CNN) were also used to construct several deep learning models, including DeepM6ASeq (29), Deep-m6A (30) and Gene2Vec (31).
Although several models with good performance have been developed, the underlying sequence determinants for m6A modifications are still limited to DRACH. Moreover, m6A modifications may be regulated dynamically and differentially in cellular processes such as stem cell differentiation, cell-state transitions and stress responses (6,32). Therefore, a cell-type-specific model can be useful for accurate prediction of RNA m6A sites. In this study, we have used CNN and recurrent neural network (RNN) to build a cell-type-specific model, named TDm6A (tissue or cell-type-dependent m6A modifications), for human m6A site prediction. The kernels/filters of CNN detect important features for m6A site prediction regardless of their positions in the transcript sequences, making CNN a useful method for motif discovery (33,34). In TDm6A, RNN is used to capture the inter-relationship between the learnt motifs. Long short-term memory (LSTM) is a variant of RNN developed to avoid gradient disappearing in a conventional RNN. By using CNN followed by LSTM, motifs and their inter-dependencies may be learnt for m6A site prediction. For cell types with low detection coverage of m6A modifications, transfer learning may be used to improve model performance. Moreover, we have utilized TDm6A to predict the possible m6A sites on human lncRNAs, providing good candidates for investigating the functional roles of m6A modifications on non-coding RNA transcripts.
MATERIALS AND METHODS
Datasets
The positive m6A data for human cell types A549, CD8T and HEK293 were collected from two studies using m6A-CLIP (3) and mi-CLIP (4), and the m6A sites conforming to the DRACH motif pattern were retained. For the negative data, we used the dataset from the SRAMP study, which included non-methylated adenosines randomly selected from the same set of m6A-methylated transcripts and also conforming to the DRACH motif pattern (24). For each cell type, the dataset had a 1:10 positive-to-negative ratio of instances since there were many more non-m6A sites than m6A sites in cells. Although the dataset had no duplicated instances, some positive and negative instances shared similar flanking sequences around the DRACH-conformed adenosines. RNA transcript sequences were extracted from the ENSEMBL GRCh38 annotation file (https://useast.ensembl.org/Homo_sapiens/Info/Index).
The whole dataset was divided randomly into training and test datasets using a ratio of 4:1, and the 1:10 positive-to-negative ratio of instances was kept in both datasets. For further model evaluation, four non-redundant test datasets (NR0.9, NR0.8, NR0.5 and NR0.3) were derived from the full test dataset. The software tool MUMmer (35) was used to analyze the nucleotide sequence similarity between the positive and negative instances, and between the training and test datasets. We tested four different thresholds of sequence identity, 0.9, 0.8, 0.5 and 0.3 to derive the four non-redundant test datasets, NR0.9, NR0.8, NR0.5 and NR0.3, respectively.
Model construction
To build a deep learning model for human m6A site prediction as shown in Figure 1, the Keras v2.2.4 in R v3.5.1 was used. Both the human pan-cell-type model, HPm6A, and cell-type-specific model, TDm6A, can be summarized as:
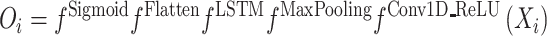 |
(1) |
Figure 1.
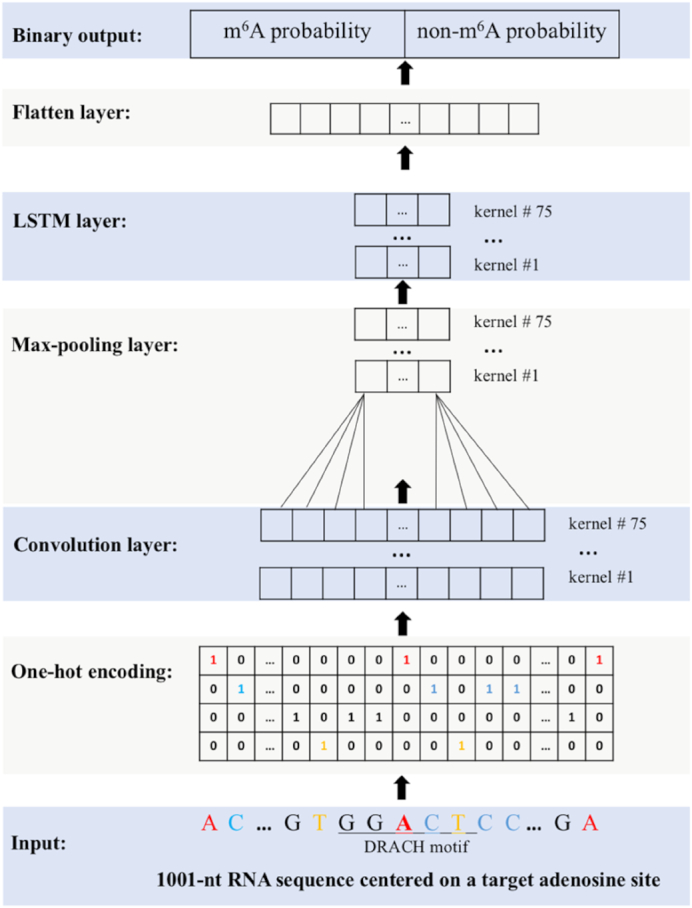
Schematic diagram of model construction for RNA m6A prediction. The input is a 1001-nt sequence centered on the target adenosine. Both m6A and non-m6A sites conform to the DRACH motif. An input sequence is converted into a 4 × 1001 matrix with 4 channels representing A, C, G, T(U). Kernels in the convolutional layer act as motif scanners. The max-pooling layer reduces the feature dimensionality. The LSTM layer learns the inter-dependencies between motifs learnt by the convolutional layer. The flatten layer combines the previous 75 kernels into a vector as the input to the fully connected binary output layer, in which the prediction probability for each class is calculated.
For the model input, we extracted l nucleotides (nt) of flanking sequence centered on the target adenosine, and l was tested from 43 to 1201 nt for the optimal input length. Since the model required the input to be of a fixed length, an input sequence less than l nt was padded with ‘N’. Each sequence was one-hot-encoded into a matrix using A (1,0,0,0), C (0,1,0,0), G (0,0,1,0), T (0,0,0,1) and N (0,0,0,0), which yielded Xi as the input matrix with dimensions of 4 × l, and was fed into the 1D convolution (Conv1D) layer. The input sequence was scanned by n kernels with size m, producing a feature map of size n * (l – m + 1) with four channels. Each kernel might be regarded as a motif scanner to identify motifs of length m nt. In this study, n was tested in the range from 16 (24) to 256 (28) and m in the range 4–20 to find the combination with the best model performance. The non-linear function, rectified linear unit (ReLU), was used to calculate the output.
The max-pooling layer with step s was used to reduce the dimensionality of the output from the preceding layer and hence the number of model parameters. The maximum value among the s values was used to form a new output matrix. The n kernels of size m nt with step s reduced the dimensionality to n * (l - m + 1)/s with four channels, and s was tested in the range from 4 to 8 for the best model performance.
The long short-term memory (LSTM) layer was used to capture the inter-dependencies between motifs learnt by the convolution layer. Each LSTM unit contained a memory cell and three gates, forget, input and output gate, to control the flow of information passing through the network. The outputs from the LSTM layer were converted into a vector and fed into a fully connected layer with the number of output nodes (Oi) equaling to the number of classification category. The non-linear sigmoid activation function calculated the prediction probability for each class. These prediction probabilities were compared to the true labels for weight-tuning through the loss function, binary cross-entropy, during the training process and for performance evaluation during the testing process.
To avoid model overfitting, a dropout layer following each of the first three layers in TDm6A was implemented. The dropout layer randomly zeroed out a fraction (r) of kernels in the inputs to the next layer, resembling the bagging technique. In this study, r was tested in the range of 0–0.9 for the best model performance.
Training strategy and model performance evaluation
The datasets in this study have a 1:10 positive-to-negative ratio of instances to mimic the cellular distribution of m6A modifications as only ∼10% of DRACH-conformed adenosines are actually m6A-methylated in cells. However, for model construction, ten balanced datasets were derived from the full training dataset with the 1:10 positive-to-negative ratio of instances. We randomly sampled the negative training instances into 10 non-overlapping parts, and combined each part with the same positive training instances to create 10 balanced datasets for training 10 sub-models. The average performance of the 10 sub-models on the same imbalanced test dataset was taken as the overall performance of HPm6A or TDm6A. Each sub-model was trained for 70 epochs to gradually tune the weights in each layer, and the validation_split argument provided by the Keras package was set to 0.2 (Supplementary Table S1). Thus, during each epoch, the sub-model was trained using 80% of the training instances, and the remaining 20% were used as the validation set for model evaluation during training. The purpose of this training strategy was to convert the original imbalanced dataset into 10 balanced datasets for model construction, which might avoid bias toward a label class. This strategy was also used by the study of SRAMP (24).
Trained models were evaluated on the test dataset using the following performance metrics (24,25):
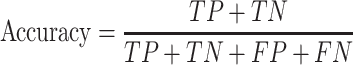 |
(2) |
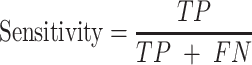 |
(3) |
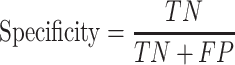 |
(4) |
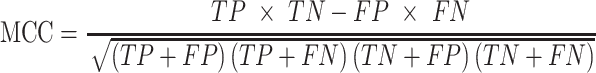 |
(5) |
Here, TP is the number of true positives; TN is the number of true negatives; FP is the number of false positives; and FN is the number of false negatives. The Matthews correlation coefficient (MCC) describes the correlation between predictions and labels (0 for random guess and 1 for a perfect model). The receiver operating characteristic curve (ROC) is the plot of the true positive rate (sensitivity) against the false positive rate (1 - specificity) with varying output thresholds. The value of the area under the ROC curve (ROC AUC) ranges between 0.5 and 1; a classifier is perfect if AUC is 1, and randomly guessing if AUC is 0.5.
Transfer learning
In recent years, transfer learning has attracted more and more attention, and can be classified into three categories: instance-based knowledge transfer, feature-based knowledge transfer and parameter-based knowledge transfer (36). In this study, transfer learning was employed to enhance the performance of a cell-type-specific model constructed with a limited amount of training instances. To simulate the low coverage of detected m6A modifications, we sampled a small fraction of m6A instances to build a cell-type-specific model, and the m6A instances from other cell types were utilized for transfer learning. For the cell type HEK293, mi-CLIP signal peak scores were provided (4), and based on the distribution of peak scores (Supplementary Figure S1), DRACH-conformed m6A sites with a peak score ≥5 were selected to form a dataset of 2756 positive instances. For the cell types A549 and CD8T, m6A sites were detected by m6A-CLIP (3), and the peak scores were not provided. We randomly sampled 20% of m6A sites from the full dataset of each cell type, resulting in 4103 and 3473 positive instances for A549 and CD8T, respectively. The ratio of positive to negative instances was also kept as 1:10 for each cell type.
For a target cell type, a TDm6A model was first initialized and pre-trained using all the available data from the other cell types. This process allowed TDm6A to capture common features for m6A modifications. Mathematically, the features learnt by a TDm6A model were the weights of kernels in each layer. We then froze the weights of the first convolutional layer of TDm6A to retain the features learnt from the other cell types, and then tuned the weights of the other layers of TDm6A using the relatively small dataset of the target cell type to build a cell-type-specific model.
Motif visualization and comparison
To visualize the position weight matrices (PWMs) learnt by TDm6A, we used the method as described in our previous study (37). Kernels of length m in the first convolutional layer of TDm6A scanned an input sequence at all positions and calculated activation scores. The sub-sequence of length m with the maximum score was selected. For each kernel, such sub-sequences collected from all the positive m6A instances in the test dataset were aligned to create a PWM in the MEME motif format, and the TOMTOM webserver (http://meme-suite.org/tools/tomtom) was used for PWM comparison and sequence logo generation (38).
Prediction of m6A sites on human lncRNAs
Human lncRNA transcript sequences (GRCh38) were downloaded from GENCODE (https://www.gencodegenes.org/human/). All adenosines conforming to the DRACH motif on each lncRNA were extracted. The three cell-type-specific TDm6A models were used to predict m6A sites on human lncRNAs. For each candidate m6A site, the average probability to be an m6A site of the 10 sub-models of TDm6A was taken as the overall score for classification with 0.5 as the threshold. To select candidate lncRNAs that might be highly m6A-methylated, two criteria were used: the total count of predicted m6A sites on a lncRNA and the frequency of predicted m6A sites (the total count normalized by the length of a lncRNA).
RESULTS AND DISCUSSION
Model construction for human m6A site prediction
Before developing cell-type-specific models, we first combined the m6A sites from all the available cell types (A549, CD8T and HEK293) to construct a pan-cell-type model, HPm6A, for permissive m6A site prediction (Figure 1). Hyper-parameters of HPm6A were tuned for the best performance (Supplementary Table S1). An HPm6A model with either the mature RNA mode or the full-transcript pre-RNA mode was constructed. Various input lengths ranging from 43 to 1201 nt were tested, and 1001-nt was selected based on the area under the receiver operating characteristic curve (ROC AUC) (Supplementary Figure S2). As shown in Table 1, HPm6A achieved comparable performance with the previous models using only sequence features as the input. Although WHISTLE had the best performance, it was mainly based on 35 genomic features collected from the transcript annotations. Interestingly, the important genomic features contributing to the superior performance of WHISTLE included long exon, being miRNA target gene, conservation score, distance to known m6A sites, and distance to UTR boundaries. However, some of these genomic features are uniquely applicable to protein-coding transcripts, but not non-coding transcripts such as lncRNAs, for which m6A modifications can be functionally important (39). In addition, since m6A modifications show overall enrichment in 3′ UTR near mRNA stop codons and long internal exons (3–5), the genomic features used by WHISTLE might make the model to be overfitted to protein-coding mRNAs and thus perform poorly on lncRNAs. In this study, we intended to utilize only RNA sequence as the model input. First, with RNA sequence as input, the kernels in the CNN layer can act as motif scanners to find motifs that may represent new sequence determinants for m6A modifications (Figure 1). Second, we are interested in the m6A modification patterns of lncRNAs, which have little annotations besides their available nucleotide sequences.
Table 1.
Performance comparison of pan-cell-type HPm6A with other previous models
| HPm6A | Gene2Vec* | SRAMP* | WHISTLE* | |
|---|---|---|---|---|
| Input features | Sequence features | Sequence features | Sequence features | Sequence and genomic features |
| Algorithm | CNN+LSTM | CNN | RF | SVM |
| Species | Human | Human | Mammals | Human |
| Cell type | Pan-cell-type | Pan-cell-type | Pan-cell-type | Cell-type-specific |
| ROC AUC (mature RNA mode) | 0.8534 | 0.8333 | 0.7970 | 0.8903 |
| ROC AUC (pre-RNA mode) | 0.8916 | / | 0.8910 | 0.9498# |
Note: *The AUC scores from the previous papers are shown since the models were constructed using the same datasets from the single-nucleotide-resolution mapping of m6A sites in the cell lines A549, CD8T and HEK293.
#According to the authors of WHISTLE (25), the predictive performance of WHISTLE on the full-transcript model may be significantly over-estimated.
As shown in Table 1, HPm6A in the full-transcript pre-RNA mode has a statistically significant improvement of ROC AUC over the mature RNA mode (P-value < 0.00001, two-sided t-test), suggesting that relevant features may be present in pre-RNA sequences. This result is consistent with the hypothesis that m6A modifications are added to exons before or soon after exon definition in the nascent pre-RNAs (40). To further evaluate the model performance, we used HPm6A to predict the m6A sites on human protein-coding transcripts in GENCODE (https://www.gencodegenes.org/human/). As shown in Figure 2, by setting the threshold for positive m6A sites at higher values from 0.5 to 0.95, the m6A sites predicted by HPm6A with higher confidence show stronger enrichment in the 3′ UTR region near the stop codon. The agreement between predictions and experimental data (3–5) suggests that HPm6A may have learnt some relevant sequence features for the control of RNA m6A modifications.
Figure 2.
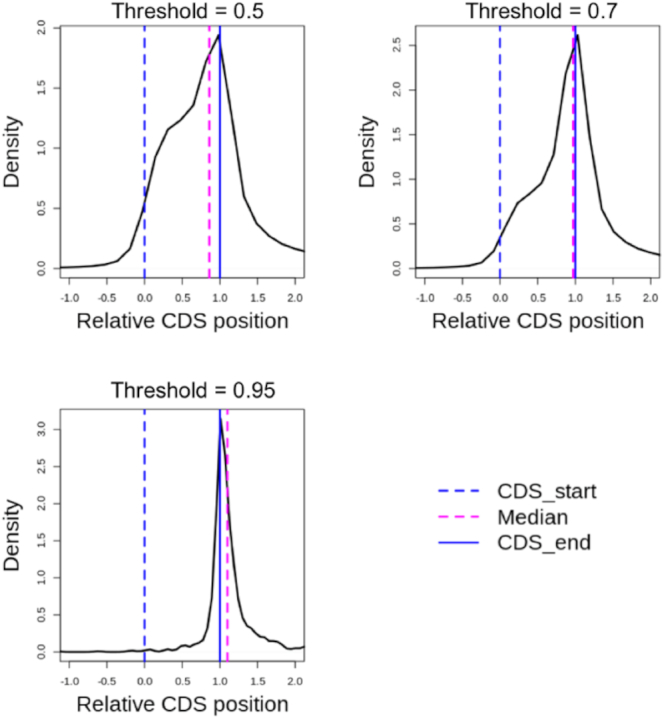
High-confidence m6A sites predicted by HPm6A show strong enrichment in the 3′ UTR region near the stop codon of human protein-coding transcripts. In the density plots, the X-axis represents the relative position of the coding sequence (CDS) region. The dashed blue line at x = 0 represents the start of the CDS, and the blue solid line at x = 1 represents the end of the CDS. Thus, the 5′ UTR region is x< 0, the CDS region is 0 < x < 1, and the 3′ UTR region is x > 1. By setting the output threshold for positive m6A predictions at higher values, TDm6A-predicted m6A sites show stronger enrichment near the stop codon. The dashed magenta line shows the statistic median of the relative CDS position of the positive m6A sites. The total numbers of predicted m6A sites are 787573, 311437 and 1576 for output thresholds of 0.5, 0.7 and 0.95, respectively.
TDm6A for cell-type-specific prediction of m6A modifications
Since m6A modifications are dynamically regulated in various cellular pathways, cell-type-specific models for m6A site prediction can be more useful and accurate than the pan-cell-type model. Thus, for each of the available cell types (A549, CD8T and HEK293), we have developed a cell-type-specific model in the full-transcript pre-RNA mode, named TDm6A (tissue or cell-type-dependent m6A modifications). As shown in Table 2 and Figure 3A, the cell-type-specific TDm6A models achieved better performance than the pan-cell-type HPm6A model on the test datasets (P-value < 0.0001, two-sided t-test). For HEK293 using the antibody Abacm, the relatively low performance was obtained by both TDm6A and other previous models (25). This might be due to the low quality of the dataset (HEK293-Abacm). Thus, for the cell type HEK293, the m6A sites detected using the antibody SySy were used in the further analysis.
Table 2.
Performance of TDm6A for cell-type-specific prediction of m6A modifications. The pre-RNA mode was used for both TDm6A and HPm6A models
| Model | TDm6A | HPm6A | |||
|---|---|---|---|---|---|
| Cell type | A549 | CD8T | HEK293 | HEK293 | A549+CD8T+HEK293 |
| Antibody used for m6A detection | SySy | SySy | SySy | Abacm | SySy + Abacm |
| Method for m6A detection | m6A-CLIP | m6A -CLIP | mi-CLIP | mi-CLIP | m6A -CLIP + mi-CLIP |
| Reference | (3) | (3) | (4) | (4) | (3,4) |
| Number of m6A sites | 20 515 | 17 365 | 5234 | 7370 | 49 618 |
| Accuracy | 0.7907 | 0.8031 | 0.7880 | 0.7230 | 0.7815 |
| Sensitivity | 0.8784 | 0.8650 | 0.8987 | 0.7714 | 0.8568 |
| Specificity | 0.7819 | 0.7969 | 0.7769 | 0.7182 | 0.7740 |
| MCC | 0.4236 | 0.4321 | 0.4304 | 0.3002 | 0.4024 |
| ROC AUC | 0.9054 | 0.9060 | 0.9094 | 0.8173 | 0.8916 |
Figure 3.

ROC curves of cell-type-specific TDm6A models in the pre-RNA mode. The ROC AUC values are given in the legend. (A) Cell-type-specific TDm6A models show better performance than the pan-cell-type model HPm6A for human m6A site prediction. For HEK293_Abacm, the low quality of the dataset might result in its poor performance. (B) Transfer learning can be used to improve cell-type-specific model performance if the available training data from a new cell type is limited. For each of the cell types A549, CD8T and HEK293 (SySy), a small fraction of m6A sites was sampled from the full dataset to build the model with transfer learning. The performance improvement in (A) and (B) was statistically significant (P-value < 0.0001) based on two-sided unpaired t-tests.
Each model was also evaluated using four non-redundant test datasets, NR0.9, NR0.8, NR0.5 and NR0.3, which were derived by removing the test instances that had similar flanking sequences with any training instances using the sequence identity thresholds 0.9, 0.8, 0.5 and 0.3, respectively (Supplementary Figure S3). Remarkably, when evaluated using the non-redundant test datasets, the TDm6A or HPm6A models performed well, actually better than on the full test datasets (Supplementary Table S2 and Supplementary Figure S4), demonstrating the robustness of our models. The TDm6A models again outperformed the HPm6A model on the non-redundant test datasets. Since the flanking sequences around the DRACH-conformed adenosines can be similar between some positive and negative instances (Supplementary Figure S5), removing the instances with similar flanking sequences may have made the positive and negative instances more distinct from each other and thus a simpler classification task. Taken together, the results suggest that our models have consistently robust performance with no sign of overfitting, and may have captured the subtle difference between the positive and negative instances with similar flanking sequences.
The available methods for single-nucleotide-resolution mapping of m6A sites may only offer limited coverage of m6A modifications with low confidence as such experiments are often expensive, laborious and difficult (25). In this situation, where researchers can only detect a limited number of m6A sites for one cell type under a specific condition, an accurate prediction model can be helpful. However, model construction with a small or low-quality training dataset can be problematic, and transfer learning may be useful in this case. Some sequence determinants for m6A modifications, such as the DRACH motif pattern, can be universal features regardless of cell types. Through transfer learning, common features may be learnt from related data and transferred to initialize a new model, and then the limited amount of training data can be used to further tune the new model and learn additional features. As shown in Figure 3B and Supplementary Table S3, for each cell type, transfer learning enhanced the performance of the TDm6A model trained with the limited amount of m6A instances as it achieved comparable accuracy with the model trained using the full dataset. The efficacy of transfer learning indicates that these models have learnt some common sequence features important for m6A modifications in all cell types. Nevertheless, the superior performance of TDm6A over HPm6A suggests that some cell-type-specific features may also need to be learnt for accurate prediction of m6A modifications.
Cell-type-specific features learnt by TDm6A
This study used data from three different cell lines: A549 from a cancerous lung tissue, CD8T from T lymphocytes and HEK293 from human embryonic kidney cells. As shown in Figure 4A, the decrease of performance for cross-cell-type m6A site prediction suggests that TDm6A may have captured some cell-type-specific sequence features. Thus, we converted the 75 kernels in the convolutional layer of TDm6A into position weight matrices (PWMs) (Supplementary Table S4) using the method as described previously (37), and compared the PWMs between cell types using TOMTOM (38). Interestingly, most of the PWMs appear to be cell-type-specific (Figure 4B). With E value ≤ 0.05, only seven distinct PWMs are shared by the three cell types (Supplementary Table S5). Particularly, as shown in Figure 4C, among the seven common PWMs learnt by TDm6A, GGACTG conforms to the known DRACH motif pattern and is enriched at the m6A modification sites, whereas the motif GGTAAG with a conserved T residue at the third position is enriched in the downstream region of non-m6A sites.
Figure 4.

Cell-type-specific features learnt by TDm6A models. (A) Cross-cell-type prediction of m6A modifications by TDm6A models. The ROC AUC value of a TDm6A model tested on each cell type is indicated. (B) Comparisons of TDm6A-learnt PWMs among cell types A549, CD8T and HEK293 using the TOMTOM server (38). E value <= 0.05 was used as the statistical threshold. The numbers of common PWMs between cell types are indicated in the overlapping regions. (C) Sequence logos and distributions of two common PWMs learnt by TDm6A. The motif GGACTG enriched at m6A sites is similar to the known DRACH pattern, and the motif GGTAAG with a conserved T residue at the third position shows enrichment at non-m6A sites. (D) Some cell-type-specific PWMs significantly match the known RNA-binding motifs in the Ray2013 Homo sapiens database. The PWM M5 of A549 matches the RNA motif of LIN28A, an m6A anti-reader protein. The PWM M41 of CD8T shows similarity with the RNA motif of HNRNPH2, an m6A reader protein. The PWM M9 of HEK293 matches the RNA motif of FXR1, which is a paralog of the m6A reader protein FMR1. Density plots of M5, M41 and M9 in the 1001-nt flanking region centered on the target adenosine are also shown.
Next, we compared the TDm6A-learnt PWMs with the known RNA motifs in the Ray2013 Homo sapiens database using TOMTOM. None of the seven common PWMs matched with any known motifs of RNA-binding proteins. However, several cell-type-specific PWMs showed similarity to the known RNA motifs (Supplementary Table S6). For instance, the PWM M5 of A549 is similar to the motif of the RNA-binding protein LIN28A (Figure 4D), which has been reported to be an m6A anti-reader that requires a non-methylated adenosine for RNA binding (41,42). The distributions of M5 in the flanking sequences of positive and negative data instances are statistically different (Figure 4D). The position of M5 appears to shift toward the upstream region of m6A sites, which may avoid the antagonistic interaction between M5 and m6A. The PWM M60 of A549 matches the RNA motif of SRSF7, which directly interacts with the nuclear m6A-binding protein YTHDC1. Together with various SR proteins, YTHDC1 is involved in pre-mRNA splicing and nuclear RNA processing (43). So far, several families of m6A readers have been identified, including the YTH domain proteins, hnRNP family and KH domain proteins (44). The hnRNP-H2 protein of the hnRNP family is an m6A reader with a binding motif similar to M41 of CD8T (Figure 4D). Moreover, FMR1, a negative regulator of translation, may preferentially interact with m6A-containing RNAs in certain sequence contexts, and its two paralogs FXR1 and FXR2 are also m6A readers (45). The PWM M9 of HEK293, matching the RNA motif of FXR1, shows enrichment in the upstream region near m6A sites, but not at m6A sites, which is consistent with the finding that FMR1 is an indirect reader of m6A sites (45). Other RNA-binding proteins with motifs matching the PWMs learnt by TDm6A are shown in Supplementary Table S6. Previous studies suggest that m6A modifications can be dynamically changed in different cell stages such as spermatogenesis (46), and their dysregulation can alter various pathways such as cytokine responses and tumorigenesis (47). In addition to METTL3/14/16 and FTO/ALKBH5 as m6A writers and erasers, respectively, about 20 other proteins have been shown to be m6A regulators. It is likely that additional cell-type-specific regulators of m6A modifications are to be identified (48). The RNA-binding proteins matching the PWMs learnt by TDm6A provide new candidates of m6A regulators, and further investigations may give insight into the cellular processes controlled by m6A modifications in a cell-type-specific manner.
Prediction of m6A sites on human lncRNAs
Protein-coding genes only comprise a small portion of the human genome, and non-coding transcripts fulfill a rich diversity of regulatory and functional roles. In particular, long non-coding RNAs (lncRNAs) are transcripts greater in length than 200 nucleotides, but not encoding proteins. LncRNAs are dynamically regulated and involved in numerous cellular activities. Although some lncRNAs have been demonstrated to be key regulators of gene expression and 3D genome organization (49–51), most of them are still uncharacterized. It has been shown that m6A modifications can be required for lncRNA functions through inducing local structural changes for protein binding (9,52). We thus utilized our TDm6A models to predict m6A modifications on human lncRNAs. The distribution of the predicted m6A sites on lncRNAs appears to be different from that on mRNAs; m6A modifications may be enriched in the middle region of lncRNA sequences (Supplementary Figure S6). Moreover, the m6A modifications on lncRNAs predicted by the cell-type-specific TDm6A models show slightly different distributions in the three cell types (A549, CD8T and HEK293).
To further evaluate the validity of TDm6A predictions, we examined two lncRNAs, XIST and MALAT1 (metastasis associated lung adenocarcinoma transcript 1), for which m6A modifications have been experimentally determined by mi-CLIP in HEK293 cells (4). As shown in Figure 5, the highly m6A-methylated regions detected by mi-CLIP were also predicted positively as m6A clusters by the TDm6A model trained with the HEK293 dataset. For MALAT1, the four sites at positions 2515, 2577, 2611 and 2720 were predicted positively by TDm6A, and also confirmed by the SCARLET technology to be consistently m6A-methylated in multiple cell lines (53). Interestingly, the m6A modifications at positions 2515 and 2577 may change the local RNA structures to facilitate the interactions with the RNA-binding proteins HNRNPG and HNRNPC, respectively (10,39,54). Compared with mi-CLIP data, TDm6A predicted more m6A sites, some of which might be false positives, but the others could be true m6A sites that were not detected by m6A-seq techniques. The m6A-seq experiments may only detect a limited amount of m6A modifications in the epitranscriptome (25), especially for lncRNAs which often have lower expression than protein-coding genes (55). The regions predicted by TDm6A to be highly m6A-methylated may be regarded as candidate m6A clusters for experimental validation, such as the region between 5000 and 10000 on XIST (Figure 5).
Figure 5.

TDm6A-predicted m6A sites for the human lncRNAs XIST and MALAT1. The X-axis indicates the nucleotide positions on a lncRNA. The Y-axis in the top panel shows the probability of a possible m6A site predicted by the HEK293 model of TDm6A, and the Y-axis in the bottom panel indicates the peak score of an m6A site detected by mi-CLIP in HEK293 cells. Of the TDm6A-predicted m6A sites, the true sites that are also detected by mi-CLIP are indicated in red. For MALAT1, four m6A sites at positions 2515, 2577, 2611 and 2720 were also detected by the SCARLET technology (53), and the m6A modifications at positions 2515 and 2577 may facilitate the interactions with the RNA-binding proteins, HNRNPG and HNRNPC, respectively (10,39,54).
Based on the predictions by TDm6A, lncRNAs may be prioritized for high-level m6A modifications in human cells through count-based or frequency-based ranking (see ‘Materials and Methods’ section). From the count-based list (Supplementary Table S7), we can select candidate lncRNAs with the most predicted m6A sites, such as ENST00000597346.1 and ENST00000604411.1. They are the non-coding transcripts produced from the genes KCNQ1OT1 (ENSG00000269821) and TSIX (ENSG00000270641), respectively. Interestingly, both KCNQ1OT1 and TSIX have been shown to play important roles in chromatin structure and gene regulation (56). The transcript of KCNQ1OT1 is a long chromatin-interacting non-coding RNA, which is moderately stable, nucleus-localized and a product of RNA polymerase II. Through the recruitment of chromatin and DNA-modifying proteins, KCNQ1OT1 can establish a repressive higher order chromatin structure to silence multiple genes in the KCNQ1 domain (57,58). The lncRNA TSIX is antisense to XIST, and negatively regulates the expression of XIST in cis through the establishment of repressive epigenetic modifications and chromatin structures at the XIST locus (59,60). Although the molecular mechanisms of the lncRNA–chromatin interactions remain unclear, the m6A modifications may play an important role as indicated by the finding that XIST has about 78 m6A sites essential for the transcriptional silencing of the future inactive X chromosome (9). The frequency-based ranking takes into account the length of a lncRNA (the total count of predicted m6A sites normalized by the length of a lncRNA), and the list can be used to select candidate lncRNAs with the highest frequency of m6A modifications. However, the top candidate lncRNAs in the frequency-based list are poorly annotated with unknown functions (Supplementary Table S8). We hope that the candidate lncRNAs ranked by TDm6A predictions in both lists can provide good targets for further investigations into m6A modifications, lncRNA functions, and chromatin structures.
CONCLUSIONS
In this study, we have developed the cell-type-specific deep learning model TDm6A for understanding RNA m6A modification patterns in human cells. The sequence-derived features allow the broad application of TDm6A to the transcriptome-wise prediction of m6A sites in both protein-coding mRNAs and non-coding transcripts such as long non-coding RNAs (lncRNAs). Several previous studies focused on the prediction of pan-cell-type m6A modifications for one or more species. In contrast, TDm6A has been developed as a cell-type-specific model for accurate prediction of human m6A modifications. The available methods for the single-nucleotide-resolution mapping of m6A sites may only offer limited coverage of m6A modifications, and these experiments can be expensive, laborious and difficult (25). It is thus likely that only a limited number of m6A sites for a cell type under a certain condition can be detected, which may not be sufficient for building an accurate model. In this study, we have demonstrated that transfer learning can be an effective method for building an accurate TDm6A model for a cell type with low coverage of m6A modifications.
Although several models have been developed for m6A site prediction, the sequence determinant for m6A modifications is still limited to the known DRACH motif pattern. In this study, sequence features learnt by TDm6A for each cell type (A549, CD8T or HEK293) were converted into position weight matrices (PWMs), most of which were found to be cell-type-specific. Common PWMs among the cell types included the known consensus motif DRACH and several new motifs. Interestingly, some PWMs learnt by TDm6A were found to match the known motifs of RNA-binding proteins, including m6A readers and anti-readers such as LIN28A, HNRNPH2 and FXR1.
Previous studies suggest that m6A modifications can be critical for the functions of some non-coding RNA transcripts. In this study, we have utilized TDm6A to predict the possible m6A sites on human lncRNAs, and found that m6A modifications might be enriched in the middle region of lncRNA sequences. Based on the predictions by TDm6A, lncRNAs have been prioritized for high-level m6A modifications in human cells. Particularly, the highly m6A-methylated candidate lncRNAs include KCNQ1OT1 and TSIX, which have been shown to be chromatin-interacting lncRNAs involved in gene regulation. Since most lncRNAs are still uncharacterized functionally, the candidate lncRNAs predicted and prioritized in this study provide good targets for further investigations into m6A modifications and lncRNA functions.
In the future, we will try to improve our models in the following two areas. First, additional features can be incorporated into our deep learning system. Besides the genomic features used by WHISTLE (25), other features such as predicted RNA secondary structures and various types of RNA modifications within the sequence region will also be examined for the prediction of m6A modifications on both protein-coding and non-coding RNAs. Our present work used only RNA sequence as the model input to discover novel motifs underlying m6A modification patterns, and achieved comparable performance with the previous models. With additional relevant features, TDm6A model performance may be enhanced. Second, more m6A datasets are needed for further development and evaluation of TDm6A models. The existing models for human m6A site prediction, including TDm6A, SRAMP (24) and WHISTLE (25), have been constructed using the limited data from two previous studies (3,4) for single-nucleotide-resolution mapping of human m6A sites in only three cell lines (A549, CD8T and HEK293). Hopefully, more high-quality m6A datasets will become available so that TDm6A models can be trained to be more robust for RNA m6A site prediction and comprehensive analyses can be performed to elucidate the determinants of m6A modifications in human cells.
DATA AVAILABILITY
Datasets and models are available in the GitHub repository (https://github.com/BioDataLearning/TDm6A). An R package, named TDm6A, is also available. Step-by-step instructions are given for using the TDm6A model to predict m6A modifications on a human RNA transcript.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
Internal funding from Clemson University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang X., He C.. Dynamic RNA modifications in posttranscriptional regulation. Mol. Cell. 2014; 56:5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lavi U., Fernandez-Mufioz R., Darnell J.E. Jr.. Content of N-6 methyl adenylic acid in heterogeneous nuclear and messenger RNA of HeLa cells. Nucleic Acids Res. 1977; 4:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ke S., Alemu E.A., Mertens C., Gantman E.C., Fak J.J., Mele A., Haripal B., Zucker-Scharff I., Moore M.J., Park C.Y. et al.. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015; 29:2037–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Linder B., Grozhik A.V., Olarerin-George A.O., Meydan C., Mason C.E., Jaffrey S.R.. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods. 2015; 12:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yue Y., Liu J., He C.. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015; 29:1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chang M., Lv H., Zhang W., Ma C., He X., Zhao S., Zhang Z.W., Zeng Y.X., Song S., Niu Y. et al.. Region-specific RNA m6A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol. 2017; 7:170166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G. et al.. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014; 505:117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C.. N6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015; 161:1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patil D.P., Chen C.K., Pickering B.F., Chow A., Jackson C., Guttman M., Jaffrey S.R.. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016; 537:369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T.. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature. 2015; 518:560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alarcón C.R., Lee H., Goodarzi H., Halberg N., Tavazoie S.F.. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015; 519:482–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A.A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y.S. et al.. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015; 347:1002–1006. [DOI] [PubMed] [Google Scholar]
- 13. Wang Y., Li Y., Toth J.I., Petroski M.D., Zhang Z., Zhao J.C.. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014; 16:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang Y., Li Y., Yue M., Wang J., Kumar S., Wechsler-Reya R.J., Zhang Z., Ogawa Y., Kellis M., Duester G. et al.. N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci. 2018; 21:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fustin J.M., Doi M., Yamaguchi Y., Hida H., Nishimura S., Yoshida M., Isagawa T., Morioka MS., Kakeya H., Manabe I. et al.. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013; 155:793–806. [DOI] [PubMed] [Google Scholar]
- 16. Zhou J., Wan J., Gao X., Zhang X., Jaffrey S.R., Qian S.B.. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. 2015; 526:591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiang Y., Laurent B., Hsu C.H., Nachtergaele S., Lu Z., Sheng W., Xu C., Chen H., Ouyang J., Wang S. et al.. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017; 543:573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deng X., Su R., Feng X., Wei M., Chen J.. Role of N6-methyladenosine modification in cancer. Curr. Opin. Genet. Dev. 2018; 48:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Csepany T., Lin A., Baldick C.J., Beemon K.. Sequence specificity of mRNA N6-adenosine methyltransferase. J. Biol. Chem. 1990; 265:20117–20122. [PubMed] [Google Scholar]
- 20. Harper J.E., Miceli S.M., Roberts R.J., Manley J.L.. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 1990; 18:5735–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. et al.. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012; 485:201–206. [DOI] [PubMed] [Google Scholar]
- 22. Chen W., Tran H., Liang Z., Lin H., Zhang L.. Identification and analysis of the N6-methyladenosine in the Saccharomyces cerevisiae transcriptome. Sci. Rep. 2015; 5:13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen W., Feng P., Ding H., Lin H., Chou K.C.. iRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem. 2015; 490:26–33. [DOI] [PubMed] [Google Scholar]
- 24. Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q.. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016; 44:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen K., Wei Z., Zhang Q., Wu X., Rong R., Lu Z., Su J., de Magalhães, J.P., Rigden D.J., Meng J.. WHISTLE: a high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 2019; 47:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. LeCun Y., Bengio Y., Hinton G.. Deep learning. Nature. 2015; 521:436–444. [DOI] [PubMed] [Google Scholar]
- 27. Angermueller C., Pärnamaa T., Parts L., Stegle O.. Deep learning for computational biology. Mol. Syst. Biol. 2016; 12:878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang Y., He N., Chen Y., Chen Z., Li L.. BERMP: a cross-species classifier for predicting m6A sites by integrating a deep learning algorithm and a random forest approach. Int. J. Biol. Sci. 2018; 14:1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y., Hamada M.. DeepM6ASeq: prediction and characterization of m6A-containing sequences using deep learning. BMC Bioinformatics. 2018; 19:524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang S.Y., Zhang S.W., Fan X.N., Meng J., Chen Y., Gao S.J., Huang Y.. Global analysis of N6-methyladenosine functions and its disease association using deep learning and network-based methods. PLoS Comput. Biol. 2019; 15:e1006663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zou Q., Xing P., Wei L., Liu B.. Gene2vec: gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from mRNA. RNA. 2019; 25:205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao B.S., Roundtree I.A., He C.. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017; 18:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831–838. [DOI] [PubMed] [Google Scholar]
- 34. Zhou J., Troyanskaya O.G.. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods. 2015; 12:931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kurtz S., Phillippy A., Delcher A.L., Smoot M., Shumway M., Antonescu C., Salzberg S.L.. Versatile and open software for comparing large genomes. Genome Biol. 2004; 5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pan S.J., Yang Q.. A survey on transfer learning. IEEE Trans. Knowl. Data Eng. 2009; 22:1345–1359. [Google Scholar]
- 37. Wang J., Wang L.. Deep learning of the back-splicing code for circular RNA formation. Bioinformatics. 2019; 35:5235–5242. [DOI] [PubMed] [Google Scholar]
- 38. Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S.. Quantifying similarity between motifs. Genome Biol. 2007; 8:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Coker H., Wei G., Brockdorff N.. m6A modification of non-coding RNA and the control of mammalian gene expression. Biochim. Biophys. Acta Gene. Regul. Mech. 2019; 1862:310–318. [DOI] [PubMed] [Google Scholar]
- 40. Ke S., Pandya-Jones A., Saito Y., Fak J.J., Vågbø C.B., Geula S., Hanna J.H., Black D.L., Darnell J.E., Darnell R.B.. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017; 31:990–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun L., Fazal F.M., Li P., Broughton J.P., Lee B., Tang L., Huang W., Kool E.T., Chang H.Y., Zhang Q.C.. RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol. 2019; 26:322–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Angela M.Y., Lucks J.B.. Tracking RNA structures as RNAs transit through the cell. Nat. Struct. Mol. Biol. 2019; 26:256–257. [DOI] [PubMed] [Google Scholar]
- 43. Xiao W., Adhikari S., Dahal U., Chen Y.S., Hao Y.J., Sun B.F., Sun H.Y., Li A., Ping X.L., Lai W.Y. et al.. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol. Cell. 2016; 61:507–519. [DOI] [PubMed] [Google Scholar]
- 44. Ji P., Wang X., Xie N., Li Y.. N6-methyladenosine in RNA and DNA: An Epitranscriptomic and Epigenetic Player Implicated in Determination of Stem Cell Fate. Stem Cells Int. 2018; 2018:3256524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Edupuganti R.R., Geiger S., Lindeboom R.G., Shi H., Hsu P.J., Lu Z., Wang S.Y., Baltissen M.P.A., Jansen P.W.T.C., Rossa M. et al.. N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 2017; 24:870–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin Z., Tong M.H.. M6A mRNA modification regulates mammalian spermatogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2019; 1862:403–411. [DOI] [PubMed] [Google Scholar]
- 47. Chang G., Leu J.S., Ma L., Xie K., Huang S.. Methylation of RNA N6-methyladenosine in modulation of cytokine responses and tumorigenesis. Cytokine. 2018; 118:35–41. [DOI] [PubMed] [Google Scholar]
- 48. Ianniello Z., Fatica A.. N6-methyladenosine role in acute myeloid leukaemia. Int. J. Mol. Sci. 2018; 19:2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quinn J.J., Chang H.Y.. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016; 17:47–62. [DOI] [PubMed] [Google Scholar]
- 50. Esteller M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011; 12:861–874. [DOI] [PubMed] [Google Scholar]
- 51. Gudenas B.L., Wang J., Kuang S.Z., Wei A.Q., Cogill S.B., Wang L.J.. Genomic data mining for functional annotation of human long noncoding RNAs. J. Zhejiang Univ. Sci. B. 2019; 20:476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhou K.I., Parisien M., Dai Q., Liu N., Diatchenko L., Sachleben J.R., Pan T.. N6-methyladenosine modification in a long noncoding RNA hairpin predisposes its conformation to protein binding. J. Mol. Biol. 2016; 428:822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu N., Parisien M., Dai Q., Zheng G., He C., Pan T.. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA. 2013; 19:1848–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu N., Zhou K.I., Parisien M., Dai Q., Diatchenko L., Pan T.. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017; 45:6051–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G. et al.. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Umlauf D., Fraser P., Nagano T.. The role of long non-coding RNAs in chromatin structure and gene regulation: variations on a theme. Biol. Chem. 2008; 389:323–331. [DOI] [PubMed] [Google Scholar]
- 57. Pandey R.R., Mondal T., Mohammad F., Enroth S., Redrup L., Komorowski J., Nagano T., Mancini-DiNardo D., Kanduri C.. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 2008; 32:232–246. [DOI] [PubMed] [Google Scholar]
- 58. Kanduri C. Kcnq1ot1: a chromatin regulatory RNA. Semin. Cell Dev. Biol. 2011; 22:343–350. [DOI] [PubMed] [Google Scholar]
- 59. Lee J., Davidow L.S., Warshawsky D.. Tsix, a gene antisense to Xist at the X-inactivation centre. Nat. Genet. 1999; 21:400–404. [DOI] [PubMed] [Google Scholar]
- 60. Sado T., Hoki Y., Sasaki H.. Tsix silences Xist through modification of chromatin structure. Dev. Cell. 2005; 9:159–165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets and models are available in the GitHub repository (https://github.com/BioDataLearning/TDm6A). An R package, named TDm6A, is also available. Step-by-step instructions are given for using the TDm6A model to predict m6A modifications on a human RNA transcript.