

Abstract
The current challenge in cancer research is to increase the resolution of driver prediction from gene-level to mutation-level, which is more closely aligned with the goal of precision cancer medicine. Improved methods to distinguish drivers from passengers are urgently needed to dig out driver mutations from increasing exome sequencing studies. Here, we developed an ensemble method, AI-Driver (AI-based driver classifier, https://github.com/hatchetProject/AI-Driver), to predict the driver status of somatic missense mutations based on 23 pathogenicity features. AI-Driver has the best overall performance compared with any individual tool and two cancer-specific driver predicting methods. We demonstrate the superior and stable performance of our model using four independent benchmarks. We provide pre-computed AI-Driver scores for all possible human missense variants (http://aidriver.maolab.org/) to identify driver mutations in the sea of somatic mutations discovered by personal cancer sequencing. We believe that AI-Driver together with pre-computed database will play vital important roles in the human cancer studies, such as identification of driver mutation in personal cancer genomes, discovery of targeting sites for cancer therapeutic treatments and prediction of tumor biomarkers for early diagnosis by liquid biopsy.
INTRODUCTION
Taken advantage of high-throughput sequencing technologies, millions of somatic mutations have been reported at base-pair resolution in the past decades (1,2). It is critical and challenging to distinguish driver missense mutations from passenger missense mutations since tumors are genetically heterogeneous populations within individual clones (3,4). The repertoire of genes affected by highly recurrent mutations is limited and there is a large collection of genes affected by mutations in <2% of cancers from a given anatomical site (5,6), which have been demonstrated by previous researches by the International Cancer Genome Consortium (ICGC) (7) and the Cancer Genome Atlas (TCGA) (8). Discriminating cancer-causing driver mutations from inconsequential passenger mutations is impeded due to the long-tail phenomenon with few common drivers and many rare drivers (9,10). Recent studies have demonstrated that some of these mutations are of functional significance and are likely to constitute bonafide drivers, therapeutic targets and therapy resistance (11–14). Identifying driver mutations based on hotspot mutations and recurrence rates has defined a partial repertoire of bonafide oncogenes and tumor suppressor genes (TSGs) which are significantly mutated in cancer (15,16). However, this strategy cannot be readily applied to the study of the genes affected by mutations in a minority of tumors and the research of personal cancer genomes. Moreover, even well-established cancer-driver genes are thought to contain a mixture of driver and passenger mutations across many patients’ tumors (10,17). However, only a few methods such as CanDrA (18) and CHASM (10,18) were designed explicitly to differentiate cancer driver mutations from passengers. Therefore, the above facts highlight the vital importance of new methods to identify putative driver missense mutations and separate them from passenger mutations even within known cancer genes.
Many computational methods have been developed to predict whether a missense mutation is generally deleterious or pathogenic, including GERP++ (19), PhastCons (pC100way) (20), PhyloP (pP100way) (21) and SiPhy (22), FATHMM (23), fitCons (24), LRT (25), MutationAssessor (26), MutationTaster (27), PolyPhen2-HDIV (28), PolyPhen2-HVAR (28), PROVEAN (29), SIFT (30) and VEST3 (31), and CADD (32), DANN (33), Eigen (34), FATHMM-MKL (35), GenoCanyon (36), M-CAP (37), MetaLR (38), MetaSVM (38) and REVEL (39). Previously, we developed an integrated database of predictive scores from 23 in silico algorithms (40) and systematically evaluated their performances (41) to consolidate and compare these functional annotations for missense variants in the human genome. Moreover, our previous studies demonstrated that combining functional evidences could improve the predictions of pathogenicity of genetic variants in human diseases (41–43). For example, weighted combination of REVEL (39) and VEST3 (31) (i.e. ReVe) could achieve better performance than individual methods though ReVe had low positive predictive value and low specificity in predicting cancer oncogenic mutations (41). Therefore, integrating these pathogenicity scores by appropriate algorithm may facilitate an automated genome-wide prioritization of functional and clinical impacts of somatic missense mutations.
Despite improvements in the understanding of pathogenic mutations, there is still no efficient and convenient tool to identify driver mutations for cancer therapeutic targets (44). Due to the above facts, we aimed to predict driver mutations more accurately by systematically integrating the large amount of newly available data and existing scores in predicting the pathogenicity. In current study, we developed a novel and efficient cancer driver annotation tool, AI-Driver, which integrates validated training data and pathogenic features to predict a driver score for each possible missense mutation (Figure 1A). We embedded and compared seven ensemble learning algorithms, and found XGBoost has the best performance. We demonstrated that AI-Driver based on XGBoost achieved better sensitivity and specificity than other tools in predicting driver mutations in four independent testing datasets. We illustrated the discriminatory abilities and applicable scenarios of the proposed models by independent datasets and case studies.
Figure 1.

Models and features included in the AI-Driver model. (A) Framework of AI-Driver showing the algorithms for detecting driver mutations. (B) Correlation among the individual features, ordered by hierarchical clustering. The heatmap illustrates the Spearman rank correlation coefficients between features for the AI-Driver training datasets. (C) Relative importance of individual features. Mean absolute SHAP value was plotted for each tool.
MATERIALS AND METHODS
The eXtreme gradient boosting model
In our model, we employed gradient tree boosting algorithm which is a special form of gradient boosting machine and predicting by combining the results of multiple weak learners. XGBoost classifier was introduced as the implementation of gradient tree boosting algorithm. XGBoost stands for eXtreme Gradient Boosting, which combines weak learners (decision trees (DTs) ) to achieve stronger overall class discrimination (45). XGBoost (XGBT) is a scalable end-to-end tree boosting system and has achieved the state-of-art performance in plenty of tasks. XGBoost learned a series of DTs to classify the labeled training data. Each DT comprises a series of rules that semi-optimally split the training data. Its sparsity-aware split search approach makes it suitable for our dataset where missing values commonly appear (46). Successive trees that ‘correct’ the errors in the initial tree were learned to improve the classification of positive and negative training examples. Briefly, by adopting second-order Taylor expansion, adding regularization, introducing sparsity-aware algorithm and supporting parallel computation, XGBoost is able to achieve better performance as well as faster training process compared with gradient boosting DT.
Model constructions and 10-fold cross-validation
XGBoost algorithm was compared with other machine learning algorithms including DT, gradient boosting tree (GBT), support vector machine (SVM), AdaBoost (ABT), multi-layer perceptron (MLP) and random forest (RF) (Supplementary File 1). These additional models were constructed by using the scikit-learn toolkit (47). Feature contribution was measured by SHapley Additive exPlanation (SHAP) approaches (48). Grid search based on 10-fold cross-validation on training set were performed in order to tune the optimal hyper-parameters. The weight of positive samples was adjusted according to the ratio of two classes, while training datasets were tuned with the unbalanced positive and negative samples.
Training features
AI-Driver incorporated a total of 23 individual pathogenicity prediction scores as predictive features (40). These 23 in-silico algorithms or tools were (i) five conservation methods: GERP++ (19), PhastCons (20), PhyloP (21), LRT (25) and SiPhy (22); (ii) nine function-prediction methods: FATHMM (23), fitCons (24), MutationAssessor (26), MutationTaster (27), PolyPhen2-HDIV (28), PolyPhen2-HVAR (28), PROVEAN (29), SIFT (30) and VEST3 (31) and (iii) nine ensemble methods: CADD (32), DANN (33), Eigen (34), FATHMM-MKL (35), GenoCanyon (36), M-CAP (37), MetaLR (38), MetaSVM (38) and REVEL (39). Missing feature values for a given variant were assigned by the average value of the non-missing values from its k = 40 nearest neighboring variants; when more than 50% of features were missing for a given variant, it was assigned to its overall mean across all variants. Feature scores of all possible single nucleotide variants were artificially generated by these 23 pathogenicity tools. Genomic positions of all variants were based on GRCh37/hg19. All scores were ranked in each set of features and normalized by PHRED-scaled score (-10*log10(rank/total)). Since lower predicting score represents higher pathogenicity for SIFT, FATHMM, LRT and PROVEAN, predicting scores of the four tools were ranked from high to low while the predicting scores for the rest of tools were ranked from low to high.
Somatic mutations from TCGA and ICGC
In total, 2 560 366 somatic mutations were collected from the entire 10 769 tumor sample dataset by harmonizing the results of seven algorithms, yielded by the uniform analysis of all TCGA exome data generated by the Multi-Center Mutation-Calling in Multiple Cancers (MC3) network (49) (https://api.gdc.cancer.gov/data/1c8cfe5f-e52d-41ba-94da-f15ea1337efc). All mutations that passed the MC3 filter criteria were included and samples marked with inconsistent pathology were excluded. Our final dataset consisted of 1651 136 missense mutations from 10 425 samples. Moreover, we curated 1 622 885 somatic missense mutations from 22 454 samples from the ICGC Data Portal (https://dcc.icgc.org/).
Neutral mutations from normal population
Over 270 million variants in the gnomAD v2.1 dataset (https://gnomad.broadinstitute.org) have been widely used as a resource for allele frequency estimates in the context of rare disease (50,51). In total, 5 292 712 missense variants were aggregated from 125 748 control-only reference individuals after the removal of mutations not passing the filtering criterion.
Training mutation datasets
First, 2616 driver and 592 passenger mutations were collected from OncoKB database (52) (Supplementary Table S1). Second, 256 driver and 475 passenger mutations were collected from FASMIC database (53) (Supplementary Table S2). Third, 15 375 neutral variants with allele frequency larger than 10% were collected from normal populations in gnomAD database (50) (Supplementary Table S3). All single nucleotide substitutions (SNSs) were selected for training when a given amino acid changes corresponded to multiple SNSs at the genetic level. For passenger training variants, all variants that curated in ICGC project (7), TCGA project (8), COSMIC database (54) and ClinVar database (55) were excluded. For both the driver and passenger training mutations, all variants that had previously been used to train individual component features in the AI-Driver model, specifically, PolyPhen-2 (28), MutationTaster (27), FATHMM (23), REVEL (39), VEST (31) and M-CAP (37), were also excluded. After the removal of the duplicates and overlaps between passengers and drivers, a dataset consisting of functionally validated 6009 cancer driver mutations (Supplementary Table S4) and 16 724 passenger and neutral variants (Supplementary Table S5) were annotated by VarCards (40). To evaluate the impact of allele frequency of neutral variants on the performance, we collected 4 489 231, 37 607, 20 607, 5388, 4707, 4622, 1001, 764, 524 neutral variants with allele frequency (AF) of <0.1%, >0.1%, 0.1–0.3%, 0.3–0.5%, 0.5–1%, 1–3%, 3–5%, 5–10% and >10% from control samples in gnomAD database (50). In this case, 4 526 838 neutral variants with all allele frequency were annotated with 23 feature scores by VarCards (40).
Construction of test datasets
First, 763 driver mutations and 48 passenger variants were curated from the ICGC database (https://dcc.icgc.org/search/m) with clinical significance of ‘pathogenic/likely pathogenic’ and ‘benign/likely benign mutations’, respectively (7) (Supplementary Table S6). Second, 2338 driver mutations were collected from Pan-Cancer Analysis of Whole Genomes (PCAWG) project (56) (Supplementary Table S7). Third, 2915 validated oncogenic mutations were sourced from CGI database (57) (Supplementary Table S8). Fourth, 849 driver mutations and 140 passenger variants were collected from a previous study (15) published in Genome Biology (GB) (Supplementary Table S9). Missense passenger variants from ICGC database and 916 somatic passenger variants (Supplementary Table S10) from dbCPM database (58) were used as negative controls. All SNSs were selected for testing when a given amino acid changes corresponded to multiple SNSs at the genetic level. The four independent test datasets that did not overlap with either the AI-Driver training data or the training data for the component features of AI-Driver were annotated with 23 feature scores by VarCards (40).
Classification of genes and mutations
All genes from four testing datasets were classified into two classes, oncogene and TSG, according to gene annotation from OncoKB database (52). We removed the genes annotated to both oncogene and TSG. To figure out the common or rare cancer genes, we collected 1 163 873 extreme missense mutations (LoF or ReVe score > 0.7) of 10 247 pancancer samples from TCGA project. Waterfall plots of pan-cancer showed that top 33 genes are highly mutated in pancancer with frequency of >4% (Supplementary Figure S1). We defined these top 33 genes as common cancer genes while the rest cancer genes are defined as rare/long-tail cancer genes. On the other hand, 2546 somatic and 369 germline driver mutations were classified according to the annotation in CGI database (57) (Supplementary Table S8). A total of 759 and 188 testing mutations of TP53 and BRCA1 were selected from four testing datasets, respectively (Supplementary Table S11).
APOBEC-generated mutations and circulating tumor DNA variants
We collected 239 APOBEC-generated mutations outside of DNA hairpin loops and 759 somatic mutations inside of DNA hairpin loops from research by Michael S. Lawrence et al. (59) (Supplementary Table S12). In addition, we collected 147, 203 and 94 circulating tumor DNA variants (ctDNA) from breast, lung and prostate metastatic cancers, respectively (60) (Supplementary Table S13). All of these mutations were annotated with 23 feature scores by VarCards (40).
Seven measurements for performance evaluation
Receiver operating characteristic curve (ROC) analysis were performed to evaluate the performance of each method, mainly by the area under the ROC curve (AUC) value. Other six criteria were also employed to further confirm the evaluation including accuracy, precision, sensitivity (recall), specificity, F1 score, Mathew correlation coefficient (MCC). These seven measurements were calculated using Scikit-learn (47), and some of the above measures were derived from the parameters of true positive (TP), true negative (TN), false positive (FP) and false negative (FN), as shown below.
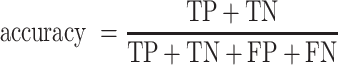 |
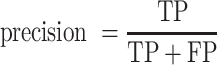 |
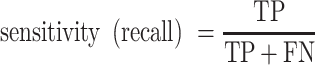 |
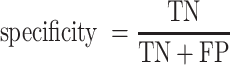 |
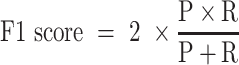 |
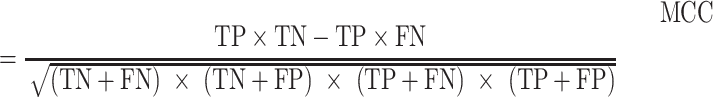 |
RESULTS
Characterization and selection of AI-Driver features
The AI-Driver likelihood score is a combination of pathogenicity predictions from 23 individual features, including 5 conservation scores, 9 functional scores and 9 ensemble scores. Figure 1B showed the correlation among individual features. The conservation scores were almost all highly (R > 0.7) to moderately correlated (0.5 < R < 0.7). Five functional scores (PolyPhen-2 HDIV, PolyPhen-2 HVAR, MutationAssessor, SIFT and PROVEAN) were almost all highly correlated (R > 0.7). Eigen and fathmm-MKL were also highly correlated with several conservation scores, such as CADD and DANN. In contrast, MuTaster had low correlations (R < 0.3) with all other prediction scores, and REVEL and VEST3 had low to moderate correlations with other scores. The five most important features of the AI-Driver model were M-CAP, MetaLR, VEST3, LRT and MetaSVM (Figure 1C). The relationships among different features were showed in clusters with Euclidean distance (Supplementary Figure S2A). We further employed SHAP (48) to measure the importance of these features for model output and the results showed individual feature had distinctly intrinsic predictive ability in AI-Driver ensemble model (Supplementary Figure S2B). Together, our results showed that many input features are strongly correlated and some of them seem to be more useful than others in AI-Driver model.
To investigate whether some features could be removed, we separated input features into three groups: conversation, function-based and ensemble. Then,we cross-validated the three independent groups by XGBoost, respectively, and found that each model has specific superiorities and defects according to different performance measurements (Figure 2A). In details, the model based on conservation features has higher specificity but has lower sensitivity and accuracy. And, the model based on function features has higher AUC value but has lower sensitivity and accuracy compared. While, the model based on ensemble features has higher accuracy, F1 score, MCC and sensitivity but has lower AUC and specificity. In general, all of the three models have comparable precision scores. Therefore, our results suggested that at least one kind of these features in each group are needed if we want to build up a robust model. Next, to investigate the suitable feature number for XGBoost model,we selected top 5, 10,15, 20 and 23 features based on the mean SHAP values (Figure 1C) for model construction. Totally, we constructed five XGBoost models and found model with 23 features has the best performance compared with the other four models according to any measurements of the accuracy, AUC value, F1 score, MCC, precision value, sensitivity and specificity (Figure 2B). We also observed that the performance increased when we added more features although this tendency was not observed in AUC values. Therefore, all 23 features were needed to be included in the model construction.
Figure 2.

AI-Driver outperforms existing pathogenicity likelihood metrics. (A) Performance plot for models based on conservation, function based and ensemble features using mutations from training datasets. (B) Performance plot for models based on different number of features using training datasets. Features were selected by top 5, top 10, top 20 and top 23 features based on SHAP values. (C) ROC for 23 popular pathogenicity metrics for training datasets shown with AUC values. (D) ROC for seven machine learning algorithms built in AI-Driver to predict driver missense variants shown with AUC values. AUC represents area under the receiver operating characteristics curve.
The effect of PHRED-scaled process on the performance
To evaluate the effect of PHRED-scaling process on the performance of training features, the AUC values before and after PHRED-scaling process were calculated. As we expected, a majority of the performances of training features were greatly improved after the PHRED-scaling process, especially for SIFT, LRT, FATHMM and PROVEAN (Figure 2C and Supplementary Figure S3A). SIFT, LRT, FATHMM and PROVEAN had very low AUC values of 0.168, 0.267, 0.186 and 0.184, respectively, before PHRED-scaled process, and strikingly, the AUC values of them were significantly improved with 0.831, 0.736, 0.814 and 0.817 after PHRED-scaling process, respectively. Moreover, we found the performance of AI-Driver was slightly improved by PHRED-scaling process, which could be considered to prevent overfitting (Supplementary Figure S3B). Therefore, we recommend users to employ PHRED-scaling process whether do training or testing with AI-Driver workflow.
AI-Driver outperforms existing pathogenicity scores
AI-Driver discriminated driver and neutral mutations well, with an overall AUC of 0.9964, which estimated using grid search method based on 10-fold cross-validation on training set. The AUC score of AI-Driver was significantly higher than any of its constituent features (maximum P < 10−15 for any pairwise comparison), such as, MetaLR (AUC = 0.959), VEST3 (AUC = 0.937), REVEL (AUC = 0.926) and MetaSVM (AUC = 0.913). AUC scores for the other individual prediction tools ranged from 0.571 to 0.860 and tended to be higher for functional predictors except MuTaster (0.766–0.937) than for conservation scores (0.736–0.780). The AUC for AI-Driver (maximum P < 10−10 for any pairwise comparison) was also significantly better than that for ensemble methods (ranging from 0.638 to 0.959) among which MetaLR (AUC = 0.959), REVEL (AUC = 0.926) and MetaSVM (AUC = 0.913) had the second highest AUCs (Figure 2C). Moreover, AI-Driver model based on XGBoost outperformed models based on DT, SVM, RF, ABT, MLP and GBT which achieved AUC values of 0.9061, 0.9771, 0.9959, 0.9907, 0.9801, 0.9946, respectively (Figure 2D and Supplementary Table S14). In addition, AI-Driver based on XGBoost had a higher accuracy value of 0.9745 than predictions based on DT, SVM, RF, ABT, MLP and GBT which achieved accuracy values of 0.8226, 0.9417, 0.9740, 0.9609, 0.9354 and 0.9734, respectively (Supplementary Table S14).
We next compared the performance of AI-Driver with that of other methods for discriminating pathogenic mutations from putatively neutral variants with AFs ranging from very rare (<0.1%) to common (>5%) variants. The result showed that all of the constituent methods tended to have a worse ability to discriminate driver mutations from rare neutral variants than from common neutral variants (Figure 3A and Supplementary Table S15). However, AI-Driver had superior discriminatory ability to neutral variants even to rare variants with AF < 0.5% (Figure 3A). In addition, AI-Driver appeared to be more robust to changes of AF of negative training variant than other methods. The AUC range was narrowest for AI-Driver (0.9677–0.9999) and widest for fathmm-MKL (0.6538–0.9140) which appeared to be the most sensitive to AF (Supplementary Table S15). For neutral variants with AF < 0.1%, AI-Driver had the lowest AUC value of 0.9944 compared with that derived from neutral variants with other AF range (Supplementary Table S16). Then, three models were constructed for the following testing based on somatic passenger mutations coupled with germline neutral variants with AF < 0.1%, 0.1% ≤ AF < 1% and all AFs from gnomAD.
Figure 3.

Performance of AI-Driver under different impact factors. (A) Performance of AI-Driver and its constituent methods using neutral mutations with different AFs of <0.1%, >0.1%, 0.1–0.3%, 0.3–0.5%, 0.5–1%, 1–3%, 3–5%, 5–10% and >10%. (B) Evaluation results for AI-Driver on mutations in common cancer genes and rare cancer genes from testing datasets. (C) Evaluation results for AI-Driver on germline and somatic mutations from CGI testing dataset. (D) Evaluation results for AI-Driver on mutations in oncogenes and TSGs defined by OncoKB from testing datasets.
Performance evaluation in four independent test datasets
The performance of AI-Driver and 23 integrated scores on four common testing datasets were evaluated, including driver mutations from ICGC, Pancancer, CGI and GB coupled with passenger mutations from ICGC and dbCPM. First of all, three models based on neutral variants with AF < 0.1%, 0.1% ≤ AF < 1% and all AFs were employed to test the four independent testing datasets. AI-Driver based on neutral variants with 0.1% ≤ AF < 1% performed best in all of the four datasets and was used for the following analysis (Supplementary Table S17). Then, we compared the performance of AI-Driver by testing it on testing mutations in common versus rare cancer genes. We found AI-Driver appeared to be more robust to identify driver mutations in rare cancer genes than common cancer genes (Figure 3B). Third, we compared the performance of AI-Driver by testing it on germline versus somatic driver mutations from CGI database (57). We found AI-Driver appeared to be more powerful to identify somatic driver mutations than germline driver mutations (Figure 3C). At last, we separated the testing mutations according to gene annotation of oncogenes and TSGs from OncoKB database. Testing and evaluation processes of four datasets from ICGC, Pancancer, CGI and GB revealed that the performances of AI-Driver in TSGs were elevated a little bit compared with these in oncogenes (Figure 3D and Supplementary Figure S4).
In test set 1 from ICGC, the relative performance of all 24 predictors was similar to that observed in the training dataset (Figure 4A and Supplementary Table S18). AI-Driver had the best performance with AUC of 0.985 while MuTaster had the worst performance with AUC of 0.572. Most of the ensemble methods, including M-CAP, REVEL, VEST3, fathmm-MKL, Eigen and MetaSVM also had great performance with AUC of >0.9 (Supplementary Table S18). In test set 2 from Pancancer, AI-Driver had the best performance with AUC of 0.942 (Figure 4B and Supplementary Table S19) and only M-CAP had great performance with AUC of 0.935. It is worth noting that FATHMM, SIFT, LRT and PROVEAN were not well performed on predicting drivers in Pancancer dataset (Supplementary Table S19). In test set 3 from CGI, AI-Driver, M-CAP and VEST3 had better performance with AUCs of 0.998, 0.952 and 0.901, respectively (Figure 4C and Supplementary Table S20). While fitCons and MuTaster had the worst performance with AUC of 0.678 and 0.55, respectively (Supplementary Table S20). In test set 4 from GB, AI-Driver performed best with AUC of 0.814, which was much higher than AUCs of other methods (Figure 4D and Supplementary Table S21). It is worth noting that most of other predictors were not well performed in GB dataset with AUC of <0.65 (Supplementary Table S21). The above results showed that the ensemble model AI-Driver outperformed individual pathogenicity predicting tools on all of the four evaluations.
Figure 4.

Evaluation of individual prediction tools on four independent testing datasets. (A) Performance on ICGC testing dataset. (B) Performance on Pancancer testing dataset. (C) Performance on CGI testing dataset. (D) Performance on GB testing dataset. AUPR, area under the precision recall curve; AUROC, area under the receiver operating characteristics curve.
Comparison of AI-Driver with other cancer-specific methods
The performance of AI-Driver, CanDrAplus (18) and CHASMplus (10) on the four testing datasets were also evaluated. AI-Driver model was based on neutral variants with 0.1% ≤ AF < 1%. Predictions of CanDrAplus were based on ‘GENERAL’ model and predictions of CanDrAplus were performed through OpenCRAVAT annotation tool with all cancer types. In test set 1 from ICGC, AI-Driver performed best with AUC of 0.985 while CanDrAplus and CHASMplus had AUC of 0.815 and 0.879, respectively (Figure 5A). In test set 2 from Pancancer, AI-Driver had the best performance with AUC of 0.942 while CanDrAplus and CHASMplus had AUC of 0.844 and 0.875, respectively (Figure 5B). In test set 3 from CGI, AI-Driver performed best with AUC of 0.998, far superior to CanDrAplus and CHASMplus which had AUC of 0.835 and 0.894, respectively (Figure 5C). In test set 4 from GB, AI-Driver had comparable AUC (0.814) with that of CanDrAplus (AUC = 0.817) though CanDrAplus surpassed AI-Driver a little bit (Figure 5D). However, CHASMplus had the worst performance with AUC of 0.668. In Conclusion, our results showed that AI-Driver outperformed two state-of-the-art tools for predicting cancer driver mutations on most of the four evaluations.
Figure 5.

Comparison of AI-Driver with three cancer-specific methods. (A) ROC and AUC of three cancer-specific methods for ICGC test dataset. (B) ROC and AUC of three cancer-specific methods for Pancancer test dataset. (C) ROC and AUC of three cancer-specific methods for CGI test dataset. (D) ROC and AUC of three cancer-specific methods for GB test dataset. ROC represents receiver operating characteristic curve and AUC represents area under the receiver operating characteristics curve.
Interpretation of AI-Driver scores
The AI-Driver scores for an individual variant ranged from zero to one, reflecting the proportion of trees, classified the mutation as oncogenic, in the all GBT. In total, 1 622 885, 1 651 136, 1 645 383 and 1882 somatic mutations from ICGC (7), TCGA (8), COSMIC (54) and ClinVar (55) database were collected, respectively. AI-Driver was applied to annotate all of the somatic mutations and the scores for each set of somatic mutations exhibited bimodal distribution in which most of them were either >0.9 or <0.1 (Figure 6). For somatic mutations from ICGC database, 72.47, 88.35 and 81.89% of the AI-Driver scores were <0.1 with minimum recurrence of 1, 5 and 10, respectively (Figure 6A and Supplementary Table S22). 9.96, 4.42 and 12.56% of the AI-Driver scores were >0.9 with minimum occurrence of 1, 5 and 10, respectively. For somatic mutations from TCGA database, 71.70, 66.82 and 29.18% of the AI-Driver scores were <0.1 with minimum occurrence of 1, 5 and 10, respectively (Figure 6B and Supplementary Table S22). And 10.27, 17.00 and 62.63% of the AI-Driver scores were >0.9 with minimum recurrence of 1, 5 and 10, respectively. For somatic mutations from COSMIC database, 69.49, 67.52 and 67.63% of the AI-Driver scores were <0.1 with minimum occurrence of 1, 5 and 10, respectively (Figure 6C and Supplementary Table S22). About 11.03, 12.66 and 13.27% of the AI-Driver scores were >0.9 with minimum recurrence of 1, 5 and 10, respectively. In summary, most of driver mutations were identified from ICGC, TCGA and COSMIC database with AI-Driver score ≥ 0.9. Moreover, we also introduced AI-Driver to predicting somatic mutations from ClinVar database and found 58.19% of these mutations had AI-Driver score ≥ 0.9 (Figure 6D and Supplementary Table S22). Our results showed that AI-Driver could clearly discriminate driver mutations from neutral mutations and its scores exhibited bimodal distribution in which most of them were either >0.9 or <0.1.
Figure 6.

Distribution of AI-Driver scores for somatic mutations from four public databases. (A). Distribution of AI-Driver scores for somatic mutations from ICGC database. (B) Distribution of AI-Driver scores for somatic mutations from TCGA database. (C) Distribution of AI-Driver scores for somatic mutations from COSMIC database. (D) Distribution of AI-Driver scores for somatic mutations from ClinVar database.
To obtain the optimized AI-Driver score for identifying driver mutations, we used three cutoffs with AI-Driver score of >0.90, >0.95 and >0.99 to investigate the overlap of the genes harboring driver mutations with known driver genes. We found AI-Driver genes harboring mutations with score > 0.99 have significant overlap with known driver genes in OncoKB database (hypergeometric P < 0.01, Supplementary Figure S5A and Table S23). Moreover, the functional enrichments by MetaScape (61) showed these driver genes are highly related to carcinogenesis (Supplementary Figure S5B). In addition, we identified 1541 and 906 new driver mutations with AI-Driver score >0.99 and recurrence >2 among patients from ICGC and TCGA datasets, covering 1072 and 635 genes, of which 602 and 335 are novel drivers and the remaining are known driver genes, from ICGC and TCGA datasets, respectively (Supplementary Table S24).
Application of AI-Driver scores
To test the usability of AI-Driver in analyzing canonical cancer driver genes, we collected and tested the driver mutations and passenger mutations of TP53 and BRCA1 from testing datasets. Performance evaluation showed that AI-Driver had great performance in both TP53 and BRCA1 genes with AUC value of 0.9645 and 0.9817, respectively (Figure 7A and B). To test the usability of AI-Driver in hotspot mutations in cancer driven by APOBEC3A (59), these APOBEC3A-driven mutations were tested by AI-Driver model built on curated drivers and passengers (canonical model). However, we achieved moderate performance according to the results of evaluation metrics (AUC = 0.7567; Supplementary Figure S6). Hence, we employed 80% the APOBEC3A-driven mutations to rebuild a new training model (APOBEC3A model) and used the rest 20% of APOBEC3A-driven mutations to do the testing. Strikingly, AI-Driver achieved superior and stable performance (AUC = 0.9692; Supplementary Figure S6). Given that even well-established cancer-driver genes are thought to contain a mixture of driver and passenger mutations, our results indicated that APOBEC3A-driven driver and passenger mutations were not well-defined though mutational hotspots at nonoptimal sites are enriched in known cancer driver genes (59). Therefore, AI-Driver could discriminate hotspot mutations from non-hotspot mutations when we rebuilt the training model by using hotspot/non-hotspot mutations instead of driver/passenger mutations. To demonstrate the usability of AI-Driver in analyzing real cancer genomics data, we collected 128, 187 and 84 biopsy-matched driver mutations of circulating tumor DNA (ctDNA) variants from patients with breast, lung and prostate metastatic cancers (60), respectively (Supplementary Table S13). Our results showed that AI-Driver had great performance in predictions of ctDNA driver mutations from breast, lung and prostate cancer with AUC values of 0.8949, 0.8997 and 0.9176, respectively (Figure 7C and D). Therefore, AI-Driver has high sensitivity and specificity in predicting driver mutations not only in canonical cancer driver genes but also in personal ctDNA variants by liquid biopsy.
Figure 7.

Performance of AI-Driver for individual genes and circulating tumor DNA variants. (A) Evaluation results for AI-Driver on TP53, BRCA1 and APOBEC3A-driven mutations. (B) ROC and AUC of AI-Driver on TP53, BRCA1 and APOBEC3A-driven mutations. (C) Evaluation results for AI-Driver on circulating tumor DNA (ctDNA) mutations in breast, lung and prostate metastatic cancers. (D) ROC and AUC of AI-Driver on ctDNA mutations in breast, lung and prostate metastatic cancers. ROC represents receiver operating characteristic curve and AUC represents area under the receiver operating characteristics curve.
DISCUSSION
Missense mutations are the most common somatic mutations found in cancer genomes and increasing number of missense mutations have been recognized as clinically actionable (16). AI-Driver is an ensemble method for predicting the driver status of missense somatic mutations and it also outperformed its individual constituent prediction tools, as expected for ensemble methods (39). We have shown that AI-Driver consistently has the best overall performance as compared to existing methods, particularly for distinguishing driver mutations from uncommon neutral missense mutations with an AF below 0.1%. AI-Driver scores for all possible missense variants have been pre-computed to facilitate convenient application in precision cancer medicine. Thus, AI-Driver addresses the need for a ‘driverness’ prediction tool with improved accuracy for interpreting somatic missense mutations in personal cancer genomes.
The AI-Driver method has several advantages compared with previous methods. First of all, AI-Driver was trained and tested on recently validated driver, passenger and neutral mutations that could closely resemble novel variants discovered by exome sequencing studies in the future. We assumed that the high frequency germline variants in human normal populations that are not known to be associated to cancer were a good representative of somatic passenger variants. However it has to be noted that common germline variants with high frequency in normal populations may also confer cancer risk, and therefore may not represent somatic passenger mutations in cancer cells. This is the reason why we also included somatic passenger mutations from OncoKB and FASMIC databases in our negative training set. Second, AI-Driver incorporated many more individual predictors than prior ensemble methods, including both M-CAP and MetaLR, which were among the most important features in the XGBoost model. Third, all variants used to train any of the component predictors in AI-Driver were carefully removed to reduce overfitting and inflated performance estimates. Fourth, the speed of AI-Driver based on XGBoost was much faster than previous ensemble learning methods. XGBoost is able to achieve better performance as well as faster training process compared with GBT by adopting second-order Taylor expansion, adding regularization, introducing sparsity-aware algorithm and supporting parallel computation.
Ensemble learning methods generally surpass unsupervised methods when high-quality training data of appropriate type and quantity are available (37). Consistent with recent study on prediction of non-coding regulatory variants (46), ensemble learning methods including XGBoost, RF and GBT exhibit better performance than conventional SVM classifier in all training datasets. Moreover, the model trained by XGBoost algorithm shows the best prediction performance. Here we show that the AI-Driver model based on XGBoost outperforms other approaches at classifying somatic missense mutations when trained with a carefully curated set of driver and neutral mutations. However, we could not perform cancer type-specific training and testing due to the limited number of validated driver mutations in a specific type of cancer. Nevertheless, AI-Driver is easily adopted to develop cancer type-specific model as the increasing of validated driver mutations in any specific tumor type.
In summary, AI-Driver is an ensemble method that outperforms existing cancer-specific methods for distinguishing driver mutations from passenger mutations. AI-Driver can be used to prioritize the most likely driver mutations in the sea of rare somatic mutations that are increasingly discovered as sequencing studies. For example, AI-Driver scores can be used to interpret somatic mutations identified by ICGC project, TCGA project and personal ctDNA variants by liquid biopsy. Pre-computed AI-Driver scores for all possible human missense variants for human hg19 genome are freely available at http://aidriver.maolab.org/. AI-Driver has the potential application in different areas of human cancer studies, such as identifying driver mutation in personal cancer genomes, discovering targeting sites for cancer therapeutic treatments and predicting tumor biomarkers for early diagnosis by liquid biopsy.
DATA AVAILABILITY
The AI-Driver models are implemented in Python. Integrated datasets, source codes, collected training/testing sets, analysis scripts for the results of this manuscript are available at https://github.com/hatchetProject/AI-Driver. The pre-computed AI-Driver scores for all genome-wide possible missense variants are available at http://aidriver.maolab.org/.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Chenghang Du in the Beijing Institutes of Life Science, Chinese Academy of Sciences for his help in maintaining the high-performance computing systems. We also thank Luoyuan Xiao from Department of Computer Science and Technology, Tsinghua University and Dr Qi Liu from Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School for their help in the website development of AI-Driver. Simultaneously, we thank the members from the Biobank of the Second Affiliated Hospital of Nanchang University for their help in the verification of the training and testing data.
Author contributions: F.B.M. and Z.S.S. designed the study and interpreted the results. H.X.W. established AI-Driver software and database. F.B.M. and T.W. collected the training and testing datasets. T.W. implemented the model and performed the evaluation. F.B.M. and X.L.Z. wrote the manuscript. H.H.W. and M.C.Y. refined the figures and revised the manuscript. Z.S.S. provided valuable suggestions on the project.
Contributor Information
Haoxuan Wang, Center of Basic Medical Research, Institute of Medical Innovation and Research, Peking University Third Hospital, Beijing 100191, China; Computer Science and Engineering Department, Shanghai Jiao Tong University, Shanghai 200240, China.
Tao Wang, Center for Medical Genetics & Hunan Key Laboratory of Medical Genetics, School of Life Sciences, Central South University, Changsha, Hunan 410083, China; Beijing Institutes of Life Science, Chinese Academy of Sciences, Beijing 100101, China.
Xiaolu Zhao, Department of Pathology, University of Michigan, Ann Arbor, MI 48109, USA.
Honghu Wu, Department of Science and Technology, Second Affiliated Hospital of Nanchang University, Nanchang 330006, China.
Mingcong You, Baiyining Medicine, Beijing 102200, China.
Zhongsheng Sun, Beijing Institutes of Life Science, Chinese Academy of Sciences, Beijing 100101, China.
Fengbiao Mao, Center of Basic Medical Research, Institute of Medical Innovation and Research, Peking University Third Hospital, Beijing 100191, China; Department of Pathology, University of Michigan, Ann Arbor, MI 48109, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
National Natural Science Foundation of China [31872237]; National Key R&D Program of China [2016YFC0900400]; National High-tech R&D Program of China [2012AA02A210].
Conflict of interest statement. None declared.
REFERENCES
- 1. Li X., Shi L., Wang Y., Zhong J., Zhao X., Teng H., Shi X., Yang H., Ruan S., Li M. et al.. OncoBase: a platform for decoding regulatory somatic mutations in human cancers. Nucleic Acids Res. 2019; 47:D1044–D1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Teng H., Wei W., Li Q., Xue M., Shi X., Li X., Mao F., Sun Z.. Prevalence and architecture of posttranscriptionally impaired synonymous mutations in 8,320 genomes across 22 cancer types. Nucleic Acids Res. 2020; 48:1192–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hiley C., de Bruin E.C., McGranahan N., Swanton C.. Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol. 2014; 15:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aran D., Sirota M., Butte A.J.. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015; 6:8971–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kandoth C., McLellan M.D., Vandin F., Ye K., Niu B., Lu C., Xie M., Zhang Q., McMichael J.F., Wyczalkowski M.A. et al.. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lawrence M.S., Stojanov P., Mermel C.H., Robinson J.T., Garraway L.A., Golub T.R., Meyerson M., Gabriel S.B., Lander E.S., Getz G.. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014; 505:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. International Cancer Genome, C. Hudson T.J., Anderson W., Artez A., Barker A.D., Bell C., Bernabe R.R., Bhan M.K., Calvo F., Eerola I. et al.. International network of cancer genome projects. Nature. 2010; 464:993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Research, N. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cho A., Shim J.E., Kim E., Supek F., Lehner B., Lee I.. MUFFINN: cancer gene discovery via network analysis of somatic mutation data. Genome Biol. 2016; 17:129–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tokheim C., Karchin R.. CHASMplus reveals the scope of somatic missense mutations driving human cancers. Cell Syst. 2019; 9:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bose R., Kavuri S.M., Searleman A.C., Shen W., Shen D., Koboldt D.C., Monsey J., Goel N., Aronson A.B., Li S.Q. et al.. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013; 3:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Toy W., Shen Y., Won H., Green B., Sakr R.A., Will M., Li Z.Q., Gala K., Fanning S., King T.A. et al.. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013; 45:1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Robinson D.R., Wu Y.M., Vats P., Su F.Y., Lonigro R.J., Cao X.H., Kalyana-Sundaram S., Wang R., Ning Y., Hodges L. et al.. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013; 45:1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Horn H., Lawrence M.S., Chouinard C.R., Shrestha Y., Hu J.X., Worstell E., Shea E., Ilic N., Kim E., Kamburov A. et al.. NetSig: network-based discovery from cancer genomes. Nat. Methods. 2018; 15:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martelotto L.G., Ng C.K.Y., De Filippo M.R., Zhang Y., Piscuoglio S., Lim R.S., Shen R.L., Norton L., Reis-Filho J.S., Weigelt B.. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome Biol. 2014; 15:484–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bailey M.H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M.C., Kim J., Reardon B. et al.. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018; 173:371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun Y., Zhou B., Mao F., Xu J., Miao H., Zou Z., Phuc Khoa L.T., Jang Y., Cai S., Witkin M. et al.. HOXA9 reprograms the enhancer landscape to promote Leukemogenesis. Cancer Cell. 2018; 34:643–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mao Y., Chen H., Liang H., Meric-Bernstam F., Mills G.B., Chen K.. CanDrA: Cancer-specific driver missense mutation annotation with optimized features. PLoS One. 2013; 8:e77945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S.. Identifying a high fraction of the human genome to be under selective constraint using GERP plus. PLoS Comput. Biol. 2010; 6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M.M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. et al.. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005; 15:1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Siepel A., Pollard K.S., Haussler D.. New methods for detecting lineage-specific selection. Lect. Notes Comput. Sci. 2006; 3909:190–205. [Google Scholar]
- 22. Garber M., Guttman M., Clamp M., Zody M.C., Friedman N., Xie X.H.. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics. 2009; 25:I54–I62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shihab H.A., Gough J., Cooper D.N., Stenson P.D., Barker G.L.A., Edwards K.J., Day I.N.M., Gaunt T.R.. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum. Mutat. 2013; 34:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gulko B., Hubisz M.J., Gronau I., Siepel A.. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat. Genet. 2015; 47:276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chun S., Fay J.C.. Identification of deleterious mutations within three human genomes. Genome Res. 2009; 19:1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reva B., Antipin Y., Sander C.. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011; 39:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jian X.Q., Boerwinkle E., Liu X.M.. In silico tools for splicing defect prediction: a survey from the viewpoint of end users. Genet. Med. 2014; 16:497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R.. A method and server for predicting damaging missense mutations. Nat. Methods. 2010; 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi Y., Chan A.P.. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015; 31:2745–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kumar P., Henikoff S., Ng P.C.. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009; 4:1073–1082. [DOI] [PubMed] [Google Scholar]
- 31. Carter H., Douville C., Stenson P.D., Cooper D.N., Karchin R.. Identifying mendelian disease genes with the variant effect scoring tool. BMC Genomics. 2013; 14:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014; 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quang D., Chen Y.F., Xie X.H.. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2015; 31:761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ionita-Laza I., McCallum K., Xu B., Buxbaum J.D.. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016; 48:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shihab H.A., Rogers M.F., Gough J., Mort M., Cooper D.N., Day I.N.M., Gaunt T.R., Campbell C.. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015; 31:1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu Q.S., Hu Y.M., Sun J.H., Cheng Y.W., Cheung K.H., Zhao H.Y.. A Statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci. Rep. 2015; 5:10576–10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jagadeesh K.A., Wenger A.M., Berger M.J., Guturu H., Stenson P.D., Cooper D.N., Bernstein J.A., Bejerano G.. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016; 48:1581–1586. [DOI] [PubMed] [Google Scholar]
- 38. Dong C.L., Wei P., Jian X.Q., Gibbs R., Boerwinkle E., Wang K., Liu X.M.. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015; 24:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ioannidis N.M., Rothstein J.H., Pejaver V., Middha S., McDonnell S.K., Baheti S., Musolf A., Li Q., Holzinger E., Karyadi D. et al.. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016; 99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li J.C., Shi L.S., Zhang K., Zhang Y., Hu S.S., Zhao T.T., Teng H.J., Li X.F., Jiang Y., Ji L.Y. et al.. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018; 46:D1039–D1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li J.C., Zhao T.T., Zhang Y., Zhang K., Shi L.S., Chen Y., Wang X.X., Sun Z.S.. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018; 46:7793–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mao F.B., Xiao L.Y., Li X.F., Liang J.L., Teng H.J., Cai W.S., Sun Z.S.. RBP-Var: a database of functional variants involved in regulation mediated by RNA-binding proteins. Nucleic Acids Res. 2016; 44:D154–D163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mao F.B., Liu Q., Zhao X.L., Yang H.N., Guo S., Xiao L.Y., Li X.F., Teng H.J., Sun Z.S., Dou Y.L.. EpiDenovo: a platform for linking regulatory de novo mutations to developmental epigenetics and diseases. Nucleic Acids Res. 2018; 46:D92–D99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Song K., Li Q., Gao W., Lu S., Shen Q., Liu X., Wu Y., Wang B., Lin H., Chen G. et al.. AlloDriver: a method for the identification and analysis of cancer driver targets. Nucleic Acids Res. 2019; 47:W315–W321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen T.Q., Guestrin C.. XGBoost: a scalable tree boosting system. Kdd'16: Proceedings of the 22nd Acm Sigkdd International Conference on Knowledge Discovery and Data Mining. 2016; 785–794. [Google Scholar]
- 46. Zhang S., He Y., Liu H., Zhai H., Huang D., Yi X., Dong X., Wang Z., Zhao K., Zhou Y. et al.. regBase: whole genome base-wise aggregation and functional prediction for human non-coding regulatory variants. Nucleic Acids Res. 2019; 47:e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. et al.. Scikit-learn: machine learning in python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 48. Lundberg S.M., Lee S.I.. A unified approach to interpreting model predictions. Adv. Neur. In. 2017; 30:1–10. [Google Scholar]
- 49. Ellrott K., Bailey M.H., Saksena G., Covington K.R., Kandoth C., Stewart C., Hess J., Ma S., Chiotti K.E., McLellan M. et al.. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018; 6:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P. et al.. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kosmicki J.A., Samocha K.E., Howrigan D.P., Sanders S.J., Slowikowski K., Lek M., Karczewski K.J., Cutler D.J., Devlin B., Roeder K. et al.. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat. Genet. 2017; 49:504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chakravarty D., Gao J., Phillips S.M., Kundra R., Zhang H., Wang J., Rudolph J.E., Yaeger R., Soumerai T., Nissan M.H. et al.. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 2017; 2017:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ng P.K., Li J., Jeong K.J., Shao S., Chen H., Tsang Y.H., Sengupta S., Wang Z., Bhavana V.H., Tran R. et al.. Systematic functional annotation of somatic mutations in cancer. Cancer Cell. 2018; 33:450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E. et al.. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019; 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W. et al.. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018; 46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Consortium, I.T.P.-C.A.o.W.G. Pan-cancer analysis of whole genomes. Nature. 2020; 578:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tamborero D., Rubio-Perez C., Deu-Pons J., Schroeder M.P., Vivancos A., Rovira A., Tusquets I., Albanell J., Rodon J., Tabernero J. et al.. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018; 10:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yue Z., Zhao L., Xia J.. dbCPM: a manually curated database for exploring the cancer passenger mutations. Brief. Bioinform. 2018; 21:1–9. [DOI] [PubMed] [Google Scholar]
- 59. Buisson R., Langenbucher A., Bowen D., Kwan E.E., Benes C.H., Zou L., Lawrence M.S.. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019; 364:eaaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Razavi P., Li B.T., Brown D.N., Jung B., Hubbell E., Shen R., Abida W., Juluru K., De Bruijn I., Hou C. et al.. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019; 25:1928–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K.. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019; 10:1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The AI-Driver models are implemented in Python. Integrated datasets, source codes, collected training/testing sets, analysis scripts for the results of this manuscript are available at https://github.com/hatchetProject/AI-Driver. The pre-computed AI-Driver scores for all genome-wide possible missense variants are available at http://aidriver.maolab.org/.