

Abstract
DNA methylation is a stable epigenetic modification, extremely polymorphic and driven by stochastic and deterministic events. Most of the current techniques used to analyse methylated sequences identify methylated cytosines (mCpGs) at a single-nucleotide level and compute the average methylation of CpGs in the population of molecules. Stable epialleles, i.e. CpG strings with the same DNA sequence containing a discrete linear succession of phased methylated/non-methylated CpGs in the same DNA molecule, cannot be identified due to the heterogeneity of the 5′–3′ ends of the molecules. Moreover, these are diluted by random unstable methylated CpGs and escape detection. We present here MethCoresProfiler, an R-based tool that provides a simple method to extract and identify combinations of methylated phased CpGs shared by all components of epiallele families in complex DNA populations. The methylated cores are stable over time, evolve by acquiring or losing new methyl sites and, ultimately, display high information content and low stochasticity. We have validated this method by identifying and tracing rare epialleles and their families in synthetic or in vivo complex cell populations derived from mouse brain areas and cells during postnatal differentiation. MethCoresProfiler is written in R language. The software is freely available at https://github.com/84AP/MethCoresProfiler/.
INTRODUCTION
DNA methylation is an inheritable epigenetic modification of the DNA. This trait is not sequence specific, and it is widely distributed along chromosomes and genes. DNA methylation patterns can be stable and invariant, such as in genomic imprinting and X inactivation, or metastable, polymorphic and highly variable, such as methylation in somatic cells (1). The polymorphism of somatic DNA methylation is due to stochastic as well as deterministic events; as a consequence, it is difficult to decode (2,3). For example, DNA damage and repair modify the status of local DNA methylation, and eventually, transcription further remodels methylation profiles increasing the polymorphism (4,5).
Bisulfite sequencing is the gold standard of DNA methylation analysis, as it uses direct sequencing of chemically treated DNA to identify methylated cytosines at the single-nucleotide level. Genome-wide sequencing of bisulfite DNA is unbiased relative to the sequence representation (excluding PCR artefacts), but limited in the coverage/single locus. A second limitation in the analysis of genome-wide methylomes is due to the fact that the DNA sequences represent a statistical collection of methylated cytosines deriving from different molecules or chromosomes.
To date, there are two main types of DNA methylation analysis: the first identifies differentially methylated cytosines (DMCs) and the second detects epialleles (epihaplotype-based analysis or EBA). DMC is used in genome-wide methylomes to quantify average methylation of each CpG from mixtures of 75–100 bp DNA fragments. EBA identifies methylated DNA molecules (epihaplotypes) generating a binary profile (0 unmethylated/1 methylated) of CpGs in DNA strings. However, these two methods cannot decipher the elevated heterogeneity of methylated molecules, mostly due to the presence of stochastic mCpGs (such as hydroxymethylated cytosines) that greatly dilute stable methylated molecules subjected to selection (6). In fact, in several cases in which the combinatorial methylation of four consecutive cytosines (grossly equivalent to 16 possible epialleles) was measured in multiple genomic loci in normal and tumour DNA, a remarkable degree of methylation polymorphism in both normal and cancer cells was found with no evidence of clonal and stable epialleles (2,7,8). Under these conditions, a considerable degree of heterogeneity is a common finding in these methylation studies as evidenced by measurements of the entropy index, i.e. the number of different species within a population of epialleles according to the formulae developed by Shannon (9) and generalized by Renyi (10). Limited sampling and variability of the average methylation of single CpGs in the populations of sequences may introduce additional bias in the analysis.
Here, we introduce a new concept in the DNA methylation analysis: methylated cores. Methylated cores are clusters of CpGs in the same methylated configuration. These signatures characterize a stable fraction of molecules in the cell population. The cores mark families of epialleles deriving from common ancestors that evolve by acquiring or losing methyl groups. Depending on the genetic makeup of the cell and the levels of expression of the methylated genomic segment, selection may amplify or reduce the number of cells carrying the specific epialleles. Independently from the selection and the function of the specific gene, each epiallele barcodes a single haploid genome or a clone longitudinally and its family identifies a cell subpopulation (11–14). In order to better understand the structure, composition and evolution of complex cell populations, we have developed MethCoresProfiler, an R-based tool that provides a simple method to track and trace stable combinations of phased mCpGs (signatures or cores) shared by all components of epiallele families. In addition, MethCoresProfiler assigns in each population a clonality index and a stability or entanglement index to each CpG in the core.
MATERIALS AND METHODS
We developed an R-based tool, MethCoresProfiler (available at https://github.com/84AP/MethCoresProfiler/), which extracts and compares methylated cores, i.e. common and stable methylated CpGs that characterize families of DNA molecules (epihaplotypes) for each given condition. MethCoresProfiler requires three types of input files: (i) a tab-delimited text file of epihaplotypes in binary format; (ii) a tab-delimited text file containing information on the CpG position in the sequence (or string); and (iii) a tab-delimited text file containing metadata (information) associated with each sample with the following columns: #SampleID, Tissue, Description, Group, Rep and ID. The first input file can be generated with available tools (14). Figure 1 shows the workflow of MethCoresProfiler.
Figure 1.

MethCoresProfiler workflow. Schematic representation of the hierarchical structure of the modules of MethCoresProfiler. The functional modules are represented as trapezes connected by arrows. Input and output files are shown as dashed and solid arrows, respectively.
MethCoresProfiler
MethCoresProfiler is formed by three main components or R modules: the MethCores_Extractor, the MethCores_Combinator and the MethCores_Analyst.
-
The MethCores_Extractor, or Module 1, calculates the average depth of the sample reads (named b4), performs and summarizes (y) repeated and iterative sampling (default y = 1000) of each experimental sample using b4 as a depth and annotates the combination(s) of two mCpGs with statistical significance, i.e. with a frequency higher than the expected frequency for independent events (chi-square for independence statistics, P-value ≤ 10−9). This module generates several tables reporting (i) all CpG methylation profiles (frequency of single mCpG), (ii) the tetrachoric correlations of CpGs, (iii) the co-occurrence of two mCpGs, (iv) the taxonomic distribution of methylated species and (v) the Shannon entropy index and the summary/sample, statistics and plots.
Significant combination(s) of mCpG pairs are computed by comparing the frequency of each mCpG pair with the expected frequency according to chi-square statistics. The expected frequency of methylation of n CpGs, assuming two or more independent events, is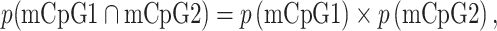
where p(mCpG1 ∩ mCpG2) represents the expected frequency of mCpG1–mCpG2. The expected frequency is computed in each sample and, depending on the methylation frequency of individual CpGs in the population of sequences, may vary for each combination.
The observed frequency is calculated as
where n(mCpG1–mCpG2) represents the number of epihaplotypes containing the specific mCpG1–mCpG2 combinations and N is the size of the sample.
The Shannon entropy and the generalized entropy (Renyi entropy) are determined as follows: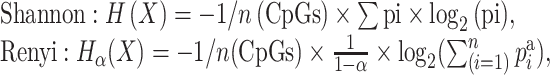
where n(CpGs) is the number of CpGs in the DNA string, pi is the frequency of each epiallele and Σpi × log2(pi) and
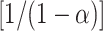 × log2(
× log2( are the Shannon and Renyi entropies, respectively. The variable α in the Renyi entropy represents the weight of the events with α = 0.5 probability. This first multiplicative term in the formula is used to normalize the values in the interval [0, 1] as reported in (15). If significant two-mCpG combinations are not found, all epialleles in the population are brought to the next step.
are the Shannon and Renyi entropies, respectively. The variable α in the Renyi entropy represents the weight of the events with α = 0.5 probability. This first multiplicative term in the formula is used to normalize the values in the interval [0, 1] as reported in (15). If significant two-mCpG combinations are not found, all epialleles in the population are brought to the next step. The MethCores_Combinator, or Module 2, analyses the complexity of the population by performing the following operations: (i) all significant two-mCpG combinations, annotated by Module 1, will be crossed; (ii) the frequency of all mCpG combinations (three mCpGs or more) will be compared to the expected frequency for independent events (chi-square for independence statistics, P-value ≤10−9) and will be annotated; (iii) the epialleles containing all significant mCpG combinations will be extracted; and (iv) the structure and frequency of significant mCpG combinations (cores) and individual epialleles will be reported (ComposCore and Tab_Epialleles, respectively). In the absence of significant mCpG combinations, all epialleles will be brought to the next step.
The MethCores_Analyst, or Module 3, performs hierarchical cluster analysis of the epialleles annotated by Module 2 generating heatmaps of their structure and frequency. In this step, the frequency of each epiallele in the sample is compared (chi-square test) to a random control (R), generated automatically by Module 1, with the same number of CpGs and depth (theoretical distribution). Individual significant epialleles are marked in a complex heatmap format (red = high frequency, blue = low frequency and white = neutral). Principal component analysis (PCA) of significant epialleles is also performed in this step. Finally, for each sample, the MethCores_Analyst extracts the most frequent mCpGs shared by significant epialleles using a decreasing percentage scale (frequency of CpGs in only significant epialleles) starting from 0.9 (minimum two mCpGs). If significant epialleles were not found at this stage, the epiallele with the highest frequency will be annotated and its structure will be considered as a stable signature or core.
The MethCores_Analyst generates three types of indices: (i) MethCores index, i.e. the frequency of the methylated cores in the population; (ii) clonality index, i.e. the frequency of methylated cores normalized to the average methylation in the population; and (iii) in-phase CpG index or stability index or entanglement index, or E, which measures the rate of coupling or association of at least two mCpGs (mCpG1 and mCpG2) in the core according to
 |
where p(mCpG1core)/p(mCpG1) and p(mCpG2 core)/p(mCpG2) are the frequencies of the first and the second (from the 5′ end) CpG in the core normalized to the frequency of the same CpG in the population. Monomethylated molecules can be excluded at this step. Note that the stability index is calculated only on extracted cores and not on the whole population. The index with a value 1 means that the mCpG1 and mCpG2 behave as a single unit and are ‘entangled’. The E index is higher when the cores maintain the mCpG constitutive elements in the same configuration in different samples or time points. The normalization step (mCpG in the core versus mCpG in the population) is essential in order to estimate the weight and the stability of the methylated core in the population, because it dissociates global methylation of the DNA in the population driven by genetic and epigenetic drift(s) from the methylation of specific sites subjected to selection pressure.
The minimum core contains at least two methylated cytosines with statistically significant frequency (chi-square test adjusted P-value ≤10−9). The workflow and the structure of the MethCoresProfiler modules are shown in Figure 1.
BS Amplicon-seq data and processing
As a proof of concept, we generated six synthetic populations, each composed of 1000 or 10 000 strings or sequences containing six randomly permutated CpGs in 1 (methylated) or 0 (non-methylated) configuration. The population, named R, is a random collection of six methylated CpGs generated automatically by Module 1 (theoretical distribution). The population, named Conditional 1 or Cond 1, contains a single string (‘1-0-1-0-0-1’) amplified to generate a specific epiallele M representing 30% of all molecules in the population. The population, named Conditional 2 or Cond 2, contains the same 30% epiallele M, as in Cond 1, with randomly permutated sites in the 0 configuration to generate divergent epialleles with a common signature or core of 1-X-1-X-X-1, where X can be 0 or 1 for the epiallele M. The whole M family accounts for 30% of the epialleles, but the individual permutated string represents <6% of the population. We also calibrated the lower limit of epiallele detectability in the Cond 1 and Cond 2 populations by lowering the representation of the epiallele M and its permutated variants to 10%. These populations are named as Cond 3 and Cond 4, respectively. We have also generated two other control populations, R1 and R2, in which we have added 30% or 10% of random sequences to maintain constant the number of sequences or strings in the R population. Note that the distribution of the epialleles in R1 and R2 is not completely random, because the 10% and 30% additional sequences skew the random distribution of some (not all) random epialleles in the R1 and R2 populations. Our synthetic populations reproduce all the possible types of clonal evolution: Cond 1 and Cond 3, and Cond 2 and Cond 4 provide models of clonal selection and clonal expansion, respectively. Conversely, R1 and R2 replicate the stochastic evolution of random clones. Supplementary Table S1 summarizes the composition and the features of stochastic populations.
As a second step to validate the MethCoresProfiler, we analysed the experimental data from the next-generation sequencing of bisulfite DNA of two mouse genes during brain differentiation (16) (GeneCards database links: DDO and DAO; protein atlas database links: DDO and DAO). The analysis was performed in brain areas of groups of three mice at different times after birth during postnatal brain differentiation. The genes are (i) DDO (d-aspartate oxidase) analysed in the brain at birth (P1), day 15 (P15), day 30 (P30) and day 60 (P60) after birth and in the small and large intestines (gut) at birth (P1) and day 90 (P90) after birth; and (ii) DAO (d-amino acid oxidase) analysed in the brain at birth (P1), day 15 (P15), day 30 (P30) and day 60 (P60) after birth. The same genes were analysed in purified astrocytes, neurons, and microglia, oligodendrocytes and endothelial cells (MOEs) derived from mouse cerebral cortex (CX) and cerebellum (CB) at birth (P1), day 7 (P7) and day 15 (P15). In total, we examined nine time points corresponding to 21 pools of DNA molecules spanning the following genomic regions: (i) DDO1 gene, from −488 to −44 bp upstream of the transcription start site (TSS), containing six CpG sites (positions −363, −330, −318, −242, −175 and −125); and (ii) DAO gene from +7 to +334 bp upstream of the TSS, containing four CpG sites (positions +7, +101, +217 and +334).
Paired-end reads in FASTQ format from ENA database (accession number: PRJEB28662) were merged using the PEAR (paired-end read merger) tool (17), setting a minimum of 40 nucleotides as the overlapping region. We retained only those reads with a mean quality score (Phred) >33 and a length between 400 and 500 nucleotides. Resulting reads were then converted in FASTA format using the PRINSEQ (preprocessing and information of sequence) tool (18). To extract mCpG configurations in single DNA molecules, reads in FASTA format were processed using ampliMethProfiler (14,19,20) (available at https://sourceforge.net/projects/amplimethprofiler/) applying several quality filters. In particular, we retained only reads characterized by (i) length ±50% compared with the reference length, (ii) at least 80% sequence similarity of the primer with the corresponding gene, (iii) at least 98% bisulfite efficiency and (iv) alignment of at least 60% of their bases with the reference sequences. The methylation status of all cytosines in the CpG sequence context is coded as methylated (1) or unmethylated (0). Reads with ambiguous calls (including gaps or A or G) at the CpG dinucleotide were removed. Supplementary Table S2 shows the features of each sample and filtered reads following the first analysis with ampliMethProfiler (14) and the sampling size used in the MethCoresProfiler. The data, in binary formats, were successively analysed with the MethCoresProfiler.
RESULTS
MethCoresProfiler strategy
MethCoresProfiler is formed by three modules that identify the basic and common elements (CpGs) present in complex populations of epialleles considering all CpGs potentially methylable and all combinations of CpGs (epialleles) as the products of independent events. Epialleles that share a specific combination of CpGs belong to the same family. The tool applies different types of normalization to identify significant combinations of CpGs: the chi-square test of independence of CpG methylation in the theoretical and experimental populations and the normalization of methylation of the CpGs in the cores to the average methylation of the same CpGs in the population.
MethCoresProfiler applies a series of analytical strategies to reduce the statistical errors and the heterogeneity of methylated DNA strings. The tool performs a (y) repeated (default y = 1000) sampling with depth N (N = average of samples reads, ≥1000) for each sample to reduce the errors derived from comparison of populations with different sizes (Supplementary Figure S1).
We first tested MethCoresProfiler on our synthetic populations of methylated molecules (see the ‘Materials and Methods’ section, Supplementary Table S1 and the ‘BS Amplicon-seq data and processing’ section). Supplementary Figure S2 shows that the average methylation (A) is comparable in all samples, whereas a lower entropy (B) characterizes Cond 1 due to the presence of 30% of the cloned M epiallele. As to the average methylation of each CpG, Cond 1 and Cond 2 show the highest levels of mCpGs at the predicted locations (1-3-6), while Cond 3 and Cond 4 display the same methylation found in random samples (R) (Supplementary Figure S2C). The correlation index shows that the populations with the cloned epiallele M at 30% and 10% are very similar (Cond 1 and Cond 3) and are different from Cond 2 and Cond 4 containing the permutated epiallele M (Supplementary Figure S2D). The interaction map or the frequency of mCpG pairs shows that only Cond 1 and Cond 2 display visible methylation signatures or cores, while Cond 3 and Cond 4 are very similar to random populations (Supplementary Figure S2E, red and grey lines). This is also shown by the co-occurrence matrix and taxonomy of all populations in Supplementary Figure S3A and B, respectively. Under these conditions, the 10% amplified epiallele(s) in Cond 3 and Cond 4 escape(s) detection by Modules 1 and 2.
Identification of rare methylation cores in complex DNA populations
The MethCores_Analyst, or Module 3, unbiasedly compares the structure and the frequency of the epialleles present in the four conditional and two R1–R2 synthetic populations to the R, random control, generated by Module 1 (theoretical distribution). Supplementary Figure S4A shows that Module 3 (MethCores_Analyst) identifies the 10% M epiallele family and the significant (red lines in the third panel on the right, P-value ≤10−9) individual epialleles in the Cond 3 and Cond 4 populations, although the PCA indicates that Cond 4, R1 and R2 are very similar (Supplementary Figure S4B). Also, the structure, the clonality index and the frequency of the cores discriminate Cond 4 from R populations (see Supplementary Figure S4C–E and the legend of Supplementary Figure S4). Software programs currently used to analyse the distribution of epialleles such as EBA, ampliMethProfiler (14) and methclone (7,8) identify frequent individual epialleles present in both R1–R2 and Cond 4 populations, which display comparable Shannon entropy and correlation coefficients (Supplementary Figure S5A and B). The programs indicated above do not distinguish random R1 and R2 epialleles from deterministic Cond 4 epialleles (Supplementary Figure S5C). In this context, MethCoresProfiler outperforms the other methods used to analyse epialleles because it distinguishes Cond 4 from R1 and R2 epialleles. The frequency of individual epialleles constituting the family in complex populations may not be statistically significant, while the frequency of the core is always significant (Supplementary Figures S4A and S5C).
Together, these data demonstrate that the method we describe is able to identify rare and stable epiallele families in complex DNA populations with a composite background and to discriminate stochastic versus deterministic epiallelic clones.
The trajectories of DDO1 and DAO epialleles mark postnatal differentiation of mouse brain cells
MethCoresProfiler extracts and identifies clones and families of epialleles with common methylation signatures in complex populations of DNA molecules (Supplementary Figures S2–S4). To validate the method in vivo, we analysed bisulfite sequencing data of two mouse genes DDO1 and DAO during brain differentiation in samples taken from groups of three mice at different time points after birth as described in the ‘Materials and Methods’ section and Supplementary Table S2. The DDO1 and DAO genes were selected in order to dissociate gene expression from selection of epialleles, because their expression profile varies significantly in different areas of mouse brain (16) (GeneCards database links: DDO and DAO; protein atlas database links: DDO and DAO).
The DDO1 epialleles present in the DNA extracted from different brain areas [CB, CX and hippocampus (HIPP)] in groups of three mice at birth (P1) or at several postnatal periods were characterized with MethCoresProfiler. The general features of DDO1 epialleles including the structure of the segment of the gene analysed (A), the average methylation (B), the Shannon entropy (C) and the methylation frequency of each CpG in the sequence (D) are shown in Figure 2A–D. The distribution of DDO1 epialleles in all samples is shown in Figure 2E. The 57 possible epiallelic configurations, excluding non- or monomethylated molecules, are present at least once in all samples analysed (Figure 2E, left panel, methylated, red; non-methylated, white bars). High- or low-frequency significant epialleles in the brain areas of each mouse during postnatal differentiation are shown in Figure 2E (central and right panels, P-value ≤10−9). The qualitative and quantitative characterizations of DDO1 epialleles in each mouse brain area are shown in Figure 2F–I. The correlation coefficient and the taxonomy of species distribution show that the epialleles in each area cluster in separate and discrete populations (Figure 2F, and Supplementary Figures S6 and S7). The average methylation of the CpGs is stable during postnatal differentiation in CX (Figure 2D), but the MethCores index and the clonality change in all brain areas (Figure 2F–H). Figure 2I shows the structure and trajectories of the methylated cores in each brain area normalized to the methylation of each CpG in the population. From these data, we can reconstruct the clonal evolution of the epialleles during postnatal mouse brain differentiation. For example, in CB and HIPP the cytosines ‘−336|−330’ are tightly and stably associated at different time points. The heterogeneous epiallele family ‘−336|−330|−318|−242|−175’ dominates in HIPP. The epialleles in CX, on the other hand, change conformation: the structure of mCpGs ‘−242|−175’ is replaced by mCpGs ‘−336|−318|−242’ (Figure 2I). Supplementary Table S3 shows the E index values of all the samples.
Figure 2.

Methylation signatures (cores) of DDO1 epialleles mark mouse brain areas during postnatal differentiation. (A) Structure of the region of mouse DDO1 gene analysed. The location of the CpGs upstream of the TSS (thick arrow and CpG nucleotide number relative to the TSS) and the oligonucleotide primers used to amplify DDO1 epialleles are shown as black circles and lines. (B) Average methylation of the six CpGs shown in (A) in the DNA extracted from mouse brain areas (CX, CB and HIPP) and the relative random control at different time points after birth (p1, p15, p30 and p60 days, colour-coded squares on the right). (C) Shannon entropy of the methylated molecules in the same brain areas and time points shown in (B). (D) Average methylation of each CpG identified by the nucleotide position shown in (A), in the populations of molecules shown in (B). (E) Structure and frequency of the epialleles containing the CpGs shown in (A). The panel on the left shows the cluster analysis of all epialleles present in the brain areas analysed (methylated, red; non-methylated, white) containing the six CpGs shown in (A). The next three panels on the right show the frequency of the epialleles in each brain area, indicated on the top of the panels (colour code at the bottom of the fourth panel from the left). The frequency of epialleles in a random control is shown on the lane on the right side of each panel (‘Random’). The three panels on the right show the significant epialleles in the brain areas indicated on the top of the panels at different time points. High- or low-frequency or neutral epialleles from the DNA of the groups of three mice at different time points are shown in red, blue or white, respectively. (F) PCA of DDO1 epialleles in the areas of mouse brain during postnatal differentiation. (G) Frequency of methylated cores (MethCores index) in the same areas of mouse brain during postnatal differentiation. (H) Frequency of the methylated cores normalized to the average methylation (clonality index) in each population. (I) The structure and the composition of the methylated cores at different time points in the populations of molecules derived from each brain area. Each CpG is labelled with a colour code shown on the right side of the panel. In Supplementary Table S3 are reported the E index values of all analysed samples. Pairwise comparison between each pair was performed with the Student’s t-test: *P < 0.05 versus P1, #P < 0.05 versus P15.
DDO1 methylated cores mark different cell populations in brain CX and CB during postnatal differentiation
Since all bisulfite DNA molecules sequenced in our samples contain the same 5′ and 3′ ends, each epiallele marks a single cell and we can associate the epialleles found in different brain areas with specific cell types. The presence of the same family of epialleles in both isolated cells and the specific brain area can further validate our analysis in vivo. To this end, we determined the distribution of DDO1 epialleles in cells isolated from mouse CX or CB brain areas. Figure 3 shows the average methylation (A), the Shannon entropy (B), the methylation frequency of each CpG (C) and the structure and the distribution (D) of the epialleles in CB and fractionated cells. There are epialleles present in all types of cells, and some are specific to neurons (marked by ∧) or astrocytes (marked by *). The trajectories of the epialleles are specific to each brain area (Supplementary Figures S6A and S7A) and reflect dynamics and clonal evolutions in isolated cells (Supplementary Figure S6A and B).
Figure 3.

DDO1 epialleles identify cells derived from mouse CB: astrocytes, MOEs and neurons during postnatal differentiation. (A) Average methylation of the DDO1 epialleles in different cell types derived from CB. (B) Shannon entropy. (C) Average methylation of each CpG in the population of molecules in the same cells as in (A). (D) The structure (first panel on the left) and distribution (panels on the right) of the DDO1 epialleles in CB and CB-derived cells during postnatal differentiation of mouse brain. The panel on the left shows the cluster analysis of all epialleles (methylated, red; non-methylated, white) containing the six CpGs shown in Figure 1A. The three panels on the right show the frequency of the same epialleles (colour code at the bottom of the second panel on the left) in the brain areas indicated on the top of the panels at different time points as in (B), relative to the random control (the lane on the right in each panel). The epialleles in red and blue represent the epialleles with a significantly higher or lower frequency relative to the controls, respectively. Neutral or non-significant epialleles are shown in white. * and ∧ mark the astrocyte- and neuron-specific epialleles, respectively. (E) PCA of mouse CB area and derived cells of DDO1 epialleles during brain postnatal differentiation. (F) Frequency of methylated cores (MethCores index) in the same populations as in (A). (G) Frequency of methylated cores normalized to the average methylation (clonality index) in each population. (H) The structure and the composition of the methylation cores in each cell population during postnatal brain differentiation. Each CpG is labelled with a colour code shown on the right side of the panel. In Supplementary Table S3 are reported the E index values of all analysed samples. Pairwise comparison between each pair was performed with the Student’s t-test: *P < 0.05 versus P1, #P < 0.05 versus P15.
The similarities (PCA) and the differences (MethCores index and clonality index) of the epiallele families in CB and isolated cells in Figure 3E–G suggest that CB astrocytes and neurons at P15 underwent a significant clonal evolution, which was also evident in whole CB. The structure and the distribution of the DDO1 epialleles in CB fractionated cells show that astrocytes and MOEs (oligodendrocytes) share a common precursor at P15, different from the epiallele family that characterizes the neurons and the whole CB, suggesting that neurons contributed significantly to the evolution of the DDO1 epiallele family found in the CB at P15 (Figure 3H). In Supplementary Table S3 are reported the E index values of analysed samples.
The same type of analysis was performed in CX and fractionated cells at various time points. CX did not show significant changes in average methylation (Figure 4A), Shannon entropy (Figure 4B) and single CpG methylation (Figure 4C) although the methylation of the six CpGs at different time points varied significantly (Figure 4C). CX-derived cells, on the other hand, displayed significant changes in all three parameters (average methylation, Shannon entropy and average methylation of CpG in the cores) and the E index (Supplementary Table S3) at the three time points analysed (Figure 4A–C). This type of analysis shows both qualitative and quantitative similarities and differences in CB and CX epialleles during postnatal brain differentiation. In CB, the trajectories of the epialleles recapitulate the trajectories of the epialleles found in purified neurons (Figure 3H), whereas in CX the trajectories of the epialleles are similar to those found in neurons and astrocytes/MOEs (Figure 4H). It is noteworthy that in the CX the appearance of the cell-specific epialleles in purified cells (P15) precedes the appearance of the same epialleles (P30) in the whole CX (Figure 4H).
Figure 4.

DDO1 epialleles in the cells derived from mouse brain CX: astrocytes, MOEs and neurons during brain postnatal differentiation. (A) Average methylation of the DDO1 epialleles in different cell types derived from CX. (B) Shannon entropy. (C) Average methylation of each CpG in the total population of molecules in the cells described in (A). (D) The structure (first panel on the left) and distribution (panels on the right) of DDO1 epialleles in the CX and CX-derived cells during postnatal differentiation of mouse brain. The panel on the left shows the cluster analysis of all epialleles (methylated, red; non-methylated, white) containing the six CpGs shown in Figure 1A. The three panels on the right show the frequency of the same epialleles described in the first panel on the left (colour code at the bottom of the second panel from the left) in the brain areas indicated on the top of the panels at different time points as in (B) or relative to the random control (the lane at the right side in each panel). The epialleles in red and blue are significantly higher or lower frequency epialleles relative to the controls, respectively. Neutral or non-significant epialleles are shown in white. (E) PCA of mouse CX cells and epiallele populations during postnatal brain differentiation. (F) Frequency of methylated core (MethCores index) in the same populations as in (A). (G) Frequency of methylated cores normalized to average methylation (clonality index) in each population. (H) The structure and the composition of the methylation cores in each cell population during postnatal mouse brain differentiation. Each CpG is labelled with a colour code shown at the right side of the panel. Supplementary Table S3 shows the E index values of all analysed samples. Pairwise comparison between each pair was performed with the Student’s t-test: *P < 0.05 versus P1, #P < 0.05 versus P15.
We wish to stress several points that confirm, validate and expand the epiallele analysis of mouse brain areas during postnatal differentiation shown here. The structure and the postnatal trajectories of the epialleles are specific to each brain area (Figures 3D and H, and 4D and H). In both the CB and CX, the epialleles with the same structure match the epialleles found in the fractionated cells (Figures 3H and 4H). The epiallele distribution in the CB or CB-derived cells is strikingly homogeneous compared to the epialleles in CX or CX-derived cells (Figures 3H and 4H, and Supplementary Figure S6B), confirming the finding that at the P8–15 postnatal differentiation period in CB, granule cells and precursors account for 78% of total cells (21). Astrocytes and MOEs share a common precursor (21) containing the epiallelic core found at P15 in CB and CX (Figures 3H and 4H). Our epiallele analysis confirms independently published data on postnatal differentiation of mouse brain cells (21,22).
To further validate the method, we performed the analysis on DDO1 epialleles in another mouse tissue: small and large intestines (gut) at birth and 90 days later. There are two major epiallele families appearing 90 days after the birth in all mice analysed (Supplementary Figure S8). The high clonality index and the structure of the core suggest that a major epigenetic family emerges during the gut postnatal differentiation (Supplementary Figure S8). This family may be represented by cells expressing a member of the SLC26 gene family of anion channels (DRA, SLC26A3). The expression of this gene marks the most abundant cell population during postnatal gut differentiation in stem cells organoids (23).
Last, we analysed the epialleles containing four CpGs at the 5′ end of the DAO gene selectively expressed in mouse CB (24). The reduced number of CpGs limits the number of possible epialleles to 16 and this may facilitate the analysis of methylated cores. Supplementary Figure S9 shows the location of the CpG relative to the TSS (A), the average methylation (B), Shannon entropy (C), the frequency of methylation of each CpG (D), the structure (E, first panel on the left) and the distribution (E, central and right panels) of DAO epialleles in the mouse brain areas during postnatal differentiation. The PCA (Supplementary Figure S9F), the MethCores index (Supplementary Figure S9G), the clonality index (Supplementary Figure S9H) and the structure of the cores (Supplementary Figure S9I) show that each brain area displays different types of DAO epiallelic trajectories, which are not correlated with the expression of the gene, which is restricted to the CB (24).
Fractionation of cells from each brain area shows that the DAO epiallele trajectories during postnatal differentiation in CB (Supplementary Figure S10) and CX (Supplementary Figure S11) recapitulate the trajectories found in the specific cell types similarly to DDO1 epialleles (Figures 2E and I, and 3D and H). The taxonomy of epiallelic species distribution and the Pearson correlation of DAO epialleles are shown in Supplementary Figures S12 and S13, respectively. As expected, the changes in the trajectories of DAO epialleles in the specific brain areas are not as evident as they appear in isolated cells, because they are the result of the algebraic sum of loss or gain of epialleles in different cell types (Figures 2 and 3, and Supplementary Figures S10 and S11). Since DDO1- and DAO-specific epialleles characterize mouse brain areas and specific cell types independently on their cellular expression (15,22), we asked whether DAO and DDO1 epialleles mark the same cell type and the same mouse brain area at the same time point during postnatal differentiation. To this end, we performed hierarchical cluster analysis of the structures and distribution of DAO and DDO1 epialleles in all samples analysed to test whether DDO1 and DAO epialleles mark the same cell type, time point and whole brain area. Strikingly, this analysis shows that both the structure of the cores and the frequency distribution of DAO and DDO1 epialleles mark the same area, the same postnatal time point and the same cell type (Figure 5). Moreover, comparing the structure and trajectories of the epiallele cores in each cell type with the frequency of progenitor cells during postnatal differentiation of mouse brain (P1–15 days after birth), we found that the trajectories of both DAO and DDO1 epialleles in each cell type overlap with trajectories of two main brain cell precursors during early mouse postnatal differentiation (1–15 days), the intermediate precursors cells (IPCs) that generate immature and mature neurons, and the radial glial cells that generate astrocytes, oligodendrocytes and IPCs during postnatal differentiation time of 0–15 days (Figure 5) (25,26) (www.cellsignal.com/neuro-atlas).
Figure 5.

DDO1 and DAO epialleles mark the same brain areas, the same cell type and the same time points during postnatal differentiation of mouse brain. Dendrogram and hierarchical clustering of the frequency (right panels) and the structure (left panels) of DDO1 and DAO epialleles in mouse brain areas (A, top panels) and fractionated cells from the CB (B, middle panels) and the CX (C, lower panels) during postnatal differentiation. The similarity of different samples is represented by the vertical distances on each branch of the dendrogram. ‘vegan’ R package (version 2.4-1) was used to perform the tests and draw correlograms. The red and blue circles mark the same brain areas, time points and cells. IP and RG represent the intermediate progenitors and the radial glial precursors, respectively. RG are upstream of the IP and generate IP and astrocytes. IP differentiate in immature neurons and, eventually, in mature neurons (19–21) (cellsignal.com/neuro-atlas).
DISCUSSION
In conclusion, the tool MethCoresProfiler identifies and tracks epiallele families in complex cell populations such as the brain and fractionated cells during postnatal differentiation. The concordance of the structure and the distribution of the methylated cores of both genes in cells and whole brain areas (Figure 5) and the same trajectories in independent mouse samples during postnatal brain differentiation (Figure 2 and Supplementary Figure S8) represent a validation of the method and a proof that DAO and DDO1 epialleles mark haploid genomes and single cells independently. This analysis can be applied to any segment of DNA containing mCpGs. The power of this tool can be further increased by also including in the cores non-methylated CpGs in phase with mCpGs.
A brief summary of the method is shown in the graphical abstract, which reports the features of the input and output data. Supplementary Table S4 shows the comparison of MethCoresProfiler with existing tools currently used for methylation analysis of bisulfite sequencing data (26) (see Supplementary Figures S4A and S5C). The majority of the software programs were designed to explicitly provide quantitative assessment of methylation of single CpG (27) or abundance of single epialleles (8,14). As shown in Supplementary Figure S5, the frequency of single epialleles analysed with ampliMethProfiler does not discriminate stochastic random clones from stable clones or families of epialleles subjected to selection. MethCoresProfiler offers three main advantages: (i) it automatically identifies and extracts significant epialleles by normalizing their distribution to the expected frequency in the experimental and theoretical populations; (ii) it automatically defines the structure of families of epialleles (cores) and provides three quantitative indices; and (iii) it works on single or multiple samples, making intersample or longitudinal comparisons.
Summarizing this method identifies and tracks single epiallelic clones (as Cond 1 or Cond 3 in the graphical abstract) or divergent clones deriving from a single ancestor (as Cond 2 or Cond 4 in the graphical abstract) with a significant reduction of epiallele heterogeneity.
Unstable or stochastic methylation might be associated with demethylation during transcription. Hydroxy-methylated dC (OHmdC) is resistant to bisulfite oxidation; it is scored as methyl dC in bisulfite reactions and is replaced by BER (base excision repair) enzymes with a non-methylated deoxycytosine (6,28–30). We have tested whether, and how, C hydroxymethylation or demethylation may alter the structure and the trajectories of epiallele cores by measuring OHmdC in DAO epialleles. Supplementary Figure S14 shows the DAO epialleles in the CB before or after oxidation of bisulfite DNA with perruthenate, which deaminates and converts OHmdC to thymine. Elimination of OHmdC does not modify the structure of the main CB DAO epiallelic cores; instead, it increases their frequency by reducing the background OHmdC. Only one epiallele core in CB disappears when OHmdC is eliminated (Supplementary Figure S14). We believe that this is an important point that may modify the interpretation of methylation profiles in complex populations of molecules with single time points.
DATA AVAILABILITY
Project name: MethCoresProfiler Project.
Home page: https://github.com/84AP/MethCoresProfiler/.
Operating system(s): Linux, MacOS X, Windows.
Programming language: R.
Other requirements: different R packages.
License: GNU GPLv3.
Supplementary Material
ACKNOWLEDGEMENTS
We wish to thank Raul Rabadan (Columbia University, NYC) and Michele Ceccarelli (University Federico II) for helpful comments.
Contributor Information
Antonio Pezone, Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università Federico II Napoli, 80131 Naples, Italy.
Alfonso Tramontano, Department of Precision Medicine, University of Campania ‘L. Vanvitelli’, 80138 Naples, Italy.
Giovanni Scala, Department of Biology, Università Federico II Napoli, 80126 Naples, Italy.
Mariella Cuomo, Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università Federico II Napoli, 80131 Naples, Italy.
Patrizia Riccio, Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università Federico II Napoli, 80131 Naples, Italy.
Sergio De Nicola, Department of Physics, Università Federico II Napoli, 80126 Naples, Italy.
Antonio Porcellini, Department of Biology, Università Federico II Napoli, 80126 Naples, Italy.
Lorenzo Chiariotti, Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università Federico II Napoli, 80131 Naples, Italy.
Enrico V Avvedimento, Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università Federico II Napoli, 80131 Naples, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
AIRC [16983 to E.V.A.]; Fondazione Medicina Molecolare e Terapia Cellulare, Università Politecnica delle Marche [to E.V.A.]; Epigenomics Flagship Project--Epigen, CNR [to E.V.A.]; Fondazione Cariplo Ricerca Biomedica sulle malattie legate all’invecchiamento [2016-1031 to E.V.A.]; MIUR-PRIN 2017 [2017237P5X-003 to E.V.A.]; PON AIM 2014–2020 [E69F19000070001 to G.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kim M., Costello J. DNA methylation: an epigenetic mark of cellular memory. Exp. Mol. Med. 2017; 49:e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Landan G., Cohen N.M., Mukamel Z., Bar A., Molchadsky A., Brosh R., Tanay A. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet. 2012; 44:1207–1214. [DOI] [PubMed] [Google Scholar]
- 3. Russo G., Landi R., Pezone A., Morano A., Zuchegna C., Romano A., Avvedimento E.V. DNA damage and repair modify DNA methylation and chromatin domain of the targeted locus: mechanism of allele methylation polymorphism. Sci. Rep. 2016; 6:33222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morano A., Angrisano T., Russo G., Landi R., Pezone A., Bartollino S., Avvedimento E.V. Targeted DNA methylation by homology-directed repair in mammalian cells. Transcription reshapes methylation on the repaired gene. Nucleic Acids Res. 2013; 42:804–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Allen B., Pezone A., Porcellini A., Muller M.T., Masternak M.M. Non-homologous end joining induced alterations in DNA methylation: a source of permanent epigenetic change. Oncotarget. 2017; 8:40359–40372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang Y., Pastor W.A., Shen Y., Tahiliani M., Liu D.R., Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010; 5:e8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li S., Garrett-Bakelman F.E., Chung S.S., Sanders M.A., Hricik T., Rapaport F., Mason C.E. Distinct evolution and dynamics of epigenetic and genetic heterogeneity in acute myeloid leukemia. Nat. Med. 2016; 22:792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li S., Garrett-Bakelman F., Perl A.E., Luger S.M., Zhang C., To B.L., Lewis J.D., Brown A.L., D’Andrea R.J., Ross M.E. et al. Dynamic evolution of clonal epialleles revealed by methclone. Genome Biol. 2014; 15:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shannon C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948; 27:379–423. [Google Scholar]
- 10. Its A.R., Korepin V.E. Generalized entropy of the Heisenberg spin chain. Theor. Math. Phys. 2010; 164:1136–1139. [Google Scholar]
- 11. Affinito O., Scala G., Palumbo D., Florio E., Monticelli A., Miele G., Cocozza S. Modeling DNA methylation by analyzing the individual configurations of single molecules. Epigenetics. 2016; 11:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tramontano A., Boffo F.L., Russo G., De Rosa M., Iodice I., Pezone A. Methylation of the suppressor gene: mechanism and consequences. Biomolecules. 2020; 10:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pezone A., Russo G., Tramontano A., Florio E., Scala G., Landi R., Avvedimento E.V. High-coverage methylation data of a gene model before and after DNA damage and homologous repair. Sci. Data. 2017; 4:170043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Scala G., Affinito O., Palumbo D., Florio E., Monticelli A., Miele G., Cocozza S. ampliMethProfiler: a pipeline for the analysis of CpG methylation profiles of targeted deep bisulfite sequenced amplicons. BMC Bioinformatics. 2016; 17:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xie H., Wang M., de Andrade A., Bonaldo M. de F., Galat V., Arndt K., Soares M.B. Genome-wide quantitative assessment of variation in DNA methylation patterns. Nucleic Acids Res. 2011; 39:4099–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cuomo M., Keller S., Punzo D., Nuzzo T., Affinito O., Coretti L., Chiariotti L. Selective demethylation of two CpG sites causes postnatal activation of the DAO gene and consequent removal of D-serine within the mouse cerebellum. Clin. Epigenetics. 2019; 11:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang J., Kobert K., Flouri T., Stamatakis A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2013; 30:614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011; 27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Caporaso J., Kuczynski J., Stombaugh J., Bittinger K., Bushman F., Costello E., Fierer N., Peña A., Goodrich J., Gordon J. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010; 7:335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T. BLAST+: architecture and applications. BMC Bioinformatics. 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valério-Gomes B., Guimarães D.M., Szczupak D., Lent R. The absolute number of oligodendrocytes in the adult mouse brain. Front. Neuroanat. 2018; 12:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cahoy J.D., Emery B., Kaushal A., Foo L.C., Zamanian J.L., Christopherson K.S., Xing Y., Lubischer J.L., Krieg P.A., Krupenko S.A. et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008; 28:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kozuka K., He Y., Koo-McCoy S., Kumaraswamy P., Nie B., Shaw K., Siegel M. Development and characterization of a human and mouse intestinal epithelial cell monolayer platform. Stem Cell Rep. 2017; 9:1976–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koga R., Miyoshi Y., Sakaue H., Hamase K., Konno R. Mouse D-amino-acid oxidase: distribution and physiological substrates. Front.Mol. Biosci. 2017; 4:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tramontin A.D. Postnatal development of radial glia and the ventricular zone (VZ): a continuum of the neural stem cell compartment. Cereb. Cortex. 2003; 13:580–587. [DOI] [PubMed] [Google Scholar]
- 26. Fuentealba L.C., Rompani S.B., Parraguez J.I., Obernier K., Romero R., Cepko C.L., Alvarez-Buylla A. Embryonic origin of postnatal neural stem cells. Cell. 2015; 161:1644–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wong N., Pope B., Candiloro I., Korbie D., Trau M., Wong S., Mikeska T., Zhang X., Pitman M., Eggers S. MethPat: a tool for the analysis and visualisation of complex methylation patterns obtained by massively parallel sequencing. BMC Bioinformatics. 2016; 17:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hahn M.A., Szabó P.E., Pfeifer G.P. 5-Hydroxymethylcytosine: a stable or transient DNA modification?. Genomics. 2014; 104:314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coppieters N., Dieriks B.V., Lill C., Faull R.L.M., Curtis M.A., Dragunow M. Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol. Aging. 2014; 35:1334–1344. [DOI] [PubMed] [Google Scholar]
- 30. Taylor K.H., Kramer R.S., Davis J.W., Guo J., Duff D.J., Xu D., Shi H. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res. 2007; 67:8511–8518. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Project name: MethCoresProfiler Project.
Home page: https://github.com/84AP/MethCoresProfiler/.
Operating system(s): Linux, MacOS X, Windows.
Programming language: R.
Other requirements: different R packages.
License: GNU GPLv3.