

Abstract
CCCTC-binding factor (CTCF) is a key regulator of 3D genome organization and gene expression. Recent studies suggest that RNA transcripts, mostly long non-coding RNAs (lncRNAs), can serve as locus-specific factors to bind and recruit CTCF to the chromatin. However, it remains unclear whether specific sequence patterns are shared by the CTCF-binding RNA sites, and no RNA motif has been reported so far for CTCF binding. In this study, we have developed DeepLncCTCF, a new deep learning model based on a convolutional neural network and a bidirectional long short-term memory network, to discover the RNA recognition patterns of CTCF and identify candidate lncRNAs binding to CTCF. When evaluated on two different datasets, human U2OS dataset and mouse ESC dataset, DeepLncCTCF was shown to be able to accurately predict CTCF-binding RNA sites from nucleotide sequence. By examining the sequence features learned by DeepLncCTCF, we discovered a novel RNA motif with the consensus sequence, AGAUNGGA, for potential CTCF binding in humans. Furthermore, the applicability of DeepLncCTCF was demonstrated by identifying nearly 5000 candidate lncRNAs that might bind to CTCF in the nucleus. Our results provide useful information for understanding the molecular mechanisms of CTCF function in 3D genome organization.
INTRODUCTION
Genomic DNA is hierarchically packaged into complex high-order structures in the nucleus. The 3D organization of a genome is highly dynamic and functionally important for gene regulation, cell differentiation and development (1–3). Disruption of 3D genome organization has been shown to be linked to human disease, such as cancer (4,5). The architectural protein, CCCTC-binding factor (CTCF), plays a critical role in orchestrating the 3D genome organization (6,7). CTCF is a ubiquitous and highly conserved zinc finger protein with a DNA-binding domain (8). It can bind to a large number of sites throughout the genome (8,9). These CTCF-binding sites are enriched in the boundaries between topologically associating domains (TADs) as well as within intra-TAD chromatin loops in mammals (10,11). Although the majority of the binding sites share a consensus DNA motif, ∼18% of the binding sites lack the motif and some DNA sites containing the motif do not bind to CTCF (12). Thus, locus-specific factors may be employed to target CTCF to these specific genomic sites.
Recent studies suggest that thousands of RNA transcripts across the genome are bound by CTCF and can serve as locus-specific factors to recruit CTCF to chromatin (13,14). Interestingly, CTCF has higher affinity for RNA over DNA (13), and the RNA-binding domain is different from its DNA-binding domain (14). Moreover, CTCF–RNA interactions have been shown to play fundamental roles in promoting CTCF self-association and clustering, and CTCF-dependent chromatin loop formation (15,16). Deletion or mutation of CTCF’s RNA-binding regions can impair the formation of chromatin loops and disturb gene expression (15,16). Although the DNA motif of CTCF has been well characterized (12), it is still unclear how CTCF recognizes its target RNAs as no consensus RNA motif has been reported for CTCF binding.
CTCF represents an increasing number of essential proteins with dual DNA- and RNA-binding capacity, which have unique structural and functional characteristics (17). RNA-binding proteins are often considered to be functionally distinct from DNA-binding proteins, but this notion has been challenged by the discovery of many long non-coding RNAs (lncRNAs), which can interact with DNA-binding proteins and play important roles in gene regulation and 3D genome organization (17–23). Particularly, many lncRNAs are found to interact with CTCF (13,14). For example, the lncRNA Wrap53, an antisense transcript of p53, directly interacts with CTCF to regulate p53 expression (14). The lncRNA Jpx activates Xist and induces the initiation of X chromosome inactivation by evicting CTCF from the Xist promoter through physical interaction with CTCF (24). Furthermore, CTCF can be recruited to specific genomic sites and mediate long-range chromosomal interactions by interacting with lncRNAs (13). For instance, the lncRNAs Tsix and Xite are necessary for X chromosome pairing by recruiting CTCF to the pairing center (13). The lncRNAs Xist and Firre, which play important roles in 3D genome organization (25,26), are shown to directly interact with CTCF in X chromosome inactivation or anchoring the inactive X chromosome to the nucleolus (13,27). Since lncRNAs are often expressed in a tissue- or cell-type-specific manner (28,29), comprehensive identification of CTCF-binding lncRNAs may provide valuable information for understanding the mechanisms of dynamic 3D genome organization.
High-throughput sequencing-based methods, such as cross-linking immunoprecipitation followed by deep sequencing (CLIP-seq), can be used to identify transcriptome-wide binding targets of CTCF (13). However, due to the low-level and cell-type-specific expression of lncRNAs (29), these methods could suffer from false negatives. Moreover, the experimental methods are expensive and time-consuming. To overcome these drawbacks, computational methods can be used to predict putative lncRNA targets of CTCF. Although machine learning models have been reported for predicting chromatin loop formation through CTCF–DNA interaction (30,31), such a predictive method has not been employed to analyze the binding of CTCF to RNA. Recently, deep learning techniques such as convolutional neural networks (CNNs) and recurrent neural networks have achieved superior performance in many biological problems (32–38). For instance, deep learning models have been developed to accurately predict 3D chromatin contacts and enhance the resolution of Hi-C data (32,39). Several deep learning approaches have also been developed for general protein–RNA binding prediction, such as DeepBind (37), DeeperBind (40) and iDeepS (41). However, no model has been trained specifically for CTCF or other proteins with dual DNA- and RNA-binding capacity. Moreover, model performance may be further improved by using advanced techniques such as the Bayesian hyperparameter optimization (42–44) and attention-based mechanisms (45,46).
In this study, we have developed a new deep learning model, DeepLncCTCF, to discover the RNA recognition patterns of CTCF and identify candidate lncRNAs that may interact with CTCF. DeepLncCTCF utilized a CNN and a bidirectional long short-term memory network (BLSTM) to predict CTCF–RNA binding from nucleotide sequence, and model performance was enhanced by the Bayesian hyperparameter optimization and using an attention-based mechanism. The model achieved accurate prediction of CTCF-binding RNA sites on two different CLIP-seq datasets. A candidate consensus RNA motif (AGAUNGGA) has been identified for the human CTCF through analyzing the learned sequence features of the convolution layer. Notably, this candidate RNA motif of CTCF is strikingly different from its DNA recognition motif (CCGCGNGGNGGCAG) (12). Moreover, by applying DeepLncCTCF to human lncRNAs, we identified 4925 candidate CTCF-binding lncRNAs, which may help elucidate how CTCF functions in 3D genome organization.
MATERIALS AND METHODS
Dataset preparation
Transcriptome-wide identification of CTCF-binding RNA sites was performed in two previous studies: one for human bone osteosarcoma epithelial cells (U2OS cells) with two biological replicates using photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation followed by deep sequencing (14), and the other for mouse embryonic stem cells (mESCs) with two biological replicates on Days 0 and 3 of cell differentiation by CLIP-seq (13). Processed CLIP-seq peak data from the two studies were downloaded from NCBI’s Gene Expression Omnibus (47) (GEO accession: GSE53554 and GSE58242). To ensure data quality for model construction, the shared regions of the two biological replicates in each study were used to derive positive instances. For mESCs, the shared regions from Days 0 and 3 were combined. The length of each shared region was normalized by obtaining the midpoint and then extending N nucleotides (nt) both upstream and downstream in the RNA transcript, yielding a positive instance of (2N + 1) nt. The negative instances were generated by randomly selecting regions of length (2N + 1) from the same RNA transcripts with the positive instances, under the constraints that they did not overlap any CLIP-seq peaks. Six different lengths of the input sequences (51, 101, 201, 301, 401 and 501 nt) were evaluated for model performance before the sequence length of 201 nt was selected. The high-confidence positive and negative instances (56 820 and 56 769 instances for the human U2OS dataset; 15 688 and 15 662 instances for the mouse ESC dataset) were randomly partitioned for training, validation and testing with proportions of 60%, 20% and 20%. To further evaluate the models for human U2OS cells or mouse ESCs, a separate test dataset with low-confidence positive instances was also compiled using the CLIP-seq peaks only from one of the two biological replicates (excluding the shared regions) with the same procedure as described earlier.
DeepLncCTCF model construction
As shown in Figure 1, a deep learning model, called DeepLncCTCF, was constructed to predict CTCF-binding RNA sites using nucleotide sequence as input. Owing to the requirement of a numerical input for a deep learning algorithm, one-hot encoding was used to convert an input sequence to a 4 × 201 binary matrix, as described in previous studies (35,48). Then, the input matrix was fed into a convolution layer to capture sequence motifs. CNNs have been shown to be powerful in image recognition (49). The encoded matrix of a sequence may be regarded as a simplified image data, and convolutional filters, the key components of the convolution layer, can be used to recognize sequence motifs, irrespective of their positions within the sequence. A filter 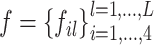 is a real number matrix with dimensions of 4 × L, where the first dimension matches the channels of the input matrix and the second dimension is the desired motif length. After the convolution layer, a max pooling layer was used to summarize the most activated presence of a motif in the sequence by computing the maximum activation value over spatially adjacent subregions. This downsampling strategy reduces the dimensionality of the feature space and thus may increase model robustness.
is a real number matrix with dimensions of 4 × L, where the first dimension matches the channels of the input matrix and the second dimension is the desired motif length. After the convolution layer, a max pooling layer was used to summarize the most activated presence of a motif in the sequence by computing the maximum activation value over spatially adjacent subregions. This downsampling strategy reduces the dimensionality of the feature space and thus may increase model robustness.
Figure 1.
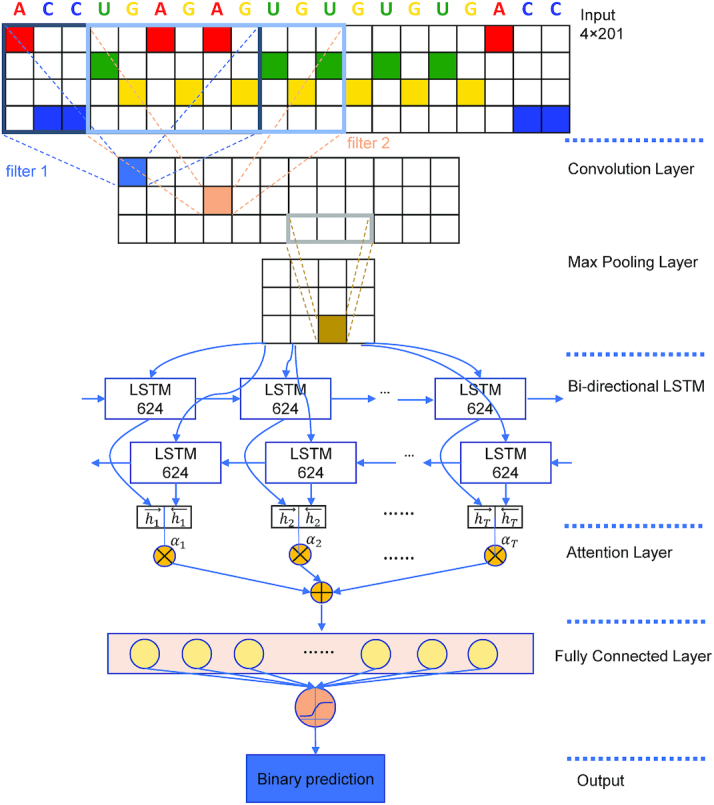
Model architecture of DeepLncCTCF. The hyperparameters shown in the figure achieved the highest model performance with the input sequence length of 201 nt on the dataset of human U2OS cells. First, the input sequence is converted into a 4 × 201 binary matrix using one-hot encoding. Second, the convolutional filters act as motif detectors to scan the input matrix. Third, max pooling is used to reduce the dimension of the representation. Fourth, a BLSTM layer is used to model the dependencies among motifs learned by previous layers in both directions. Fifth, an attention layer is added to capture the most informative features. Finally, the fully connected layers produce a binary output to predict a CTCF-binding site on the input RNA.
A BLSTM layer was then used to model the long-distance dependencies among learned motifs in both forward and backward directions. Our rationale for including the BLSTM layer is that the specificity and affinity of CTCF binding may be determined by multiple related motifs in the target RNA sequence. LSTM is a type of recurrent neural network that can overcome the vanishing gradient problem (50). The capability of LSTM to remember information for a long duration enables it to capture the combinations or dependencies among sequence motifs. Each LSTM unit typically consists of four components: three gates (input, forget and output) and a single cell. The cell memorizes values over arbitrary intervals and the gates regulate the information flow into and out of the cell (51). Specifically, suppose the LSTM takes a sequence 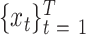 as input, and at each position t, denote the hidden state as
as input, and at each position t, denote the hidden state as 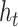 , cell state as
, cell state as  , forget gate as
, forget gate as 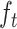 , input gate as
, input gate as 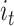 and output gate as
and output gate as 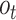 , then the information flow can be summarized as follows:
, then the information flow can be summarized as follows:
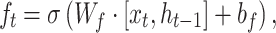 |
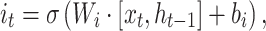 |
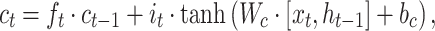 |
 |
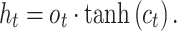 |
Following the BLSTM layer, an attention layer was used to capture the most important sequence motifs to enhance model performance. Since the most discriminating motifs may be located somewhere in the input sequence, the attention mechanism can be used to retrieve more information by accessing the whole hidden state sequence of the BLSTM and then assign more weights to the important positions (52). Mathematically, the attention layer takes the hidden states  from preceding BLSTM layer as inputs, and returns the output vector
from preceding BLSTM layer as inputs, and returns the output vector 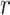 as shown below:
as shown below:
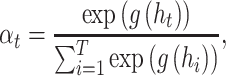 |
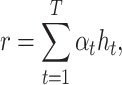 |
where 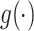 is a neural network with one fully connected layer that returns a scalar importance value.
is a neural network with one fully connected layer that returns a scalar importance value.
Lastly, a fully connected layer was used to integrate the information from the attention layer and learn the nonlinear relationships. Dropout and L2 regularization were used to regularize the model and thus avoid overfitting. Dropout is a process to randomly set the activation to zero for a proportion of units, which can reduce overfitting (53). L2 regularization can be used to generalize the model by adding penalty to the sum of the square of weights (54).
Model training and evaluation
DeepLncCTCF was trained using the Adam optimization algorithm with minibatches (https://arxiv.org/abs/1412.6980). All model parameters were learned to minimize the binary cross-entropy loss function, which captures the difference between the target values and predicted values. After each epoch of training, the validation loss was monitored. The model was continuously trained until the validation loss stopped to decrease for five consecutive epochs. After training, the model was evaluated using a test dataset and the performance was measured by several metrics, including accuracy, sensitivity, specificity, Matthews correlation coefficient (MCC) and the area under the receiver operating characteristic (ROC) curve (AUC).
The hyperparameters for model construction were tuned using Bayesian optimization via Hyperopt (55) with the training and validation data from human U2OS cells. Bayesian optimization has been shown to be more time-efficient for hyperparameter tuning than grid or random search, especially for a large hyperparameter space (42–44). It optimizes the hyperparameters by constructing a probability model (surrogate function) based on the past evaluation results of the objective function. The probability model is updated after each evaluation of the objective function by incorporating new results, and is relatively easier to optimize than the objective function. Tree-structured Parzen estimator was employed as the surrogate function in Hyperopt, and (1 − AUC) was used as the objective function to optimize the ROC AUC values. The hyperparameters that achieved the best performance on the validation data for each input length are detailed in Supplementary Table S1. DeepLncCTCF was implemented in Python using Keras 2.2.4 (https://github.com/fchollet/keras). In this study, model training and testing were performed using a high-performance computing cluster with 20 CPU cores and 100 GB memory. It took ∼5.5 h to fully train the DeepLncCTCF model using the data from human U2OS cells and ∼2 min to make the predictions for the high-confidence test dataset. By comparison, iDeepS (41) took ∼5 h for model training and ∼40 min to make the predictions (owing to secondary structure prediction). DeeperBind (40) made the predictions after each epoch of training, and the total time was ∼16 h. For DeepBind (37), the training process could be speeded up using parallel implementation on a GPU.
Motif analysis
Filters in a convolution layer act as motif detectors to scan input sequences. Therefore, the filters in the convolution layer were converted to position weight matrices (PWMs) as described for DeepBind and Basset (37,56). Specifically, for each positive sequence s in the test set, the activation value 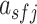 of the filter f at position j was calculated. If
of the filter f at position j was calculated. If 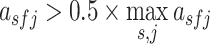 , where
, where  denotes the maximum activation value of the filter f across all the sequences in the dataset, the subsequence with length L and starting at the position j was selected. The selected subsequences were aligned to obtain PWMs using WebLogo (57). The PWMs learned from the human U2OS and mouse ESC datasets were compared using Tomtom 4.11.4 of MEME Suite (http://meme-suite.org/tools/tomtom) (58).
denotes the maximum activation value of the filter f across all the sequences in the dataset, the subsequence with length L and starting at the position j was selected. The selected subsequences were aligned to obtain PWMs using WebLogo (57). The PWMs learned from the human U2OS and mouse ESC datasets were compared using Tomtom 4.11.4 of MEME Suite (http://meme-suite.org/tools/tomtom) (58).
The PWMs captured by DeepLncCTCF served as relevant features to distinguish between positive and negative instances. To identify the PWMs that may represent specific RNA motifs in positive instances, two analyses were conducted. First, enrichment analysis on the PWMs was used to measure their overrepresentations in the positive instances of the test data using hypergeometric test. Second, for each PWM, the Kolmogorov–Smirnov (KS) test was used to detect any significant difference in its position distributions between positive and negative instances.
The PWMs were grouped by hierarchical clustering based on Spearman’s correlations between the activities of filters from which the PWMs were obtained. The activity of a filter f over a sequence s was computed as the weighted sum of the activation values of filter f across all positions over sequence s, 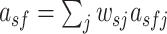 , as described for DeepCpG (35). The weight
, as described for DeepCpG (35). The weight 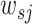 was assigned the highest value if j is the central position of the sequence s, and linearly decreased as the distance between position j and the central position increases.
was assigned the highest value if j is the central position of the sequence s, and linearly decreased as the distance between position j and the central position increases.
Motif discovery using MEME
Many computational methods such as MEME (58), LDDMS (59) and EMS3 (60) have been developed to efficiently explore the putative patterns hidden in biological sequences. In this study, the widely used and readily accessible MEME (http://meme-suite.org/tools/meme) with the differential enrichment mode (58) was applied to the identification of candidate RNA motifs enriched in the positive instances relative to the negative instances in the human U2OS dataset. The motif length was set to range from 8 to 25 nt. Motifs discovered by MEME were compared with the PWMs captured by DeepLncCTCF using Tomtom (58).
RNA secondary structure prediction
In addition to nucleotide sequences, RNA secondary structure profiles were also examined for model construction. RNA secondary structures were predicted using a modified version of RNAplfold (61,62), and the probabilities for each position of a sequence to be in a hairpin loop, inner loop, multi-loop, external regions and paired regions were calculated. The probabilities of all positions in a sequence were represented as a real number matrix with dimensions of 5 × 201.
Conventional machine learning algorithms
DeepLncCTCF was compared with several conventional machine learning algorithms, including support vector machine (SVM), random forest (RF) and gradient boosting (XGB, implemented using XGBoost). To derive sequence-based features for these models, we calculated the frequencies of all subsequences of length k, known as k-mers, for each data instance. A set of 340 k-mer features was obtained with k equal to 1–4 (41 + 42 + 43 + 44 = 340). The parameters for SVM, RF and XGB were tuned using grid search with the training data from human U2OS cells. The training parameters are shown in Supplementary Table S2.
Identification of candidate CTCF-binding lncRNAs
To identify candidate lncRNAs that bind to CTCF, human lncRNA transcripts were downloaded from GENCODE version 29 (63). DeepLncCTCF was used to predict potential CTCF-binding sites on the lncRNAs that were not included in the training dataset. All subsequences of 201 nt were fed into DeepLncCTCF to predict whether they could bind to CTCF. If the predicted probability of a subsequence to be a CTCF-binding RNA site was ≥0.8, it was regarded as a high-confidence CTCF-binding RNA site. The CTCF-binding sites that overlapped with each other were combined into a single one. To reduce the false positive rate, the lncRNAs with at least two high-confidence CTCF-binding RNA sites were selected as candidate CTCF-binding lncRNAs.
RESULTS
DeepLncCTCF for accurate prediction of CTCF-binding sites on RNAs
We have developed DeepLncCTCF to discover the RNA recognition pattern of CTCF using nucleotide sequence as input (Figure 1). As described in the ‘Materials and Methods’ section, high-confidence positive and negative instances for model construction were obtained from human U2OS cells and mouse ESCs (13,14). Since the input size may affect model performance, we examined six different input sequence lengths, ranging from 51 to 501 nt. As shown in Figure 2A, model performance increased steeply from 51 to 201 nt, but began to level off after 201 nt. Since the risk of model overfitting as well as computational cost generally increases with the size of input, we selected the input length of 201 nt for model construction in this study.
Figure 2.

Accurate prediction of CTCF-binding RNA sites by DeepLncCTCF. (A) The AUC of DeepLncCTCF using six different input sequence lengths in nucleotides. The trend line was fitted using polynomial regression. The shaded area indicates the 95% confidence intervals. (B) Comparison of model performance based on accuracy, sensitivity, specificity, MCC and AUC using the high-confidence test dataset of human U2OS cells. The models are DeepLncCTCF, DeepLncCTCF_struct (using both RNA sequences and secondary structure information as input), DeepLncCTCF_no_attn (without the attention layer), iDeepS (41), DeeperBind (40), DeepBind (37), XGB and SVM. (C) ROC curves of the models on the high-confidence test dataset of human U2OS cells. (D) ROC curves of the models on the separate, low-confidence test dataset of human U2OS cells (see the ‘Materials and Methods’ section).
DeepLncCTCF achieved an AUC of 0.863 for human U2OS cells and 0.861 for mouse ESCs on the high-confidence test datasets (Table 1, Figure 2B and C, and Supplementary Figure S1A and B). It outperformed conventional SVM, RF and XGB models as indicated by AUC, accuracy, sensitivity, specificity and MCC (Table 1, Figure 2B and C, and Supplementary Figure S1A and B). The results suggest that DeepLncCTCF can learn relevant features from nucleotide sequence for accurate prediction of CTCF-binding RNA sites. However, model performance was not further improved by adding RNA secondary structure information to the input with the human U2OS dataset (Table 1 and Figure 2B and C), and might be slightly enhanced with the mouse ESC dataset (Supplementary Figure S1A and B), suggesting that DeepLncCTCF could capture the structural information from the nucleotide sequence. Notably, when an attention layer was used, a statistically significant increase in prediction accuracy was achieved with both datasets (Table 1, Figure 2B and C, and Supplementary Figure S1A and B; P-value = 0.0002 for the human U2OS dataset and P-value = 0.001 for the mouse ESC dataset, one-sided Wilcoxon rank-sum test), indicating the effectiveness of emphasizing on the most important features to enhance model performance.
Table 1.
Predictive performance of various models on the high-confidence test dataset of human U2OS cells
| Model | Accuracy | Sensitivity | Specificity | MCC | AUC |
|---|---|---|---|---|---|
| DeepLncCTCF | 0.775 ± 0.004 | 0.733 ± 0.031 | 0.817 ± 0.033 | 0.553 ± 0.008 | 0.863 ± 0.005 |
| DeepLncCTCF_struct | 0.773 ± 0.007 | 0.727 ± 0.038 | 0.818 ± 0.036 | 0.549 ± 0.015 | 0.860 ± 0.008 |
| DeepLncCTCF_no_attn | 0.761 ± 0.004 | 0.739 ± 0.028 | 0.784 ± 0.032 | 0.524 ± 0.009 | 0.848 ± 0.003 |
| iDeepS | 0.737 ± 0.006 | 0.709 ± 0.087 | 0.765 ± 0.086 | 0.483 ± 0.008 | 0.818 ± 0.002 |
| DeeperBind | 0.725 ± 0.009 | 0.676 ± 0.080 | 0.773 ± 0.076 | 0.457 ± 0.012 | 0.803 ± 0.006 |
| DeepBind | 0.670 ± 0.004 | 0.678 ± 0.029 | 0.663 ± 0.027 | 0.324 ± 0.009 | 0.744 ± 0.004 |
| SVM | 0.696 ± 0.002 | 0.734 ± 0.005 | 0.658 ± 0.004 | 0.393 ± 0.004 | 0.768 ± 0.003 |
| RF | 0.680 ± 0.002 | 0.702 ± 0.003 | 0.658 ± 0.004 | 0.360 ± 0.004 | 0.750 ± 0.002 |
| XGB | 0.696 ± 0.003 | 0.716 ± 0.005 | 0.676 ± 0.005 | 0.392 ± 0.006 | 0.771 ± 0.002 |
Model performance is measured by the mean accuracy, sensitivity, specificity, MCC and AUC for 10 repetitions. The highest value for each performance metric is shown in bold. The standard deviation for each metric value is also shown. The models include DeepLncCTCF, DeepLncCTCF_struct (using both RNA sequence and secondary structure information as input), DeepLncCTCF_no_attn (without the attention layer), iDeepS (41), DeeperBind (40), DeepBind (37), SVM, RF and XGB.
Furthermore, we compared the performance of DeepLncCTCF with the other deep learning models, including iDeepS (41), DeeperBind (40) and DeepBind (37). We demonstrated that DeepLncCTCF with or without an attention layer significantly outperformed iDeepS, DeeperBind and DeepBind on the same test datasets (Figure 2B and C, and Supplementary Figure S1A and B). Moreover, we noted that DeepLncCTCF achieved comparable performance on the human U2OS and mouse ESC datasets, whereas the other three models did not perform as well on the mouse ESC dataset (Supplementary Figure S1A and B) as they did on the human U2OS dataset (Figure 2B and C). When the models were further evaluated with the separate, low-confidence test datasets (see the ‘Materials and Methods’ section), DeepLncCTCF also achieved better performance (Figure 2D and Supplementary Figure S1C). Taken together, the results suggest the superior performance of DeepLncCTCF for identifying CTCF-binding RNA sites from the primary sequence.
Identification of CTCF-binding RNA motifs
Convolutional filters can recognize motif patterns in the input sequences. The filters in the convolution layer of DeepLncCTCF constructed with the human U2OS dataset were thus converted into 128 PWMs (Supplementary Figure S2). To identify the PWMs that may represent candidate motifs in CTCF-binding RNA sites, their enrichments in positive instances and differences of position distributions between positive and negative instances were analyzed using hypergeometric and KS tests, respectively. Among the 128 PWMs, 65 were found to be significantly enriched in the positive data with the hypergeometric false discovery rate (FDR) ≤0.01 and show significantly different position distributions between positive and negative instances (KS P-value ≤0.01) (Supplementary Table S3), including the six most significant PWMs shown in Figure 3 and Supplementary Figure S3 (M35, M48, M71, M75, M96 and M123; KS P-value <1e-40 and hypergeometric FDR <1e-180). These six PWMs appear to fall into two groups based on their sequence logos (Figure 3A; M48, M71, M75 and M96 in group 1; M35 and M123 in group 2), and each group may represent one consensus motif as multiple filters can learn the shifted and truncated versions of a single motif. Interestingly, the sequence sites matching the 6 PWMs or the 65 selected PWMs showed a position bias with the highest frequency near the center of CTCF-binding RNA regions, whereas these sites appeared to be evenly distributed along the negative instances (Supplementary Figures S3 and S4). The results suggest that CTCF may have a specific recognition pattern for RNA binding.
Figure 3.

Selected DeepLncCTCF PWMs that may represent CTCF-binding RNA motifs. (A) Sequence logos of the PWMs. (B) Significant enrichment of the PWMs in positive data instances (hypergeometric FDR ≤1e-180).
To further reveal the consensus RNA motifs of CTCF, clustering analysis of the 128 PWMs was performed by first calculating pairwise Spearman’s correlations between filter activities and then applying hierarchical clustering on the correlation matrix (see the ‘Materials and Methods’ section). As shown in Figure 4 and Supplementary Table S4, we have empirically identified six clusters, including the majority (66.2%) of the 65 PWMs that may represent specific RNA motifs in positive instances. PWMs in the same cluster tend to have similar core motifs (Figure 4). In particular, the PWMs in clusters 2 and 4 may represent candidate motifs in CTCF-binding RNA sites. Clusters 2 and 4 include the most significant PWMs in group 1 (M48, M71, M75 and M96) and group 2 (M35 and M123), respectively (Figure 3). The core motifs with the consensus sequences AGAU (cluster 2) and AGAUNGGA (cluster 4) are likely to be involved in the RNA recognition of CTCF. The multiple occurrences of the two consensus motifs in the two clusters indicate their importance for predicting CTCF-binding RNA sites. Interestingly, the consensus sequence AGAU appears to be part of the larger motif AGAUNGGA, suggesting the latter as the possible consensus RNA motif of CTCF. Remarkably, this consensus RNA motif (AGAUNGGA) is strikingly different from CTCF’s DNA consensus sequence (CCGCGNGGNGGCAG) (12). The result is consistent with the previous finding that CTCF’s RNA-binding domain is different from its DNA-binding domain (14).
Figure 4.

Clustering analysis of the PWMs captured by DeepLncCTCF. Six clusters were identified by inspecting the dendrogram, and PWMs with similar core motifs tended to cluster together. The core motifs of clusters 2 and 4 appear to have the consensus sequences AGAU and AGAUNGGA, respectively.
To our knowledge, specific RNA motifs for CTCF binding have not been demonstrated in previous studies. To further assess the candidate RNA motifs identified by DeepLncCTCF, we also performed motif discovery in the same dataset using MEME with the differential enrichment mode (58). Eleven enriched motifs were discovered (Supplementary Figure S5), which were further compared with the 128 PWMs learned by DeepLncCTCF using the Tomtom algorithm (58). As shown in Supplementary Table S5, 38 of the DeepLncCTCF PWMs were significantly matched to 8 MEME motifs (E-value ≤0.05). In particular, 22 and 15 DeepLncCTCF PWMs were matched to the MEME motifs ME4 and ME0, respectively. More importantly, all PWMs in cluster 2 and six of the seven PWMs in cluster 4 were matched to ME4, further suggesting the consensus sequence, AGAUNGGA, as a CTCF-binding RNA motif. However, besides the DeepLncCTCF PWMs matched with MEME motifs, 30 of the 65 PWMs selected for candidate RNA motifs were not discovered by MEME, indicating that DeepLncCTCF learned additional informative motifs. Collectively, our results suggest that CTCF clearly has a specific recognition pattern for RNA binding and DeepLncCTCF is able to recognize the complex pattern.
Comparison of human and mouse motifs learned by DeepLncCTCF
The CTCF protein and its DNA-binding motif are highly conserved between humans and mice (12). It is thus interesting to examine whether CTCF-binding RNA motifs may also share any similarity between these two species. To this end, we have compared the PWMs learned by the DeepLncCTCF model using the human U2OS dataset with the PWMs of the model using the mouse ESC dataset. The mouse model discovered 126 PWMs (Supplementary Figure S6), 40 of which may represent candidate RNA motifs in positive instances based on KS and hypergeometric tests (Supplementary Figure S7 and Supplementary Table S6; KS P-value ≤0.01 and hypergeometric FDR ≤0.01). Interestingly, 10 mouse PWMs were significantly matched to human PWMs (Supplementary Table S7; E-value ≤0.05). In particular, the mouse PWM M2 (1 of the 40 PWMs selected for candidate RNA motifs, KS P-value = 0.006 and hypergeometric FDR = 4.35e−7) significantly matched to the human PWM M121 (1 of the 65 selected PWMs, KS P-value = 9.63e−43 and hypergeometric FDR = 7.22e−21) (Figure 5). If the threshold of Tomtom E-value was changed to 0.2, additional 23 mouse PWMs were also found to match with human PWMs. For instance, the mouse PWMs, M36 and M93 (2 of the 40 PWMs for candidate RNA motifs), matched to human M65 and M32 (2 of the 65 PWMs for candidate RNA motifs), respectively (Figure 5). The results suggest that the RNA-binding patterns of CTCF in humans and mice may share some similarities.
Figure 5.

Sequence logos of some similar PWMs learned from human U2OS and mouse ESC datasets. The PWMs learned by DeepLncCTCF from the human U2OS dataset were compared with the PWMs of the mouse ESC model using the Tomtom algorithm (58). The E-values of the matches are shown above the sequence logos.
Identification of candidate lncRNAs that may bind to CTCF
One potential application of DeepLncCTCF is to identify CTCF-binding lncRNAs. As described in the ‘Materials and Methods’ section, DeepLncCTCF was applied to 13 997 human lncRNA genes that were not included in the training dataset, and 4925 were predicted as candidate CTCF-binding lncRNAs (Supplementary Table S8). Notably, the predicted candidate lncRNAs include XIST, TSIX and MYCNOS, which were previously shown to directly interact with CTCF (13,64). Moreover, of the 23 conserved lncRNAs that were previously shown to bind CTCF in mice, 18 were included in the human training dataset and the remaining 5 were predicted as CTCF-binding lncRNAs. The results demonstrate the applicability of DeepLncCTCF for identifying candidate lncRNAs that may bind to CTCF.
CTCF–lncRNA interactions play important roles in 3D genome organization (13,24,27). Particularly, XIST and FIRRE have been shown to directly interact with CTCF during X chromosome inactivation (13,27), and several lncRNAs such as NEAT1 and MALAT1 are known to function as nuclear organization factors to shape 3D genome architecture (25,65–70). Moreover, the candidate CTCF-binding lncRNAs were found to be significantly enriched in a set of nuclear lncRNAs identified in a previous study (71) (Supplementary Figure S8A). Interestingly, ∼63% of the lncRNAs transcribed using bidirectional promoters were predicted as candidate CTCF-binding lncRNAs, which is relatively high when compared with the candidate lncRNAs of other biotypes (Supplementary Figure S8B). These lncRNAs may function as enhancer RNAs to facilitate the formation of enhancer–promoter loops as CTCF is known to be enriched at the loop boundaries to regulate gene expression (72). These findings provide additional evidence for the involvement of CTCF–lncRNA interactions in 3D genome organization.
Recent studies suggest that disruption of 3D genome architecture is involved in human disease, including cancer (73,74). We have thus examined whether CTCF-binding lncRNAs play roles in cancer development. Interestingly, 763 of 1619 known cancer-associated lncRNAs in the Lnc2Cancer database (version 2.0) (75) were identified as candidate CTCF-binding lncRNAs (Supplementary Table S8) or included in the training dataset. In particular, the lncRNAs XIST, NEAT1 and MALAT1 have been shown to be dysregulated in multiple cancers (Table 2) (28,76–81). Many other well-known cancer-associated lncRNAs, such as MEG3, GAS5, SNHG1 and CCAT2 (28,76,77,82–88), may also interact with CTCF (Table 2). The interactions between these cancer-associated lncRNAs and CTCF provide useful information for understanding their roles in cancer development.
Table 2.
List of selected lncRNAs that may interact with CTCF
| LncRNA | Function | CTCF-binding RNA sites | Nuclear/cytosolic (log2 fold change) | References |
|---|---|---|---|---|
| XIST | X chromosome inactivation; promote cell proliferation, migration and invasion and induce apoptosis | Predicted | 6.73 | (66,78,79) |
| MALAT1 | Function in nuclear speckle; promote cell proliferation and metastasis | Experimental | 5.35 | (69,77,80) |
| NEAT1 | Formation and maintenance of nuclear paraspeckle; promote cell growth and metastasis | Experimental | 3.34 | (28,70,81) |
| FIRRE | Formation of interchromosomal regulatory domain | Experimental | 0.77 | (25,68) |
| GAS5 | Inhibit cell proliferation, migration and invasion | Experimental | 6.55 | (77,84–85) |
| MEG3 | Inhibit cell proliferation and induce apoptosis | Predicted | 5.96 | (76,86) |
| SNHG1 | Promote cell proliferation, cell cycle progression and inhibit cell apoptosis | Experimental | 5.27 | (82,87) |
| CCAT2 | Promote tumor growth, metastasis and chromosomal instability | Predicted | 4.02 | (76,88) |
The CTCF-binding sites on the lncRNAs are either used for model training (‘Experimental’) or predicted by DeepLncCTCF. These lncRNAs are involved in 3D genome organization and/or cancer development. The nuclear to cytosolic log2 fold change from a previous study (71) is shown to indicate their enrichment in the nucleus.
DISCUSSION
In this study, we have developed DeepLncCTCF to discover the RNA recognition pattern of CTCF and identify candidate CTCF-binding lncRNAs. CTCF exemplifies a new class of dual DNA- and RNA-binding proteins, which play essential roles in transcriptional regulation and chromatin organization (22,23). We demonstrated that DeepLncCTCF could accurately predict CTCF-binding RNA sites based on nucleotide sequence alone, and significantly outperformed iDeepS (41), DeeperBind (40) and DeepBind (37). Notably, by examining the sequence features learned by the convolution layer of DeepLncCTCF, we have identified a candidate consensus RNA motif, AGAUNGGA, for CTCF binding in humans. To our knowledge, this is the first RNA motif reported so far for CTCF binding. Furthermore, we have applied DeepLncCTCF to the transcriptome-wide prediction of candidate CTCF-binding lncRNAs, some of which were previously known to interact with CTCF.
When compared with the other deep learning methods for protein–RNA binding prediction, such as iDeepS (41), DeeperBind (40) and DeepBind (37), DeepLncCTCF utilized the Bayesian optimization for hyperparameter tuning and an attention-based mechanism to capture the most important features in the input sequence. The optimized DeepLncCTCF model, even without the attention layer, significantly outperformed iDeepS (41), DeeperBind (40) and DeepBind (37), suggesting the robustness of our approach for RNA motif modeling and discovery. The attention layer further enhanced the predictive performance of DeepLncCTCF as indicated by several metrics, including AUC, accuracy, sensitivity, specificity and MCC. Besides the model architecture and hyperparameter optimization, efforts were made to improve the quality of training data. As pointed out by a previous study (89), generating negative data instances by shuffling the positive data instances or randomly selecting from transcripts without positive data instances could lead to overoptimistic performance or introduce false negatives. In this study, the negative data instances were compiled by randomly selecting regions within the same transcripts as for the positive data instances, enabling the model to learn the informative RNA motifs for CTCF binding.
Although DeepLncCTCF learned certain sequence features to distinguish between CTCF-binding and non-binding sites, the underlying RNA motif pattern only became clear after further analyses. The filters in the convolution layer of DeepLncCTCF were converted into PWMs, and statistical analyses were conducted to identify the PWMs that might represent candidate RNA motifs for CTCF binding. Since multiple filters could be employed to represent a single motif, clustering analysis was thus applied to the PWMs. To this end, we have discovered the candidate CTCF-binding RNA motif with a consensus sequence of AGAUNGGA in humans. Interestingly, this RNA motif is strikingly different from CTCF’s DNA consensus sequence (CCGCGNGGNGGCAG) (12). The result is consistent with the previous finding that CTCF’s RNA-binding domain is different from its DNA-binding domain (14). However, further experimental verification is required to confirm the prediction of this RNA motif for CTCF binding.
Recent studies suggest that CTCF–lncRNA interactions can play important roles in shaping 3D genome organization (17–23). Considering the cell-type-specific expression of most lncRNAs (28,29), the interactions between CTCF and lncRNAs may provide a regulatory mechanism for establishing dynamic 3D genome organization. We thus utilized the DeepLncCTCF model to identify a list of candidate CTCF-binding lncRNAs, which could provide useful information for further elucidating the RNA-dependent mechanism of CTCF function in 3D genome organization. In addition, we noted that many well-known cancer-associated lncRNAs might interact with CTCF. Since the disruption of 3D genome architecture occurs in cancer cells (73,74), the possible interactions between these lncRNAs and CTCF might help understand their roles in cancer development.
In conclusion, our work provides a powerful method for exploring the RNA recognition patterns of CTCF and identifying candidate CTCF-binding lncRNAs. The deep learning method has been used to analyze human and mouse datasets in this study, and can be applied to other species when datasets become available in the future. The DeepLncCTCF model may also be used to predict the functional impact of sequence variations on CTCF–RNA interaction. Genome-wide association studies have identified a large number of disease-associated variants with a majority of them located in the non-coding regions (90). Computational methods are needed to predict which of these variants may disrupt CTCF–RNA interaction and thus affect 3D genome organization. The prediction results can be used to annotate and prioritize the disease-associated variants for further experimental studies.
DATA AVAILABILITY
The source code and datasets used in this study for model construction and downstream analyses are freely available at https://github.com/BioDataLearning/DeepLncCTCF.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
No external funding for this work.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bonev B., Cavalli G.. Organization and function of the 3D genome. Nat. Rev. Genet. 2016; 17:661–678. [DOI] [PubMed] [Google Scholar]
- 2. Gomez-Diaz E., Corces V.G.. Architectural proteins: regulators of 3D genome organization in cell fate. Trends Cell Biol. 2014; 24:703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pombo A., Dillon N.. Three-dimensional genome architecture: players and mechanisms. Nat. Rev. Mol. Cell Biol. 2015; 16:245–257. [DOI] [PubMed] [Google Scholar]
- 4. Norton H.K., Phillips-Cremins J.E.. Crossed wires: 3D genome misfolding in human disease. J. Cell Biol. 2017; 216:3441–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Corces M.R., Corces V.G.. The three-dimensional cancer genome. Curr. Opin. Genet. Dev. 2016; 36:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Phillips-Cremins J.E., Sauria M.E., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S., Ong C.T., Hookway T.A., Guo C., Sun Y. et al.. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013; 153:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S. et al.. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010; 467:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phillips J.E., Corces V.G.. CTCF: master weaver of the genome. Cell. 2009; 137:1194–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen H., Tian Y., Shu W., Bo X., Wang S.. Comprehensive identification and annotation of cell type-specific and ubiquitous CTCF-binding sites in the human genome. PLoS One. 2012; 7:e41374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Handoko L., Xu H., Li G., Ngan C.Y., Chew E., Schnapp M., Lee C.W., Ye C., Ping J.L., Mulawadi F. et al.. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011; 43:630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim T.H., Abdullaev Z.K., Smith A.D., Ching K.A., Loukinov D.I., Green R.D., Zhang M.Q., Lobanenkov V.V., Ren B.. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007; 128:1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kung J.T., Kesner B., An J.Y., Ahn J.Y., Cifuentes-Rojas C., Colognori D., Jeon Y., Szanto A., del Rosario B.C., Pinter S.F. et al.. Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol. Cell. 2015; 57:361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saldana-Meyer R., Gonzalez-Buendia E., Guerrero G., Narendra V., Bonasio R., Recillas-Targa F., Reinberg D.. CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Gene Dev. 2014; 28:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hansen A.S., Hsieh T.-H.S., Cattoglio C., Pustova I., Saldaña-Meyer R., Reinberg D., Darzacq X., Tjian R.. Distinct classes of chromatin loops revealed by deletion of an RNA-binding region in CTCF. Mol. Cell. 2019; 76:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saldaña-Meyer R., Rodriguez-Hernaez J., Escobar T., Nishana M., Jácome-López K., Nora E.P., Bruneau B.G., Tsirigos A., Furlan-Magaril M., Skok J.. RNA interactions are essential for CTCF-mediated genome organization. Mol. Cell. 2019; 76:412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonasio R., Shiekhattar R.. Regulation of transcription by long noncoding RNAs. Annu. Rev. Genet. 2014; 48:433–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Engreitz J.M., Ollikainen N., Guttman M.. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat. Rev. Mol. Cell Biol. 2016; 17:756–770. [DOI] [PubMed] [Google Scholar]
- 19. Rinn J.L., Chang H.Y.. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012; 81:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vance K.W., Ponting C.P.. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014; 30:348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hung T., Wang Y., Lin M.F., Koegel A.K., Kotake Y., Grant G.D., Horlings H.M., Shah N., Umbricht C., Wang P.. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011; 43:621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xiao R., Chen J.-Y., Liang Z., Luo D., Chen G., Lu Z.J., Chen Y., Zhou B., Li H., Du X.. Pervasive chromatin–RNA binding protein interactions enable RNA-based regulation of transcription. Cell. 2019; 178:107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hudson W.H., Ortlund E.A.. The structure, function and evolution of proteins that bind DNA and RNA. Nat. Rev. Mol. Cell Biol. 2014; 15:749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sun S., Del Rosario B.C., Szanto A., Ogawa Y., Jeon Y., Lee J.T.. Jpx RNA activates Xist by evicting CTCF. Cell. 2013; 153:1537–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hacisuleyman E., Goff L.A., Trapnell C., Williams A., Henao-Mejia J., Sun L., McClanahan P., Hendrickson D.G., Sauvageau M., Kelley D.R. et al.. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014; 21:198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Splinter E., de Wit E., Nora E.P., Klous P., van de Werken H.J., Zhu Y., Kaaij L.J., van Ijcken W., Gribnau J., Heard E. et al.. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Gene Dev. 2011; 25:1371–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang F., Deng X., Ma W., Berletch J.B., Rabaia N., Wei G., Moore J.M., Filippova G.N., Xu J., Liu Y. et al.. The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 2015; 16:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huarte M. The emerging role of lncRNAs in cancer. Nat. Med. 2015; 21:1253–1261. [DOI] [PubMed] [Google Scholar]
- 29. Gloss B.S., Dinger M.E.. The specificity of long noncoding RNA expression. Biochim. Biophys. Acta. 2016; 1859:16–22. [DOI] [PubMed] [Google Scholar]
- 30. Kai Y., Andricovich J., Zeng Z., Zhu J., Tzatsos A., Peng W.. Predicting CTCF-mediated chromatin interactions by integrating genomic and epigenomic features. Nat. Commun. 2018; 9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang R., Wang Y., Yang Y., Zhang Y., Ma J.. Predicting CTCF-mediated chromatin loops using CTCF-MP. Bioinformatics. 2018; 34:i133–i141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li W., Wong W.H., Jiang R.. DeepTACT: predicting 3D chromatin contacts via bootstrapping deep learning. Nucleic Acids Res. 2019; 47:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Singh R., Lanchantin J., Robins G., Qi Y.. DeepChrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics. 2016; 32:i639–i648. [DOI] [PubMed] [Google Scholar]
- 34. Leung M.K., Xiong H.Y., Lee L.J., Frey B.J.. Deep learning of the tissue-regulated splicing code. Bioinformatics. 2014; 30:i121–i129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Angermueller C., Lee H.J., Reik W., Stegle O.. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017; 18:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Quang D., Xie X.. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016; 44:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831–838. [DOI] [PubMed] [Google Scholar]
- 38. Luo X., Chi W., Deng M.. Deepprune: learning efficient and interpretable convolutional networks through weight pruning for predicting DNA–protein binding. Front. Genet. 2019; 10:1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu T., Wang Z.. HiCNN: a very deep convolutional neural network to better enhance the resolution of Hi-C data. Bioinformatics. 2019; 35:4222–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hassanzadeh H.R., Wang M.D.. DeeperBind: enhancing prediction of sequence specificities of DNA binding proteins. 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2016; Piscataway: IEEE; 178–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pan X., Rijnbeek P., Yan J., Shen H.B.. Prediction of RNA–protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics. 2018; 19:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Snoek J., Larochelle H., Adams R.P.. Practical Bayesian optimization of machine learning algorithms. Advances in Neural Information Processing Systems. 2012; 2951–2959. [Google Scholar]
- 43. Bergstra J.S., Bardenet R., Bengio Y., Kégl B.. Algorithms for hyper-parameter optimization. Advances in Neural Information Processing Systems. 2011; 2546–2554. [Google Scholar]
- 44. Bergstra J., Yamins D., Cox D.D.. Making a science of model search: hyperparameter optimization in hundreds of dimensions for vision architectures. Proceedings of the 30th International Conference on Machine Learning. 2013; 28:I115–I123. [Google Scholar]
- 45. Hong Z., Zeng X., Wei L., Liu X.. Identifying enhancer–promoter interactions with neural network based on pre-trained DNA vectors and attention mechanism. Bioinformatics. 2019; 36:1037–1043. [DOI] [PubMed] [Google Scholar]
- 46. Chen H., Gao M., Zhang Y., Liang W., Zou X.. Attention-based multi-NMF deep neural network with multimodality data for breast cancer prognosis model. Biomed. Res. Int. 2019; 2019:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Edgar R., Domrachev M., Lash A.E.. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang M., Tai C., E W., Wei L.. DeFine: deep convolutional neural networks accurately quantify intensities of transcription factor–DNA binding and facilitate evaluation of functional non-coding variants. Nucleic Acids Res. 2018; 46:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. LeCun Y., Bengio Y., Hinton G.. Deep learning. Nature. 2015; 521:436–444. [DOI] [PubMed] [Google Scholar]
- 50. Hochreiter S., Schmidhuber J.. LSTM can solve hard long time lag problems. Advances in Neural Information Processing Systems. 1997; 473–479. [Google Scholar]
- 51. Greff K., Srivastava R.K., Koutník J., Steunebrink B.R., Schmidhuber J.. LSTM: a search space odyssey. IEEE Trans. Neural Netw. Learn. Syst. 2016; 28:2222–2232. [DOI] [PubMed] [Google Scholar]
- 52. Zhou P., Shi W., Tian J., Qi Z., Li B., Hao H., Xu B.. Attention-based bidirectional long short-term memory networks for relation classification. Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics. 2016; 2:207–212. [Google Scholar]
- 53. Srivastava N., Hinton G., Krizhevsky A., Sutskever I., Salakhutdinov R.. Dropout: a simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014; 15:1929–1958. [Google Scholar]
- 54. Ng A.Y. Feature selection, L1 vs. L2 regularization, and rotational invariance. Proceedings of the 21st International Conference on Machine Learning. 2004; 78. [Google Scholar]
- 55. Bergstra J., Yamins D., Cox D.D.. Hyperopt: a Python library for optimizing the hyperparameters of machine learning algorithms. Proceedings of the 12th Python in Science Conference. 2013; 13–20. [Google Scholar]
- 56. Kelley D.R., Snoek J., Rinn J.L.. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016; 26:990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xiao P., Schiller M., Rajasekaran S.. Novel algorithms for LDD motif search. BMC Genomics. 2019; 20:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xiao P., Cai X., Rajasekaran S.. Efficient algorithms for finding edit-distance based motifs. International Conference on Algorithms for Computational Biology. 2019; 212–223. [Google Scholar]
- 61. Lorenz R., Bernhart S.H., Honer Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kazan H., Morris Q.. RBPmotif: a web server for the discovery of sequence and structure preferences of RNA-binding proteins. Nucleic Acids Res. 2013; 41:W180–W186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Harrow J., Denoeud F., Frankish A., Reymond A., Chen C.K., Chrast J., Lagarde J., Gilbert J.G., Storey R., Swarbreck D. et al.. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006; 7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhao X., Li D., Pu J., Mei H., Yang D., Xiang X., Qu H., Huang K., Zheng L., Tong Q.. CTCF cooperates with noncoding RNA MYCNOS to promote neuroblastoma progression through facilitating MYCN expression. Oncogene. 2016; 35:3565–3576. [DOI] [PubMed] [Google Scholar]
- 65. Mao Y.S., Sunwoo H., Zhang B., Spector D.L.. Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat. Cell Biol. 2011; 13:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Engreitz J.M., Pandya-Jones A., McDonel P., Shishkin A., Sirokman K., Surka C., Kadri S., Xing J., Goren A., Lander E.S. et al.. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013; 341:1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rinn J., Guttman M.. RNA and dynamic nuclear organization: long noncoding RNAs may function as organizing factors that shape the cell nucleus. Science. 2014; 345:1240–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Quinodoz S., Guttman M.. Long noncoding RNAs: an emerging link between gene regulation and nuclear organization. Trends Cell Biol. 2014; 24:651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tripathi V., Ellis J.D., Shen Z., Song D.Y., Pan Q., Watt A.T., Freier S.M., Bennett C.F., Sharma A., Bubulya P.A. et al.. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell. 2010; 39:925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Clemson C.M., Hutchinson J.N., Sara S.A., Ensminger A.W., Fox A.H., Chess A., Lawrence J.B.. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell. 2009; 33:717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gudenas B.L., Wang L.. Prediction of lncRNA subcellular localization with deep learning from sequence features. Sci. Rep. 2018; 8:16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kadauke S., Blobel G.A.. Chromatin loops in gene regulation. Biochim. Biophys. Acta. 2009; 1789:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Valton A.L., Dekker J.. TAD disruption as oncogenic driver. Curr. Opin. Genet. Dev. 2016; 36:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Achinger-Kawecka J., Clark S.J.. Disruption of the 3D cancer genome blueprint. Epigenomics. 2017; 9:47–55. [DOI] [PubMed] [Google Scholar]
- 75. Ning S., Zhang J., Wang P., Zhi H., Wang J., Liu Y., Gao Y., Guo M., Yue M., Wang L.. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2015; 44:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schmitt A.M., Chang H.Y.. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016; 29:452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang R., Xia L.Q., Lu W.W., Zhang J., Zhu J.S.. LncRNAs and cancer. Oncol. Lett. 2016; 12:1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wei W., Liu Y., Lu Y., Yang B., Tang L.. LncRNA XIST promotes pancreatic cancer proliferation through miR‐133a/EGFR. J. Cell. Biochem. 2017; 118:3349–3358. [DOI] [PubMed] [Google Scholar]
- 79. Ma L., Zhou Y., Luo X., Gao H., Deng X., Jiang Y.. Long non-coding RNA XIST promotes cell growth and invasion through regulating miR-497/MACC1 axis in gastric cancer. Oncotarget. 2017; 8:4125–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jiao F., Hu H., Yuan C., Wang L., Jiang W., Jin Z., Guo Z., Wang L.. Elevated expression level of long noncoding RNA MALAT-1 facilitates cell growth, migration and invasion in pancreatic cancer. Oncol. Rep. 2014; 32:2485–2492. [DOI] [PubMed] [Google Scholar]
- 81. Sun C., Li S., Zhang F., Xi Y., Wang L., Bi Y., Li D.. Long non-coding RNA NEAT1 promotes non-small cell lung cancer progression through regulation of miR-377-3p-E2F3 pathway. Oncotarget. 2016; 7:51784–51814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang M., Wang W., Li T., Yu X., Zhu Y., Ding F., Li D., Yang T.. Long noncoding RNA SNHG1 predicts a poor prognosis and promotes hepatocellular carcinoma tumorigenesis. Biomed. Pharmacother. 2016; 80:73–79. [DOI] [PubMed] [Google Scholar]
- 83. You J., Fang N., Gu J., Zhang Y., Li X., Zu L., Zhou Q.. Noncoding RNA small nucleolar RNA host gene 1 promote cell proliferation in nonsmall cell lung cancer. Indian J. Cancer. 2014; 51:e99–e102. [DOI] [PubMed] [Google Scholar]
- 84. Qiao H.P., Gao W.S., Huo J.X., Yang Z.S.. Long non-coding RNA GAS5 functions as a tumor suppressor in renal cell carcinoma. Asian Pac. J. Cancer Prev. 2013; 14:1077–1082. [DOI] [PubMed] [Google Scholar]
- 85. Hu L., Ye H., Huang G., Luo F., Liu Y., Liu Y., Yang X., Shen J., Liu Q., Zhang J.. Long noncoding RNA GAS5 suppresses the migration and invasion of hepatocellular carcinoma cells via miR-21. Tumour Biol. 2016; 37:2691–2702. [DOI] [PubMed] [Google Scholar]
- 86. Lu K.H., Li W., Liu X.H., Sun M., Zhang M.L., Wu W.Q., Xie W.P., Hou Y.Y.. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013; 13:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cui Y., Zhang F., Zhu C., Geng L., Tian T., Liu H.. Upregulated lncRNA SNHG1 contributes to progression of non-small cell lung cancer through inhibition of miR-101-3p and activation of Wnt/β-catenin signaling pathway. Oncotarget. 2017; 8:17785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ling H., Spizzo R., Atlasi Y., Nicoloso M., Shimizu M., Redis R.S., Nishida N., Gafa R., Song J., Guo Z. et al.. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013; 23:1446–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pan X., Shen H.-B.. Predicting RNA–protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics. 2018; 34:3427–3436. [DOI] [PubMed] [Google Scholar]
- 90. Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L.. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2013; 42:D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code and datasets used in this study for model construction and downstream analyses are freely available at https://github.com/BioDataLearning/DeepLncCTCF.