

Abstract
Osteoclasts are bone-resorbing cells that play an essential role in the remodeling of bone. Defects in osteoclasts thus result in unbalanced bone remodeling, leading to numerous pathological conditions such as osteoporosis, bone metastasis, and inflammatory bone erosion. Metabolism is any process a cell utilizes to fulfill its energetic demand for biological functions. Along with signaling pathways and osteoclast-specific gene expression programs, osteoclast differentiation activates metabolic programs. The energy generated from metabolic reprogramming in osteoclasts not only supports the phenotypic changes from mononuclear precursor cells to multinuclear osteoclasts, but also facilitates bone resorption, a major function of terminally differentiated, mature osteoclasts. While oxidative phosphorylation is studied as a major metabolic pathway that fulfills the energy demands of osteoclasts, all metabolic pathways are closely interconnected. Therefore, it remains important to understand the various aspects of osteoclast metabolism, including the roles and effects of glycolysis, glutaminolysis, fatty acid synthesis, and fatty acid oxidation. Targeting the pathways associated with metabolic reprogramming has shown beneficial effects on pathological conditions. As a result, it is clear that a deeper understanding of metabolic regulation in osteoclasts will offer broader translational potential for the treatment of human bone disorders.
Keywords: Osteoclasts, Metabolism, Metabolic reprogramming
Introduction
As bone-resorbing cells, osteoclasts differentiate from myeloid lineage cells, a process during which osteoclast precursor cells fuse with one another to produce giant, multinucleated cells[1, 2]. At the same time, mononuclear preosteoclasts generate variety proteins to induce fusion and promote osteoclast differentiation, which demands increased bioenergetics[3]. When differentiation is complete, mature osteoclasts are tightly attached to bone, forming sealing zones that isolate a resorptive compartment; ruffled boarders are developed in the mature osteoclast membranes within these zones. The proton pumps and chloride channels in ruffled boarders facilitate the massive transport of transcellular acid, which acidifies the resorptive compartment. As a result, when bone matrix is released, it is subsequently resorbed by acid proteases released from osteoclasts, such as cathepsin K[4]. Osteoclasts are also highly motile[5]. Thus, although energy metabolism in osteoclasts has not been extensively investigated, both osteoclast formation and bone resorption are viewed as energy-intensive steps that require active metabolic reprograming.
Given the rapid development of metabolic studies, the important functions and drivers of metabolic reprogramming in osteoclast differentiation have been increasingly highlighted and uncovered in recent years. However, understanding of metabolic reprogramming in osteoclasts remains in an early stage, with incomplete characterizations. In this review, we identify recent insights on metabolic reprogramming during osteoclastogenesis and bone resorption, examining how cellular metabolism shape osteoclasts, and offering questions that must be addressed by the scientific community going forward, in order to target the interplay between osteoclasts and their pathological environments.
Mitochondria biogenesis in osteoclasts
Cellular processes are driven by the hydrolysis of adenosine triphosphate (ATP), a key factor for energy transfer and storage in cells, as well as a building block for DNA, RNA, and proteins. Cellular ATP levels are correlated with cell viability, and the depletion of ATP leads to the necrosis or apoptosis of cells[6]. Mitochondria are unique, doublemembrane organelles responsible for energy generation in cells, converting oxygen and nutrients into ATP [7]. Beyond ATP production, mitochondria also produce metabolic precursors and reactive oxygen species, and maintain ion homeostasis and sense stressors[8]. The proper functioning of mitochondria is critical for the successful differentiation of osteoclasts.
Cells with higher levels of mitochondria are considered to have higher capacities for energy generation. RANKL-induced osteoclastogenesis results in increases in mitochondria size and number, with mature osteoclasts containing an abundance of mitochondria[9]. Due to the direct increase in mitochondria, osteoclast differentiation has long been considered a process of active metabolic reprogramming and adaptation. Consistently, several factors related to mitochondrial biogenesis and functions, such as peroxisome proliferator–activated receptor-gamma coactivator 1β (PGC1β), peroxisome proliferator–activated receptor y (PPARy), and estrogen-related receptor α (ERRα), have been shown to play a fundamental role in osteoclast differentiation and function. Such factors govern key metabolic processes through the transcriptional regulation of distinct metabolic genes during osteoclast differentiation undertaken as a part of bone remodeling [10–13]. Among them, PGC1β, a known regulator of mitochondrial biogenesis in a wide array of cells, has been shown to stimulate mitochondrial biogenesis in osteoclasts[10]. This link between PGC1β and osteoclasts is well-established. PGC1β-deficient mice exhibit increased bone mass with impaired osteoclast function, and the deletion of PGC1β suppresses in vitro osteoclastogenesis[10, 11]. RANKL can induce PGC1β in many ways, such as via ROS and pCREB activation[10]. Wei et al show that PGC-1β can be induced by β-catenin when cells are treated with rogiglitazone, a PPARy agonist[11]. PGC1β is also induced by alternative NF-kB downstream pathways of RANKL[14]. However, the overexpression of PGC1β does not restore impaired osteoclastogenesis and defective mitochondrial biogenesis of RelB cKO osteoclasts. Furthermore, overexpression of PGC1β has been observed to be incapable of rescuing defects in mitochondrial biogenesis in ASXL2-deficient cells[15]. These results suggest that mitochondrial biogenesis during osteoclast differentiation is regulated in both PGC1β-dependent and PGC1β –independent manners[16], and that PGC1β regulates osteoclastogenesis by controlling other factors in addition to the generation of mitochondria (Figure 1A). Therefore, while RANKL stimulation induces mitochondrial biogenesis through complicated signaling cascades, it is unclear whether mitochondrial biogenesis is a prerequisite for osteoclast differentiation and function.
Figure 1. Mitochondria play an important role in osteoclast differentiation.
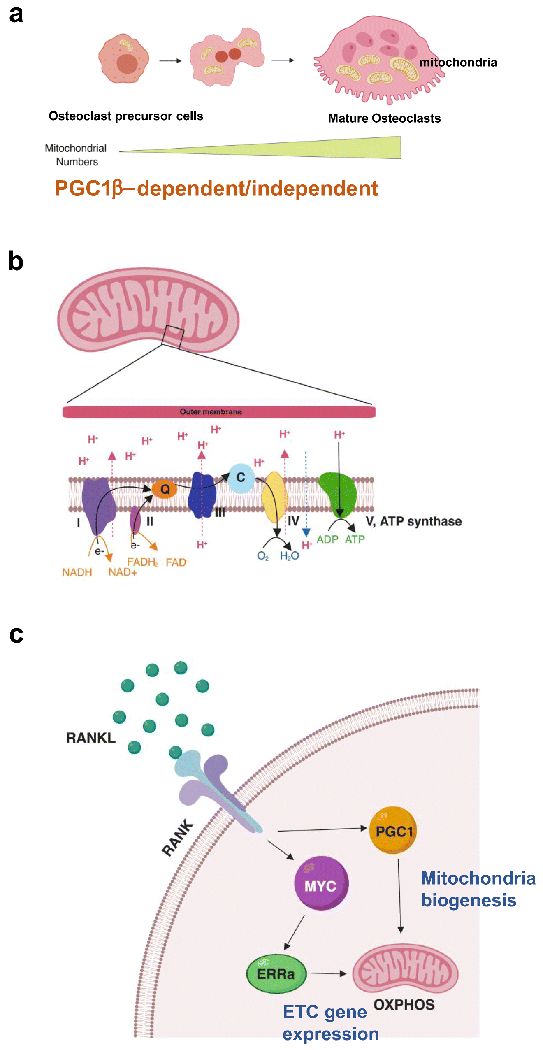
(A) Osteoclasts are formed by the fusion of monocytic osteoclast precursors after precursor cell exposure to osteoclastogenic signals such as RANKL. During differentiation, osteoclast precursor cells undergo rapid and extensive changes in shape and size to generate multinuclear cells. In fully differentiated mature osteoclasts, mitochondria are formed in a complex tubular network, increasing in size and full of cristae. Mitochondria biogenesis is induced by PGC1β-dependent and independent mechanism. (B) Electro transport chain (ETC) reactions occur in the inner membrane of mitochondria and ETC is the site of oxidative phosphorylation. Electron [58] transfer couples with the transfer of protons (H+), leading to generating proton gradient. NADH and FADH2 : reduced electro carriers; Q: ubiquinone; C; cytochrome C. (C) RANKL signaling induces PGC1β, which is involved in mitochondria biogenesis, and the MYC-ERRα axis to regulate ETC gene expression and OXPHOS.
Mitochondria have their own mitochondrial DNA (mtDNA), which encode genes of the respiratory chain. The mitochondrial transcription factor A (Tfam,) regulates mtDNA transcription and repair, and controls mtDNA copy numbers[17]. Myeloid cell-specific Tfam deficiency causes the depletion of mtDNA and reduces ATP production in osteoclasts[18]. However, despite significantly decreased osteoclast and mitochondrial biogenesis levels, Tfam cKO mice exhibit similar bone mass compared to control mice, increased resorption activity, and accelerated apoptosis, which results in decreased osteoclast numbers in vivo. These results suggest that mitochondrial biogenesis is not a key determining factor for osteoclast formation and activity.
Oxidative phosphorylation
Mitochondrial structure is uniquely built with two membrane layers known as the outer and inner membranes (Figure 1B). Mitochondria generate energy mainly through oxidative phosphorylation (OXPHOS). OXPHOS generates ATP through reactions by the electron transport chain (ETC) located in the inner membrane. Although the production of most mitochondrial ATP takes place through a series of reactions known as the citric acid or Krebs cycle, OXPHOS has been identified as the primary bioenergetic source for osteoclast formation[19–21]. The treatment of ETC inhibitors blocks osteoclastogenesis and cells lacking complex I subunits fail to differentiate into osteoclasts[22]. The RANKL program increases mitochondrial respiratory chain proteins and MYC induces ERRα and the expression of ETC genes, and plays a critical role in activating OXPHOS[19], suggesting MYC as a key upstream regulator for metabolic reprogramming in osteoclasts Subsequently, DNMT3A-mediated regulation of methylation increases S adenosylmethionine (SAM), which accompanies oxidative phosphorylation[20]. Osteoclast-specific MYC-deficient mice exhibit increased bone mass due to defective osteoclast development, and osteoporosis-induced bone loss is protected in MYC-deficient mice[19]. Thus, disturbing oxidative phosphorylation in osteoclasts results in changes in the bone phenotype by decreasing osteoclast numbers. In contrast to osteoclast differentiation, bone resorption is enhanced when OXPHOS is low. Treatment cells with rotenone, an inhibitor of the mitochondrial complex I, has been shown to enhance osteoclast activity[21]. Therefore, the metabolic switch between OXPHOS and glycolysis fine-tunes osteoclast state transitions.
Glycolysis
Cells provide ATP through the consumption of fuels, including carbohydrates (sugars), fats, and proteins[23]. Carbohydrates are indigested into smaller components known as monosaccharaides: glucose, fructose, and galactose. Glucose is processed by cells through a sequentially linked enzyme cascade to produce ATP (Figure 2) Pyruvate can be utilized to fuel OXPHOS or converted into acetyl CoA in mitochondria, feeding into the tricarboxylic acid (TCA) cycle. As a final product of glycolysis, pyruvate enters mitochondria in aerobic conditions and converts into lactate in anaerobic conditions.
Figure 2. Osteoclasts increase glycolytic flux.
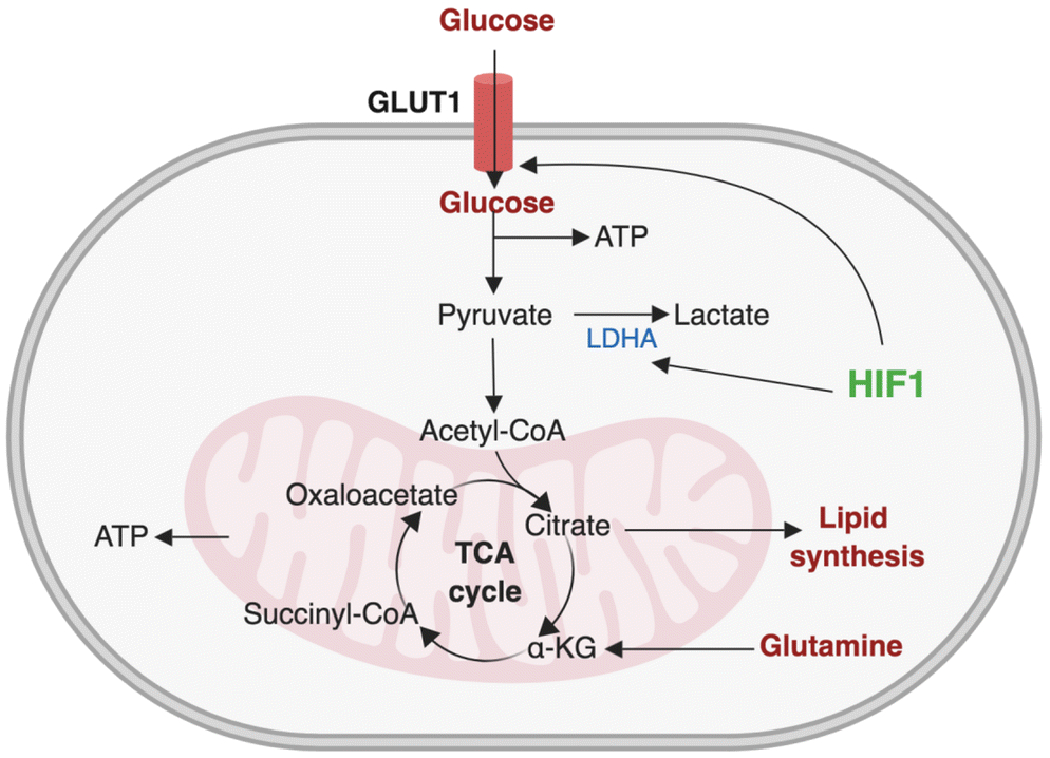
Extracellular glucose crosses plasma membrane via GLUT1. Glycolysis converts glucose to pyruvate, producing two molecules of ATP in the process. Pyruvate then enters mitochondria and the production of most mitochondrial ATP takes place through a series of reactions known as TCA cycle (tricarboxylic acid cycle). HIF1 regulates the expression of glycolytic genes including GLUT1 and LDHA. TCA cycle intermediate citrate is used for lipid synthesis and acetyl-CoA is converted into fatty acid by fatty acid synthesis and cholesterol. Glutamine is converted into alpha-ketoglutarate (α-KG).
Additionally, glucose serves as a major source for bone development and growth, and plays a crucial role in the skeletal system [24]. Treatment with inhibitors that block the glycolysis pathway or the depletion of glucose in culture media have demonstrated inhibited osteoclastogenesis, implying that glycolysis plays a key role in osteoclast differentiation[25]. During osteoclast differentiation, the glycolytic rate is not increased relative to resting precursor cells, but mature osteoclasts have been shown to have higher glycolytic rates. Glycolysis is currently viewed as an influential factor in the bone resorption of mature osteoclasts[21]. When mature osteoclasts are exposed to glucose-only media, Type I collagen degradation activity is significantly enhanced while glycolytic efficiency in producing lactate is significantly diminished[21]. Interestingly, immunohistochemistry analysis has found PKM2 and GAPDH, enzymes associated with the glycolytic pathway, to be located close to the sealing zones of mature osteoclasts, where bone resorption occurs. These results suggest glycolysis to be a driving energetic pathway for bone resorption.
Moreover, glucose uptake is mediated by glucose transporters[26]. Glucose transporters such as facilitative glucose transporters (GLUTs) or sodium coupled glucose transporters (SGLTs) mediate glucose transfer across cell membranes.. GLUTs are a member of the SLC2A protein family and comprise 14 isoforms. Class I GLUTs are GLUT1-4; GLUT1, a ubiquitous glucose transporter, is responsible for basal glucose uptake. GLUT2 and GLUT3 are identified as high affinity neuronal glucose transporters and GLUT3 is crucial for embryonic development. GLUT4, an insulin-sensitive glucose transporter expressed in adipose tissue and skeletal and cardiac muscle, identifies changes in circulating glucose.
Hypoxia increases the expression of hypoxia-inducible factors (HIFs) and their target genes, which are important for glycolytic metabolism such as GLUT1, lactate dehydrogenase (LDH), and angiogenesis, including VEGF[27]. The HIF-1 transcription factor consists of an oxygen-regulated alpha subunit–HIF1α–and a stable subunit, HIF1β. When oxygen is present, HIF1α is hydroxylated by prolyl hydroxylases and the following ubiquitylation of hydroxylated HIF1α by the von Hippel-Lindau (VHL) protein leads to rapid proteasomal degradation of HIF1α. Reduced oxygen inhibits proline hydroxylation, allowing HIF1α stabilization and the formation of an active transcriptional complex with HIF1β. Hypoxia augments osteoclastogenesis by increasing glycolysis via a COMMD1/E2F1 pathway[28]. During osteoclastogenesis, RANKL activates HIF-1α and induces the expression of GLUT1 and glycolytic enzymes—even under normoxia conditions[25]—with increased glycolysis appearing to be mediated by HIF-1α [27, 29]. The role of HIF-1α in osteoclasts has been addressed in the context of hypoxia and ovariectomy-induced osteoporosis[25, 30], and HIF-1α has been shown to regulate osteoclast activity [31–35]. However, the connections between RANKL signaling and HIF-1 have not been fully elucidated. Glucose conversion to lactate is also enhanced in hypoxic environments[36]. LDH, which catalyzes pyruvate into lactate, is increased during osteoclast differentiation; the knock-down of LDHA or LDHB decreases differentiation[37]. Therefore, basal glycolytic activity is required for osteoclast differentiation, and increased glycolysis fuels bone resorption.
mTOR and osteoclasts
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase and well-established regulator of metabolic processes[38], mTOR exists in two structurally and functionally distinct complexes: mTORC1 and mTORC2. mTOR coordinates metabolic/anabolic pathways to provide macromolecules and the energy needed for cell survival and growth. mTORC1 is comprised of mTOR, RAPTOR (regulatory-associated protein of mTOR), mLST8 (mammalian lethal with Sec13 protein 8), PRAS40 (proline-rich AKT substrate 40 kDa), and Deptor (DEP-domain-containing mTOR-interacting protein). When mTORC1 is activated, mTORC1 phosphorylates and is released from PRAS40 and Deptor for further activation. The activation of mTORC1 is required during the development, differentiation, and activation of diverse cell types to provide macromolecules such as lipids and proteins. mTORC1 promotes metabolic/anabolic pathways and protein synthesis in addition to controlling autophagy formation, and has been linked to mitochondrial biogenesis and metabolism. However, aberrant or persistent activation of mTORC1 is connected to various diseases, including inflammation, type 2 diabetes, and cancers. Rapamycin is an inhibitor of mTOR and interacts with the FKBP12-rapamycin binding domain (FRB) of mTOR by binding to the FK506-binding protein of 12 kDa (FKBP12); rapamycin has been increasingly used to treat various pathological conditions. mTORC2 consists of mTOR, Rictor (rapamycin-insensitive companion of mTOR), mSIN1 (mammalian stress-activated protein kinase interacting protein), Protor-1 (protein observed with Rictor-1, mLST8), and Deptor. mTORC2 regulates actin cytoskeletal organization and cell morphology, in addition to fully activating AKT signaling molecules.
mTOR regulates both osteoclasts and osteoblasts, playing an important role in skeletal development and disease[39]. mTOR signaling is required for osteoclast formation and survival[40]. mTORC1, mTOR/raptor axis contributes to cytoplasm growth while mTORC2, mTOR/rictor/Akt axis regulates the fusion of precursor cells[41]. mTORC1 activity is highest in osteoclast precursor cells but decreases as osteoclasts mature, correlating with the expression pattern of nutrient transporters and metabolic enzymes[42]. Raptor deficiency in early stages of hematopoiesis and osteoclast lineages (Cathepsin K cre) leads to increased bone mass and impaired osteoclastogenesis. Remarkably, all effects mediated by Raptor deficiency can be rescued by overexpressing constitutively active S6K1 or rapamycin treatment, suggesting a dependence on mTOR signaling[43]. In contrast, Raptor deficiency in myeloid cell lineages (Lysozyme M cre) enhance osteoclastogenesis[42]. TSC1 is a suppressor of mTOR. Mice with a deficiency of TSC1 in myeloid lineage cells or osteoclasts have constitutively high mTOR signals but exhibit higher bone mass than control mice[44], supporting the negative role of mTOR activation in osteoclasts. Studies of tuberous sclerosis complex patients with mutations in TSC1/2 demonstrate a negative correlation between bone metabolism and mTOR signaling. In addition, mTOR activation in mesenchymal stem cells (MSCs) promotes bone mineral accretion and inhibits osteoclast differentiation and activity directly or via coupling with MSCs. Thus, mTOR biophysically regulates osteoclast differentiation and bone quality by suppressing catabolic activities of osteoclasts[44].
Lipid metabolism
Cells also utilize lipid species as an energy source. Numerous lipid species exist in cells, infused into different cellular metabolic pathways and networks. The skeleton is a metabolically dynamic tissue; bone cells sensitively respond to environmental changes. Changes in cellular environments such as inflammation, diets, exercise, and other metabolic disease conditions or genetic mutations of key proteins in lipid metabolism alter cellular metabolic pathways and lead to the metabolic adaptation of bone cells. For example, hyperlipidemia increases bone resorption and osteoclasatogenesis[45]. A positive correlation between increased fracture risk and high cholesterol or hyperlipidemia has been suggested by many studies.
Accumulated in the skeleton, fatty acids are required for the maintenance of normal bone structure in mice[46]. Human bone marrow contains 28~84% of neural lipids—including triglyceride, cholesterol, and free fatty acids—and less than 3% of phospholipids[47]. Fatty acids in osteoclasts can be derived from de novo lipogenesis or taken up through passive diffusion derived from adipocyte lipolysis or triacylglycerol lipolysis. When fatty acids enter the mitochondria matrix, they are used as substrates for fatty acid oxidation (FAO), which provides and maintains energy homeostasis. Moreover, fatty acids can also be stored as lipid droplets or make up cell membranes.
Long chain polyunsaturated fatty acids (LCPUFAs) are essential fatty acids with a minimum of 18 carbons and 2 double bonds[27]. Anti-inflammatory ω-3 LCPUFAs are derived from α-linolenic acid while pro-inflammatory ω-6 LCPUFAs are derived from linoleic acid (LA). LCPUFAs, which cannot be synthesized in the body and must be supplied in diets, directly suppress osteoclastogenesis and osteoclast activity[48, 49]. Nonetheless, the effect of LCPUFAs on bone health is inconclusive[50]. Saturated fatty acids (SFA) promote osteoclast survival by preventing apoptosis in a TLR4-dependent way[51]. Although several studies demonstrate osteoclastogenesis suppression after treatment of SFA in the early phase, adding SFA in the late phase promotes osteoclast differentiation. Short chain fatty acids (SCFA) and other microbial metabolites can be produced in the gut from the microbial fermentation of dietary fiber[52]. SCFA modulates systemic immune functions[53] and suppress in vitro and in vivo osteoclastogenesis[54]. SCFA C3 and C4 enhance glycolysis at the expense of OXPHOS and downregulate osteoclast-related genes such as NFATc1 Additionally, SCFA and related products protect mice form ovariectomy-induced bone loss. The supplementation of the right probiotics, SCFA, or diets that increase the endogenous production of SCFA may assist in balancing osteoclast-mediated bone resorption and bone formation. Therefore, diets and microbiomes greatly contribute to the supply and processing of fatty acids, leading to changes in bone.
Cholesterol plays an important role in many cellular functions and is an essential component of lipid bilayers[55]. However, excess accumulation of cholesterol is highly deleterious to cells and underlies the pathogenesis of a number of metabolic diseases. High cholesterol may increase bone turnover. Statins such as simvastatin, lovastatin, atorvastatin, pravastatin, and mevastatin are the most prescribed cholesterol-lowering drugs2, and suppresses osteoclastogenesis and promotes osteogenesis[56]. However, the correlation between statins and bone remains inconclusive. Cellular cholesterol can be supplied through imports from lipoproteins or synthesis via the tightly regulated mevalonate pathway. Lipoproteins are particles that contain triacylglycerol, cholesterol, phospholipids and amphipathic proteins known as apolipoproteins. Diacylglycerol acyl transferase 1 (Dgat1) controls fatty acid absorption, lipoprotein assembly, and regulation of plasma TAG (Triacylglycerol) concentrations[57]. Dgat1 knockout mice have low levels of TAG and decreased bone mass[58]. The depletion of low density lipoprotein (LDL) suppresses osteoclast formation[59]. Consistently, LDLR-deficient osteoclasts display fusion and survival defects[60]; impaired osteoclastogenesis in LDLR-deficient osteoclasts is rescued by the addition of cholesterol[61]. Removing cholesterol through treatment with cyclodextrin or high-density lipoprotein (HDL) has also been demonstrated to increase apoptosis via ABCG1 and, consequently, suppress osteoclastogenesis[61]. The depletion of cholesterol has been further found to suppress V-ATPase activity by disturbing lipid rafts and affecting bone resorption[62]. Therefore, different lipid species may have distinct role in osteoclast differentiation and activity.
Conclusions
This review outlines the metabolic properties of osteoclasts and the mechanisms that control energy substrate utilization and bioenergetics in osteoclast differentiation and activity. The studies discussed here have identified major metabolic pathways in osteoclasts and the role of key checkpoint proteins in their differentiation and function. Moreover, while several, currently available drugs diminish osteoclast differentiation or activity, there remains room for improvement, particularly in the areas of patient side effects and target specificity. Targeting metabolism has proven to be an effective therapy for certain diseases, including many cancers, and full comprehension of the metabolic networks of osteoclasts may lead to the development of novel therapeutic interventions for pathological bone loss that target the metabolic differences between overly active and normal osteoclasts.
These bioenergetic changes in osteoclasts for physiological and pathological conditions have recently garnered increased interest. However, metabolism remains a relatively new area of research in osteoclast biology, and little is known about the metabolic changes and programs that play crucial roles in osteoclast differentiation and bone resorption. Given the importance of metabolic regulation in osteoclasts, several considerations should be addressed in future studies. First, the relationship between osteoclasts and their metabolic environment must be carefully examined. Nutritional availability and diet types all contribute to individual metabolic conditions, which change frequently and rapidly, and metabolic deregulation—such as diabetes mellitus—also affects systemic and local metabolite availability and bone health. Therefore, investigating how osteoclasts sense and interact with nutrients will significantly affect the ability to design better, more specific therapeutics that can control the fates and functions of osteoclasts in diverse metabolic environments. Second, the cellular metabolic status of osteoclasts in different diseases settings requires characterization. While pathological bone loss results from various factors, including aging and menopause, it can also occur as a result of metabolic deregulation. The modification of an individual’s surrounding environment dynamically affects the metabolic status of his or her osteoclasts, which further leads to epigenetic regulation in chromatin or protein modifications. Thus, establishing the metabolic differences between healthy and disease-conditioned osteoclasts will provide more expansive knowledge regarding the changes caused by bone diseases, and assist in their treatment. Third, the impacts of anti-resorptive therapies on metabolic networks in osteoclasts need to be revisited. Although overly active osteoclasts have prevailing energy requirements for differentiation and function, how anti-resorptive therapies influence the interlinked metabolic network in osteoclasts remains unexplored. Finally, since metabolites can directly regulate gene expression through post-transcriptional modifications[63], going forward, integrated studies combining metabolism with transcriptomics will be necessary.
As examined, the identification of the metabolic characteristics of osteoclast generation and bone resorption will provide deeper scientific insight into the changes that occur during the pathogenesis of osteoclast-mediated bone diseases. With this improved understanding of the human osteoclast metabolism, we may also see the rise of innovative therapeutic interventions that specifically target osteoclast activity.
Acknowledgements:
Figures are created with BioRender.com.
Funding: This work was supported by grants from the N.I.H. (R01AR069562 and R01 AR073156-01), by Weill Cornell CTSC, and by support for the Rosensweig Genomics Center from The Tow Foundation.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- [1].Ikeda K, Takeshita S, The role of osteoclast differentiation and function in skeletal homeostasis, J Biochem 159(1) (2016) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nakashima T, Hayashi M, Takayanagi H, New insights into osteoclastogenic signaling mechanisms, Trends Endocrinol Metab 23(11) (2012) 582–90. [DOI] [PubMed] [Google Scholar]
- [3].Boyce BF, Advances in osteoclast biology reveal potential new drug targets and new roles for osteoclasts, J Bone Miner Res 28(4) (2013) 711–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fuller K, Kirstein B, Chambers TJ, Regulation and enzymatic basis of bone resorption by human osteoclasts, Clin Sci (Lond) 112(11) (2007) 567–75. [DOI] [PubMed] [Google Scholar]
- [5].Teitelbaum SL, Ross FP, Genetic regulation of osteoclast development and function, Nat Rev Genet 4(8) (2003) 638–49. [DOI] [PubMed] [Google Scholar]
- [6].Feldenberg LR, Thevananther S, del Rio M, de Leon M, Devarajan P, Partial ATP depletion induces Fas- and caspase-mediated apoptosis in MDCK cells, Am J Physiol 276(6) (1999) F837–46. [DOI] [PubMed] [Google Scholar]
- [7].Spinelli JB, Haigis MC, The multifaceted contributions of mitochondria to cellular metabolism, Nat Cell Biol 20(7) (2018) 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Eisner V, Picard M, Hajnoczky G, Mitochondrial dynamics in adaptive and maladaptive cellular stress responses, Nat Cell Biol 20(7) (2018) 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dudley HR, Spiro D, The Fine Structure of Bone Cells, J Biophys Biochem Cytol 11(3) (1961) 627–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, Aburatani H, Taketani S, Lelliott CJ, Vidal-Puig A, Ikeda K, Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation, Nat Med 15(3) (2009) 259–66. [DOI] [PubMed] [Google Scholar]
- [11].Wei W, Wang X, Yang M, Smith LC, Dechow PC, Sonoda J, Evans RM, Wan Y, PGC1beta mediates PPARgamma activation of osteoclastogenesis and rosiglitazone-induced bone loss, Cell Metab 11(6) (2010) 503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wan Y, PPARgamma in bone homeostasis, Trends Endocrinol Metab 21(12) (2010) 722–8. [DOI] [PubMed] [Google Scholar]
- [13].Wei W, Schwaid AG, Wang X, Wang X, Chen S, Chu Q, Saghatelian A, Wan Y, Ligand Activation of ERRalpha by Cholesterol Mediates Statin and Bisphosphonate Effects, Cell Metab (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zeng R, Faccio R, Novack DV, Alternative NF-kappaB Regulates RANKL-Induced Osteoclast Differentiation and Mitochondrial Biogenesis via Independent Mechanisms, J Bone Miner Res 30(12) (2015) 2287–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Izawa T, Rohatgi N, Fukunaga T, Wang QT, Silva MJ, Gardner MJ, McDaniel ML, Abumrad NA, Semenkovich CF, Teitelbaum SL, Zou W, ASXL2 Regulates Glucose, Lipid, and Skeletal Homeostasis, Cell Rep 11(10) (2015) 1625–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wilson L, Yang Q, Szustakowski JD, Gullicksen PS, Halse R, Pyruvate induces mitochondrial biogenesis by a PGC-1 alpha-independent mechanism, Am J Physiol Cell Physiol 292(5) (2007) C1599–605. [DOI] [PubMed] [Google Scholar]
- [17].Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM, Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA, Nat Genet 31(3) (2002) 289–94. [DOI] [PubMed] [Google Scholar]
- [18].Miyazaki T, Iwasawa M, Nakashima T, Mori S, Shigemoto K, Nakamura H, Katagiri H, Takayanagi H, Tanaka S, Intracellular and extracellular ATP coordinately regulate the inverse correlation between osteoclast survival and bone resorption, J Biol Chem 287(45) (2012) 37808–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bae S, Lee MJ, Mun SH, Giannopoulou EG, Yong-Gonzalez V, Cross JR, Murata K, Giguere V, van der Meulen M, Park-Min KH, MYC-dependent oxidative metabolism regulates osteoclastogenesis via nuclear receptor ERRalpha, J Clin Invest 127(7) (2017) 2555–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nishikawa K, Iwamoto Y, Kobayashi Y, Katsuoka F, Kawaguchi S, Tsujita T, Nakamura T, Kato S, Yamamoto M, Takayanagi H, Ishii M, DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S-adenosylmethionine-producing metabolic pathway, Nat Med 21(3) (2015) 281–7. [DOI] [PubMed] [Google Scholar]
- [21].Lemma S, Sboarina M, Porporato PE, Zini N, Sonveaux P, Di Pompo G, Baldini N, Avnet S, Energy metabolism in osteoclast formation and activity, Int J Biochem Cell Biol 79 (2016) 168–180. [DOI] [PubMed] [Google Scholar]
- [22].Jin Z, Wei W, Yang M, Du Y, Wan Y, Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization, Cell Metab 20(3) (2014) 483–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lunt SY, Vander Heiden MG, Aerobic glycolysis: meeting the metabolic requirements of cell proliferation, Annu Rev Cell Dev Biol 27 (2011) 441–64. [DOI] [PubMed] [Google Scholar]
- [24].Karner CM, Long F, Glucose metabolism in bone, Bone 115 (2018) 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Indo Y, Takeshita S, Ishii KA, Hoshii T, Aburatani H, Hirao A, Ikeda K, Metabolic regulation of osteoclast differentiation and function, J Bone Miner Res 28(11) (2013) 2392–9. [DOI] [PubMed] [Google Scholar]
- [26].Chen LQ, Cheung LS, Feng L, Tanner W, Frommer WB, Transport of sugars, Annu Rev Biochem 84 (2015) 865–94. [DOI] [PubMed] [Google Scholar]
- [27].Palazon A, Goldrath AW, Nizet V, Johnson RS, Review HIF Transcription Factors , Inflammation , and Immunity, Immunity 41 (2014) 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Murata K, Fang C, Terao C, Giannopoulou EG, Lee YJ, Lee MJ, Mun SH, Bae S, Qiao Y, Yuan R, Furu M, Ito H, Ohmura K, Matsuda S, Mimori T, Matsuda F, Park-Min KH, Ivashkiv LB, Hypoxia-Sensitive COMMD1 Integrates Signaling and Cellular Metabolism in Human Macrophages and Suppresses Osteoclastogenesis, Immunity 47(1) (2017) 66–79 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Indo Y, Takeshita S, Ishii K-A, Hoshii T, Aburatani H, Hirao A, Ikeda K, Metabolic regulation of osteoclast differentiation and function., Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 28 (2013) 2392–9. [DOI] [PubMed] [Google Scholar]
- [30].Miyauchi Y, Sato Y, Kobayashi T, Yoshida S, Mori T, Kanagawa H, Katsuyama E, Fujie A, Hao W, Miyamoto K, Tando T, Morioka H, Matsumoto M, Chambon P, Johnson RS, Kato S, Toyama Y, Miyamoto T, HIF1alpha is required for osteoclast activation by estrogen deficiency in postmenopausal osteoporosis, Proc Natl Acad Sci U S A 110(41) (2013) 16568–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Knowles HJ, Athanasou NA, Acute hypoxia and osteoclast activity: A balance between enhanced resorption and increased apoptosis, Journal of Pathology 218 (2009) 256–264. [DOI] [PubMed] [Google Scholar]
- [32].Knowles HJ, Cleton-Jansen A-M, Korsching E, Athanasou NA, Hypoxia-inducible factor regulates osteoclast-mediated bone resorption: role of angiopoietin-like 4, The FASEB Journal 24 (2010) 4648–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Morten KJ, Badder L, Knowles HJ, Differential regulation of HIF-mediated pathways increases mitochondrial metabolism and ATP production in hypoxic osteoclasts, Journal of Pathology 229 (2013) 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Miyauchi Y, Sato Y, Kobayashi T, Yoshida S, Mori T, Kanagawa H, Katsuyama E, Fujie A, Hao W, Miyamoto K, Tando T, Morioka H, Matsumoto M, Chambon P, Johnson RS, Kato S, Toyama Y, Miyamoto T, HIF1 α is required for osteoclast activation by estrogen de fi ciency in postmenopausal osteoporosis, Proceedings of the National Academy of Sciences 110 (2013) 16568–16573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Leger AJ, Altobelli A, Mosquea LM, Belanger AJ, Song A, Cheng SH, Jiang C, Yew NS, Inhibition of osteoclastogenesis by prolyl hydroxylase inhibitor dimethyloxallyl glycine, J Bone Miner Metab 28(5) (2010) 510–9. [DOI] [PubMed] [Google Scholar]
- [36].Denko NC, Hypoxia, HIF1 and glucose metabolism in the solid tumour, Nat Rev Cancer 8(9) (2008) 705–13. [DOI] [PubMed] [Google Scholar]
- [37].Ahn H, Lee K, Kim JM, Kwon SH, Lee SH, Lee SY, Jeong D, Accelerated Lactate Dehydrogenase Activity Potentiates Osteoclastogenesis via NFATc1 Signaling, PLoS One 11(4) (2016) e0153886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Saxton RA, Sabatini DM, mTOR Signaling in Growth, Metabolism, and Disease, Cell 168(6) (2017) 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen J, Long F, mTOR signaling in skeletal development and disease, Bone Res 6 (2018) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Glantschnig H, Fisher JE, Wesolowski G, Rodan GA, Reszka AA, M-CSF, TNFalpha and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase, Cell Death Differ 10(10) (2003) 1165–77. [DOI] [PubMed] [Google Scholar]
- [41].Tiedemann K, Le Nihouannen D, Fong JE, Hussein O, Barralet JE, Komarova SV, Regulation of Osteoclast Growth and Fusion by mTOR/raptor and mTOR/rictor/Akt, Front Cell Dev Biol 5 (2017) 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Huynh H, Wan Y, mTORC1 impedes osteoclast differentiation via calcineurin and NFATc1, Commun Biol 1 (2018) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dai Q, Xie F, Han Y, Ma X, Zhou S, Jiang L, Zou W, Wang J, Inactivation of Regulatory-associated Protein of mTOR (Raptor)/Mammalian Target of Rapamycin Complex 1 (mTORC1) Signaling in Osteoclasts Increases Bone Mass by Inhibiting Osteoclast Differentiation in Mice, J Biol Chem 292(1) (2017) 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wu H, Wu Z, Li P, Cong Q, Chen R, Xu W, Biswas S, Liu H, Xia X, Li S, Hu W, Zhang Z, Habib SL, Zhang L, Zou J, Zhang H, Zhang W, Li B, Bone Size and Quality Regulation: Concerted Actions of mTOR in Mesenchymal Stromal Cells and Osteoclasts, Stem Cell Reports 8(6) (2017) 1600–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tintut Y, Morony S, Demer LL, Hyperlipidemia promotes osteoclastic potential of bone marrow cells ex vivo, Arterioscler Thromb Vasc Biol 24(2) (2004) e6–10. [DOI] [PubMed] [Google Scholar]
- [46].Kim SP, Li Z, Zoch ML, Frey JL, Bowman CE, Kushwaha P, Ryan KA, Goh BC, Scafidi S, Pickett JE, Faugere MC, Kershaw EE, Thorek DLJ, Clemens TL, Wolfgang MJ, Riddle RC, Fatty acid oxidation by the osteoblast is required for normal bone acquisition in a sex- and diet-dependent manner, JCI Insight 2(16) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].LUND DMAPK, and MATHI JC, Lipid composition of normal human bone marrow as determined by column chromatography, Journal of Lipid Resesrch 3(1) (1962). [Google Scholar]
- [48].Kasonga AE, Deepak V, Kruger MC, Coetzee M, Arachidonic acid and docosahexaenoic acid suppress osteoclast formation and activity in human CD14+ monocytes, in vitro, PLoS One 10(4) (2015) e0125145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sun D, Krishnan A, Zaman K, Lawrence R, Bhattacharya A, Fernandes G, Dietary n-3 fatty acids decrease osteoclastogenesis and loss of bone mass in ovariectomized mice, J Bone Miner Res 18(7) (2003) 1206–16. [DOI] [PubMed] [Google Scholar]
- [50].Mangano KM, Sahni S, Kerstetter JE, Kenny AM, Hannan MT, Polyunsaturated fatty acids and their relation with bone and muscle health in adults, Curr Osteoporos Rep 11(3) (2013) 203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Oh SR, Sul OJ, Kim YY, Kim HJ, Yu R, Suh JH, Choi HS, Saturated fatty acids enhance osteoclast survival, J Lipid Res 51(5) (2010) 892–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Li JY, Chassaing B, Tyagi AM, Vaccaro C, Luo T, Adams J, Darby TM, Weitzmann MN, Mulle JG, Gewirtz AT, Jones RM, Pacifici R, Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics, J Clin Invest 126(6) (2016) 2049–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Correa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MA, Regulation of immune cell function by short-chain fatty acids, Clin Transl Immunology 5(4) (2016) e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lucas S, Omata Y, Hofmann J, Bottcher M, Iljazovic A, Sarter K, Albrecht O, Schulz O, Krishnacoumar B, Kronke G, Herrmann M, Mougiakakos D, Strowig T, Schett G, Zaiss MM, Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss, Nat Commun 9(1) (2018) 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS, Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance, Cell Metab 8(6) (2008) 512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mundy G, Garrett R, Harris S, Chan J, Chen D, Rossini G, Boyce B, Zhao M, Gutierrez G, Stimulation of bone formation in vitro and in rodents by statins, Science 286(5446) (1999) 1946–9. [DOI] [PubMed] [Google Scholar]
- [57].Smith SJ, Cases S, Jensen DR, Chen HC, Sande E, Tow B, Sanan DA, Raber J, Eckel RH, Farese RV Jr., Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking Dgat, Nat Genet 25(1) (2000) 87–90. [DOI] [PubMed] [Google Scholar]
- [58].Drosatos-Tampakaki Z, Drosatos K, Siegelin Y, Gong S, Khan S, Van Dyke T, Goldberg IJ, Schulze PC, Schulze-Spate U, Palmitic acid and DGAT1 deficiency enhance osteoclastogenesis, while oleic acid-induced triglyceride formation prevents it, J Bone Miner Res 29(5) (2014) 1183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sato T, Morita I, Murota S, Involvement of cholesterol in osteoclast-like cell formation via cellular fusion, Bone 23(2) (1998) 135–40. [DOI] [PubMed] [Google Scholar]
- [60].Okayasu M, Nakayachi M, Hayashida C, Ito J, Kaneda T, Masuhara M, Suda N, Sato T, Hakeda Y, Low-density lipoprotein receptor deficiency causes impaired osteoclastogenesis and increased bone mass in mice because of defect in osteoclastic cell-cell fusion, J Biol Chem 287(23) (2012) 19229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Luegmayr E, Glantschnig H, Wesolowski GA, Gentile MA, Fisher JE, Rodan GA, Reszka AA, Osteoclast formation, survival and morphology are highly dependent on exogenous cholesterol/lipoproteins, Cell Death Differ 11 Suppl 1 (2004) S108–18. [DOI] [PubMed] [Google Scholar]
- [62].Ryu J, Kim H, Chang EJ, Kim HJ, Lee Y, Kim HH, Proteomic analysis of osteoclast lipid rafts: the role of the integrity of lipid rafts on V-ATPase activity in osteoclasts, J Bone Miner Metab 28(4) (2010) 410–7. [DOI] [PubMed] [Google Scholar]
- [63].Wellen KE, Thompson CB, A two-way street: reciprocal regulation of metabolism and signalling, Nat Rev Mol Cell Biol 13(4) (2012) 270–6. [DOI] [PubMed] [Google Scholar]