

ABSTRACT
Macrophage Activation Syndrome (MAS) is a potentially fatal inflammatory condition that can rapidly lead to multi-organ failure if inadequately treated. Also, known as secondary Hemophagocytic Lymphohistiocytosis (sHLH), MAS is commonly seen as a complication of systemic inflammatory disorders, like systemic juvenile idiopathic arthritis (sJIA) and systemic lupus erythematosus (SLE). However, MAS can also present as a complication of malignancies and infections, particularly viral infections like Epstein-Barr virus (EBV), cytomegalovirus (CMV) and HIV. Here we describe a patient with an underlying history of SLE and Sjogren’s disease who was found have both EBV and CMV infections, presented to our facility with fever, lymphadenopathy, pneumonia and pancytopenia. Patient was treated in line with sepsis in the intensive care unit but rapidly developed multigrain failure despite early aggressive treatment. As will be discussed below, patient had characteristic signs and symptoms of MAS with biochemical parameters pointing towards the same.
KEYWORDS: Macrophage activation syndrome, hemophagocytic lymphohistiocytosis, juvenile idiopathic arthritis, hemophagocytosis
1. Introduction
Macrophage activation syndrome (MAS) has been commonly reported in pediatric population as a complication sJIA, but cases in adults have emerged with the association of MAS with a variety of inflammatory conditions, like malignancy, infection (i.e., Epstein-Barr virus), and primary immunodeficiencies [1]. Hemophagocytosis is defined as the engulfment of blood cells, including red blood cells, white blood cells, or platelets by phagocytic cells. This can be detected by serum laboratory tests like soluble interleukin 2 receptor alpha chain (sCD25) and soluble CD163 (sCD163), a high-affinity scavenger receptor for hemoglobin-haptoglobin complexes [1]. This clinical syndrome was first described in the 1980s in children with severe cases of sJIA [1]. Due to rapid development of multiorgan failure most patients are admitted to the Intensive Care unit (ICU). The frequency of ICU admission from MAS has been reported as 43.7%, with the mortality rate as high as 11.4% [2]. Clinical and laboratory features of MAS include sustained fever, hyperferritinemia, pancytopenia, fibrinolytic coagulopathy, and liver dysfunction [1,3].
A broad variety of infections have been identified as triggers of HLH/MAS, either in isolation or in addition to an underlying inflammatory disease state. Certain infections, particularly by members of the herpesvirus family, are the most notorious triggers of HLH/MAS. Treatment for infection-triggered MAS requires therapy for both the underlying infection and dampening of the hyperactive immune response [2]. Early diagnosis is key in MAS given its high mortality. Our patient was admitted with an initial diagnosis of pneumonia, rapidly deteriorated and was admitted to the ICU within hours, developed multiorgan failure the next day, was intubated and treated with broad-spectrum antibiotics, antifungals and antivirals with poor response. After several days of treatment, a diagnosis of MAS was considered. Suffice it to say, it is usually difficult in these cases to differentiate between a true sepsis, rheumatological disease flare-ups, disseminated intravascular coagulation (DIC) or MAS. In most cases, the laboratory abnormalities are similar to those of a DIC, which shows pancytopenia, coagulopathy, hypofibrinogenemia, and an elevated d-dimer test, but it can also be a late stage of MAS [4].
Poor survival and frequent relapse are common not only in patients with malignancy-associated HLH, but also in those with active Epstein-Barr virus (EBV) infection and in some high-risk patients with HLH of unknown cause [5]. Retrospective studies in sJIA, Kawasaki’s disease and SLE show that MAS is under-recognized, highlighting a need for increased vigilance, understanding and education for this condition, enabling timely delivery of life-saving treatment [1]
2. Case description
A 50-year-old woman with history of SLE and Sjogren’s disease presented to the emergency room with fever and malaise for 2 weeks. She had been on oral antibiotics for suspected pneumonia as an outpatient but failed to improve, thus presented to the hospital. She appeared very ill, able to follow commands but easily fatigued and had a temperature of 104.7 F. Examination revealed palpable cervical lymph nodes. A chest x-ray revealed infiltrates suggestive of pneumonia (Figure 1) and she was admitted for sepsis likely secondary to community acquired pneumonia and started on Ceftriaxone and Azithromycin. The next day patient became lethargic, developed severe shortness of breath with respiratory rate of 40, heart rate of 169 and continued to have high-grade fevers. With suspicion of aspiration pneumonia, patient was upgraded to the ICU and antibiotics were broadened with vancomycin, meropenem and metronidazole. Her blood work showed a white blood cell count of 7000, Red blood cell count of 3000, hemoglobin of 9 mg/dl and hematocrit of 28 mg/dl and platelet counts of 249. The chemistry panel showed metabolic acidosis, elevated liver enzymes and normal renal function. After failing to improve, the patient underwent a CT scan of her chest, abdomen and pelvis which revealed extensive lymphadenopathy in the cervical, axillary, retroperitoneal and inguinal area (Figure 2a-2d), along with enlarged liver and spleen. Further blood work revealed Ferritin of 5098, Erythrocyte Sedimentation Rate (ESR) of 138, C-Reactive Protein (CRP) 44, Lactate Dehydrogenase (LDH) 1116, lactic acid of 11 and normal Completement levels (C3 and C4). A right axillary Lymph node biopsy was done with suspicion of lymphoproliferative disorder, which showed extensive degeneration and necrosis with positive Epstein-Barr virus (EVB) cells without any evidence of malignant cells. A Viral Polymerase Chain Reaction (PCR) test was conducted where a positive viral load for both EBV (more than 20,00000 copies/ml) and Cytomegalovirus (more than 300,000 copies/ml) was seen. Patient tested negative for Human Immunodeficiency virus (HIV-1), Respiratory Syncytial Virus (RSV), Influenza A and B, Streptococcal Pneumonia Antigen and Legionella Antigen. The patient was started on antiviral treatment with Ganciclovir.
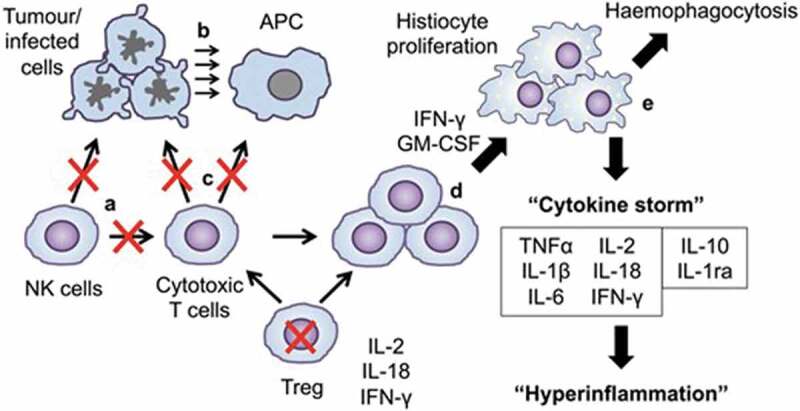
Figure 1.
Pathogenesis of MAS/sHLH. Cytotoxic function of NK cells fails to clear tumor or infected cells and cytotoxic T cells. (b) Persistent tumor-infected cells cause persistent stimulation by persistent antigen presentation. (c) Cytotoxic function of CTLs fail to clear tumor cells and APCs, and Tregs are overwhelmed. (d) Proliferation of the population of activated CTLs induce activation and proliferation of tissue macrophages (histiocytes). (e) Activated histiocytes haemophagocytose and produce a cytokine storm, due to which imbalance of pro- and anti-inflammatory cytokines induces fever and hyperinflammatory haemophagocytic syndrome. MAS: macrophage activation syndrome; sHLH: secondary haemophagocytic lymphocytosis; APC: antigen-presenting cell; CTLs: cytotoxic T cells [3].
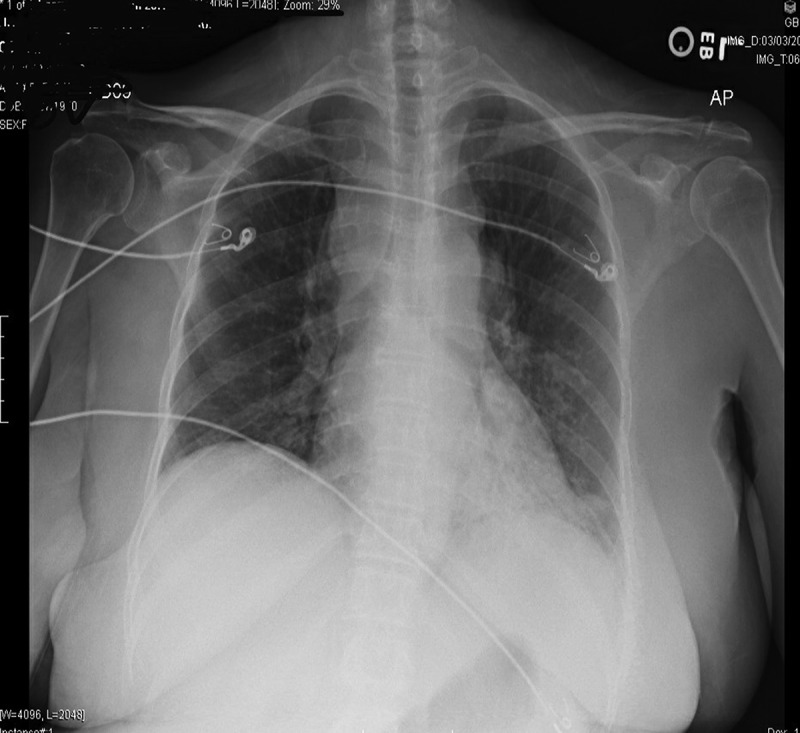
Figure 1.
CXR showing b/l hilar adenopathy with multifocal infiltrate involving left lower lobe and right middle lobe.
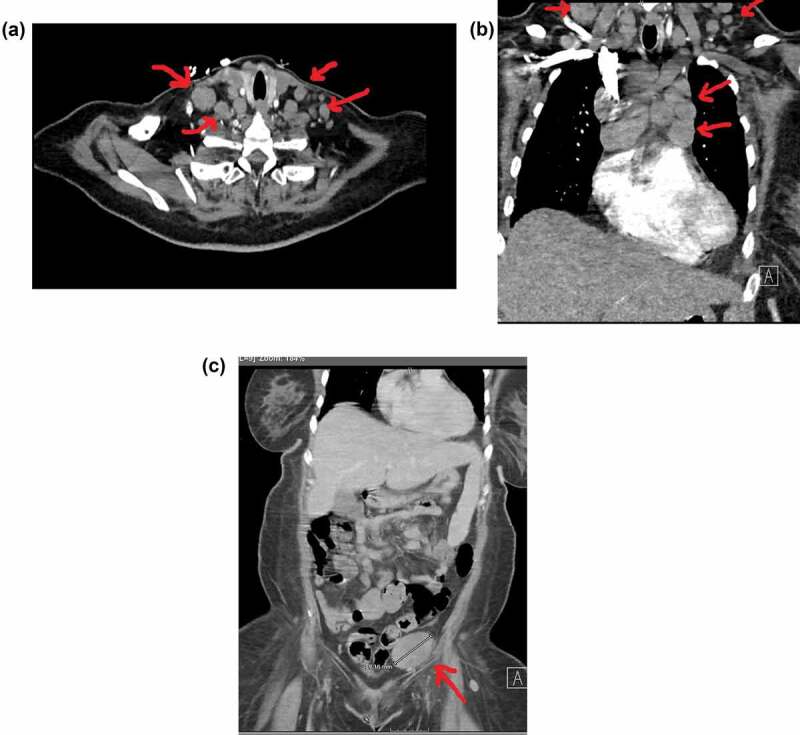
Figure 2.
A. Bulky Cervical lymphadenopathy B. Mediastinal, Hilar and Supraclavicular lymphadenopathy C. Large bulky inguinal LypLarge bulky inguinal Lymph node D. Retroperitoneal lymphadenopathy.
One week since initial presentation, patient continued to have high-grade fevers, remained hypoxic with increasing lactic acid levels. Her white blood cells dropped to 3000, platelets dropped to 100,000, hemoglobin to 6.9 with negative blood and urine cultures. Antibiotics were broadened further with three antibacterial drugs, micafungin for possible fungal infection and an antiviral agent. Other than Viremia from EBV and CMV, no other source of infection was found. After reviewing the case, history and clinical progress, MAS was suspected, and patient was started on high dose steroids and Intravenous Immunoglobulin (IVIG). Immunological workup sent which revealed positive Antinuclear Antigen (ANA, titer 1:1280), positive Sjogren’s Antibodies (SSA and SSB), positive Sm/RNP antibodies and highly positive Interleukin-2 Receptor Alpha Chain (IL-2Ra/CD25) of 24885 pg/mL (reference range: 532–1891 pg/ml). With high index of suspicion for MAS, patient was urgently transferred to a tertiary care center, where she was started on chemo-immunotherapy.
3. DISCUSSION
MAS is a potentially fatal inflammatory condition that can lead to multiorgan failure if inadequately treated. Due to the variation in presentation and wide differential diagnoses to consider, recognition is often delayed. The diagnosis of MAS or sHLH can be made based on past medical history or underlying medical conditions, clinical signs and symptoms supported by laboratory parameters. A firm understanding of the pathogenesis of MAS can guide diagnosis and direct therapy toward target-specific treatment [1]. Inability to clear antigen from infection, or from autoimmune processes leads to inappropriate immune stimulation and a self-perpetuating hyperinflammatory state known as the cytokine storm (Figure 3). This in turn results in sudden activation of macrophages, causing hemophagocytosis as well as contributing to multi-organ dysfunction [3].
MAS is common in children, therefore has well-defined treatment algorithms as it occurs in ∼10% of patients with sJIA. In adults, MAS is more prevalent in patients with Adult Onset Still’s Disease (AOSD), with 10–15% prevalence rate [5] followed by SLE-associated MAS which is reported in the medical literature, with overall prevalence estimated between 0.9% and 9% [3]. One large retrospective study applied the 2016 sJIA PRINTO Classification Criteria for MAS to patients with SLE admitted to hospital with fever, and found one-third were classified as having MAS, of whom 35% died, compared with 3% without MAS, indicating that the classification criteria could be used in patients with SLE and fever to identify MAS, and facilitate effective treatment [3]
Malignancies associated with sHLH are mostly T cell and NK-cell leukemias or lymphomas, diffuse large B cell lymphoma and Hodgkin lymphoma, of which a significant proportion may be driven by EBV [3]. In the USA and Asia, EBV infection is notably the most common viral infection causing MAS [3]. Precise mechanism of EBV-induced MAS has not been recognized yet, but it is speculated that EBV-infected B cells stimulate cytotoxic T lymphocytes leading to cytokine storm and stimulation of histolytic cells [6]. Other members of the herpes virus family including CMV, HSV and varicella zoster virus are common infectious triggers. HIV, influenza, Dengue and Ebola virus are other notable examples [3]
A commonly used diagnostic criteria of MAS or sHLH are shown in Table 1 [7].
3.1. A diagnosis is consistent with HLH if 5/8 of the below criteria are met
1. Fever ≥38.5°C
2. Splenomegaly
3. Cytopenias (affecting at least 2 lineages)
Hemoglobin <9 g/dL (in infants <4 weeks: hemoglobin <10 g/dL)
Platelets <100 × 103/mL
Neutrophils <1 × 103/mL
4. Hypertriglyceridemia (fasting, >265 mg/dL) and/or hypofibrinogenemia (<150 mg/dL)
5. Hemophagocytosis in bone marrow, spleen, lymph nodes, liver, or other tissue
6. Low or absent NK cell activity
7. Ferritin >500 ng/mL
8. Elevated sCD25 (soluble IL-2 receptor): >2,400 U/mL or elevated based on the laboratory-defined normal range.
Table 1 Commonly used diagnostic criteria for HLH, adapted from Henter et al.
In early sHLH, the absolute values of laboratory results may be less helpful than a review of the trend of results, particularly when considering cytopenias and fibrinogen. Suffice it to say that normal levels of these markers are inappropriate in the context of active inflammatory disease and should prompt the clinician to consider MAS, and emphasizes the need for close monitoring in both trends in clinical status and laboratory parameters [1]
Our case presented with unremitting fever, hepatosplenomegaly, pancytopenia with very high levels ferritin and sCD25 (soluble IL-2 receptor), meeting the criteria for sHLH. Patient did not have high Triglycerides or low fibrinogen levels and a bone marrow biopsy was not done. Furthermore, to support the diagnosis, there was a strong evidence of co-infection with Epstein-Barr Virus and Cytomegalovirus. The presence of underlying poorly controlled SLE/Sjogren’s disease, rapidly deteriorating clinical course and failure to improve with broad-spectrum antimicrobial treatment strongly hinted towards MAS.
In addition to treating any underlying infection, the mainstay of MAS treatment is glucocorticoid therapy. An intravenous methylprednisolone 30 mg/kg/dose (maximum 1 g) for 1–3 days followed by 2–3 mg/kg/day in a divided dose is commonly used. If not responding to steroids, an additional therapy with cyclosporine A 2–7 mg/kg/day is recommended. For patients who are refractory to the high dosages of corticosteroids and cyclosporine A, an HLH-2004 protocol treatment should be considered which includes chemo-immunotherapy with etoposide, dexamethasone, and Cyclosporine A. IVIG can be used particularly in infection-associated cases, but has to be given early to be effective. For refractory cases, bone marrow transplant is now being considered [2,4,5,8]
Some reports suggested a role of acyclovir in the management of EBV-associated disease, but no relevant data are available on the additional effects of antiviral therapy [5]
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Crayne CB, Albeituni S, Nichols KE, et al. The immunology of macrophage activation syndrome. Front Immunol. 2019;10:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Eloseily EM, Cron RQ.. Macrophage activation syndrome. In: Ragab G, Atkinson T, Stoll M, editors. The microbiome in rheumatic diseases and infection. Cham: Springer; 2018;151–168. DOI: 10.1007/978-3-319-79026-8_14 [DOI] [Google Scholar]
- [3].Carter SJ, Tattersall RS, Ramanan AV.. Macrophage activation syndrome in adults: recent advances in pathophysiology, diagnosis and treatment. Rheumatology. 2019;58(1):5–17. [DOI] [PubMed] [Google Scholar]
- [4].Lerkvaleekul B, Vilaiyuk S.. Macrophage activation syndrome: early diagnosis is key. Open Access Rheumatol. 2018;10:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yoon J-H, Park -S-S, Jeon Y-W, et al. Treatment outcomes and prognostic factors in adult patients with secondary hemophagocytic lymphohistiocytosis not associated with malignancy. Haematologica. 2019;104(2):269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Goudarzipour K, Kajiyazdi M, Mahdaviyani A. Epstein-barr virus-induced hemophagocytic lymphohistiocytosis. Int J Hematol Oncol Stem Cell Res. 2013;7(1):42–45. PMID: 24505517; PMCID: PMC3913132 [PMC free article] [PubMed] [Google Scholar]
- [7].Marsh RA. Epstein–barr virus and hemophagocytic lymphohistiocytosis. Front Immunol. 2018;8:1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Henter J-I, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. [DOI] [PubMed] [Google Scholar]