

The interaction of host cells with mycobacteria is complex and can lead to multiple outcomes ranging from bacterial clearance to progressive or latent infection. Autophagy is recognized as one component of host cell responses that has an essential role in innate and adaptive immunity to intracellular bacteria. Many microbes, including Mycobacterium tuberculosis, have evolved to evade or exploit autophagy, but the precise mechanisms and virulence factors are mostly unknown. Through a loss-of-function screening of an M. tuberculosis transposon mutant library, we identified 16 genes that contribute to autophagy inhibition, six of which encoded the PE/PPE protein family.
KEYWORDS: autophagy, high-throughput screen, Mycobacterium tuberculosis, PE/PPE proteins, host-pathogen interactions, innate immunity, intracellular growth
ABSTRACT
The interaction of host cells with mycobacteria is complex and can lead to multiple outcomes ranging from bacterial clearance to progressive or latent infection. Autophagy is recognized as one component of host cell responses that has an essential role in innate and adaptive immunity to intracellular bacteria. Many microbes, including Mycobacterium tuberculosis, have evolved to evade or exploit autophagy, but the precise mechanisms and virulence factors are mostly unknown. Through a loss-of-function screening of an M. tuberculosis transposon mutant library, we identified 16 genes that contribute to autophagy inhibition, six of which encoded the PE/PPE protein family. Their expression in Mycobacterium smegmatis confirmed that these PE/PPE proteins inhibit autophagy and increase intracellular bacterial persistence or replication in infected cells. These effects were associated with increased mammalian target of rapamycin (mTOR) activity and also with decreased production of tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β). We also confirmed that the targeted deletion of the pe/ppe genes in M. tuberculosis resulted in enhanced autophagy and improved intracellular survival rates compared to those of wild-type bacteria in the infected macrophages. Differential expression of these PE/PPE proteins was observed in response to various stress conditions, suggesting that they may confer advantages to M. tuberculosis by modulating its interactions with host cells under various conditions. Our findings demonstrated that multiple M. tuberculosis PE/PPE proteins are involved in inhibiting autophagy during infection of host phagocytes and may provide strategic targets in developing therapeutics or vaccines against tuberculosis.
INTRODUCTION
Mycobacterium tuberculosis is an extraordinarily successful pathogen that is responsible for an estimated 2 million deaths annually and has latently infected nearly one-quarter of humanity (1). As an intracellular pathogen, it can modulate its environment within infected phagocytic cells to survive within the host. A well-known aspect of the M. tuberculosis intracellular survival mechanism is its ability to block phagolysosome maturation and acidification (2–4). In the last few years, cumulative evidence has revealed that autophagy, a ubiquitous and fundamental cellular process in eukaryotic cells, is a powerful tool in host cell defense that bacteria must overcome upon cell invasion. Autophagy can initiate phagosome maturation and enhance the processing and presentation of antigens to T cells, thus increasing bacterial clearance through both innate and adaptive immunity (5). This targeted, selective degradation of intracellular pathogens by autophagosomes for clearance and antigen presentation has been termed xenophagy (6). Regulation and modulation of autophagy are complex and are affected by many feedback mechanisms, not all of which are fully elucidated. Stimulation of autophagy by starvation, or by inhibition of its critical regulator mammalian target of rapamycin (mTOR) by drugs such as rapamycin, results in colocalization of mycobacteria to autophagosomes and consequently increased bacterial clearance (7–10).
Autophagy also plays critical roles in inflammation by influencing the development, homeostasis, and survival of inflammatory cells, as well as their transcription, processing, and secretion of cytokines. Autophagy can also be regulated by cytokines (11, 12), with gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), interleukin-1α (IL-1α), and IL-1β all known to be inducers of autophagy in macrophages (7, 13, 14), while IL-4, IL-13, and IL-10 have all been shown to inhibit autophagy in macrophages (11, 15–17). Along with cytokines, other bacterial-specific effectors involved in the inhibition of autophagy have been previously identified as virulence determinants in mycobacteria. These include the secreted proteins Eis (enhanced intracellular survival) (18, 19) and several members of the PE_PGRS family of virulence-associated proteins in M. tuberculosis (20–22). In addition, the ESX-1 type VII secretion system and its secreted substrates are required for the maturation of M. tuberculosis-containing autophagosomes into autolysosomes (23–25). This process involves damage to the phagosome membrane, which triggers the ubiquitination of M. tuberculosis and its DNA to recruit autophagic adaptors, thereby linking this system to the autophagic machinery (26). The Eis protein of M. tuberculosis inhibits autophagy through multiple mechanisms, including the downmodulation of reactive oxygen species (ROS) generation by NADPH oxidase and mitochondria and by upregulating IL-10 via effects on acetylation of histone H3 and activation of the Akt/mTOR/p70S6K pathway (19).
The PE and PPE proteins are acidic, glycine-rich proteins that are encoded by two related large families of genes that together constitute approximately 10% of the coding capacity of the M. tuberculosis genome. The names for these families refer to a characteristic proline-glutamic acid (PE) or proline-proline-glutamic acid (PPE) motif in the N termini of these proteins. Phylogenetic analysis has demonstrated several classes of PE proteins, one of which, the PE_PGRS family, includes C-terminal polymorphic GC-rich sequence (PGRS) domains of variable length. The genome of M. tuberculosis strain H37Rv encodes 61 PE_PGRS proteins and 68 PPE proteins (27). The PE and PPE proteins are found most abundantly in slow-growing pathogenic mycobacteria like M. tuberculosis, Mycobacterium bovis, and Mycobacterium ulcerans. This gene family has seemingly coevolved with the ESX or type VII secretion systems found in mycobacteria, which appear to play a significant role in their secretion (28, 29). Three members of the PE_PGRS family have been implicated in the regulation of autophagy in M. tuberculosis infection. For example, the deletion of the gene encoding PE_PGRS47 (rv2741) implicated this protein in the inhibition of autophagy in infected host phagocytes. The PE_PGRS47 mutant also led to attenuated growth of M. tuberculosis in vitro and in vivo and showed enhanced major histocompatibility complex (MHC) class II-restricted antigen presentation (20). Also, when expressed in Mycobacterium smegmatis, PE_PGRS41 confers autophagy inhibition resulting in decreased bacterial clearance from macrophages (21). Conversely, the PE_PGRS29 protein has been shown to interact with many autophagy receptors, resulting in increased autophagy and bacterial clearance through a unique host antimicrobial autophagy pathway (22).
The ability to inhibit autophagy varies substantially among mycobacterial species (30). For example, M. smegmatis and Mycobacterium marinum induce prominent autophagosome formation (31, 32), while M. tuberculosis and M. bovis BCG markedly inhibit autophagy (18, 25). It has been previously demonstrated that autophagy induction and mTOR signaling take place concurrently during mycobacterial infection and that host autophagy responses to any given mycobacterium stem from multiple factors, including the presence of activating macromolecules and inhibitory mechanisms (30). This conclusion is evidenced by the fact that different mycobacteria elicit variable levels of infection-induced autophagy, yet all mycobacterial species tested are potent activators of mTOR (9, 30). Thus, exploration of the mechanisms by which mycobacteria modulate autophagy is an essential topic for further research, which may present opportunities for new therapeutics or vaccine candidates against tuberculosis. In this study, we identified M. tuberculosis genes encoding PE and PPE proteins that inhibit infection-induced autophagy using genome-wide high-throughput screening of an arrayed library of random transposon (Tn) mutants. We demonstrated that five of the proteins identified were able to inhibit autophagy through increased mTOR signaling when expressed in M. smegmatis, which generally is a potent autophagy inducer. Specific gene deletion mutants generated in M. tuberculosis confirmed that these PE/PPE proteins regulate infection-induced autophagy. Macrophages infected with recombinant M. smegmatis expressing these PE or PPE proteins also showed reduced secretion of proinflammatory cytokines from infected macrophages, consistent with associated defects in host cell innate immune responses.
RESULTS
Identification of M. tuberculosis genes inhibiting autophagy.
It has been previously demonstrated that virulent M. tuberculosis induces deficient levels of autophagy, while the nonpathogenic M. smegmatis strongly induces this response in infected phagocytes (30). This observation suggested that M. tuberculosis may actively produce factors that block host cell autophagy. To test this, we carried out an experiment in which RAW 264.7 macrophages were first infected with M. tuberculosis overnight and then reinfected with M. smegmatis for 3 h. Preinfection with M. tuberculosis significantly inhibited autophagy induction by M. smegmatis compared to cells that had not been preinfected with M. tuberculosis (Fig. 1A), suggesting that M. tuberculosis expresses dominant autophagy-inhibiting factors that are absent in M. smegmatis.
FIG 1.
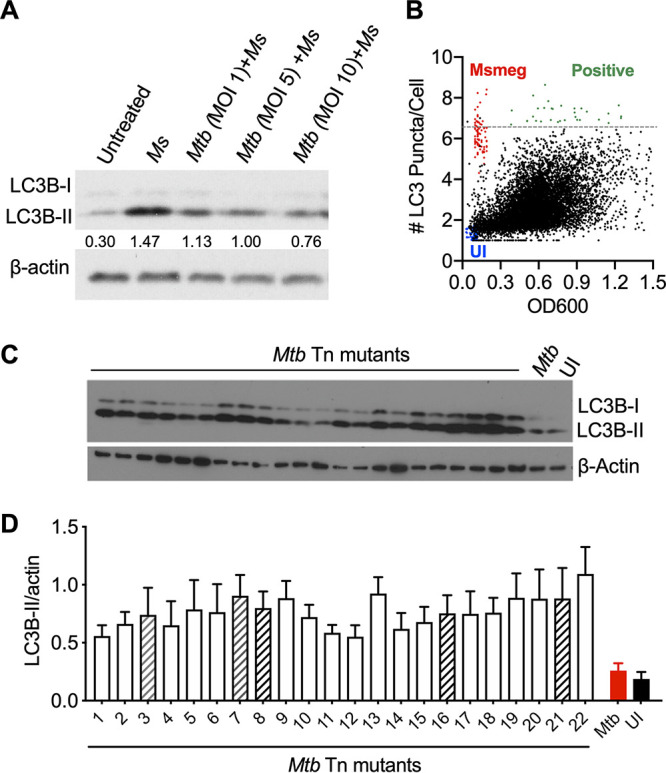
Loss-of-function screening using M. tuberculosis (Mtb) transposon library and RAW 264.7 LC3B-GFP macrophages. (A) LC3B immunoblot of RAW264.7 macrophages is shown. Macrophages were pretreated with M. tuberculosis (24 h) at an MOI of 1, 5, and 10 and then treated with M. smegmatis (Ms) (MOI, 10) for 3 h. The ratio of LC3B-II compared to β-actin is shown below LC3B-II immunoblot. (B) Single experimental plot of 10,165 mutants generated by Cellomics ArrayScan identifying mutants showing increased puncta formation (green dots). M. smegmatis (red dots) was used as a high-autophagy control, and uninfected cells were negative controls (blue dots). Significant induction of autophagy was determined as the mean number of puncta plus three times the standard deviation, calculated from 10,165 transposons infected with GFP-LC3 cells. The horizontal dashed line (gray) shows where this threshold value lies. (C) LC3B immunoblot confirmation with 22 autophagy-inducing mutants identified in high-throughput screening at 24 h postinfection in RAW 264.7 macrophages. (D) Summary densitometric analyses were calculated based on duplicate blots from three separate experiments. Data is mean ± standard error of the mean (SEM). Hatched bars indicate five mutants studied further.
To identify M. tuberculosis genes that directly or indirectly encode putative autophagy-regulating factors, we performed a genome-wide loss-of-function screen. A library of 10,165 M. tuberculosis random transposon (Tn) insertion mutants, arrayed as individual clones in 96-well plates, were used to infect an autophagy reporter cell line, LC3B- green fluorescent protein (GFP) RAW 264.7 macrophages. After 24 h of infection with an average multiplicity of infection (MOI) of 10, autophagy induction was quantitated by measuring the number, size, and intensity of LC3B-GFP puncta with automated high-throughput fluorescence microscopy (Fig. 1B; see also Fig. S1 in the supplemental material). Significant induction of autophagy was determined by mutants showing higher than the mean number of puncta greater than 3 standard deviations above the mean for the entire pool of 10,165 transposon mutants. This screen was repeated twice, yielding consistent positive signals for 22 transposon mutants that were selected for further analysis. The reduced capacity of these mutants to inhibit autophagy was confirmed by LC3B-II immunoblot (Fig. 1C and D). Sixteen of these mutants displayed stable growth and evidence of a single Tn insertion site in identifiable open reading frames upon sequence analysis, consistent with unique clonal populations. The genes interrupted by Tn insertions based on sequencing of these 16 mutants are summarized in Table 1.
TABLE 1.
Summary of 16 autophagy-inducing mutants from loss-of-function screen
| Protein | Function | M. smegmatis orthologues |
|---|---|---|
| Rv0464c | Conserved protein | MSMEG_0905 |
| Rv0020c | FHA domain-containing protein, FhaA | MSMEG_0035 |
| Rv3857c | Hypothetical membrane protein | Not found |
| Rv1179c | Hypothetical protein | Not found |
| Rv1804c | Hypothetical protein | Not found |
| Rv2466c | Hypothetical protein | MSMEG_4688 |
| Rv2065 | Iron complex transporter substrate-binding protein, CobH | MSMEG_3872 |
| Rv1510 | Membrane protein | MSMEG_6140 |
| Rv1162 | Respiratory nitrate reductase beta chain, NarH | MSMEG_5139 |
| Rv2214c | Short-chain type dehydrogenase, EphD | MSMEG_4280 |
| Rv1068c | PE_PGRS family protein, PE_PGRS20 | Not found |
| Rv1087 | PE_PGRS family protein, PE_PGRS21 | Not found |
| Rv1651c | PE_PGRS family protein, PE_PGRS30 | Not found |
| Rv2741 | PE_PGRS family protein, PE_PGRS47 | Not found |
| Rv2770c | PPE family protein, PPE44 | Not found |
| Rv3136 | PPE family protein, PPE51 | Not found |
In order to prioritize the candidate autophagy-regulating genes from our screen, we focused on the M. tuberculosis genes that did not have identifiable orthologues in M. smegmatis. This sequence analysis highlighted nine genes, three of which encoded hypothetical proteins without known functions. However, the remaining six genes encoded members of the M. tuberculosis PE and PPE protein families, which are widely accepted to be virulence factors in pathogenic mycobacteria (33, 34). This was noteworthy given that several PE and PPE proteins, including PE_PGRS47 (20), PE_PGRS41 (21), MMAR_0242 (35), and PE_PGRS29 (22), have previously been shown to be involved in modulating autophagy in infected cells. In particular, the identification of a Tn insertion mutant in the gene encoding PE_PGRS47 in our screen was consistent with our previously published detailed studies on the effect of this protein in inhibition of autophagy in M. tuberculosis-infected cells (20). The other five PE or PPE proteins identified were not, to our knowledge, known to be involved in the regulation of autophagy, although PE_PGRS30 has a previously demonstrated role in phagolysosome formation and cell death in host cells (36).
Analysis of PE and PPE proteins by expression in M. smegmatis.
The variability in autophagy induction by different mycobacterial species provides useful opportunities for assessing if and how specific mycobacterial proteins are responsible for controlling autophagy during infection (21, 30). The nonpathogenic mycobacterium M. smegmatis strongly induces autophagy while maintaining high levels of mTOR activation (30). Previous studies have shown that expression of the M. tuberculosis genes encoding PE_PGRS47 or PE_PGRS41 in M. smegmatis can significantly suppress autophagy induction and promote bacterial survival in phagocytic cells (20, 21). We adopted this approach for analysis of five of the six PE and PPE proteins identified through our loss-of-function Tn mutant screen. Recombinant M. smegmatis strains expressing each of these proteins (PE_PGRS20 [Rv1068c], PE_PGRS21 [Rv1087], PE_PGRS47 [Rv2741], PPE44 [Rv2770c], and PPE51 [Rv3136]) were generated using an episomal plasmid containing a constitutive Hsp60 promoter and maintained by antibiotic selection. PE_PGRS30 was not further characterized due to technical difficulties in the creation of the expression construct. The expression of these proteins did not affect M. smegmatis growth in vitro (Fig. 2A). The secretion of these proteins is believed to be at least partially dependent on a specialized set of type VII secretion systems (ESX-1 to ESX-5) (29, 37), and most PE and PPE proteins contain a highly conserved ESX secretion signal, the YXXXD/E motif, within their PE or PPE domain (38). The PPE and PE_PGRS domains of the PE/PPE proteins studied here were aligned using Clustal Omega (39) to determine if the conserved ESX secretion signal is present. PE_PGRS20, PE_PGRS21, PE_PGRS47, and PPE51 all contain this conserved secretion signal (Fig. 2B), indicating their secretion by an ESX system. PPE44 did not contain an identifiable secretion signal.
FIG 2.
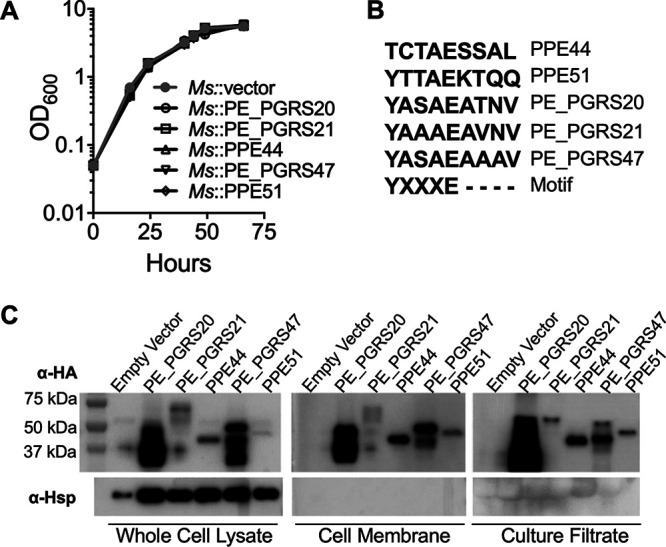
PE/PPE proteins are secreted by M. smegmatis (Ms). (A) Growth curves of recombinant M. smegmatis expressing PE/PPE in 7H9 are shown. The mean ± standard deviation (SD) of three cultures shown as a representative experiment of three independent assays. (B) Clustal Omega alignment of PE/PPE proteins and ESX secretion signal (motif). PPEs aligned from residue 77, and PE_PGRSs aligned from residue 87. (C) The immunoblot is showing the expression and subcellular localization of HA-tagged PE/PPE proteins in M. smegmatis. The expected protein sizes are as follows: PE_PGRS20, 39.3 kDa; PE_PGRS21, 62.5 kDa; PPE44, 37.4 kDa; PE_PGRS47, 44.2 kDa; and PPE51, 40.0 kDa. Representative immunoblots of three independent experiments.
Analysis by immunoblotting of bacterial cell lysates showed that all five proteins were expressed at varying levels in the corresponding M. smegmatis transformants (Fig. 2C; see also Fig. S2 in the supplemental material), with PE_PGRS20 and PE_PGRS47 at much higher levels than PE_PGRS21, PPE44, and PPE51. Many PE and PPE proteins have been shown to be either secreted or presented on the surface of bacterial cells where they are thought to interact with components of the host cell environment, potentially having an impact on host immune responses (34). Using the recombinant M. smegmatis strains overexpressing M. tuberculosis PE or PPE proteins, we examined the bacterial cell membrane fraction and culture filtrate for expression of the proteins by immunoblotting. All five proteins were detected in both the culture filtrate and cell membrane fraction, consistent with their secretion during bacterial growth in culture (Fig. 2C).
PE and PPE proteins inhibit autophagy during M. tuberculosis and M. smegmatis infection.
The role of M. tuberculosis PE or PPE proteins in autophagy regulation was measured by immunoblot analysis for LC3B from RAW 264.7 macrophages infected with recombinant M. smegmatis strains expressing the PE or PPE proteins. Significantly decreased levels of LC3B were observed during infection with recombinant M. smegmatis strains at an MOI of 10 (Fig. 3A and B; see also Fig. S3 in the supplemental material). The accumulation of LC3B was confirmed using the reporter cell line utilized for the loss-of-function screening in Fig. 1, which revealed significantly reduced GFP intensity by flow cytometry with infection by all recombinant M. smegmatis strains at both 4 and 20 h postinfection (Fig. 3C and D). At 24 h postinfection, infected macrophages were also examined for puncta formation by confocal microscopy (Fig. 3E). Significantly decreased puncta formation was observed in LC3B-GFP macrophages infected with recombinant M. smegmatis strains (Fig. 3F). Increased intracellular bacteria were also observed in RAW 264.7 macrophages infected at an MOI of 10, with several of the recombinant M. smegmatis strains expressing M. tuberculosis PE or PPE proteins compared with those of the empty vector control at both 4 and 20 h postinfection (Fig. 3G).
FIG 3.

Expression of PE/PPE genes in M. smegmatis (Ms) inhibits autophagy, increasing bacterial survival in RAW264.7 macrophages. (A) Immunoblot of LC3B accumulation in RAW 264.7 macrophages infected with M. smegmatis expressing PE/PPE proteins at an MOI of 10 at 20 h postinfection. The ratio of LC3B-II compared to β-actin is shown below LC3B-II immunoblot. Due to strong autophagy induction by M. smegmatis, Western blots were exposed for a short duration to observe LC3B-II accumulation (longer exposures of blots to examine LC3B-I are shown in Fig. S3 in the supplemental material). (B) The densitometric summary analysis was calculated by LC3B-II normalized to β-actin, and then the fold change ratio was calculated compared to the uninfected control for each assay. The mean ± SD of 3 independent assays is shown. Significance was calculated by two-way ANOVA corrected by Dunnett’s Test for multiple comparisons. (C) Example of flow cytometry histograms for LC3B-GFP RAW 264.7 macrophages infected at an MOI of 10 at 20 h postinfection. (D) GFP mean fluorescence intensity (MFI) of LC3B-GFP RAW 264.6 macrophages as measured by flow cytometry. The mean ± SD of a representative assay is shown. Significance was calculated by two-way ANOVA corrected by Dunnett’s Test for multiple comparisons. (E) Puncta formation in LC3B-GFP RAW 264.7 macrophages was determined after infection at an MOI of 10 at 24 h postinfection. Green, LC3B-GFP; blue, DAPI. (F) Mean ± SD of average puncta per cell was counted from 200 to 250 cells (from panel E). Significance is calculated by one-way ANOVA corrected by Dunnett’s test for multiple comparisons. (G) M. smegmatis survival was determined in RAW 264.7 macrophages (MOI, 10) at 4 and 20 h postinfection. The mean ± SD of 3 independent assays is shown. Significance was calculated by two-way ANOVA corrected by Dunnett’s test for multiple comparisons. (H) Summary densiometric analysis of the ratio between LC3B-II levels from RAW 264.7 macrophages infected at an MOI of 10 for 4 h with and without bafilomycin (Bafil) A1 treatment (autophagy flux). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
To exclude a possibility that the recombinant M. smegmatis strains block the late stage of autophagy, RAW 264.7 macrophages were infected with the recombinant M. smegmatis::PE/PPE strains with or without bafilomycin A1 (BafA1) that blocks degradation of LC3B-II in lysosomes. Similarly, increased levels of lipidated LC3B upon BafA1 treatment in both control and recombinant strains suggested that maturation of autophagosomes was not inhibited during recombinant M. smegmatis infection (Fig. 3H; see also Fig. S4 in the supplemental material). Since recent work has defined a macrophage pathway called “LC3-associated phagocytosis” (LAP) that can eliminate microbes including M. tuberculosis, we evaluated if LAP is responsible for the decreased LC3B level and inefficient clearance of intracellular recombinant M. smegmatis strains. We assessed this by immunoblotting lysates of infected macrophages with an antibody against Rubicon, a host cell protein which inhibits autophagy and is required for LAP (40). We found that the Rubicon protein level was not affected by expression of any of the PE/PPE proteins in M. smegmatis, suggesting that LAP was not augmented by these PE/PPE proteins (see Fig. S5 in the supplemental material).
To confirm the results of the Tn library screen and analyze the functions of the identified PE/PPE proteins in M. tuberculosis directly, the relevant pe/ppe genes were specifically deleted by specialized transduction in M. tuberculosis strain H37Rv (see Fig. S6 in the supplemental material). The deletion of these genes did not have any effect on M. tuberculosis growth in vitro (Fig. 4A), suggesting that the increased autophagy observed during the screen was not likely due to increased bacterial replication. To confirm that autophagy is manipulated by the M. tuberculosis PE/PPE proteins, Δpe/ppe mutant strains were used to infect RAW 264.7 macrophages at an MOI of 10 for 24 h. Immunoblot analysis for LC3B-II showed increased levels with infection by the M. tuberculosis Δpe/ppe strains compared to those of the M. tuberculosis wild-type (WT) control (Fig. 4B and C). Our studies also showed that cells infected with pe/ppe mutant bacteria had lower bacterial load at 24 and 48 h postinfection than cells infected with wild-type H37Rv (Fig. 4D). These data provided further evidence that these PE/PPE proteins play essential roles in the survival and multiplication of M. tuberculosis bacilli during infection and suggest that enhanced autophagy is detrimental to bacterial survival.
FIG 4.

Expression of pe/ppe genes in M. tuberculosis (Mtb) is induced in response to some stress conditions and inhibits autophagy. (A) Growth curves of M. tuberculosis Δpe/ppe strains in 7H9. The mean ± SD of three cultures shown as a representative experiment of 3 independent assays. (B) Immunoblot of LC3B in RAW 264.7 macrophages infected with M. tuberculosis Δpe/ppe at an MOI of 10 at 24 h postinfection. The representative immunoblot is shown of three independent assays. (C) The densiometric summary analysis was calculated by LC3B-II normalized to β-actin, and then the fold change ratio was calculated compared to uninfected control for each assay. The mean ± SD of 3 independent assays is shown. Significance was calculated by one-way ANOVA corrected by Dunnett’s test for multiple comparisons. (D) The survival of M. tuberculosis deletion mutants was determined in RAW 264.7 macrophages (MOI, 10) at 24 and 48 h postinfection. The mean ± SD of 2 independent assays is shown. Significance was calculated by two-way ANOVA corrected by Dunnett’s test for multiple comparisons. (E) Relative gene expression of pe/ppe genes in M. tuberculosis (H37Rv) in response to oxidative stress, acidic stress (pH 5.4), and starvation after 3 h. Relative gene expression normalized to the housekeeping gene rpoB determined by ΔΔCT method. Shown is the mean ± SD of 3 independent assays. Significance was calculated by two-way ANOVA corrected by Dunnett’s test for multiple comparisons. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Since PE/PPE proteins are shown to be differentially regulated under various stress conditions (33), quantitative reverse transcription-PCR (qRT-PCR) was used to determine the expression levels of pe_pges20, pe_pgrs21, pe_pgrs47, ppe44, and ppe51 relative to the housekeeping gene rpoB. Three in vitro stress conditions were chosen to emulate the intracellular macrophage environment. Treatment of M. tuberculosis with hydrogen peroxide (oxidative stress) significantly increased expression of pe_pgrs20, pe_pgrs21, and ppe44 and moderately increased expression of pe_pgrs47 (Fig. 4D). While the exposure of M. tuberculosis to low pH (acidic stress) demonstrated increased expression of pe_pgrs47, ppe44, and ppe51, no differential regulation of pe/ppe genes in M. tuberculosis was observed under low nutrient/starvation conditions (Fig. 4D). The differential upregulation of these PE/PPE proteins in response to different stress conditions may be advantageous to M. tuberculosis in order for the bacteria to modulate interactions with host cells, such as autophagy, under a variety of stress conditions to facilitate bacterial survival during infection.
Modulation of cytokine secretion by RAW 264.7 macrophages by PE and PPE proteins.
Mycobacteria are efficient inducers of host proinflammatory cytokines and chemokines that may promote autophagic responses (34). Autophagy modulates the transcription, processing, and secretion of several proinflammatory cytokines, acting as an essential negative feedback mechanism for the control of inflammatory responses. To assess the potential effects of autophagy inhibition by PE/PPE proteins on proinflammatory cytokine secretion, we quantified TNF-α and IL-1β in RAW 264.7 cells infected with recombinant M. smegmatis strains. Expression of PE/PPE proteins in M. smegmatis inhibited the production of TNF-α and IL-1β (Fig. 5A and B). In addition, secretion of IL-10, which has been previously shown to inhibit autophagy during mycobacterial infection (12, 37, 38), was not increased in recombinant M. smegmatis-infected macrophages (Fig. 5C). In spite of the relatively strong TNF-α release from macrophages infected with M. smegmatis, we did not observe significant levels of macrophage cell death based on lactate dehydrogenase (LDH) release in our culture model following infections with either unmodified M. smegmatis or the recombinant strains expressing PE/PPE proteins (Fig. 5D).
FIG 5.

PE/PPE proteins modulate cytokine secretion by RAW 264.7 macrophages. Cytokine concentration in RAW 264.7 macrophage culture supernatants was measured after infection with M. smegmatis (Ms) or M. smegmatis expressing PE/PPE proteins after 20 h for TNF-α (A), IL-1β (B), and 4 h for IL-10 (C). (D) Percentage LDH release from RAW 264.7 macrophages 20 h postinfection with M. smegmatis::empty vector or M. smegmatis expressing PE/PPE proteins. All data shown are mean ± SD of 3 independent assays. Significance was calculated by one-way ANOVA corrected by Dunnett’s Test for multiple comparisons. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. LPS, lipopolysaccharide.
Activation of mTOR by PE and PPE proteins.
The mTOR complex coordinates eukaryotic cell growth and metabolism with environmental inputs, including nutrients and growth factors. It has a well-established central role in regulating many fundamental cell processes, from protein synthesis to autophagy. Protein synthesis is promoted by mTOR primarily through the phosphorylation of two key effectors, p70S6 kinase 1 (S6K1) and eIF4E binding protein (4EBP) (41). When cells undergo nutrient depletion or are treated with rapamycin, mTOR signaling is inhibited, resulting in autophagosome formation marked by LC3B-II (42). In order to determine if M. tuberculosis PE and PPE proteins inhibit autophagy by regulating mTOR signaling, we evaluated mTOR signaling in macrophages infected with recombinant M. smegmatis expressing M. tuberculosis PE or PPE proteins.
All recombinant M. smegmatis bacteria expressing the PE/PPE proteins demonstrated higher levels of at least one of three proteins associated with mTOR-dependent regulation than the M. smegmatis expressing empty vector. At 60 min postinfection, M. smegmatis::PE_PGRS20 and M. smegmatis::PPE51 showed increased phosphorylation of S6; however, no significant difference in the phosphorylation of mTOR or S6K1 was observed for these two strains (Fig. 6B). Recombinant M. smegmatis expressing PE_PGRS21 and PE_PGRS47 proteins significantly increased mTOR, pS6K, and S6 phosphorylation compared to M. smegmatis::empty vector at 60 min postinfection. Infection with M. smegmatis::PPE44 also led to significantly increased phosphorylation of mTOR but not activation of S6 (Fig. 6B). The phosphorylation of ULK1 and 4EBP1, other downstream effectors of mTOR, was also examined at these early time points for these infections and was found to mimic the results of S6K1 and S6 (data not shown). None of the PE or PPE proteins affected the total levels of these phosphorylated proteins (see Fig. S7 in the supplemental material). Different phosphorylation levels of mTOR-dependent downstream effectors suggest that expression and secretion of these M. tuberculosis PE/PPE proteins in M. smegmatis are varied in infected cells. This result may also suggest that these PE/PPE proteins are induced at different stages of infection to modulate autophagy through regulation of the mTOR pathway.
FIG 6.

PE/PPE proteins induce mTOR activation. (A) Immunoblot of phospho-mTOR, phospho-p70S6K, and phospho-S6 were evaluated in RAW 264.7 macrophages infected with M. smegmatis (Ms) or M. smegmatis overexpressing PE/PPE proteins at an MOI of 10 for 15, 30, and 60 min postinfection. The ratio of phospho-S6 against actin is shown below the phospho-S6 blot. Representative immunoblots of three independent assays are shown. (B) Summary densitometric analysis at 60 min was determined from immunoblots for phosphorylated proteins. Phosphoproteins normalized to β-actin, and then ratio increase was calculated compared to uninfected control at each time point. The mean ± SD of a representative assay of 3 independent assays is shown. Significance was calculated by one-way ANOVA. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Effects on lipid composition and antibiotic sensitivity.
The expression of some PE or PPE proteins in M. smegmatis has been shown to modify the lipid composition of the complex mycobacterial cell wall and have significant effects on antibiotic sensitivity (21, 43, 44). Considering that mycobacterial lipids are able to induce autophagy (30), changes in lipid profile due to the expression of PE or PPE proteins could be related to effects on autophagy during recombinant M. smegmatis infection (30). The polar (Fig. 7A) and apolar (Fig. 7B) lipid fractions of M. smegmatis and M. smegmatis expressing PE/PPE proteins were extracted and visualized using thin-layer chromatography (TLC). No apparent differences were observed in any of the PE or PPE protein-expressing strains when compared to wild-type M. smegmatis. Resazurin reduction assays, which measure the viability of living cells, including mycobacteria, were used to determine the MIC90 for rifampicin, isoniazid, gentamycin, amoxicillin, SDS, and H2O2 (Fig. 7C). Other than a modest increase in MIC90 for amoxicillin with M. smegmatis expressing PPE51, no significant effects on susceptibility to antibiotics, detergent, or oxidative stress were observed. While a modest increase in resistance to amoxicillin in the M. smegmatis::PPE51 strain indicates some modification of the cell wall or outer membrane, the PPE51 protein is secreted, enabling its interaction with host immune modulators.
FIG 7.

PE/PPE proteins do not affect M. smegmatis (Ms) lipid composition or antibiotic sensitivity. (A) Thin-layer chromatography of polar lipid extraction from M. smegmatis::empty vector or M. smegmatis::PE/PPE overexpressing strains. TDM, trehalose dimycolate; TMM, trehalose monomycolate; PE, phosphatidylethanolamine; PDIM, phthiocerol dimycocerosates. Polar lipids were analyzed using the solvent system chloroform/methanol/water (20:4:0.5, vol/vol/vol) and visualized by charring with 10% copper sulfate in 8% phosphoric acid. (B) Two-dimensional thin-layer chromatography of apolar lipid extraction from M. smegmatis::empty vector or M. smegmatis::PE/PPE. Apolar lipids were analyzed using the solvent system petroleum ether/ethyl acetate (98:2, vol/vol, 3×) in the first direction and petroleum ether/acetone (98:2, vol/vol) in the second direction. Lipids were visualized by charring with ceric ammonium molybdate in 6% acid. (C) MIC90 of M. smegmatis::empty vector and M. smegmatis::PE/PPE overexpressing strains to various antibiotics as was determined by resazurin reduction assay. The mean ± SD of three independent experiments is shown. Statistical significance was calculated by two-way ANOVA corrected for multiple comparisons by Dunnett’s test. ***, P ≤ 0.001.
Amoxicillin is a beta-lactam antibiotic that mycobacteria are resistant to due to the presence of mycobacterial beta-lactamase. Although some studies have shown that the permeability of the cell wall is responsible for beta-lactam resistance (44), the M. tuberculosis Δppe51 strain exhibited no change in susceptibility to beta-lactam antibiotics (data not shown), indicating that decreased cell wall permeability is not causing the increased resistance of M. smegmatis::PPE51.
DISCUSSION
Using high-throughput loss-of-function screening, we have identified 16 genes of M. tuberculosis that are required for the inhibition of autophagy during macrophage infection. Significantly, one-third of these genes belong to the PE and PPE protein families, which are prominent expanded families of virulence-related proteins in M. tuberculosis and other pathogenic mycobacteria. One of the proteins identified by our screen, PE_PGRS47, has previously been identified to inhibit autophagy and MHC class II antigen presentation (20) and was independently identified in this study along with PE_PGRS20, PE_PGRS21, PE_PGRS30, PPE44, and PPE51. PE_PGRS30 has previously been shown to play a role in phagolysosome formation and cell death in host cells (36). This protein was also identified in our Tn screen but was not further studied because of our inability to create the mycobacterial expression construct in M. smegmatis for reasons that have not been determined.
M. smegmatis is a potent inducer of autophagy and carries 10 PE or PPE proteins. None of the six PE and PPE proteins identified in this study had orthologous genes in M. smegmatis, so we hypothesized that this subset of PE and PPE genes might inhibit autophagy when expressed in M. smegmatis. Similarly, previous studies described recombinant M. smegmatis strains expressing PE_PGRS41 or PE_PGRS47 as having the ability to inhibit autophagy during infection as presented here (20, 21). Considering that our screening did not identify PE_PGRS41 or other PE/PPE proteins known to be involved in autophagy regulation other than PE_PGRS47, a broader subset of PE and PPE proteins may be able to modulate autophagy.
Pathogen-host interactions are mediated in part by secreted microbial proteins capable of exploiting host cells for their survival. Some bacteria secrete effector proteins that impair the functions of lysosomes or inhibit bacteria-containing autophagosome fusion with lysosomes to block the lysosomal degradation of the bacteria (23, 25). Importantly, all five PE and PPE proteins analyzed in this study were found to be secreted into the culture filtrate and also found in the membrane fraction of recombinant M. smegmatis lysates. These five proteins are likely secreted through one of three mycobacterial type VII secretion systems (ESX1, ESX3, or ESX4) present in M. smegmatis (29). PE_PGRS20, PE_PGRS21, PE_PGRS47, and PPE51 all contain the motif YXXXD/E, which is known to constitute a signal sequence for secretion by the type VII systems. PPE44 does not contain this motif, indicating an alternative signal sequence or a separate secretion pathway for this protein.
Many PPE proteins are thought to function with paired PE proteins to facilitate secretion. PPE51 contains the YXXXD/E signal sequence and was found to be secreted in M. smegmatis, indicating that a PE protein partner is not needed for its secretion. While PPE44 does not contain the secretion motif, it was still secreted by M. smegmatis with no apparent need for the introduction of a paired PE protein, although it should be noted that the genes encoding PPE44 and PE27 are adjacent in the M. tuberculosis genome. Among this group of proteins, only PE_PGRS47 has previously been experimentally identified as being secreted (20), while PPE51 has been identified in the cell wall and cell membrane fractions of M. tuberculosis (45, 46).
PPE51 has been known to have other functions in mediating sugar and nutrient transport (46, 47) and arresting M. tuberculosis growth at acidic pH (48). A suggested mechanism of starvation at low pH depend on the PPE51 protein as a cotransporter of H+/glucose/glycerol, which uses the energy of the transmembrane electrochemical ion gradient to drive the accumulation of a substrate against its concentration gradient into the cell. In this mechanism, the binding of the cation (low pH) increases the affinity of the transporter for sugar. In contrast, the involvement of PPE51 in the uptake of disaccharides seems to be cation independent. Although we have not directly evaluated how the two functions mentioned above affect the PPE51 protein as an autophagy regulator, we believe that PPE51 directly modulates autophagy. The reason is that we did not observe any growth defect of the recombinant M. smegmatis::PPE51 or Δppe51 M. tuberculosis mutant strain in liquid culture (Fig. 3A and 4A). M. smegmatis::PPE51 survived better in infected cells like the other recombinant M. smegmatis strains that we tested, probably at least in part due to decreased autophagy (Fig. 3G).
In recent years, autophagy has gained attention as an essential cellular mechanism in response to mycobacterial infection (7, 10, 20, 26, 49). Many studies have shown that induction of autophagy by starvation or pharmacological means, such as high doses of rapamycin, enhances mycobacterial clearance and, in some cases, antigen presentation. Our findings are consistent with previous findings that inhibition of autophagy leads to increased bacterial burden within macrophages. Similar to autophagy, LAP can clear microorganisms through a lysosomal-trafficking pathway. The ability of M. tuberculosis to inhibit LAP, survive, and subsequently cause disease depends upon CpsA, a member of the LytR-CpsA-Psr (LCP) protein family (40). However, M. tuberculosis PE/PPE proteins did not contribute to the inhibition of LAP (see Fig. S5 in the supplemental material). Although we have not yet extended our studies to examine in vivo effects in mouse models, it is known from previous studies that PE_PGRS47 is required for the maximal in vivo persistence of M. tuberculosis (20). Models that employ the deletion of autophagy-related genes will be useful to confirm that the PE and PPE proteins identified in the current study have a significant role in vivo, indirectly targeting the autophagy machinery.
An increased proinflammatory cytokine profile has been associated with autophagy inhibition during mycobacterial infection in some models (10, 18, 21, 50, 51). However, we observed that expression of autophagy inhibiting PE or PPE proteins in M. smegmatis tended to reduce the secretion of TNF-α and IL-1β by macrophages infected with these bacteria. This suggests a more generalized effect of these M. tuberculosis PE and PPE proteins on inhibiting the innate immune response of host phagocytes to intracellular infection. It remains to be determined whether the effects that we observed on cytokine secretion are directly related to the inhibition of autophagy in infected cells as opposed to other direct or indirect effects of the expressed PE or PPE proteins.
Overall, our results provide further evidence that the PE and PPE families expressed by pathogenic mycobacteria have a role in host-microbe interactions, specifically in the inhibition of autophagy and subsequent increased bacterial survival within macrophages. The expression of PE or PPE proteins in M. smegmatis resulted in significantly increased mTOR signaling, a possible mechanism for inhibiting mycobacteria-induced autophagy. While further work is needed to determine the detailed mechanisms by which the PE and PPE proteins of M. tuberculosis are able to modulate autophagy, as well as their role in vivo, this study suggests that M. tuberculosis proteins inhibit autophagy as a critical strategy to interfere with the ability of the macrophages to control infection. Our understanding of the role of autophagy during mycobacterial infection will allow for much-needed advances in both drug development and vaccine design to help fight the global tuberculosis problem.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
M. tuberculosis (strain H37Rv) or M. smegmatis (strain mc2155) were cultured at 37°C with shaking in Middlebrook 7H9 supplemented with 10% OADC (oleic acid, albumin, dextrose, catalase), 0.5% glycerol, and 0.02% tyloxapol. Middlebrook 7H10 agar medium was supplemented with 10% OADC and 0.5% glycerol without or with antibiotics as per requirements (hygromycin, 50 μg/ml, and kanamycin, 25 μg/ml). The targeted pe/ppe genes were deleted from M. tuberculosis H37Rv by allelic exchange via the previously described specialized phage transduction method (60). Briefly, allelic exchange substrates (AES) were constructed with an approximately 1,000-bp region upstream and downstream of the pe/ppe genes, which was amplified by PCR and directionally cloned into a PacI-containing E. coli cosmid, flanking a hygromycin cassette. The AES was ligated into the temperature-sensitive mycobacteriophage derived from TM4. Mycobacteriophage-packaged shuttle phasmids were transduced to M. tuberculosis H37Rv. Colonies were selected for hygromycin resistance following transduction and screened for gene deletion by PCR. Escherichia coli strains DH5α and Stbl3 were used for cloning and were grown on LB agar or broth at 37°C with the addition of hygromycin (50 μg/ml) as required.
Construction of recombinant M. smegmatis strains.
Full-length pe/ppe genes were amplified from M. tuberculosis H37Rv genomic DNA using gene-specific primers. The amplified genes were cloned in-frame into pMV261, an episomal mycobacterial vector (52) with a 3′ His6-hemagglutinin (HA) tag. The expression constructs were electroporated into M. smegmatis and selected for kanamycin resistance. The expression of PE and PPE proteins was confirmed by immunoblot using an anti-HA antibody.
Cell culture.
RAW 264.7 macrophages and RAW GFP-LC3 macrophages (30) were maintained in Dulbecco modified Eagle medium (DMEM) complete (high glucose DMEM supplemented with 1% nonessential amino acids, 10% heat-inactivated fetal bovine serum (Corning), and 50 μM β-mercaptoethanol) at 37°C with 5% CO2. All macrophage assays were conducted in 12-well plates, seeded at 2.5 × 105 cells/well. M. smegmatis and M. tuberculosis strains were grown to an optical density at 600 nm (OD600) of 0.6 to 0.8 and were infected at an MOI of 10 unless otherwise stated. Infection was carried out for 3 h and followed by phosphate-buffered saline (PBS) wash and treatment with 50 μg/ml gentamycin in DMEM complete for 1 h to kill extracellular bacteria. Assays were conducted in DMEM, complete with 20 μg/ml gentamycin. For M. tuberculosis and M. smegmatis coinfection, RAW 264.7 cells were first infected with M. tuberculosis at an MOI of 1, 5, or 10 for 3 h, treated with 50 μg/ml gentamycin for 1 h, and then incubated for 24 h with 20 μg/ml gentamycin. After overnight infection with M. tuberculosis, the cells were then coinfected with M. smegmatis at an MOI of 10 for 3 h. Cells were harvested at indicated time points in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1% NP-40 or Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 8.0, 20 mM Tris-HCl, pH 7.5) for plating for intracellular survival or analysis of autophagy induction by immunoblotting. For CFU enumeration, lysates were serially diluted and plated on 7H10 with 25 μg/ml kanamycin.
High-throughput screening of Tn mutants.
LC3B-GFP RAW 264.7 macrophages were utilized for the high-throughput loss-of-function screening. An M. tuberculosis Mariner transposon library, previously described (53), was grown as individual clones arrayed in 96-well plates in 7H9 medium. After 5 days, the OD600 was determined for each well. These values were averaged to determine an average MOI for macrophage infections. LC3B-GFP macrophages were seeded at 1 × 105 cells per well in 96-well plates in 150 μl of complete medium (DMEM supplemented with 1% nonessential amino acids, 10% heat-inactivated fetal bovine serum [Corning] and 50 μM β-mercaptoethanol) and incubated overnight at 37°C with 5% CO2. Macrophages were then infected with individual clones from the M. tuberculosis transposon library by adding 10 μl of each Tn mutant culture adjusted to give an average MOI of 10. The average OD was calculated from all mutants. An uninfected control and an M. smegmatis positive control were included in each plate. Other transposon mutants served as a control for the parental strain since most Tn mutants should induce a similar level of autophagy as WT parental M. tuberculosis. Macrophages were infected for 3 h, and cells were then washed 3 times with PBS and incubated for 24 h in complete medium. Counting of autophagosomes (GFP-LC3) was done on a Thermo Scientific Cellomics ArrayScan high-content screening (HCS) reader (20× objective) using the Spot detector V3 Cellomics BioApplication. For identification of cells, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) and detected in the primary channel by a Hoechst-associated filter. Autophagosomes were identified on secondary channels using fluorescein isothiocyanate (FITC)-associated filters, dependent upon size, shape, and intensity thresholding. The ArrayScan software calculated the mean number of autophagosomes per cell (object) as the total number of autophagosomes per field divided by the total number of objects per field. One thousand cells were counted per well, and the analysis was repeated twice. Significant induction of autophagy was determined by the mean number of puncta plus three times the standard deviation, which was calculated from 10,165 transposon-infected GFP-LC3 cells. Initial positive Tn mutants from the primary screen were confirmed by LC3B Western blotting.
Immunoblotting.
Cellular protein was prepared in 1× RIPA buffer, and its concentration was determined by bicinchoninic acid (BCA) assay (Pierce). One to ten micrograms of protein was resolved on 12% SDS-PAGE gels at 170 V for 40 min. Proteins were transferred to 0.2 μm polyvinylidene difluoride (PVDF) using a Bio-Rad transblot turbo at 2.5 A and 25 V for 5 to 10 min, depending on molecular weight. PVDF membranes were blocked in 5% nonfat dry milk in 1× Tris-buffered saline (TBS) plus 0.2% Tween 20 (TBST) or OneBlock Western-CL blocking buffer (Genesee Scientific) for LC3B blots at 4°C overnight. Primary antibodies at 1:5,000 dilution were incubated for 2 h at room temperature in TBST. Anti-rabbit IgG-horseradish peroxidase (HRP) antibody (Cell Signaling Technology) (1:10,000) was added to membranes for 45 min in TBST. Proteins of interest were revealed using Clarity ECL (Bio-Rad) according to manufacturer’s instructions. Films were scanned and densitometric analysis conducted by ImageJ software (https://imagej.nih.gov/ij). LC3B-II was normalized to β-actin loading control to calculate levels of autophagy (54).
Antibodies and other reagents.
Antibodies were purchased from Cell Signaling Technology unless indicated otherwise and are cataloged in Table S1 in the supplemental material. Anti-HA-peroxidase conjugated antibody (final concentration, 25 mU/ml) was purchased from Sigma-Aldrich (clone 3F10). Anti-HSP65 (clone J105G) was purchased from Thermo Fisher Scientific and used at 1:5,000 dilution. All reagents and media were purchased from Sigma-Aldrich unless otherwise stated.
Gene expression.
M. tuberculosis (H37Ra) was incubated under an in vitro stress medium for the indicated time points. PBS was used to induce starvation stress, 7H9 was adjusted to pH 5.4 for acidic stress, and 7H9 with 10 mM H2O2 was used for oxidative stress. RNA was extracted as previously described (55). RNA was confirmed to be free from mycobacterial genomic DNA (gDNA) contamination by PCR for rpoB, and cDNA was prepared using the Tetro RT cDNA synthesis kit as per the manufacturer’s instructions (Bioline, London). qRT-PCR was conducted using SYBR green master mix (Applied Biosystems), as per manufacturer’s instructions, in a 10-μl final volume containing 5 ng cDNA. PCR was conducted on a CFX Connect real-time PCR detection system (Bio-Rad). Primers were used at a final concentration of 0.3 μM, and sequences can be found in Table S2 in the supplemental material. A comparative threshold cycle (CT) method was used to determine relative gene expression compared to an endogenous control (rpoB) as previously described (56).
Flow cytometry analysis.
Cells for flow cytometry analysis were collected at indicated time points by trypsinization. Data were acquired using a BD Accuri 6 Plus flow cytometer. All data were analyzed using FlowJo software (Tree Star), and statistical analysis was conducted using GraphPad Prism.
Confocal microscopy imaging.
Macrophage assays were carried out as described above with RAW-LC3B-GFP cells adhered to glass coverslips. At 24 h postinfection, macrophages were washed with PBS and fixed in 4% paraformaldehyde for 24 h. Coverslips were then mounted in ProLong Gold with DAPI (Cell Signaling Technology) at 4°C overnight. Images were taken on a Zeiss laser scanning microscope 880 and analyzed with ImageJ software.
ELISA.
At indicated time points, cell culture supernatants were collected for cytokine analysis. BioLegend enzyme-linked immunosorbent assay (ELISA) Max Deluxe kits were used for IL-10, TNF-α, and IL-1β analysis as per manufacturer’s instructions. Briefly, plates were coated in capture antibody for 16 h and blocked in 5% nonfat milk for 1 h at room temperature. A total of 100 μl of standards and culture supernatant was added and incubated for 2 h at room temperature. The detection antibody was incubated for 1 h, followed by avidin-HRP for 30 min. TMB (3,3′,5,5′-tetramethylbenzidine) substrate was incubated for 20 min, and sulfuric acid was added to stop the reaction. Absorbance was read at 450 nm, and background absorbance (570 nm) was subtracted.
LDH assay.
At 20 h postinfection, cell culture supernatants were collected for LDH analysis. Supernatants were stored at −80°C until analysis. BioLegend LDH-Cytox assay kit was used as per the manufacturer’s instructions. Briefly, 50 μl of culture supernatants was added to 50 μl PBS in a 96-well plate. A total of 100 μl of working solution was added to wells and incubated at room temperature for 30 min. Fifty microliters of stop solution was added and each well and absorbance read to 490 nm. Cytotoxicity was calculated by subtracting the absorbance of untreated cells normalized to the absorbance of 100% lysed cells.
Mycobacterial fractionation and secretion assays.
Recombinant M. smegmatis strains were cultured in 7H9 medium to an OD600 of 0.6 to 0.8. Cultures were diluted to an OD600 of 0.3 and allowed to replicate once (5 h). Bacteria were resuspended in Sauton’s medium after washing 2 times with PBS and cultured for 16 h. Culture supernatant was collected, filtered (0.2 μm), and then concentrated 10 times by ultrafiltration in Amicon filters with a 10-kDa cutoff (Amicon). Bacterial cells were lysed by bead-beating with 1 mM silica zirconium beads in 0.05 M potassium phosphate and 0.02% β-mercaptoethanol. Cells were fractioned as previously described (57) and subcellular fractions analyzed by immunoblot for HA-tagged PE/PPE proteins.
Lipid extraction.
Polar and apolar lipid extractions were conducted as previously described (58). Briefly, the mid-exponential phase (OD600, 0.4 to 0.8) of bacterial cells was grown in 7H9 to extract apolar lipids by resuspending bacterial pellets in methanol/0.3% NaCl (10:1) and emulsifying with petroleum ether. Separated top layers (containing apolar lipids) were collected, and emulsions were repeated. Both petroleum ether fractions were combined and dried at 50°C. From the remaining lower layer, polar lipids were extracted by the addition of chloroform/methanol/0.3% NaCl (9:10:3) with vigorous shaking for 1 h. The supernatant was resuspended in chloroform/methanol/0.3% NaCl (5:10:4). Cells were pelleted and supernatant combined with first polar lipid extraction, and an equal volume of chloroform/0.3% NaCl (1:1) was added. The lower phase was recovered and dried at 60°C. Apolar and polar lipids were resuspended in chloroform at 5 mg/ml. Lipids were analyzed by TLC using aluminum-backed Silica 60 plates (Merck Millipore). Polar lipids were resolved in chloroform/methanol/water (20:4:0.5, vol/vol/vol) and visualized by charring with 10% copper sulfate in 8% phosphoric acid. Apolar lipids were resolved in petroleum ether/ethyl acetate (98:2, vol/vol, 3×) in the first dimension of the 2-dimensional TLC analysis and petroleum ether/acetone (98:2, vol/vol) as the solvent for the second dimension. Lipids were visualized by charring with ceric ammonium molybdate in 6% phosphoric acid.
Resazurin reduction microplate assay.
M. smegmatis was grown to mid-exponential phase (OD600, 0.4 to 0.8) in the presence of kanamycin and used to determine MIC90 by resazurin reduction as previously described (59). Briefly, M. smegmatis was diluted to an OD600 of 0.002 (4 × 105 bacilli/ml) in 7H9S (7H9 supplemented with 10% ADC (Albumin, Dextrose, Catalase), 0.05% glycerol, 0.05% Tween 80, and 1% tryptone, without kanamycin). Two-fold serial dilutions of the drug in 7H9S were added to each well of a 96-well microtiter plate. Diluted bacteria were added to each well, and plates were incubated for 48 h at 37°C. A total of 30 μl of 0.02% resazurin and 12.5 μl of 20% Tween 80 were added to each well and incubated at 37°C for 6 h. Sample fluorescence was measured using a BioTek Synergy H1 plate reader with an excitation of 530 nm and emission at 590 nm. Changes in fluorescence relative to wells with no inhibitors minus background fluorescence (media only wells) were plotted to determine MIC90.
Statistical analysis.
GraphPad Prism 8 was used for all analyses. Analysis of variance (ANOVA) was used to determine significance with Dunnett correction for multiple comparisons. A P value of <0.05 was considered to be significant.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institute of Allergy and Infectious Diseases (R01 AI127711). The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
We thank Maria Gonzalez Orozco, Jia Wang, and Rakel Arrazuria Fernandez (University of Texas Medical Branch, Galveston, Texas) for manuscript advice and helpful discussions.
E.J.S., K.L.J.S., and S.L. conceived the study. E.J.S., K.L.J.S., N.K.S., and T.W.N. designed and conducted experimental procedures. S.A.P. and S.L. interpreted data and supervised the study. E.J.S., S.A.P., and S.L. wrote the manuscript, which was read, edited, and approved in its final version by all of the authors.
We declare no competing financial interests.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.World Health Organization. 2019. Global tuberculosis report 2018. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Atesoh Awuh J, Flo TH. 2017. Molecular basis of mycobacterial survival in macrophages. Cell Mol Life Sci 74:1625–1648. doi: 10.1007/s00018-016-2422-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanley SA, Cox JS. 2013. Host-pathogen interactions during Mycobacterium tuberculosis infections. Curr Top Microbiol Immunol 374:211–241. doi: 10.1007/82_2013_332. [DOI] [PubMed] [Google Scholar]
- 4.Smith I. 2003. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin Microbiol Rev 16:463–496. doi: 10.1128/cmr.16.3.463-496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jagannath C, Lindsey DR, Dhandayuthapani D, Xu Y, Hunter RL Jr, Eissa NT. 2009. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med 15:267–276. doi: 10.1038/nm.1928. [DOI] [PubMed] [Google Scholar]
- 6.Knodler LA, Celli J. 2011. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol 13:1319–1327. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gutierrez MG, Master SS, Sing SB, Taylor GA, Colombo MI, Deretic V. 2004. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 8.Alonso S, Pethe K, Russell DG, Purdy GE. 2007. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc Natl Acad Sci U S A 104:6031–6036. doi: 10.1073/pnas.0700036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zullo AJ, Jurcic Smith KL, Lee S. 2014. Mammalian target of rapamycin inhibition and mycobacterial survival are uncoupled in murine macrophages. BMC Biochem 15:4. doi: 10.1186/1471-2091-15-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, Hutt J, Lyons CR, Dobos KM, Deretic V. 2012. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci U S A 109:E3168–E3176. doi: 10.1073/pnas.1210500109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris J. 2011. Autophagy and cytokines. Cytokine 56:140–144. doi: 10.1016/j.cyto.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 12.Harris J. 2013. Autophagy and IL-1 family cytokines. Front Immunol 4:83. doi: 10.3389/fimmu.2013.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris J, Keane J. 2010. How tumour necrosis factor blockers interfere with tuberculosis immunity. Clin Exp Immunol 161:1–9. doi: 10.1111/j.1365-2249.2010.04146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi C-S, Shenderov K, Huang N-N, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. 2012. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS. 2010. HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS One 5:e11733. doi: 10.1371/journal.pone.0011733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ní Cheallaigh C, Keane J, Lavelle EC, Hope JC, Harris J. 2011. Autophagy in the immune response to tuberculosis: clinical perspectives. Clin Exp Immunol 164:291–300. doi: 10.1111/j.1365-2249.2011.04381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park H-J, Lee SJ, Kim S-H, Han J, Bae J, Kim SJ, Park C-G, Chun T. 2011. IL-10 inhibits the starvation induced autophagy in macrophages via class I phosphatidylinositol 3-kinase (PI3K) pathway. Mol Immunol 48:720–727. doi: 10.1016/j.molimm.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 18.Shin D-M, Jeon B-Y, Lee H-M, Jin HS, Yuk J-M, Song C-H, Lee S-H, Lee Z-W, Cho S-N, Kim J-M, Friedman RL, Jo E-K. 2010. Mycobacterium tuberculosis Eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog 6:e1001230. doi: 10.1371/journal.ppat.1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan L, Yi M, Chen J, Li S, Chen W. 2016. Mycobacterium tuberculosis EIS gene inhibits macrophage autophagy through up-regulation of IL-10 by increasing acetylation of histone H3. Biochem Biophys Res Commun 473:1229–1234. doi: 10.1016/j.bbrc.2016.04.045. [DOI] [PubMed] [Google Scholar]
- 20.Saini NK, Baena A, Ng TW, Venkataswamy MM, Kennedy SC, Kunnath-Velayudhan S, Carreño LJ, Xu J, Chan J, Larsen MH, Jacobs WR Jr, Porcelli SA. 2016. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat Microbiol 1:16133. doi: 10.1038/nmicrobiol.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng W, Long Q, Zeng J, Li P, Yang W, Chen X, Xie J. 2017. Mycobacterium tuberculosis PE_PGRS41 enhances the intracellular survival of M. smegmatis within macrophages via blocking innate immunity and inhibition of host defense. Sci Rep 7:46716. doi: 10.1038/srep46716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chai Q, Wang X, Qiang L, Zhang Y, Ge P, Lu Z, Zhong Y, Li B, Wang J, Zhang L, Zhou D, Li W, Dong W, Pang Y, Gao GF, Liu CH. 2019. A Mycobacterium tuberculosis surface protein recruits ubiquitin to trigger host xenophagy. Nat Commun 10:1973. doi: 10.1038/s41467-019-09955-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu J, Laine O, Masciocchi M, Manoranjan J, Smith J, Du SJ, Edwards N, Zhu X, Fenselau C, Gao L-Y. 2007. A unique Mycobacterium ESX-1 protein co-secretes with CFP-10/ESAT-6 and is necessary for inhibiting phagosome maturation. Mol Microbiol 66:787–800. doi: 10.1111/j.1365-2958.2007.05959.x. [DOI] [PubMed] [Google Scholar]
- 24.MacGurn JA, Cox JS. 2007. A genetic screen for Mycobacterium tuberculosis mutants defective for phagosome maturation arrest identifies components of the ESX-1 secretion system. Infect Immun 75:2668–2678. doi: 10.1128/IAI.01872-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romagnoli A, Etna MP, Giacomini E, Pardini M, Remoli ME, Corazzari M, Falasca L, Goletti D, Gafa V, Simeone R, Delogu G, Piacentini M, Brosch R, Fimia GM, Coccia EM. 2012. ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 8:1357–1370. doi: 10.4161/auto.20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watson RO, Manzanillo PS, Cox JS. 2012. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry IIC, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream M-A, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 28.Bottai D, Brosch R. 2009. Mycobacterial PE, PPE and ESX clusters: novel insights into the secretion of these most unusual protein families. Mol Microbiol 73:325–328. doi: 10.1111/j.1365-2958.2009.06784.x. [DOI] [PubMed] [Google Scholar]
- 29.Abdallah AM, Gey van Pittius NC, DiGiuseppe Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CMJE, Appelmelk BJ, Bitter W. 2007. Type VII secretion - mycobacteria show the way. Nat Rev Microbiol 5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 30.Zullo AJ, Lee S. 2012. Mycobacterial induction of autophagy varies by species and occurs independently of mammalian target of rapamycin inhibition. J Biol Chem 287:12668–12678. doi: 10.1074/jbc.M111.320135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lerena MC, Colombo MI. 2011. Mycobacterium marinum induces a marked LC3 recruitment to its containing phagosome that depends on a functional ESX-1 secretion system. Cell Microbiol 13:814–835. doi: 10.1111/j.1462-5822.2011.01581.x. [DOI] [PubMed] [Google Scholar]
- 32.Cardenal-Muñoz E, Arafah S, López-Jiménez AT, Kicka S, Falaise A, Bach F, Schaad O, King JS, Hagedorn M, Soldati T. 2017. Mycobacterium marinum antagonistically induces an autophagic response while repressing the autophagic flux in a TORC1- and ESX-1-dependent manner. PLoS Pathog 13:e1006344. doi: 10.1371/journal.ppat.1006344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. 2006. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol 6:95. doi: 10.1186/1471-2148-6-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sampson SL. 2011. Mycobacterial PE/PPE proteins at the host-pathogen interface. Clin Dev Immunol 2011:497203. doi: 10.1155/2011/497203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh VK, Berry L, Bernut A, Singh S, Carrère-Kremer S, Viljoen A, Alibaud L, Majlessi L, Brosch R, Chaturvedi V, Geurtsen J, Drancourt M, Kremer L. 2016. A unique PE_PGRS protein inhibiting host cell cytosolic defenses and sustaining full virulence of Mycobacterium marinum in multiple hosts. Cell Microbiol 18:1489–1507. doi: 10.1111/cmi.12606. [DOI] [PubMed] [Google Scholar]
- 36.Iantomasi R, Sali M, Cascioferro A, Palucci I, Zumbo A, Soldini S, Rocca S, Greco E, Maulucci G, De Spirito M, Fraziano M, Fadda G, Manganelli R, Delogu G. 2012. PE_PGRS30 is required for the full virulence of Mycobacterium tuberculosis. Cell Microbiol 14:356–367. doi: 10.1111/j.1462-5822.2011.01721.x. [DOI] [PubMed] [Google Scholar]
- 37.Brennan MJ. 2017. The enigmatic PE/PPE multigene family of mycobacteria and tuberculosis vaccination. Infect Immun 85:e00969-16. doi: 10.1128/IAI.00969-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daleke MH, Ummels R, Bawono P, Heringa J, Vandenbroucke-Grauls CMJE, Luirink J, Bitter W. 2012. General secretion signal for the mycobacterial type VII secretion pathway. Proc Natl Acad Sci U S A 109:11342–11347. doi: 10.1073/pnas.1119453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Köster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, Grigsby SJ, Mittal E, Park HS, Jones V, Hsu F-F, Jackson M, Sassetti CM, Philips JA. 2017. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc Natl Acad Sci U S A 114:E8711–E8720. doi: 10.1073/pnas.1707792114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levine B, Mizushima N, Virgin HW. 2011. Autophagy in immunity and inflammation. Nature 469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deretic V. 2014. Autophagy in tuberculosis. Cold Spring Harb Perspect Med 4:a018481. doi: 10.1101/cshperspect.a018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh P, Rao RN, Reddy JRC, Prasad R, Kotturu SK, Ghosh S, Mukhopadhyay S. 2016. PE11, a PE/PPE family protein of Mycobacterium tuberculosis is involved in cell wall remodeling and virulence. Sci Rep 6:21624. doi: 10.1038/srep21624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delogu G, Pusceddu C, Bua A, Fadda G, Brennan MJ, Zanetti S. 2004. Rv1818c‐encoded PE_PGRS protein of Mycobacterium tuberculosis is surface exposed and influences bacterial cell structure. Mol Microbiol 52:725–733. doi: 10.1111/j.1365-2958.2004.04007.x. [DOI] [PubMed] [Google Scholar]
- 45.Målen H, De Souza GA, Pathak S, Søfteland T, Wiker HG. 2011. Comparison of membrane proteins of Mycobacterium tuberculosis H37Rv and H37Ra strains. BMC Microbiol 11:18. doi: 10.1186/1471-2180-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Q, Boshoff HIM, Harrison JR, Ray PC, Green SR, Wyatt PG, Barry IIC. 2020. PE/PPE proteins mediate nutrient transport across the outer membrane of Mycobacterium tuberculosis. Science 367:1147–1151. doi: 10.1126/science.aav5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korycka-Machała M, Pawełczyk J, Borówka P, Dziadek B, Brzostek A, Kawka M, Bekier A, Rykowski S, Olejniczak AB, Strapagiel D, Witczak Z, Dziadek J. 2020. PPE51 is involved in the uptake of disaccharides by Mycobacterium tuberculosis. Cells 9:603. doi: 10.3390/cells9030603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baker JJ, Abramovitch RB. 2018. Genetic and metabolic regulation of Mycobacterium tuberculosis acid growth arrest. Sci Rep 8:4168. doi: 10.1038/s41598-018-22343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Budzik JM, Swaney DL, Jimenez-Morales D, Johnson JR, Garelis NE, Repasy T, Roberts AW, Popov LM, Parry TJ, Pratt D, Ideker T, Krogan NJ, Cox JS. 2020. Dynamic post-translational modification profiling of M. tuberculosis-infected primary macrophages. Elife 9:e51461. doi: 10.7554/eLife.51461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. 2015. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528:565–569. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleinnijenhuis J, Oosting M, Plantinga TS, van der Meer JWM, Joosten LAB, Crevel RV, Netea MG. 2011. Autophagy modulates the Mycobacterium tuberculosis-induced cytokine response. Immunology 134:341–348. doi: 10.1111/j.1365-2567.2011.03494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stover CK, De La Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR Jr, Bloom BR. 1991. New use of BCG recombinant vaccines. Nature 351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 53.Jurcic Smith KL, Lee S. 2016. Inhibition of apoptosis by Rv2456c through nuclear factor κB extends the survival of Mycobacterium tuberculosis. Int J Mycobacteriol 5:426–436. doi: 10.1016/j.ijmyco.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klionsky D, Abeliovich H, Agostinis P, Agrawal D, Aliev G, Askew D, Baba M, Baehrecke E, Bahr B, Ballabio A, Bamber B, Bassham D, Bergamini E, Bi X, Biard-Piechaczyk M, Blum J, Bredesen D, Brodsky J, Brumell J, Brunk U, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin L, Choi A, Chu C, Chung J, Clarke P, Clark R, Clarke S, Clavé C, Cleveland J, Codogno P, Colombo M, Coto-Montes A, Cregg J, Cuervo A, Debnath J, Demarchi F, Dennis P, Dennis P, Deretic V, Devenish R, Di Sano F, Dice J, Difiglia M, Dinesh-Kumar S, Distelhorst C, et al. . 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autphagy 4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venkataraman B, Gupta N, Gupta A. 2013. A robust and efficient method for the isolation of DNA-free, pure and intact RNA from Mycobacterium tuberculosis. J Microbiol Methods 93:198–202. doi: 10.1016/j.mimet.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 56.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 57.Rezwan M, Lanéelle MA, Sander P, Daffé M. 2007. Breaking down the wall: fractionation of mycobacteria. J Microbiol Methods 68:32–39. doi: 10.1016/j.mimet.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 58.Slayden RA, Barry CE. 2001. Analysis of the lipids of Mycobacterium tuberculosis, vol 54 Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 59.Tran AT, Watson EE, Pujari V, Conroy T, Dowman LJ, Giltrap AM, Pang A, Wong WR, Linington RG, Mahapatra S, Saunders J, Charman SA, West NP, Bugg TDH, Tod J, Dowson CG, Roper DI, Crick DC, Britton WJ, Payne RJ. 2017. Sansanmycin natural product analogues as potent and selective anti-mycobacterials that inhibit lipid I biosynthesis. Nat Commun 8:14414. doi: 10.1038/ncomms14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bardarov S, Bardarov S Jr, Pavelka MS Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR Jr.. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.