

Abstract
Introduction
The triple-combination (TC) cystic fibrosis transmembrane conductance regulator (CFTR) modulator regimen elexacaftor, tezacaftor, and ivacaftor was shown to be safe and efficacious in phase 3 trials of people with cystic fibrosis (pwCF) ≥ 12 years of age with ≥ 1 F508del-CFTR allele. Here, a simulation study predicted ivacaftor, tezacaftor, and elexacaftor exposures and impacts on CFTR modulation following transition from ivacaftor [a cytochrome P450 3A (CYP3A) substrate], lumacaftor (a CYP3A inducer)/ivacaftor, or tezacaftor/ivacaftor to TC.
Methods
Physiologically based pharmacokinetic (PBPK) modeling was used to evaluate plasma exposures during transition from mono- or dual-combination CFTR modulator regimens to TC. PBPK models were parameterized using data from human hepatocytes to account for CYP3A induction by lumacaftor and validated to match clinical data from healthy volunteers and pwCF. Using dosing regimens for pwCF ≥ 12 years of age, simulations were performed for ivacaftor, lumacaftor/ivacaftor, and tezacaftor/ivacaftor dosing for 14 days followed by immediate transition to elexacaftor/tezacaftor/ivacaftor dosing for 14 days. Drug exposures during transitions were compared with respective half-maximal effective concentrations (EC50) estimated from efficacy endpoint data from clinical studies.
Results
In simulations of immediate transition from ivacaftor or tezacaftor/ivacaftor to TC, the preceding treatment had no impact on ivacaftor, tezacaftor, or elexacaftor exposures. In simulations of immediate transition from lumacaftor/ivacaftor to TC, ivacaftor exposure decreased to 64% of maximum effective concentration (EC), due to reduction in ivacaftor dose and residual CYP3A4 induction, then returned to 90–95% of maximum EC. Lumacaftor-mediated CYP3A induction resolved within approximately 2 weeks. In all simulations, ivacaftor, tezacaftor, and elexacaftor exposures approached steady state within 2 weeks following transition and, at all times, ivacaftor and ≥ 1 CFTR corrector remained above EC50.
Conclusion
PBPK modeling indicates that immediate transition to the elexacaftor/tezacaftor/ivacaftor regimen from an ivacaftor, lumacaftor/ivacaftor, or tezacaftor/ivacaftor regimen results in sustained CFTR modulation in pwCF ≥ 12 years of age.
Electronic supplementary material
The online version of this article (10.1007/s41030-020-00124-7) contains supplementary material, which is available to authorized users.
Keywords: CFTR modulator, Cystic fibrosis, Elexacaftor, Ivacaftor, Lumacaftor, Physiologically based pharmacokinetic modeling, Tezacaftor, Transition modeling, Triple combination
Key Summary Points
| Why carry out this study? |
| Some people with cystic fibrosis (pwCF) currently receiving cystic fibrosis transmembrane conductance regulator (CFTR) modulator regimens of ivacaftor, lumacaftor/ivacaftor, or tezacaftor/ivacaftor are transitioning to the triple-combination (TC) regimen of elexacaftor/tezacaftor/ivacaftor. |
| The impact of these transitions on CFTR modulator exposures, and whether adequate exposures to achieve clinical efficacy are maintained during transition, have not been directly addressed in clinical trials. |
| We used physiologically based pharmacokinetic (PBPK) modeling to evaluate whether CFTR modulation is sustained during the transition from ivacaftor, lumacaftor/ivacaftor, or tezacaftor/ivacaftor to the TC regimen; this study tested the hypotheses that (1) lumacaftor induction of cytochrome P450 3A (CYP3A) would resolve within 14 days after transitioning from lumacaftor/ivacaftor to TC and (2) that during all three transitions, the exposure of each CFTR modulator with ongoing or newly initiated dosing would stay above its half-maximal effective concentration (EC50) value during the transition. |
| What was learned from the study? |
| Lumacaftor-mediated CYP3A induction resolved within approximately 2 weeks; in all simulations, ivacaftor, tezacaftor, and elexacaftor exposures approached steady state within 2 weeks following transition and, at all times, ivacaftor and ≥ 1 CFTR corrector remained above EC50. |
| PBPK modeling indicates that immediate transition to the elexacaftor/tezacaftor/ivacaftor regimen from an ivacaftor, lumacaftor/ivacaftor, or tezacaftor/ivacaftor regimen results in sustained CFTR modulation in pwCF ≥ 12 years of age. |
Introduction
Cystic fibrosis (CF) is a life-shortening, multisystem disease caused by mutations in the CF transmembrane conductance regulator (CFTR) gene that lead to reduced quantity or function of the CFTR protein [1]. Small-molecule CFTR modulators include correctors (e.g., lumacaftor, tezacaftor, and elexacaftor) that improve CFTR processing and trafficking [2–4] and potentiators (e.g., ivacaftor) that increase CFTR channel-open probability [5]. In studies of participants with CF, clinical benefit was observed with ivacaftor in those 6 months of age and older with CFTR gating mutations [6–10] and in those 12 years of age and older heterozygous for the F508del-CFTR mutation and a residual function CFTR mutation (F/RF) [11]. Clinical benefit was observed with lumacaftor/ivacaftor in studies of participants with CF 2 years of age and older homozygous for the F508del-CFTR mutation (F/F genotype) [12–14]. Tezacaftor/ivacaftor also showed clinical benefit in studies of participants 6 years of age and older with F/F or F/RF genotypes [11, 15, 16].
The triple-combination (TC) regimen of elexacaftor, tezacaftor, and ivacaftor was shown to be highly efficacious in clinical studies of participants 12 years of age and older who were heterozygous for the F508del-CFTR mutation and a minimal function CFTR mutation (F/MF) [17] or who had the F/F genotype [18]. In participants with the F/F genotype, this TC regimen showed superior efficacy across all endpoints over tezacaftor/ivacaftor [18]. Results from the study of participants with F/MF genotypes showed that one copy of the F508del-CFTR allele is sufficient for TC to show strong efficacy [17]. Elexacaftor/tezacaftor/ivacaftor was approved by the US Food and Drug Administration in October 2019 to treat people with CF (pwCF) 12 years of age and older with ≥ 1 copy of the F508del-CFTR mutation [19].
Some pwCF currently receiving ivacaftor, lumacaftor/ivacaftor, or tezacaftor/ivacaftor are transitioning to this TC regimen. However, the impact of these transitions on CFTR modulator exposures, and whether adequate exposures to achieve clinical efficacy are maintained during transition, have not been directly addressed in clinical trials. Transition from lumacaftor/ivacaftor is of particular interest, as lumacaftor is a strong cytochrome P450 3A (CYP3A) inducer [20] and may lead to reduced exposures of drugs that are CYP3A substrates. Ivacaftor, tezacaftor, and elexacaftor are CYP3A substrates, with ivacaftor being a particularly sensitive CYP3A substrate [19, 21, 22]. Here, we used modeling and simulation to evaluate whether CFTR modulation is sustained during the transition period. This approach required integrating results from multiple models. First, qualified physiologically based pharmacokinetic (PBPK) models were used to simulate ivacaftor, lumacaftor, tezacaftor, and elexacaftor exposures during immediate transitions between these treatment regimens. Exposures during the transition periods were then compared to half-maximal effective concentration (EC50) values, as estimated from efficacy endpoint data (sweat chloride or percent predicted forced expiratory volume in 1 s) from clinical studies (data on file, unpublished) to determine impacts on CFTR modulation. This study tested the hypotheses that lumacaftor induction of CYP3A would resolve within 14 days after transitioning and that the exposure of each CFTR modulator with ongoing or newly initiated dosing would stay above its EC50 value during the transition.
Methods
Ivacaftor PBPK Model
The physiochemical properties of ivacaftor [e.g., permeability, blood-to-plasma ratio, plasma protein binding, logarithm of acid dissociation constant (pKa)] were obtained from internal sources. All ivacaftor PBPK model parameters are available in Supplementary Table S1. The PBPK base model for oral absorption of ivacaftor was developed using an advanced dissolution, absorption, and metabolism model (ADAM) in Simcyp™ version 16 (Certara). The absorption rate constant (ka) was predicted by the Simcyp™ built-in simulator using Caco-2 permeability values measured in vitro and further validated to capture clinical data. For distribution, an initial steady-state volume of distribution (Vss) of 1.89 L/kg was estimated by allometric scaling from three species (mouse, rat, and dog) and later optimized to 1.74 L/kg. Using this initial value, the Vss, single adjusted compartment volume of distribution (Vsac) and blood flow (SAC Q) were estimated by fitting clinical study data. Because ivacaftor is a sensitive substrate of CYP3A, and only approximately 3% parent ivacaftor was found in fecal samples in an in vivo human absorption, distribution, metabolism, and excretion (ADME) study, a fraction metabolism (Fm) value of greater than 95% was assigned to CYP3A. A bottom-up approach was used to model elimination of ivacaftor in Simcyp™. Mechanistic human liver microsome (HLM) kinetics data [maximum rate (Vmax); Michaelis–Menten constant (Km)] were estimated and further optimized to capture the clinical oral pharmacokinetic (PK) profile. Drug interaction parameters Ki (inhibitory constant) and fumic (fraction unbound in microsome) for CYP3A inhibition were obtained from in vitro experiments. The base model was validated against phase 1 clinical data (data on file, unpublished).
Lumacaftor PBPK Model
The physiochemical properties of lumacaftor (e.g., permeability, blood-to-plasma ratio, plasma protein binding, pKa) were obtained from internal sources. All lumacaftor PBPK model parameters are available in Supplementary Table S2. First-order absorption was used to model oral absorption of lumacaftor. Absorption parameters [fraction absorbed (Fa), ka, and lag time (Tlag)] were optimized based on clinical PK data. For distribution, Vss, Vsac, and SAC Q were estimated by fitting clinical trial data. Lumacaftor is not extensively metabolized, and the majority is excreted unchanged in feces [20]. Intrinsic clearance (CLint), additional hepatic clearance (HLM CLint), and additional systemic clearance were captured in the PBPK model using a built-in retrograde calculator. Lumacaftor is an inducer of CYP3A. Enzyme induction parameters used in Simcyp™ [maximum fold change in CYP3A4 mRNA (Indmax) and concentration at half-maximum induction (IndC50)] were obtained from in vitro experiments. PBPK models were parameterized using data from human hepatocytes to account for CYP3A induction by lumacaftor. The base model was validated against phase 1 clinical data (data on file, unpublished).
Tezacaftor PBPK Model
The physiochemical properties of tezacaftor (e.g., permeability, blood-to-plasma ratio, plasma protein binding, pka) were obtained from internal sources. Tezacaftor PBPK model parameters are available in Supplementary Table S3. The tezacaftor absorption model was developed similarly to the lumacaftor absorption model. Fa and ka were estimated based on clinical PK data for tezacaftor. For distribution, a minimal PBPK model was used, and Vss, Vsac, and SAC Q were estimated by fitting clinical study data. The Fm by CYP3A for tezacaftor was estimated to be 73.2% from the human ADME study. Therefore, the CYP3A CLint and HLM CLint were calculated using the built-in Simcyp™ retrograde calculator assuming that CYP3A accounts for 73.2% of the systemic oral clearance. The interaction parameters Ki and fumic were obtained from in vitro experiments. The base model was validated against phase 1 clinical data (data on file, unpublished).
Elexacaftor PBPK Model
The physiochemical and ADME properties of elexacaftor (e.g., permeability, blood-to-plasma ratio, plasma protein binding), as well as relevant clinical data, were obtained from internal sources. Elexacaftor PBPK model parameters are available in Supplementary Table S4. First-order absorption parameters ka, Fa, and Tlag were estimated using clinical PK data. The drug distribution parameters of the minimal PBPK model were first estimated by optimizing three distribution parameters [Vss, Vsac, and intercompartmental clearance (Q)] based on available clinical PK data obtained following intravenous dosing. Elexacaftor is primarily metabolized by CYP3A. The contribution of CYP3A metabolism (Fm) to the overall elimination of elexacaftor was estimated to be 67% for CYP3A; 33% of elexacaftor elimination occurs via other pathways. With this information, CYP3A CLint was back-calculated in Simcyp™ using a built-in retrograde calculator. Enzyme interaction parameters Ki and fumic were obtained from in vitro experiments. The base model was validated against phase 1 clinical data (data on file, unpublished).
Simulation Design
PK simulations were performed for the transition from ivacaftor, lumacaftor/ivacaftor, and tezacaftor/ivacaftor to the TC regimen using the previously developed and validated PBPK models for the four CFTR modulators (Supplementary Fig. 1). All simulations were performed with the default Sim-Healthy Volunteers population from the Simcyp™ virtual population library; the population was Caucasian and between 20 and 50 years of age. Information for each simulation is reported in Tables 1, 2, and 3. All simulations used ivacaftor, lumacaftor/ivacaftor, or tezacaftor/ivacaftor dosing regimens for pwCF 12 years of age and older for 14 days followed by the TC dosing regimen for pwCF 12 years of age and older for 14 days. PK parameters for independent PBPK modeling are reported in Supplementary Table S5. See Supplementary Methods for additional information.
Table 1.
Transition modeling simulation design for ivacaftor to elexacaftor/tezacaftor/ivacaftor
| Compound type | Substrate | Inhibitor 1 | Inhibitor 2 |
|---|---|---|---|
| Compound name | Ivacaftor | Elexacaftor | Tezacaftor |
| Route | Oral | Oral | Oral |
| Dose | 150 mg | 200 mg | 100 mg |
| Time of administration | 9:00 AM and 9:00 PM (days 1–28) | 9:00 AM (days 15–28) | 9:00 AM (days 15–28) |
| Regimen | Once every 12 h | Once daily | Once daily |
| Fasting/fed | Fed | Fed | Fed |
| Metabolite | NA | NA | NA |
| Population | Sim-healthy volunteersa | ||
| Trial number | 3 | ||
| Subject number | 3 | ||
| Gender ratio (female proportion) | 0.5 | ||
| Age (years) | 20–50 | ||
NA not applicable
aSimcyp™ version 16 (Certara)
Table 2.
Transition modeling simulation design for lumacaftor/ivacaftor to elexacaftor/tezacaftor/ivacaftor
| Compound type | Substrate | Inhibitor 1 | Inhibitor 2 | Inhibitor 3 | |
|---|---|---|---|---|---|
| Compound name | Ivacaftor | Lumacaftor | Elexacaftor | Tezacaftor | |
| Route | Oral | Oral | Oral | Oral | |
| Dose | 250 mg | 150 mg | 400 mg | 200 mg | 100 mg |
| Time of administration | 9:00 AM and 9:00 PM (days 1–14) | 9:00 AM and 9:00 PM (days 15–28) | 9:00 AM (days 1–14) | 9:00 AM (days 15–28) | 9:00 AM (days 15 to 28) |
| Regimen | Once every 12 h | Once every 12 h | Once daily | Once daily | |
| Fasting/fed | Fed | Fed | Fed | Fed | |
| Metabolite | NA | NA | NA | NA | |
| Population | Sim-healthy volunteersa | ||||
| Trial number | 3 | ||||
| Subject number | 3 | ||||
| Gender ratio (female proportion) | 0.5 | ||||
| Age (years) | 20–50 | ||||
NA not applicable
aSimcyp™ version 16 (Certara)
Table 3.
Transition modeling simulation design for tezacaftor/ivacaftor to elexacaftor/tezacaftor/ivacaftor
| Compound type | Substrate | Inhibitor 1 | Inhibitor 2 |
|---|---|---|---|
| Compound name | Ivacaftor | Elexacaftor | Tezacaftor |
| Route | Oral | Oral | Oral |
| Dose | 150 mg | 200 mg | 100 mg |
| Time of administration | 9:00 AM and 9:00 PM (days 1–28) | 9:00 AM (days 15–28) | 9:00 AM (days 1–28) |
| Regimen | Once every 12 h | Once daily | Once daily |
| Fasting/fed | Fed | Fed | Fed |
| Metabolite | NA | NA | NA |
| Population | Sim-healthy volunteersa | ||
| Trial number | 3 | ||
| Subject number | 3 | ||
| Gender ratio (female proportion) | 0.5 | ||
| Age (years) | 20–50 | ||
NA not applicable
aSimcyp™ version 16 (Certara)
Software
The PBPK analyses were performed using Simcyp™. All plots were generated by GraphPad Prism software, version 8.1.2.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Results
Ivacaftor to TC Transition
No change in the plasma drug exposure profile of ivacaftor during the transition from ivacaftor to the TC regimen was predicted (Fig. 1a); tezacaftor and elexacaftor exposures increased and reached steady state within 1 week (Fig. 1b, c).
Fig. 1.
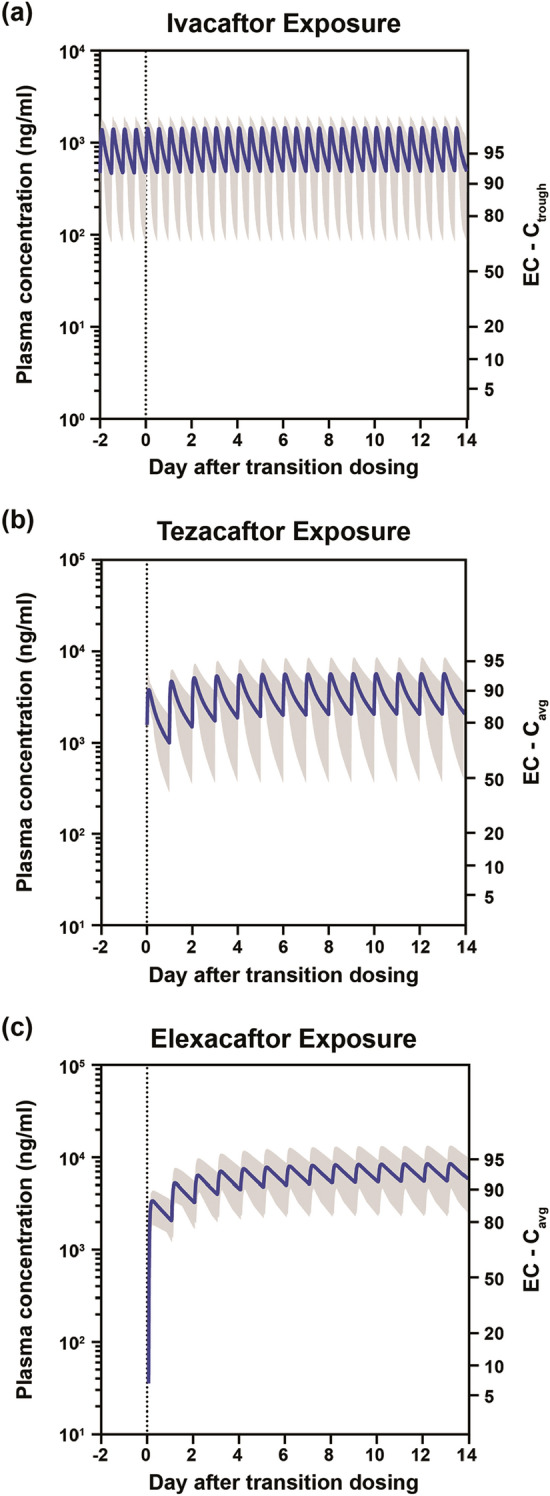
Plasma exposure of CFTR modulators after transition from ivacaftor to elexacaftor/tezacaftor/ivacaftor. a Ivacaftor exposure. b Tezacaftor exposure. c Elexacaftor exposure. In all figures, shaded area indicates 5th–95th percentile. Cavg average concentration, Ctrough trough concentration, EC effective concentration
Lumacaftor/Ivacaftor to TC Transition
Simulations showed that when transitioning from lumacaftor/ivacaftor (lumacaftor 400 mg and ivacaftor 250 mg once every 12 h) to the TC regimen (elexacaftor 200 mg once daily, tezacaftor 100 mg once daily, and ivacaftor 150 mg once every 12 h), ivacaftor plasma exposure transiently decreased upon immediate transition and then gradually increased to steady state on day 14 after the transition (Fig. 2a), decreasing to a nadir of 64% of maximum effective concentration (EC) and then increasing to 90–95% EC (EC90–95) within approximately 3 days. The exposure of lumacaftor decreased slowly and was completely eliminated on day 8 after discontinuation of lumacaftor/ivacaftor (Fig. 2b); CYP3A induction due to lumacaftor was predicted to resolve within approximately 2 weeks. The plasma exposures of tezacaftor and elexacaftor increased over time, reaching steady state on day 14 after transition (Fig. 2c, d); these exposures were maintained above EC50 at all times. Exposures of tezacaftor and elexacaftor were not greatly impacted by lumacaftor-mediated CYP3A induction during this transition. At all times during the transition period, exposures of ≥ 1 corrector (lumacaftor, tezacaftor, or elexacaftor) remained above their respective EC50.
Fig. 2.

Plasma exposures of CFTR modulators after transition from lumacaftor/ivacaftor to elexacaftor/tezacaftor/ivacaftor. a Ivacaftor exposure. b Lumacaftor exposure. c Tezacaftor exposure. d Elexacaftor exposure. In all figures, shaded area indicates 5th–95th percentile. Cavg average concentration, Ctrough trough concentration, EC effective concentration
Tezacaftor/Ivacaftor to TC Transition
When transitioning from tezacaftor/ivacaftor to the TC regimen, ivacaftor and tezacaftor exposures were maintained (Fig. 3a, b). The exposure of elexacaftor increased and reached steady state within 1 week (Fig. 3c).
Fig. 3.
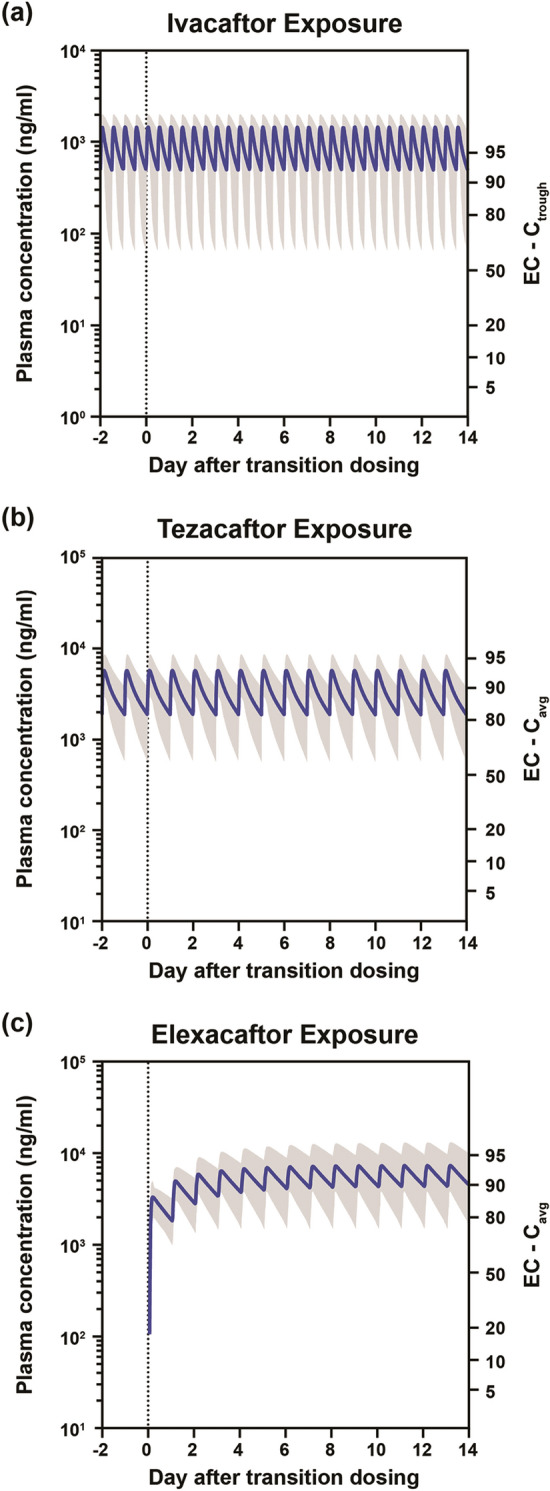
Plasma exposures of CFTR modulators after transition from tezacaftor/ivacaftor to elexacaftor/tezacaftor/ivacaftor. a Ivacaftor exposure. b Tezacaftor exposure. c Elexacaftor exposure. In all figures, shaded area indicates 5th–95th percentile. Cavg average concentration, Ctrough trough concentration, EC effective concentration
Discussion
Our PBPK models of ivacaftor, lumacaftor, tezacaftor, and elexacaftor were developed and validated in an adult healthy Caucasian volunteer population between 20 and 50 years of age. This virtual population was the most appropriate Simcyp™ population to use compared with other available virtual populations of differing races or functional renal or hepatic status because most pwCF are white [23, 24], and PK parameters are not expected to be substantially different between healthy volunteers and pwCF. Protein expression and enzyme activity of CYP3A are expected to be similar for all people 12 years of age and older, including those with CF [25]. Moreover, the differences between simulated plasma exposures in the virtual healthy population and those observed in pwCF 12 years of age and older were negligible, and the extent of drug–drug interactions is similar between adults and adolescents [26], indicating that the developed PBPK relationship can be applied to pwCF 12 years of age and older.
Using the PBPK modeling approach, we simulated transitions from ivacaftor, lumacaftor/ivacaftor, and tezacaftor/ivacaftor to the TC regimen elexacaftor/tezacaftor/ivacaftor to evaluate whether CFTR modulation was maintained during transitions between these CFTR modulator regimens. This question is particularly germane when transitioning from lumacaftor/ivacaftor, because lumacaftor is a strong CYP3A inducer and ivacaftor is a sensitive CYP3A substrate whose exposure is impacted by lumacaftor [20]. When transitioning from lumacaftor/ivacaftor to TC, pwCF lower their ivacaftor dose from 250 mg once every 12 h to 150 mg once every 12 h and discontinue lumacaftor. Following this switch in therapies, lumacaftor exposure is predicted to decrease and ivacaftor exposure is predicted to increase, reaching steady state on day 14 after the transition. During the transition, tezacaftor and elexacaftor were minimally impacted and remained above their respective EC50 throughout. Based on the simulations in this study, ivacaftor exposure is predicted to remain above EC50 throughout the transition, decreasing to a nadir of 64% of maximum EC and then increasing to EC90-95 within approximately 3 days. Lumacaftor-mediated CYP3A induction is predicted to resolve within approximately 2 weeks.
Tezacaftor and elexacaftor have low potential to inhibit or induce CYP3A [19]. In all simulated transitions, no change in ivacaftor exposure due to addition of tezacaftor and/or elexacaftor was predicted. Similarly, no change in tezacaftor exposure during the transition from tezacaftor/ivacaftor to TC was predicted. Finally, no change in the plasma exposure profile was predicted for elexacaftor after transition in any simulation.
Each individual CFTR modulator in the TC regimen has its own target EC. In these simulations, EC50 was chosen as the threshold above which CFTR modulator exposures should remain during the transition period; however, after transitioning, all modulators reached or returned to their target EC. EC50 is commonly used when analyzing drug exposure and was used in a previous analysis of simulated transitions between lumacaftor/ivacaftor and tezacaftor/ivacaftor [27]; during those simulated transitions, the CFTR modulators remained above EC50 and CFTR modulation was sustained. Keeping exposures above EC50 is important during transitions between treatments, because the more time that is spent above EC50, the more likely it is that the drug will show efficacy.
A limitation of this study is that all the simulations were based on available clinical data from study participants 12 years of age and older. Additional simulations will be needed to address transitions in the pediatric CF population due to the impact of CYP3A enzyme ontogeny, which typically occurs between 0 and 2 years of age, as well as potential differences in pediatric dosing regimens.
Conclusions
The PBPK modeling approach is useful for integrating all available in vitro and in vivo ADME and PK data to predict the impact of an immediate transition from ivacaftor, lumacaftor/ivacaftor, or tezacaftor/ivacaftor to elexacaftor/tezacaftor/ivacaftor on the exposures of those CFTR modulators in pwCF 12 years of age and older. Given the CYP3A-inducing effects of lumacaftor, these simulations predict that during all three of these transitions to the TC regimen, exposures of ivacaftor, tezacaftor, and elexacaftor will reach steady state within 2 weeks of the transition and CFTR modulation will be sustained.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Funding
This study was funded by Vertex Pharmaceuticals Incorporated, Boston, MA, USA. The journal’s Rapid Service Fee was funded by Vertex Pharmaceuticals Incorporated, Boston, MA, USA.
Medical Writing, Editorial, and Other Assistance
Editorial coordination and support were provided by Morgan Deng, PharmD, of Vertex Pharmaceuticals Incorporated; Morgan Deng may own stock or stock options in that company. Medical writing and editorial support were provided under the direction of the authors by Christopher Edwards, PhD, and Karen Kaluza Smith, PhD, CMPP, with support from Vertex Pharmaceuticals Incorporated.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Alice Tsai, Shu-Pei Wu, Eric Haseltine, Sanjeev Kumar, Samuel M. Moskowitz, Paul Panorchan, and Kushal Shah are employees of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. Alice Tsai, Shu-Pei Wu, Eric Haseltine, Sanjeev Kumar, Samuel M. Moskowitz, Paul Panorchan, and Kushal Shah have received nonfinancial support (assistance with manuscript preparation) from ArticulateScience LLC, which received funding from Vertex Pharmaceuticals Incorporated. Eric Haseltine: Pending patent for Compositions and Methods for Treatment of Cystic Fibrosis (WO 2019/010092, PCT/US2018/040427). Samuel M. Moskowitz: Pending patent for Methods of Treatment for Cystic Fibrosis; pending patent for Methods of Treatment of Cystic Fibrosis; pending patent for Pharmaceutical Compositions for Treating Cystic Fibrosis.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. Vertex Pharmaceuticals Incorporated is committed to advancing medical science and improving the health of people with cystic fibrosis. This includes the responsible sharing of clinical trial data with qualified researchers. Proposals for the use of these data will be reviewed by a scientific board. Approvals are at the discretion of Vertex Pharmaceuticals Incorporated and will be dependent on the nature of the request, the merit of the research proposed, and the intended use of the data. Please contact CTDS@vrtx.com if you would like to submit a proposal or need more information.
Open Access
This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
Footnotes
The original version of this article was revised: In section “Methods”, the word “physiochemical” was incorrectly changed to “physicochemical” in four instances.
Digital Features
To view digital features for this article go to 10.6084/m9.figshare.12666467.
Change history
8/27/2020
The original version of this article unfortunately contained a mistake.
References
- 1.Elborn JS. Cystic fibrosis. Lancet. 2016;388:2519–2531. doi: 10.1016/S0140-6736(16)00576-6. [DOI] [PubMed] [Google Scholar]
- 2.Van Goor F, Hadida S, Grootenhuis PDJ, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA. 2011;108:18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Goor F, Grootenhuis P, Hadida S, et al. Nonclinical profile of the CFTR corrector VX-661. Pediatr Pulmonol. 2016;51 [abstract 217].
- 4.Keating D, Marigowda G, Burr L, et al. VX-445–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379:1612–1620. doi: 10.1056/NEJMoa1807120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Goor F, Hadida S, Grootenhuis PD, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci USA. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies J, Wang LT, Panorchan P, et al. Ivacaftor (IVA) treatment in patients 6 to < 12 months old with cystic fibrosis with a CFTR gating mutation: results of a 2-part, single-arm, phase 3 study. J Cyst Fibros. 2019;18:S11 [abstract WS06-4].
- 7.Rosenfeld M, Wainwright CE, Higgins M, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to less than 24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med. 2018;6:545–553. doi: 10.1016/S2213-2600(18)30202-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies JC, Cunningham S, Harris WT, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med. 2016;4:107–115. doi: 10.1016/S2213-2600(15)00545-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365:1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Boeck K, Munck A, Walker S, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13:674–680. doi: 10.1016/j.jcf.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Rowe SM, Daines C, Ringshausen FC, et al. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med. 2017;377:2024–2035. doi: 10.1056/NEJMoa1709847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McNamara JJ, McColley SA, Marigowda G, et al. Safety, pharmacokinetics, and pharmacodynamics of lumacaftor and ivacaftor combination therapy in children aged 2–5 years with cystic fibrosis homozygous for F508del-CFTR: an open-label phase 3 study. Lancet Respir Med. 2019;7:325–335. doi: 10.1016/S2213-2600(18)30460-0. [DOI] [PubMed] [Google Scholar]
- 13.Ratjen F, Hug C, Marigowda G, et al. Efficacy and safety of lumacaftor and ivacaftor in patients aged 6–11 years with cystic fibrosis homozygous for F508del-CFTR: a randomised, placebo-controlled phase 3 trial. Lancet Respir Med. 2017;5:557–567. doi: 10.1016/S2213-2600(17)30215-1. [DOI] [PubMed] [Google Scholar]
- 14.Wainwright CE, Elborn JS, Ramsey BW, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373:220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PubMed] [Google Scholar]
- 15.Walker S, Flume P, McNamara J, et al. A phase 3 study of tezacaftor in combination with ivacaftor in children aged 6 through 11 years with cystic fibrosis. J Cyst Fibros. 2019;18:708–713. doi: 10.1016/j.jcf.2019.06.009. [DOI] [PubMed] [Google Scholar]
- 16.Taylor-Cousar J, Munck A, McKone EF, et al. Tezacaftor–ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377:2013–2023. doi: 10.1056/NEJMoa1709846. [DOI] [PubMed] [Google Scholar]
- 17.Middleton PG, Mall MA, Drevinek P, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381:1809–1819. doi: 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heijerman HGM, McKone EF, Downey DG, et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. 2019;394:1940–1948. doi: 10.1016/S0140-6736(19)32597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vertex Pharmaceuticals Incorporated . Trikafta (elexacaftor/tezacaftor/ivacaftor) [package insert] Boston: Vertex Pharmaceuticals Incorporated; 2019. [Google Scholar]
- 20.Vertex Pharmaceuticals Incorporated . Orkambi (lumacaftor/ivacaftor) [package insert] Boston: Vertex Pharmaceuticals Incorporated; 2018. [Google Scholar]
- 21.Vertex Pharmaceuticals Incorporated . Kalydeco (ivacaftor) [package insert] Boston: Vertex Pharmaceuticals Incorporated; 2019. [Google Scholar]
- 22.Vertex Pharmaceuticals Incorporated . Symdeko (tezacaftor/ivacaftor) [package insert] Boston: Vertex Pharmaceuticals Incorporated; 2019. [Google Scholar]
- 23.Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry: 2017 annual data report. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2017-Patient-Registry-Annual-Data-Report.pdf. Accessed June 17, 2020.
- 24.O'Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891–1904. doi: 10.1016/S0140-6736(09)60327-5. [DOI] [PubMed] [Google Scholar]
- 25.Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45:931–956. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 26.Salem F, Johnson TN, Barter ZE, Leeder JS, Rostami-Hodjegan A. Age related changes in fractional elimination pathways for drugs: assessing the impact of variable ontogeny on metabolic drug-drug interactions. J Clin Pharmacol. 2013;53:857–865. doi: 10.1002/jcph.100. [DOI] [PubMed] [Google Scholar]
- 27.Wu S, Garg V, Tsai A, et al. Sustained CFTR correction and potentiation is predicted during transitions between lumacaftor/ivacaftor and tezacaftor/ivacaftor-based regimens. Pediatr Pulmonol. 2017;52 [abstract 253].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. Vertex Pharmaceuticals Incorporated is committed to advancing medical science and improving the health of people with cystic fibrosis. This includes the responsible sharing of clinical trial data with qualified researchers. Proposals for the use of these data will be reviewed by a scientific board. Approvals are at the discretion of Vertex Pharmaceuticals Incorporated and will be dependent on the nature of the request, the merit of the research proposed, and the intended use of the data. Please contact CTDS@vrtx.com if you would like to submit a proposal or need more information.