

Abstract
G protein-coupled receptors (GPCRs) are the most common class of therapeutic targets, accounting for ~35% of all FDA-approved drugs. Cancer patients receive numerous medications not only to combat cancer but also to alleviate pain, nausea, and anxiety, many of which target GPCRs. Emerging evidence has implicated GPCRs as drivers of cancer progression, therapeutic resistance, and metastasis. Therefore, the effects of commonly prescribed GPCR-targeted drugs must be reevaluated in the context of cancer. Epidemiological and experimental evidence indicate that widely used GPCR-targeted drugs may promote or inhibit cancer progression. It is crucial that we more fully understand the indirect effects of GPCR-targeted drugs on the cancer phenotype. This review summarizes recent advances in characterizing these interactions and highlights future research opportunities.
GPCR Signaling and the Cancer Phenotype
GPCRs are a diverse class of seven-transmembrane proteins (Box 1) that regulate a myriad of biological processes, including immune response and neurotransmission, and are frequently dysregulated in disease states. Approximately 35% of drugs approved by the US Food and Drug Administration (FDA) target GPCRs for indications as varied as pain, allergy, hypertension, and neuropsychiatric disorders [1]. Therapeutic evaluation of these drugs was performed to determine the efficacy for these indications, where the putative effect on cancer progression was not a primary consideration. However, GPCR modulation can significantly impact on the cancer phenotype.
Box 1. How Drugs Modulate GPCR Signaling.
GPCRs are commonly expressed on the cell surface but can also signal from intracellular membranes. These receptors activate intracellular signaling pathways when ligand binds to the receptor. When an agonist (Figure I) binds to a GPCR this produces a conformational change that promotes the conversion of guanine nucleotides bound to linked heterotrimeric G proteins from GDP to GTP, leading to activation of the G protein. The Gα subunit of the G protein then dissociates from the β and γ subunits (Figure II, modeled after [2]). Depending on the type of Gα subunit (Gαs, Gαi, Gαq/11, Gα12/13) linked to a GPCR, this elicits a specific downstream signaling pathway. Gαs activates adenylyl cyclase, increasing the levels of the second messenger cAMP, thus promoting the activity of protein kinase A (PKA). Gαi inhibits adenylyl cyclase, decreasing cAMP levels and PKA activity. Gαq/11 activates phospholipase C (PLC), increasing levels of the second messengers diacylglycerol (DAG), inositol triphosphate (IP3), and calcium, thus promoting the activity of protein kinase C (PKC). Gα12/13 promotes the recruitment of Rho guanine nucleotide exchange factors (Rho-GEFs) [2]. The βγ dimer can also form protein–protein interactions and promote downstream signaling independently of the Gα subunit. β-Arrestins are recruited to active GPCRs and elicit distinct downstream signals. Drugs that are partial agonists activate a signaling pathway, but not as effectively as a full agonist (Figure I). Conversely, when an antagonist drug binds to a GPCR, this suppresses the signaling pathway promoted by the agonist, often by preventing the agonist from binding. Biased agonists promote activation of an alternative downstream signaling pathway, often in a β-arrestin-dependent manner. Inverse agonists bind to the agonist binding site but elicit the opposite response. Owing to the complex nature of GPCR signaling, small-molecule drugs can modulate a diverse array of downstream signaling pathways, many of which are altered in disease states.
Figure I. Drug–Receptor Activity Terms.
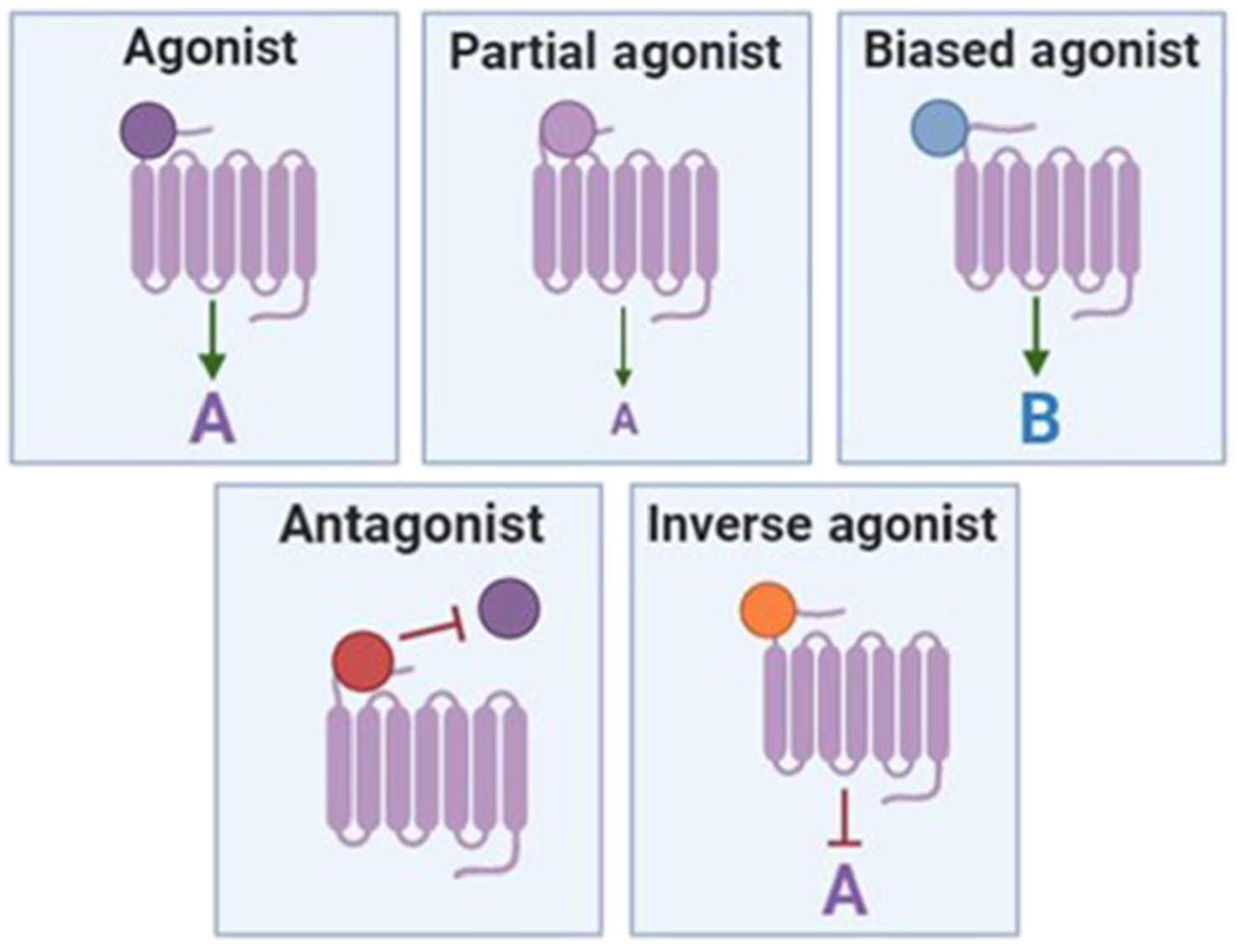
GPCR-targeted drugs can modulate GPCR signaling by activating the canonical signaling pathway (agonists, partial agonists) or alternative downstream signaling pathways (biased agonists). Antagonists and inverse agonists inhibit GPCR signaling. Abbreviation: GPCR, G protein-coupled receptor.
Figure II. GPCR Signaling.
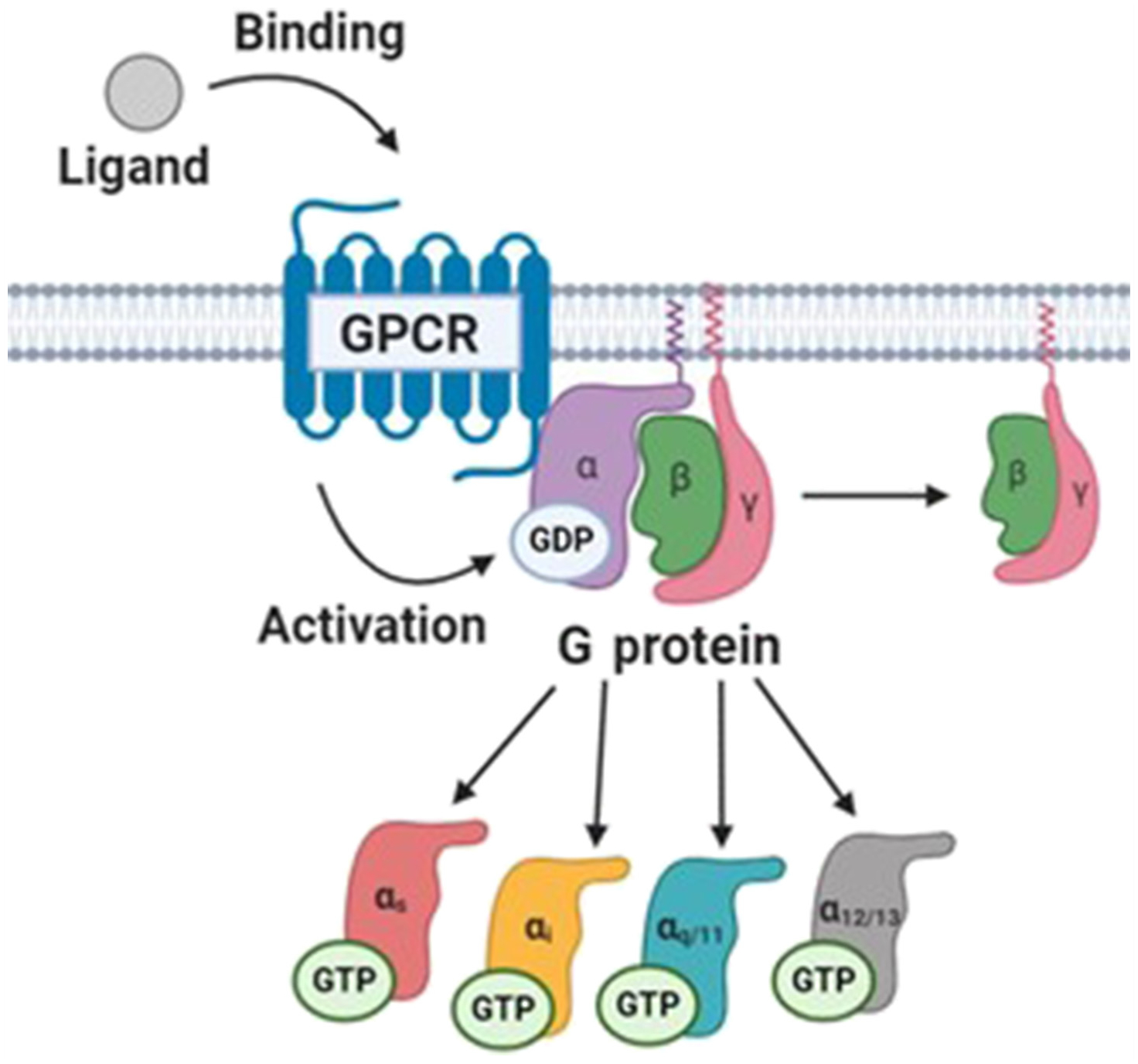
GPCR signaling occurs when a ligand binds to the receptor and produces a conformational change that either activates or inhibits the activity of heterotrimeric G proteins. G proteins then regulate specific downstream signaling pathways depending on the type of Gα subunit linked to a GPCR. Abbreviation: GPCR, G protein-coupled receptor.
Cancer patients are prescribed many medications for cancer treatment and palliative care, as well as to treat comorbid conditions, many of which target GPCRs. Recent reviews have highlighted the broad roles of GPCR/G-protein signaling in cancer (Box 2) [2–4]. However, there have been no attempts to integrate how commonly prescribed drugs targeting these pathways impact on cancer patient outcome. This review summarizes recent literature regarding how specific GPCR-targeted drugs commonly prescribed to cancer patients, and the receptors they target, influence the cancer phenotype (Table 1). Owing to space constraints, we highlight recent illustrative examples, most often in the context of pancreatic cancer, and discuss their broader implications. The goal of this review is to encourage the reader to consider the unintended effects of commonly prescribed medications and to spur efforts to undertake related clinically relevant research questions (Figure 1, Key Figure). We focus on four major drug classes commonly prescribed to cancer patients: cardiovascular, antiemetic and analgesic, anti-inflammatory, and neuropsychiatric drugs.
Box 2. Role of GPCRs in Cancer.
GPCRs and G proteins, that mediate the downstream signaling of GPCRs, are frequently dysregulated in cancer as a result of mutations, copy-number alterations, or modifications of methylation or gene expression [3]. GPCRs can directly affect cancer-related processes by upregulating oncogenic signaling pathways, such as PI3K–AKT–mTOR, Hippo, and MAPK, or indirectly by altering the function of oncogenes or tumor-suppressors [109]. GPCRs also play important roles in the transactivation of other cell-surface receptors that have established roles in cancer, such as the epidermal growth factor receptor [3]. GPCRs and GPCR-targeting drugs can modify a multitude of cancer-related processes such as tumor growth, immunity, angiogenesis, metastasis, and tumor–stroma crosstalk [1]. Thousands of papers have been published regarding the role of GPCRs in cancer (Figure I). As a result, GPCRs are emerging as potential therapeutic targets in cancer, and several anticancer agents that target GPCRs are in clinical development [110]. For example, ONC201 inhibits the GPCR dopamine receptor D2, and is currently in Phase II clinical trials for various solid cancers such as endometrial cancer (NCT03099499, NCT03485729) and neuroendocrine tumors (NCT03034200). In addition, GPCR-targeted drugs have been FDA-approved as anticancer therapeutics, including the Smoothened inhibitors vismodegib and sonidegib. Patients with basal cell carcinoma are prescribed these drugs to block Hedgehog signaling, a crucial pathway driving this malignancy. Hormonal and endocrine cancers are also commonly treated with a variety of GPCR-targeted drugs [111]. Cabergoline, a dopamine 1 receptor agonist, is used to treat neuroendocrine and pituitary cancers. Degarelix, a gonadotropin-releasing hormone receptor antagonist, is used to treat prostate cancer. As our understanding of GPCR action in cancer improves, there will likely be an increase in the number of GPCR-targeted drugs that are approved as anticancer agents.
Figure I. PubMed Search Query Results for Each GPCR and Cancer from 1977 to 2020.
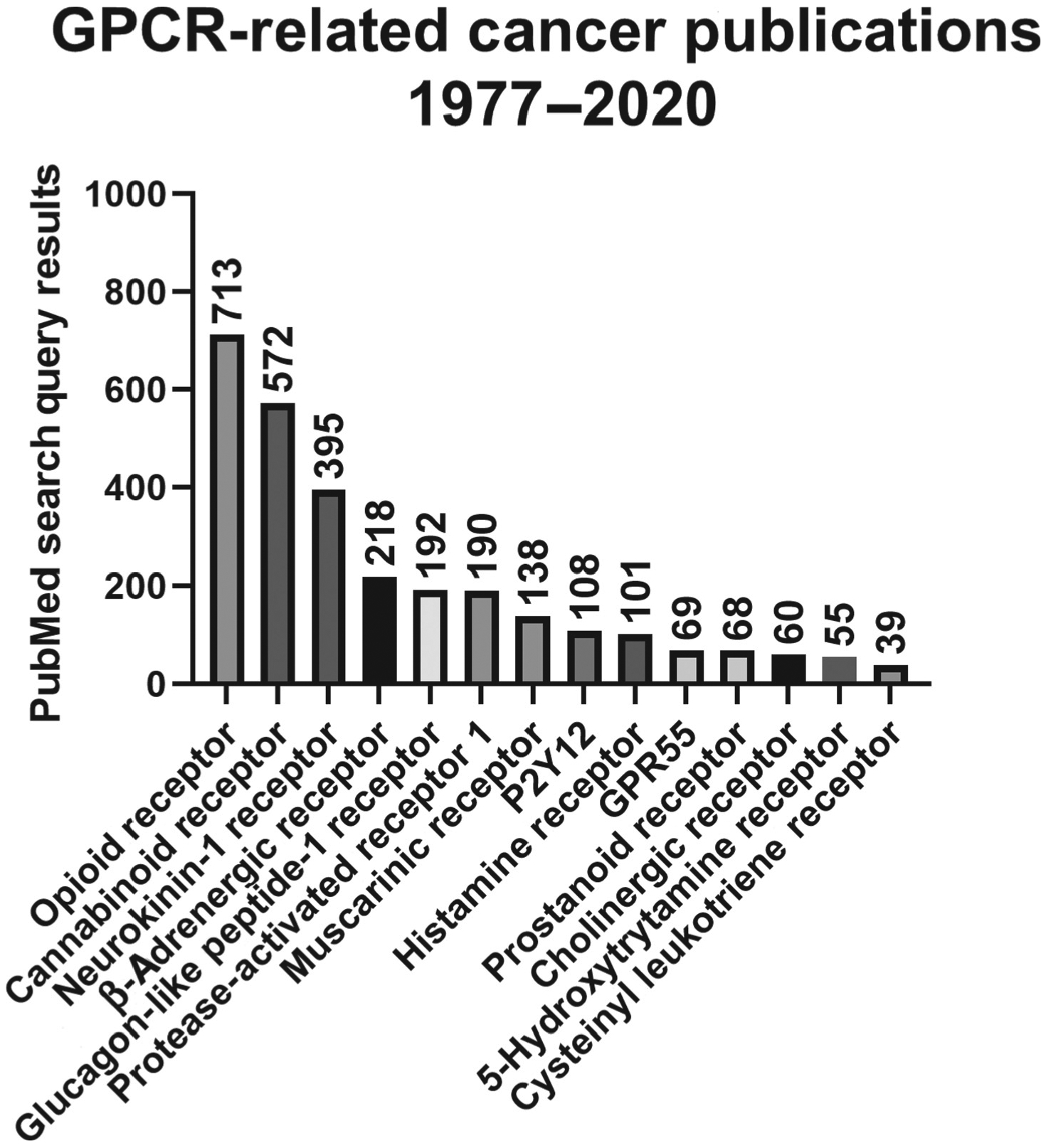
Some GPCRs have been heavily researched in the context of cancer, whereas the role of others is newly established. Abbreviation: GPCR, G protein-coupled receptor.
Table 1.
Classes of GPCR-Targeted Drugs Discussed in This Review
| Class | Medication | Primary indications | Primary GPCR target | Number of cancer-related PubMed search results (August 2020) | Mostly antitumorigenic (+), or both pro- and antitumorigenic (+/−) |
|---|---|---|---|---|---|
| Cardiac | β-Blockers | Hypertension, arrhythmia, anxiety, migraine headaches | β-Adrenergic receptors | 334 | + |
| Vorapaxar | Thrombosis, history of myocardial infarction or peripheral arterial disease | Protease-activated receptor 1 (PAR1) | 15 | + | |
| Clopidogrel | Risk of myocardial infarction or stroke | P2Y12 | 241 | + | |
| Aspirin | Chemoprevention, fever, arthritis, risk of myocardial infarction or stroke | Prostanoid receptors | 4375 | + | |
| Antiemetic/analgesic | Neurokinin-1 receptor antagonists | Nausea, vomiting | Neurokinin 1 (NK-1R) receptors | 131 | + |
| Dronabinol/THC | Nausea, vomiting, anorexia, appetite stimulant | GPR55, cannabinoid receptors 1 and 2 (CB1/CB2) | 344/361 | +/− | |
| Cannabinoids | Anxiety, nausea, appetite stimulant | GPR55, cannabinoid receptors 1 and 2 (CB1/CB2) | 1009 | +/− | |
| Opioids | Moderate to severe pain | μ, δ, and κ opioid receptors | 8286 | +/− | |
| Anti-Inflammatory | Leukotriene receptor antagonists (LTRAs) | Asthma, allergies | Cysteinyl leukotriene receptors | 12 | + |
| Antihistamines | Allergies, cold and flu symptoms, motion sickness Acid reflux, stomach ulcers | Histamine H1 receptor (HRH1) HRH2 | 152 | +/− | |
| Antidiabetic | Glucagon-like peptide 1 agonists | Type II diabetes mellitus | Glucagon-like peptide 1 (GLP-1) receptors | 20 | +/− |
| Parasympathetic | Muscarinic/cholinergic agonist/antagonists | Glaucoma, bladder/gastrointestinal disorders, chronic obstructive pulmonary disease | Cholinergic receptor muscarinic 1–5 (CHRM 1–5) | 27/31 | +/− |
| Neuropsychiatric | Selective serotonin reuptake inhibitors (SSRIs) | Depression, anxiety, post-traumatic stress disorder, obsessive compulsive disorder | 5-Hydroxytryptamine (5-HT) receptors | 110 | +/− |
| Antiemetic dopamine antagonists | Nausea, schizophrenia, bipolar disorder | Dopamine receptors 1–5 (D1R–D5R) | 9 | +/− |
Figure 1. Key Figure. GPCR-Targeted Drugs Indirectly Modulate the Cancer Phenotype.
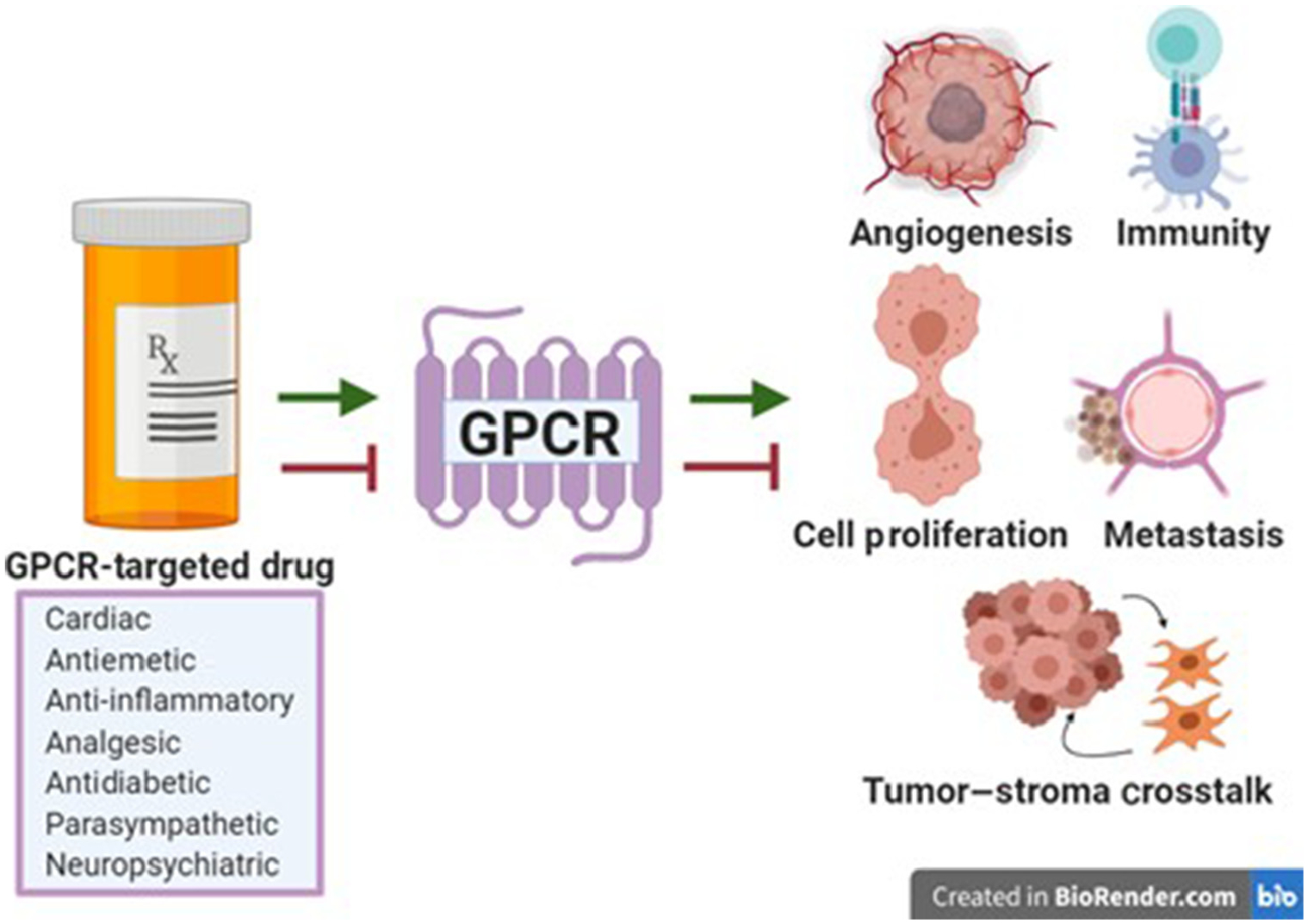
Many drug classes target GPCRs. This review addresses cardiac, antiemetic, anti-inflammatory, analgesic, antidiabetic, parasympathetic, and neuropsychiatric drugs that either promote or inhibit GPCR signaling, and subsequently influence aspects of the cancer phenotype, including angiogenesis, immunity, cell proliferation, metastasis, and tumor–stroma crosstalk. Abbreviation: GPCR, G protein-coupled receptor.
Cardiac Medications
10–30% of cancer patients have pre-existing cardiovascular disease, and this number is projected to increase owing to rising obesity rates and an aging population [5,6]. In addition, cancer patients frequently develop cardiovascular complications as a direct result of the cancer or as a side effect of cancer treatment [7–9]. Consequently, these patients are prescribed cardiac medications such as β-adrenergic receptor antagonists (β-blockers) and antiplatelet medications.
β-Blockers
β-Blockers are commonly prescribed to treat conditions such as hypertension, tachycardia, anxiety, and migraine headaches [10]. These drugs are antagonists of β-adrenergic receptors (β1AR, β2AR, β3AR) – GPCRs activated by catecholamines (see Glossary) that regulate the sympathetic nervous system. In multiple cancer types, stress promotion stimulates β2-adrenergic signaling, resulting in alterations to the tumor microenvironment and profoundly impacting on therapeutic response. Under chronic stress conditions, pancreatic cancer, which is highly innervated, activates the sympathetic nervous system and subsequently increases catecholamine production, leading to stimulation of β-adrenergic receptors and increasing cell proliferation, invasiveness, and proangiogenic signaling [11–13]. Recent evidence indicates that inhibition of β2-adrenergic signaling can improve chemotherapeutic efficacy and inhibit pancreatic cancer development and progression. Increased β2-adrenergic signaling promoted the overexpression of nerve growth factor, resulting in increased nerve density and accelerated disease progression [11]. Pharmacological blockade of β2-adrenergic signaling improved the efficacy of gemcitabine chemotherapy treatment in patient-derived tumor organoids and in genetically engineered tumor-bearing KPC mice [11]. In addition, pharmacological inhibition and knockout of β2AR in KC (without a p53 mutation) mice reduced the incidence of pancreatic ductal adenocarcinoma in the presence of chronic restraint stress [11].
Similarly, in prostate cancer mouse models, cancer growth and angiogenesis were dependent on crosstalk between β2-adrenergic receptors on endothelial cells and noradrenaline secreted by adrenergic nerves. Growth of orthotopically implanted PC-3 cells in immunocompromised mice with whole body β2AR and β3AR receptor knockout resulted in significantly smaller tumors with decreased blood vessel density, branching, and length [14]. These studies suggest that non-selective β-blockers may improve prostate and pancreatic cancer survival. In fact, a retrospective cohort study using the Swedish Cancer Register indicated that β-blocker usage is associated with decreased pancreatic cancer-specific mortality relative to non-users [15]. However, β-blockers may not be ideal anticancer drugs in pancreatic cancer owing to the high prevalence of diabetes in these patients [16]. A 2018 prospective cohort study found that all-cause mortality was significantly increased in diabetic individuals taking β-blockers [17]. Information regarding the role of GPCR-targeted antidiabetic medications in cancer is given in Box 3.
Box 3. GPCR-Targeting Diabetic Drugs Impact on the Cancer Phenotype.
Approximately 9% of the US population have diabetes mellitus and are prescribed antidiabetic medications [112]. Diabetes is a chronic illness characterized by insufficient insulin production (type 1) or insulin responsiveness (type 2), resulting in elevated levels of glucose in the bloodstream. Many cancer types undergo alterations in metabolism, known as the Warburg effect, resulting in increased dependence on glucose for energy production. Increased glucose levels in the bloodstream of diabetics may aid cancer growth. Epidemiological studies indicate that diabetes is associated with increased cancer risk and cancer progression. For example, ~50% of pancreatic cancer patients develop new-onset diabetes before receiving a pancreatic cancer diagnosis [16]. Antidiabetic medications such as glucagon-like peptide 1 (GLP-1) receptor agonists alter cancer metabolism, but the impact of these medications on the cancer phenotype is still being elucidated [113].
The GLP-1 receptor agonists liraglutide and exenatide agonize the GLP-1 receptor, a GPCR that regulates blood sugar and insulin levels. GLP-1 modulation can alter cancer growth and early cancer development. Exenatide treatment inhibits the development of diet-induced and chemically induced murine models of HCC by promoting apoptosis and decreasing diet-induced inflammation, fibrosis, and lipid accumulation [114]. Thus, diabetic patients with HCC may benefit from receiving GLP-1 receptor agonists, especially if they are obese or consume high-fat diets. Similarly, the GLP-1 receptor agonist exendin-4 decreased the growth of prostate and ovarian cancer cells injected subcutaneously into immunocompromised mice [115,116]. Additional studies will be necessary to assess alterations in cancer growth relative to other antidiabetic drugs. This is crucial because in the context of some cancer types GLP-1 agonists play protumorigenic roles. For example, exendin-4 treatment increased intestinal polyp formation in a genetically engineered colon cancer mouse model, suggesting caution in GLP-1 receptor agonist use in diabetic individuals with a genetic predisposition to develop colon cancer [117]. In humans, some clinical studies suggest that these drugs may increase the risk of pancreatitis and pancreatic cancer [118]. However, a recent meta-analysis of 12 GLP-1 receptor agonist trials did not observe an increased risk of pancreatic cancer in GLP-1 receptor agonist users [119]. In addition, long-term treatment of mice, rats, and monkeys with liraglutide did not promote pancreatitis or pancreatic cancer [120]. Although GLP-1 receptor agonists have pleiotropic roles in cancer, the cause of these differences is unknown. Differences in GLP-1 receptor expression and metabolism profiles between tumor types may drive the observed differences [121].
β-Blockers are likely promising anticancer agents in other cancer types. In stress-induced murine breast cancer and melanoma immunocompetent xenograft models, β2-adrenergic signaling blockade, using both pharmacological and genetic methods, reduced tumor growth. In addition, the non-selective β-blocker propranolol improved the efficacy of the immune checkpoint inhibitor, anti-PD-1, in these xenograft models by promoting CD8+ T cell activity in a β-adrenergic receptor-dependent manner [18]. This was further substantiated by a retrospective study of clinical data, which found that pan-β-blocker use was associated with improved survival in metastatic melanoma patients treated with immunotherapy [19]. In breast cancer, the impact of β-blockers on immunotherapeutic efficacy in the clinic is unknown, but retrospective studies indicate that β-blockers improve relapse-free survival on chemotherapy and reduce the risk of metastasis, recurrence, and cancer-specific mortality [20–22]. The survival benefits of breast cancer patients receiving chemotherapy can likely be attributed to tumor-specific actions of these drugs and to a reduction in cardiac-related side effects [23].
Overall, these findings suggest that blocking β2-adrenergic signaling leads to a more favorable tumor microenvironment that is more responsive to both chemotherapy and immunotherapy. However, a 2017 meta-analysis of breast cancer prognosis with β-blockers indicated no survival benefit [24]. This is likely due to the inability to control for comorbid conditions in retrospective studies; for example, β-blocker users are likely to suffer from higher rates of cardiac-related fatalities. It has also been speculated that the positive effect of β-blockers on cancer patient survival is an artifact of immortal time bias [25]. Prospective studies with placebo-controlled conditions are underway to determine the anticancer efficacy of β-blockers (clinical trial numbers NCT02013492, NCT04005365; https://clinicaltrials.gov/).
Antiplatelet Medications
Antiplatelet medications such as vorapaxar, clopidogrel, and aspirin prevent blood coagulation and are prescribed to individuals with a history or risk of developing coronary artery disease, stable angina, or acute ischemic stroke [26]. The role of antiplatelet drugs in cancer prevention, including clinical trials, has been extensively reviewed [27]. This section focuses on the impact of antiplatelet drugs vorapaxar and clopidogrel on the cancer phenotype. Owing to the indirect role of aspirin, an irreversible cyclooxygenase 1/2 inhibitor, in inhibiting signaling by GPCR prostanoid receptors, it was excluded from this review.
Experimental and clinical evidence indicate that crosstalk between platelets and cancer cells promotes proliferative signaling, angiogenesis, immune evasion, and metastasis [28]. Recent studies indicate that vorapaxar, an antagonist of the GPCR protease-activated receptor 1 (PAR1), can decrease metastasis, protumorigenic tumor–stroma crosstalk, and cancer cell proliferation. PAR1 knockout in KPC-derived tumor cells significantly impaired tumor growth (orthotopic and subcutaneous) and metastatic spread (tail vein lung metastasis assay) following tumor cell injection into C57/BL6 mice. This was due to alterations in tumor–immune cell crosstalk, particularly by promoting CD8+ T cell activity via a PAR1/thrombin-dependent signaling pathway [29]. Similarly, colon cancer tumors grew significantly slower in PAR1 knockout mice, suggesting that stromal PAR1 aids colon cancer growth [30]. PAR1 expression in breast cancer cells induced a hormone-refractory, metastatic breast cancer phenotype similar to advanced basal-like breast cancer. In fact, tail vein injection of MCF-7 cell lines stably expressing PAR1 promoted metastatic colonization to the lungs of nude mice, whereas PAR1 null cells did not [31]. Factors secreted from platelets such as thrombin, can also modify PAR1 signaling on tumor cells. Vorapaxar decreased the proliferation of epithelial ovarian cancer cells in a thrombin-dependent manner [32]. PAR1 may also modulate metastatic potential by altering stem-like cell activity. In vitro and in vivo studies indicated that PAR1 increases stem-like cell proliferation of glioma cells and early engraftment of acute myeloid leukemia cells [33,34]. Overall, these studies indicate that crosstalk between tumor cells and platelets, or the clotting factor thrombin, can promote tumor cell proliferation, stem cell activity, and metastasis, and can also modify tumor–immune cell crosstalk in a PAR-1 dependent manner.
Clopidogrel also plays important roles in altering tumor growth and the immune landscape. The active metabolite of clopidogrel covalently binds to the adenosine diphosphate (ADP) binding site on the purinergic GPCR P2Y12 located on platelets, preventing the activation of a glycoprotein complex essential for platelet aggregation [35]. Both pharmacological inhibition of P2Y12 and P2Y12 knockout in an immunocompetent murine ovarian cancer model decreased tumor growth in the peritoneum in a platelet-dependent manner [36]. This finding supports previous research indicating that platelets aid ovarian cancer proliferation [37]. In melanoma, P2Y12 receptor expression on CD163+ tumor-associated macrophages (TAMs) is associated with increased TAM migration and chemokine secretion. In addition, ADP, the ligand for P2Y12, is secreted during melanoma cell death, and likely functions as a homing signal to direct the migration of TAMs to tumor tissue. Targeting TAMs will aid in promoting a more favorable immune microenvironment, and future studies should therefore explore the consequences of therapeutic inhibition of P2Y12 on immunotherapeutic response [38]. Collectively, these findings indicate that inhibition of platelet coagulation is therapeutically beneficial for cancer patients, and that GPCR-targeted antiplatelet drugs can decrease metastasis, angiogenesis, and cancer cell growth while promoting a more favorable immune microenvironment [28].
Antiemetic and Analgesic Medications
Pain, nausea, and vomiting are common symptoms experienced by cancer patients as a direct or indirect result of the cancer, treatment side effects, or comorbid conditions. Nearly 70% of cancer patients experience nausea and vomiting, and up to 96% experience pain [39]. Antiemetic/analgesic medications such as neurokinin-1 (NK-1) receptor (NK-1R) antagonists, dronabinol, cannabinoids, and opioids are frequently prescribed to cancer patients and can significantly impact on the cancer phenotype.
NK-1R Antagonists
Antagonists of the GPCR NK-1R, including aprepitant and rolapitant, block the action of the protumorigenic neuropeptide, substance P (SP), reducing nausea. NK-1R activation on both tumor and endothelial cells has protumorigenic activity in multiple cancer types. NK-1R antagonists inhibit angiogenesis [40], tumor growth [41], perineural invasion and metastatic signaling [42], and decrease the proliferation of many cancer cell lines by promoting apoptosis and cell-cycle arrest [43,44].
The role of NK-1R in hematological malignancies is not well explored. NK-1R is significantly overexpressed in acute myeloid leukemia (AML), and an NK-1R antagonist promoted apoptosis in cell line and xenograft models [43]. In addition, NK-1R antagonists decreased the proliferation of acute lymphoblastic leukemia cells, supporting the possibility that NK-1R antagonists may be repurposed to treat hematological malignancies [45]. There is extensive evidence for upregulation of the SP/NK-1R signaling pathway in solid cancers. SP/NK-1R signaling promotes cancer cell development, proliferation, angiogenesis, and metastasis via autocrine, paracrine, or endocrine mechanisms [46,47]. In addition, NK-1R antagonism attenuates early tumorigenesis and inhibits angiogenesis. Precursor lesions of pancreatic cancer, a cancer with a highly innervated tumor microenvironment, overexpress NK-1R, thus promoting nerve–tumor cell crosstalk and aiding early tumorigenesis [48]. In mouse models of hepatoblastoma, NK-1R antagonists were effective in decreasing tumor volume and inhibiting angiogenesis [40]. Moreover, NK-1R antagonists were effective antinociceptive agents in a chronic myeloid leukemia bone pain mouse model, suggesting that these agents could reduce multiple side effects of cancer in addition to slowing tumor growth [43]. Overall, these studies support a broad role for NK-1R antagonists as anticancer agents across multiple cancer types. Epidemiological studies will be necessary to determine whether patients taking NK-1R antagonists have better outcomes in the clinic.
Dronabinol/Cannabinoids
Dronabinol is an FDA-approved drug used as an antiemetic and an appetite stimulant. This drug is a synthetic form of Δ9-tetrahydrocannabinol (THC) and is an agonist of the GPCR cannabinoid receptors 1 and 2 (CBD-1, CBD-2) and GPR55 [49]. In addition to this FDA-approved drug, the use of cannabis and cannabinoids in the treatment of pain, nausea, anxiety, and appetite loss is a topic of debate in oncology. Equally debated is the role of these drugs in impacting on the cancer phenotype. A 2016 review of all preclinical mouse models studying the impact of the endocannabinoid system on cancer found that the majority of cancer types, including glioma, colon cancer, and skin cancer, demonstrated either a reduction in tumor size, decreased metastasis, or improved survival [50]. For example, cannabinoid use in colon cancer mouse models decreased polyp formation, aberrant crypt foci formation, tumor growth, angiogenesis, and chemotherapy-induced cardiotoxicity. In pancreatic cancer, cannabidiol (CBD) had no effect as a single agent, but significantly improved the efficacy of gemcitabine treatment in immunocompetent tumor-bearing KPC mice [51]. This response was probably because of CBD antagonism of GPR55, a receptor that is known to promote pancreatic cancer growth and metastasis [51]. In gastric cancer, CBD slowed cancer cell growth and proliferation by promoting apoptosis in immunocompromised mice and gastric cancer cell lines, respectively [52].
However, several studies in animal models have observed a protumorigenic role for cannabinoids. THC promoted the growth of human papilloma virus (HPV)-positive head and neck squamous cell carcinoma cells subcutaneously implanted into nude mice. Inverse agonists of CBD-1 and CBD-2 as well as shRNA knockdown of these receptors inhibited the growth of this xenograft model, suggesting that cannabinoid receptors promote the proliferative phenotype [53]. In liver cancer, immunodeficient mouse models indicated that cannabinoids reduced tumor growth by activating either peroxisome proliferator-activated receptor γ (PPARγ), GPR55, or autophagy-dependent pathways. However, in chemically induced mouse models of hepatocellular carcinoma (HCC), tumor development in CBD-1 knockout mice and pharmacological inhibition of CBD-1 significantly inhibited tumor incidence and progression, respectively, suggesting that, in the context of an intact immune system, cannabinoids may be harmful for HCC patients [54]. Collectively, these studies indicate that cannabinoids play both tumor-suppressive and tumor-promoting roles in cancer, and may improve chemotherapeutic efficacy, depending on the type of cannabinoid prescribed, cannabinoid receptor expression within the tumor, dosing scheme, and subsequent immune modulation.
Unfortunately, no clinical study has been completed to determine the anticancer activity of cannabinoids. Lessons from the weight-loss drug rimonabant, a CBD-1 antagonist, provide an excellent example why further research is needed before these drugs continue to be widely accepted by the oncology community. Rimonabant was released in Europe as an antiobesity drug; despite being effective, it was banned by the FDA as a result of unintended increases in suicidal thoughts, nausea, depression, and anxiety [55]. Similarly in cancer, the desirable effects in palliative care should be balanced by the potentially negative effects that these drugs could have on cancer progression. Because these drugs are already in widespread use [56], both experimental and clinical studies are of crucial importance.
Opioids
Opioids are the primary treatment for moderate to severe cancer pain [39]. Morphine, codeine, and fentanyl are agonists or partial agonists of the GPCR μ-opioid receptors (MORs). Opioid usage may negatively or positively impact on cancer patients by altering immune function, cancer progression, metastasis, angiogenesis, and recurrence [57]. These contradictory findings can be attributed to numerous factors, including peripheral versus central actions of the drug, the duration of administration, dose-dependent differences in activity, the route of administration, and differences in opioid metabolism between mice and humans [58]. In addition, opioids can act on numerous cell types in the tumor microenvironment, including immune cells, endothelial cells, cancer stem cells, and tumor cells. Opioids have established roles in modifying many aspects of innate and adaptive immunity in both humans and experimental models, including a reduction in T cell viability, an increase in anti-inflammatory cytokine levels, and decreases in macrophage number and phagocytic activity. However, the impact of the combination of these factors on cancer patients is difficult to predict [59]. In addition, the impact of morphine usage on immunotherapy response in cancer patients is important to decipher and needs to be evaluated both experimentally and epidemiologically.
Opioids may negatively impact on HCC patients. MOR is overexpressed on HCC cancer stem-like cells and is associated with increased tumor growth and promotion of cancer stem cell activity via nuclear factor of activated T cells (NFAT) signaling, and its expression is associated with decreased survival in the clinic. In addition, knockdown of MOR in patient-derived HCC cells implanted subcutaneously into nude mice slowed tumor growth [60]. Similarly, morphine enhanced the stemness and invasiveness of BT-549 breast cancer cells, and subsequently promoted breast cancer growth in immunocompromised mice [61]. However, in breast cancer, morphine has been shown to play positive and negative roles depending on the cellular context. In vitro studies suggest that morphine may favorably modulate macrophage activity, resulting in decreased production of the proinvasive matrix metalloprotease MMP-9 and decreased secretion of proangiogenic factors [62,63]. The apparent differential role of morphine in breast cancer and other cancers may be due to the immunocompetence of the selected models and differences in dosing schemes.
In addition to modulating tumor cells and macrophages, morphine promotes fibrosis and inflammatory signaling [64,65]. Morphine treatment after caerulein induction increases the severity of acute pancreatitis in C57BL/6 mice by promoting necrosis and inflammation, and delaying tissue regeneration in a MOR-dependent manner [66]. Alterations in inflammation and fibrosis, as well as the promotion of acute pancreatitis, suggest that these medications could influence pancreatic cancer progression. Pancreatic cancers are highly innervated, thus promoting significant levels of pain, and the impact of these drugs on pancreatic cancer patients needs to be directly assessed.
Collectively, these findings indicate that opioids can influence the cancer phenotype, but the actual impact of these drugs on cancer patients is unclear. There is also a lack of consensus regarding clinical studies to assess the impact of opioids on cancer patient survival. In some retrospective studies, reduction of opioid usage in the perioperative period through the use of alternative analgesia has been associated with improved cancer patient survival [67]. However, opioid sparing or alternative analgesia in other retrospective studies did not impact on patient survival [68,69]. Extensive research has been conducted regarding the impact of medications taken in the perioperative period on cancer patient outcomes, which is outside the scope of this review [70]. In addition, a retrospective analysis found that high MOR expression and higher doses of opioids were associated with significantly shortened overall survival and progression-free survival of metastatic prostate cancer patients [71]. These results may be confounded by the fact that terminally ill patients are more likely to have higher pain and to be prescribed higher doses of opioids [57]. It is imperative that additional clinical trials are conducted to dissect the effects of opioids on cancer patient outcome. Unfortunately, unrelieved pain can also worsen patient outcome; these studies can therefore only be performed in comparison to other forms of analgesia, and there are few alternative pain relief options with similar efficacy to opioids [70].
Anti-Inflammatory Medications
Cancer development and progression are highly influenced by inflammation. Approximately 15–20% of all cancer development is promoted by inflammatory conditions [72]. For example, pancreatitis significantly increases the risk of developing pancreatic cancer [73]. Consequently, one of Hanahan and Weinberg’s hallmarks of cancer is tumor-promoting inflammation [74]. After cancer has developed, inflammation continues to play important roles in remodeling the tumor microenvironment to favor cancer growth, inhibit antitumorigenic immune responses, and aid metastasis [72]. Therefore, the use of anti-inflammatory drugs as anticancer and chemopreventive agents has been heavily researched.
Anti-inflammatory medications are commonly prescribed to treat conditions such as allergies, asthma, and atopic dermatitis. In the USA, ~8% of adults aged 57–85 years, an age group with an increased risk of cancer development, are prescribed allergy medications and 8% are prescribed asthma medications [75]. Intriguingly, epidemiological studies suggest that individuals with asthma or nasal allergies have a reduced risk of developing pancreatic cancer independently of the medications prescribed, suggesting innate differences in the immune systems of these patients [76]. This section summarizes the roles of the anti-inflammatory drugs, leukotriene receptor antagonists, and antihistamines on the cancer phenotype.
Leukotriene Receptor Antagonists (LTRAs)
LTRAs, including montelukast and zafirlukast, block the GPCR cysteinyl leukotriene (CysLT1) receptor 1, reducing proinflammatory signaling pathways associated with asthma. The ability of LTRAs to reduce inflammatory signaling suggests that these drugs may be repurposed for chemoprevention or anticancer therapies. The role of LTRAs in chemoprevention was recently assessed in a retrospective nationwide epidemiological study in Taiwan. LTRA use was associated with a significant decrease in cancer incidence in a dose-dependent manner (hazard ratio 0.31; 95% CI 0.24–0.39) [77]. In addition, inhibitors of other components of the leukotriene pathway, such as 5-lipoxygenase inhibitors, reduced the development and progression of chemically induced murine lung cancer [78]. 5-Lipoxygenase inhibitors also inhibited the development of chemically induced oral squamous cell carcinoma in hamsters, and they had an additive effect when combined with COX-2 inhibitors, indicating that targeting multiple pathways involved in arachidonic acid metabolism may inhibit cancer development [79]. The role of LTRAs in cancer progression is not well characterized, but recent studies indicate that these drugs play antitumorigenic roles by decreasing stemness, angiogenic signaling, and tumor growth. Montelukast downregulated cancer stem-cell marker expression and inhibited the growth of colon cancer cells subcutaneously injected into nude mice [80]. A novel CysLT1 antagonist, Q8, was recently identified as a potent angiogenic inhibitor in zebrafish. Q8 improved the efficacy of the antiangiogenic drug bevacizumab by decreasing endothelial cell migration and tube formation in vitro [81]. In medulloblastoma cells, leukotriene levels are upregulated and promote hedgehog pathway signaling, a known driver of this malignancy. Conversely, MK886, a 5-lipoxygenase inhibitor, selectively inhibited the growth of murine medulloblastoma cells subcutaneously injected into CB17/SCID mice, supporting the potential role of therapeutically targeting leukotriene biosynthesis in medulloblastoma patients [82]. Additional studies will be necessary to determine the impact of LTRAs on cancer progression in immunocompetent mouse models. Overall, LTRAs, although relatively unexplored, may attenuate cancer development and progression.
Antihistamines
There are two classes of antihistamine drugs. Antihistamines such as diphenhydramine block the GPCR histamine H1 receptor (HRH1) by either antagonism or inverse agonism. HRH1 is expressed on endothelial cells, immune cells, nerves, and epithelial cells where these receptors regulate vasodilation, bronchoconstriction, swelling, and inflammation [83]. The second class of antihistamines, including cimetidine, antagonize the histamine H2 receptor (HRH2). HRH2 antagonists regulate gastric acid secretion and are used to treat heartburn and ulcers. The role of histamine signaling in influencing the cancer phenotype remains a topic of debate. Recent research indicates that blockade of HRH1 or HRH2 decreases fibrotic signaling, inhibits metastasis, and slows cancer progression. H1- and H2-targeted antihistamines decreased fibrosis and hepatic stellate cell activation in Mdr2−/− mice, a model of primary sclerosing cholangitis (PSC), supporting the use of antihistamines as a chemopreventive agent in individuals with PSC. Furthermore, athymic nude mice subcutaneously injected with cholangiocarcinoma cells treated with either H1- or H2-targeted antihistamines displayed decreased tumor growth, angiogenic marker expression, and epithelial to mesenchymal transition (EMT) marker expression [84]. Similarly, hepatocellular carcinomas overexpress HRH1, and this correlates with decreased patient survival. In vitro knockdown and in vivo pharmacological inhibition of HRH1 attenuated hepatocellular carcinoma cell growth and metastasis [85]. However, HRH1/2 antagonism may promote a more unfavorable immune tumor microenvironment in some cancer types. Histamine significantly improved responsiveness to anti-PD-1 and anti-PD-L1 combination therapy by depleting myeloid-derived suppressor cells in subcutaneous colon cancer and lymphoma cell allograft mouse models [86]. The protumorigenic and antitumorigenic roles of histamine may reflect different expression levels of histamine receptors in different cancer types, as well as in other cell types within the tumor microenvironment.
Antihistamines are therapeutically beneficial in the clinic. For example, some populations of colorectal cancer patients have benefitted from post-surgical HRH2 antagonists. In a randomized clinical trial, colorectal cancer patients undergoing curative surgical resection receiving ranitidine intraoperatively and for 5 years post-surgery had improved survival relative to non-users if they did not receive a perioperative blood transfusion or develop complications from a post-surgical infection [87]. Collectively, these findings indicate that antihistamines could improve patient survival, but additional studies will be necessary to better understand the protumorigenic and antitumorigenic roles of different histamine receptors in each cancer type [83].
Neuropsychiatric Medications
Neuropsychiatric medications are commonly prescribed to cancer patients to treat depression and to relieve chemotherapy-induced nausea and vomiting. Depression affects approximately one in five patients with cancer, and these patients are frequently prescribed antidepressants [88]. Depression in cancer patients is related to poor prognosis, cancer treatment, or tumor-intrinsic aspects of the disease such as altered cytokine pools [88]. In addition, antipsychotic drugs used for treating schizophrenia and bipolar disorder are used as antiemetics in the cancer setting [89].
Antidepressants
Many antidepressants frequently prescribed to cancer patients, such as selective serotonin reuptake inhibitors (SSRIs), modulate serotonin GPCR signaling. SSRIs, including sertraline, increase brain serotonin levels by blocking serotonin reuptake and subsequent degradation. In addition to the impact of these drugs on serotonin signaling in the brain, these medications can alter serotonin signaling in tumors, impacting on patient survival.
A recent population-based cohort study in the UK assessed the impact of SSRIs on breast cancer patient survival. They found that individuals using SSRIs had 27% higher mortality relative to non-users [90]. The degree of risk may have been inflated by confounding factors such as the use of other antidepressants and prior history of depression. Mechanistically, SSRIs such as paroxetine can increase levels of the protumorigenic factor prolactin in human plasma, promoting breast cancer progression [91]. Similarly, SSRI use by epithelial ovarian cancer patients was associated with shorter time to disease progression [92]. The authors further found that sertraline promoted the growth of ovarian cancer cells orthotopically injected into athymic nude mice. It should be noted, however, in the context of other cancer types, that SSRIs may be protective owing to the role of these drugs in decreasing stress levels. The impact of stress reduction on cancer progression is a rapidly growing area of research [93,94] that is outside the scope of this review.
In liver cancer and pancreatic cancer, serotonin appears to promote disease progression. Serum levels of serotonin are higher in patients with HCC versus patients with cirrhosis [95]. In addition, serotonin is a hepatocyte mitogen to aid liver regeneration, and serotonin receptor inhibition with an 5HTR2B antagonist slowed the growth of HCC cells subcutaneously injected into athymic mice, suggesting that SSRIs may play an antitumorigenic role in HCC [96–98]. However, it is unknown how SSRIs impact on serotonin levels in tumor tissue. If SSRIs increase serotonin levels within tumors, these medications would likely be protumorigenic. Similarly, pancreatic cancer tissue has increased serotonin expression relative to normal pancreatic tissue. Increasing serotonin levels in the media of pancreatic cancer cell lines promoted cell proliferation and glycolysis, and inhibited apoptosis, in part by promoting PI3K–Akt–mTOR signaling [99]. More broadly, serotonin promotes profibrotic, angiogenic, and anti-inflammatory signaling in macrophages [100,101]. Studies in tryptophan hydrolase 1 knockout mice, a model of serotonin deficiency, indicate that serotonin signaling in macrophages promotes subcutaneous colon cancer cell growth by blocking the production of angiostatin, an angiogenic inhibitor, thus promoting tumor vascularization [101]. By continuing to bridge the gap between the epidemiological and experimental evidence regarding the impact of serotonin signaling and SSRIs on the cancer phenotype, it will be possible to determine the global implications of SSRIs on patient outcome. In addition, it is vital that the influence of SSRIs on the tumor microenvironment, particularly in modulating serotonin levels, is evaluated. The impact of SSRIs on chemotherapeutic drug metabolism must also be considered [102].
Antipsychotics
Dopamine receptor antagonists are a class of antipsychotic drugs that have antiemetic properties. These drugs, including thioridazine, fluphenazine, and prochlorperazine, primarily target the GPCR D2 dopamine receptor (D2R). In various cancer types, D2R antagonists decrease cancer stemness and promote autophagy. An unbiased screen of small-molecule drugs on normal and human pluripotent stem cells (hPSCs) identified thioridazine as a drug which specifically differentiated neoplastic hPSCs [103]. In a follow-up screen, thioridazine, fluphenazine, and prochlorperazine reduced the proliferation of AML blast cells without impacting on healthy hPSCs, suggesting that D2R antagonists could be repurposed to treat AML and that D2R may serve as a biomarker for cancer stem cells [103]. Similarly, thioridazine inhibited the growth of glioblastoma cells subcutaneously injected into NOD/SCID mice by depleting glioblastoma stem cells and inducing autophagy [104]. The D2R antagonists penfluridol and sertindole suppressed cancer growth and cell proliferation by promoting autophagy-mediated apoptosis in pancreatic cancer and breast cancer, respectively [105,106]. In addition, sertindole inhibited the migration and invasion of breast cancer cells in vitro [106]. These studies support a protective role for these medications in cancer patients. In fact, ONC201, a small-molecule D2R antagonist, is currently undergoing Phase II clinical trials for various solid cancers (Box 2). Epidemiological studies and further clinical testing with ONC201 will be necessary to determine the relationship between D2R antagonists and cancer patient survival.
Concluding Remarks and Future Perspectives
This review has summarized important interactions between several classes of GPCR-targeted drugs commonly prescribed to cancer patients and the impact these drugs have on the cancer phenotype. These effects include directly modulating tumor cell growth, interactions with the microenvironment, and altering the therapeutic efficacy of immunotherapies, chemotherapies, and targeted therapies. Owing to space constraints, many GPCR-targeted therapies whose primary targets are known to influence the cancer phenotype were excluded from this review, including parasympathetic medications (Box 4) and the HIV antiviral drug maraviroc, an antagonist of the chemokine receptor CCR5 [1]. Because GPCRs regulate a multitude of cancer-associated processes, there has been intense interest in repurposing GPCR-targeted drugs as anticancer agents. However, this may come with undesirable effects related to the primary drug indication. For example, β-blockers can cause cardiac-related side effects in individuals without cardiac conditions such as hypotension and bradycardia [107]. In addition, prolonged use of GPCR agonists can cause receptor desensitization, complicating dosing strategies [108]. Thus, it is crucial to more fully explore the effects of GPCR-targeted drugs currently being taken by cancer patients with the goal of improving survival and treatment efficacy (see Outstanding Questions). By understanding common GPCR pathways that are known to alter the growth and development of particular cancer types, new therapeutic targets may be identified that more specifically target cancer cells. Finally, by continuing to bridge the gap between epidemiological studies that assess the impact of these drugs on patient outcome, and experimental studies that assess the impact of these drugs on the cancer phenotype, we can begin to develop a comprehensive understanding of how commonly prescribed GPCR-targeted drugs impact on cancer etiology and treatment response.
Box 4. GPCR-Targeting Parasympathetic Medications and the Cancer Phenotype.
Cancer patients may receive either muscarinic agonists or muscarinic antagonists (anticholinergic) that modulate the parasympathetic nervous system. These drugs are used to treat a wide array of comorbid conditions such as bladder disorders and gastrointestinal disorders. These drug classes target muscarinic and cholinergic GPCRs.
Muscarinic agonists and cholinergic agonists either selectively or non-selectively activate muscarinic acetylcholine receptors (CHRM1–5). Conversely, muscarinic and cholinergic antagonists inhibit the activity of these receptors. Activation of muscarinic receptors can promote or inhibit tumorigenesis by modulating cancer stem cells, cellular signaling, and tumor–nerve crosstalk in a tissue-specific manner. In pancreatic cancer, muscarinic agonist drugs decreased stemness, slowed pancreatic cell/organoid proliferation, and improved survival in both tumor-bearing KPC mice and a PANC02 cell line liver metastasis model, in a CHRM1-dependent manner. Muscarinic antagonists such as scopolamine produced the opposite effect [122].
In other cancer types, muscarinic agonists are protumorigenic, probably because of tissue-specific differences in tumor–nerve crosstalk [122]. In prostate cancer, muscarinic agonists promote a more aggressive disease phenotype. CHRM1 and CHRM3 are overexpressed in prostate cancer and are associated with diminished progression-free survival. Muscarinic agonists promote a castrate-recurrent phenotype in prostate cancer cell lines by activating a FAK–YAP signaling pathway [123]. These findings support earlier work in Hi-Myc transgenic mice where CHRM1 knockout slowed disease progression and metastasis [124]. Similarly, gastric cancer and colon cancer growth are dependent on parasympathetic signaling. The cholinergic agonist, carbachol, promoted nerve growth factor expression and gastric cancer organoid growth in a CHRM3-dependent fashion [125]. In a chemically induced murine model of colon cancer, CHRM3 knockout decreased tumor burden, corroborating earlier findings indicating that cholinergic agonists promote colon cancer cell proliferation [126]. These studies indicate that muscarinic drugs impact on cancer progression, supporting the drive to fully dissect the role of parasympathetic signaling in innervated cancers.
Outstanding Questions.
Is it feasible to alter the medications cancer patients are taking for other indications to improve survival and therapeutic efficacy?
Can researchers identify new therapeutic targets by better understanding how different GPCR-targeted drugs alter the cancer phenotype?
Do GPCR-targeted drugs that modulate the cancer phenotype have different responses in cancer patients taking the drugs to treat a specific condition relative to those taking the drugs purely as anticancer agents?
How does the use of cannabinoids in palliative care impact cancer patient survival?
Do NK-1R or D2R antagonists improve cancer patient survival in the clinic?
How do opioids alter immunotherapy?
Highlights.
GPCRs and G proteins are frequently dysregulated in cancer.
Alterations in GPCR/G-protein signaling modulate many aspects of the cancer phenotype, including metastasis, angiogenesis, tumor–stroma crosstalk, and tumor growth.
GPCRs are the most common receptor class targeted by FDA-approved drugs.
Drugs that target GPCRs can directly or indirectly alter therapeutic efficacy and cancer patient survival.
Cancer patients take many medications for palliative care and to treat comorbid conditions that target GPCRs. Medications to alleviate pain, nausea, anxiety, depression, heart conditions, diabetes, and inflammation all target GPCRs.
Acknowledgments
This work was supported by National Cancer Institute grant P30 CA016056. We thank Dr Paul Insel and the members of the laboratory of M.E.F. for helpful comments on the manuscript.
Glossary
- Acute myeloid leukemia (AML) blast cells
abnormal immature white blood cells that are present in the blood of AML patients
- Anti-PD-1
a monoclonal antibody that blocks the activity of the programmed cell death 1 (PD-1) receptor, an immune checkpoint molecule expressed on T cells
- Castrate-recurrent phenotype
a highly aggressive, innervated form of prostate cancer
- Catecholamines
neurotransmitters such as epinephrine and norepinephrine that regulate stress responses
- Chemopreventive agents
compounds that prevent, inhibit, or reverse cancer development
- Cholangiocarcinoma
bile duct cancer
- Cysteinyl leukotriene (CysLT1) 1 receptors
GPCRs that mediate inflammation by activating and attracting neurotrophils and eosinophils
- Desensitization
decreased response to an agonist often as a result of overstimulation
- Epithelial to mesenchymal transition (EMT)
a process by which cancer cells lose epithelial markers and become more invasive
- GPR55
cannabinoid GPCR with known roles in obesity, inflammation, angiogenesis, neuropathic pain, osteoclast formation, and cancer
- Hallmarks of cancer
a list of cancer-associated processes that drive tumor initiation and progression
- Hi-Myc transgenic mice
mice expressing human c-Myc in prostatic tissue and spontaneously develop prostate cancer
- Histamine H1 receptor (H1HR)
a GPCR that regulates inflammation, bronchoconstriction, itching, and vasodilation
- Histamine H2 receptor (H2HR)
GPCR that regulates gastric acid secretion
- Immortal time bias
Inaccurate epidemiological outcome because there is a timeframe in which the primary outcome (excluding death from cancer) cannot occur in at least one of the cohorts owing to the exposure (β-blocker use) definition
- KPC mice
a LSL–KrasG12D, LSL–Tp53R172H, Pdx1–cre genetically engineered mouse model of pancreatic cancer that closely mimics human pancreatic cancer progression
- MK886
a 5-lipoxygenase inhibitor that antagonizes leukotriene biosynthesis
- Neurokinin-1 (NK-1) receptor
GPCR that regulates stress, pain, and inflammatory signals
- μ-Opioid receptors (MORs)
GPCRs that modulate pain response and addictive behavior
- Parasympathetic nervous system
branch of the autonomic nervous system that promotes relaxation and slows highly energetic processes
- Peroxisome proliferator-activated receptor γ (PPARγ)
a nuclear receptor that regulates glucose metabolism, adipocyte differentiation, and fatty acid storage
- Primary sclerosing cholangitis (PSC)
a precursor condition to cholangiocarcinoma
- Prostanoid receptors
receptors that respond to prostaglandins – hormone-like molecules that regulate inflammation, blood pressure, and smooth muscle contraction
- Protease-activated receptor 1 (PAR1)
a GPCR that mediates thrombin signaling and has been implicated in protumorigenic signaling pathways
- P2Y12
a GPCR that regulates blood coagulation and hemostasis
- Selective serotonin reuptake inhibitors (SSRIs)
the most commonly prescribed antidepressants; they work by increasing serotonin levels in the brain
- Sympathetic nervous system
regulates the involuntary response of the body to stress and danger
- Tumor microenvironment
a combination of cell types (fibroblasts, immune cells, neurons, etc.), secreted factors (cytokines, metabolites, etc.), and extracellular matrix proteins (collagen, fibronectin, etc.) that comprise the tumor volume
- Tumor–stroma crosstalk
the interactions between cancer cells and various cell types, as well as with extracellular matrix proteins surrounding cancer cells
- Warburg effect
the preference of cancer cells to undergo glycolysis to produce energy even in the presence of abundant oxygen
References
- 1.Sriram K and Insel PA (2018) G protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol. Pharmacol 93, 251–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu J et al. (2012) Dissection of aberrant GPCR signaling in tumorigenesis – a systems biology approach. Cancer Genomics Proteomics 9, 37–50 [PubMed] [Google Scholar]
- 3.Feigin ME (2013) Harnessing the genome for characterization of G-protein coupled receptors in cancer pathogenesis. FEBS J. 280, 4729–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu V et al. (2019) Illuminating the Onco-GPCRome: novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem 294, 11062–11086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janssen-Heijnen ML et al. (2010) Cancer patients with cardiovascular disease have survival rates comparable to cancer patients within the age-cohort of 10 years older without cardiovascular morbidity. Crit. Rev. Oncol. Hematol 76, 196–207 [DOI] [PubMed] [Google Scholar]
- 6.Okura Y et al. (2019) Future projection of cancer patients with cardiovascular disease in Japan by the year 2039: a pilot study. Int. J. Clin. Oncol 24, 983–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khakoo AY and Yeh ET (2008) Therapy insight: management of cardiovascular disease in patients with cancer and cardiac complications of cancer therapy. Nat. Clin. Pract. Oncol 5, 655–667 [DOI] [PubMed] [Google Scholar]
- 8.Stone RL et al. (2012) Paraneoplastic thrombocytosis in ovarian cancer. N. Engl. J. Med 366, 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khorana AA and Fine RL (2004) Pancreatic cancer and thromboembolic disease. Lancet Oncol. 5, 655–663 [DOI] [PubMed] [Google Scholar]
- 10.Frishman WH (2016) Beta-adrenergic receptor blockers in hypertension: alive and well. Prog. Cardiovasc. Dis 59, 247–252 [DOI] [PubMed] [Google Scholar]
- 11.Renz BW et al. (2018) β2 adrenergic–neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 33, 75–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim-Fuchs C et al. (2014) Chronic stress accelerates pancreatic cancer growth and invasion: a critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav. Immun 40, 40–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo K et al. (2009) Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncol. Rep 22, 825–830 [DOI] [PubMed] [Google Scholar]
- 14.Zahalka AH et al. (2017) Adrenergic nerves activate an angiometabolic switch in prostate cancer. Science 358, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Udumyan R et al. (2017) Beta-blocker drug use and survival among patients with pancreatic adenocarcinoma. Cancer Res. 77, 3700–3707 [DOI] [PubMed] [Google Scholar]
- 16.Sharma A et al. (2018) Model to determine risk of pancreatic cancer in patients with new-onset diabetes. Gastroenterology 155, 730–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsujimoto T et al. (2018) Risk of all-cause mortality in diabetic patients taking β-blockers. Mayo Clin. Proc 93, 409–418 [DOI] [PubMed] [Google Scholar]
- 18.Bucsek MJ et al. (2017) β-Adrenergic signaling in mice housed at standard temperatures suppresses an effector phenotype in CD8+ T cells and undermines checkpoint inhibitor therapy. Cancer Res. 77, 5639–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kokolus KM et al. (2018) Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology 7, e1405205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melhem-Bertrandt A et al. (2011) Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J. Clin. Oncol 29, 2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Powe DG et al. (2010) Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget 1, 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choy C et al. (2016) Inhibition of β2-adrenergic receptor reduces triple-negative breast cancer brain metastases: the potential benefit of perioperative β-blockade. Oncol. Rep 35, 3135–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gulati G et al. (2017) Neurohormonal blockade and circulating cardiovascular biomarkers during anthracycline therapy in breast cancer patients: results from the PRADA (prevention of cardiac dysfunction during adjuvant breast cancer therapy) study. J. Am. Heart Assoc 6, e006513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HY et al. (2017) Is beta-blocker use beneficial in breast cancer? A meta-analysis. Oncology 92, 264–268 [DOI] [PubMed] [Google Scholar]
- 25.Weberpals J et al. (2016) Beta blockers and cancer prognosis – the role of immortal time bias: a systematic review and meta-analysis. Cancer Treat. Rev 47, 1–11 [DOI] [PubMed] [Google Scholar]
- 26.Ahmed A and Majeed A (2019) Antiplatelet drug management In Patient Blood Management in Cardiac Surgery (von Heynmann C and Boer C, eds), pp. 51–60, Springer [Google Scholar]
- 27.Frere C et al. (2019) Antiplatelet agents for cancer prevention: current evidences and continuing controversies. Cancers 11, 1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu XR et al. (2018) Cancer and platelet crosstalk: opportunities and challenges for aspirin and other antiplatelet agents. Blood 131, 1777–1789 [DOI] [PubMed] [Google Scholar]
- 29.Yang Y et al. (2019) Thrombin signaling promotes pancreatic adenocarcinoma through PAR-1-dependent immune evasion. Cancer Res. 79, 3417–3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adams GN et al. (2015) Colon cancer growth and dissemination relies upon thrombin, stromal PAR-1, and fibrinogen. Cancer Res. 75, 4235–4243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang E et al. (2016) Dysregulated protease activated receptor 1 (PAR1) promotes metastatic phenotype in breast cancer through HMGA2. Oncogene 35, 1529–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chanakira A et al. (2017) Tissue factor–factor VIIa complex triggers protease activated receptor 2-dependent growth factor release and migration in ovarian cancer. Gynecol. Oncol 145, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Auvergne R et al. (2016) PAR1 inhibition suppresses the self-renewal and growth of A2B5-defined glioma progenitor cells and their derived gliomas in vivo. Oncogene 35, 3817–3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goyama S et al. (2017) Protease-activated receptor-1 inhibits proliferation but enhances leukemia stem cell activity in acute myeloid leukemia. Oncogene 36, 2589–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Savi P et al. (2006) The active metabolite of clopidogrel disrupts P2Y12 receptor oligomers and partitions them out of lipid rafts. Proc. Natl. Acad. Sci 103, 11069–11074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho MS et al. (2017) Role of ADP receptors on platelets in the growth of ovarian cancer. Blood 130, 1235–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cho MS et al. (2012) Platelets increase the proliferation of ovarian cancer cells. Blood 120, 4869–4872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kloss L et al. (2019) ADP secreted by dying melanoma cells mediates chemotaxis and chemokine secretion of macrophages via the purinergic receptor P2Y12. Cell Death Dis. 10, 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henson LA et al. (2020) Palliative care and the management of common distressing symptoms in advanced cancer: pain, breathlessness, nausea and vomiting, and fatigue. J. Clin. Oncol 38, 905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berger M et al. (2014) Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J. Hepatol 60, 985–994 [DOI] [PubMed] [Google Scholar]
- 41.Palma C et al. (2000) Anti-tumour activity of tachykinin NK 1 receptor antagonists on human glioma U373 MG xenograft. Br. J. Cancer 82, 480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang C et al. (2018) MMP1/PAR1/SP/NK1R paracrine loop modulates early perineural invasion of pancreatic cancer cells. Theranostics 8, 3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ge C et al. (2019) Neurokinin-1 receptor is an effective target for treating leukemia by inducing oxidative stress through mitochondrial calcium overload. Proc. Natl. Acad. Sci 116, 19635–19645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y-X et al. (2017) β-Arrestin 1 has an essential role in neurokinin-1 receptor-mediated glioblastoma cell proliferation and G2/M phase transition. J. Biol. Chem 292, 8933–8947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muñoz M et al. (2012) The NK-1 receptor is expressed in human leukemia and is involved in the antitumor action of aprepitant and other NK-1 receptor antagonists on acute lymphoblastic leukemia cell lines. Investig. New Drugs 30, 529–540 [DOI] [PubMed] [Google Scholar]
- 46.Javid H et al. (2019) The emerging role of substance P/neurokinin-1 receptor signaling pathways in growth and development of tumor cells. J. Physiol. Biochem 75, 415–421 [DOI] [PubMed] [Google Scholar]
- 47.Muñoz M et al. (2015) The substance P/NK-1 receptor system: NK-1 receptor antagonists as anti-cancer drugs. J. Biosci 40, 441–463 [DOI] [PubMed] [Google Scholar]
- 48.Sinha S et al. (2017) PanIN neuroendocrine cells promote tumorigenesis via neuronal cross-talk. Cancer Res. 77, 1868–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.May MB and Glode AE (2016) Dronabinol for chemotherapy-induced nausea and vomiting unresponsive to antiemetics. Cancer Manag. Res 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ladin DA et al. (2016) Preclinical and clinical assessment of cannabinoids as anti-cancer agents. Front. Pharmacol 7, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferro R et al. (2018) GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene 37, 6368–6382 [DOI] [PubMed] [Google Scholar]
- 52.Jeong S et al. (2019) Cannabidiol promotes apoptosis via regulation of XIAP/Smac in gastric cancer. Cell Death Dis. 10, 846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu C et al. (2020) Cannabinoids promote progression of HPV positive head and neck squamous cell carcinoma via p38 MAPK activation. Clin. Cancer Res 26, 2693–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mukhopadhyay B et al. (2015) Cannabinoid receptor 1 promotes hepatocellular carcinoma initiation and progression through multiple mechanisms. Hepatology 61, 1615–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sam AH et al. (2011) Rimonabant: from RIO to ban. J. Obes 2011, 432607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pergam SA et al. (2017) Cannabis use among patients at a comprehensive cancer center in a state with legalized medicinal and recreational use. Cancer 123, 4488–4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wigmore T and Farquhar-Smith P (2016) Opioids and cancer: friend or foe? Curr. Opin. Support. Palliat. Care 10, 109–118 [DOI] [PubMed] [Google Scholar]
- 58.Afsharimani B et al. (2011) Morphine and tumor growth and metastasis. Cancer Metastasis Rev. 30, 225–238 [DOI] [PubMed] [Google Scholar]
- 59.Al-Hashimi M et al. (2013) Opioids and immune modulation: more questions than answers. Br. J. Anaesth 111, 80–88 [DOI] [PubMed] [Google Scholar]
- 60.Li Y et al. (2019) The μ-opioid receptor (MOR) promotes tumor initiation in hepatocellular carcinoma. Cancer Lett. 453, 1–9 [DOI] [PubMed] [Google Scholar]
- 61.Niu D-G et al. (2015) Morphine promotes cancer stem cell properties, contributing to chemoresistance in breast cancer. Oncotarget 6, 3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khabbazi S et al. (2015) Morphine modulates interleukin-4-or breast cancer cell-induced pro-metastatic activation of macrophages. Sci. Rep 5, 11389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khabbazi S et al. (2016) Morphine decreases the proangiogenic interaction between breast cancer cells and macrophages in vitro. Sci. Rep 6, 31572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Minicis S et al. (2008) Role of endogenous opioids in modulating HSC activity in vitro and liver fibrosis in vivo. Gut 57, 352–364 [DOI] [PubMed] [Google Scholar]
- 65.Xie N et al. (2018) Effect of perioperative opioids on cancer-relevant circulating parameters: mu opioid receptor and toll-like receptor 4 activation potential, and proteolytic profile. Clin. Cancer Res 24, 2319–2327 [DOI] [PubMed] [Google Scholar]
- 66.Barlass U et al. (2018) Morphine worsens the severity and prevents pancreatic regeneration in mouse models of acute pancreatitis. Gut 67, 600–602 [DOI] [PubMed] [Google Scholar]
- 67.Exadaktylos AK et al. (2006) Can anesthetic technique for primary breast cancer surgery affect recurrence or metastasis? Anesthesiology 105, 660–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cata JP et al. (2014) Effects of postoperative epidural analgesia on recurrence-free and overall survival in patients with nonsmall cell lung cancer. J. Clin. Anesth 26, 3–17 [DOI] [PubMed] [Google Scholar]
- 69.Gottschalk A et al. (2010) Association between epidural analgesia and cancer recurrence after colorectal cancer surgery. Anesthesiology 113, 27–34 [DOI] [PubMed] [Google Scholar]
- 70.Dubowitz JA et al. (2018) Implicating anaesthesia and the perioperative period in cancer recurrence and metastasis. Clin. Exp. Metastasis 35, 347–358 [DOI] [PubMed] [Google Scholar]
- 71.Zylla D et al. (2013) Opioid requirement, opioid receptor expression, and clinical outcomes in patients with advanced prostate cancer. Cancer 119, 4103–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Greten FR and Grivennikov SI (2019) Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 51, 27–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rayburn ER et al. (2009) Anti-inflammatory agents for cancer therapy. Mol. Cell. Pharmacol 1, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 75.Ajmani GS et al. (2017) Allergy and asthma medication use in home-dwelling US older adults. Int. Forum Allergy Rhinol 7, 192–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gomez-Rubio P et al. (2017) Reduced risk of pancreatic cancer associated with asthma and nasal allergies. Gut 66, 314–322 [DOI] [PubMed] [Google Scholar]
- 77.Tsai M-J et al. (2016) Cysteinyl leukotriene receptor antagonists decrease cancer risk in asthma patients. Sci. Rep 6, 23979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gunning WT et al. (2002) Chemoprevention by lipoxygenase and leukotriene pathway inhibitors of vinyl carbamate-induced lung tumors in mice. Cancer Res. 62, 4199–4201 [PubMed] [Google Scholar]
- 79.Li N et al. (2005) Overexpression of 5-lipoxygenase and cyclooxygenase 2 in hamster and human oral cancer and chemopreventive effects of zileuton and celecoxib. Clin. Cancer Res 11, 2089–2096 [DOI] [PubMed] [Google Scholar]
- 80.Bellamkonda K et al. (2018) Montelukast, a CysLT1 receptor antagonist, reduces colon cancer stemness and tumor burden in a mouse xenograft model of human colon cancer. Cancer Lett. 437, 13–24 [DOI] [PubMed] [Google Scholar]
- 81.Butler CT et al. (2017) A quininib analogue and cysteinyl leukotriene receptor antagonist inhibits vascular endothelial growth factor (VEGF)-independent Angiogenesis and exerts an additive antiangiogenic response with bevacizumab. J. Biol. Chem 292, 3552–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Du F et al. (2019) Leukotriene synthesis is critical for medulloblastoma progression. Clin. Cancer Res 25, 6475–6486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Faustino-Rocha AI et al. (2017) Antihistamines as promising drugs in cancer therapy. Life Sci. 172, 27–41 [DOI] [PubMed] [Google Scholar]
- 84.Kennedy L et al. (2018) Blocking H1/H2 histamine receptors inhibits damage/fibrosis in Mdr2−/− mice and human cholangiocarcinoma tumorigenesis. Hepatology 68, 1042–1056 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 85.Zhao J et al. (2019) Upregulation of histamine receptor H1 promotes tumor progression and contributes to poor prognosis in hepatocellular carcinoma. Oncogene 39, 1724–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wiktorin HG et al. (2019) Histamine targets myeloid-derived suppressor cells and improves the anti-tumor efficacy of PD-1/PD-L1 checkpoint blockade. Cancer Immunol. Immunother 68, 163–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nielsen HJ et al. (2002) Ranitidine as adjuvant treatment in colorectal cancer. Br. J. Surg 89, 1416–1422 [DOI] [PubMed] [Google Scholar]
- 88.Pitman A et al. (2018) Depression and anxiety in patients with cancer. BMJ 361, k1415. [DOI] [PubMed] [Google Scholar]
- 89.Navari RM and Aapro M (2016) Antiemetic prophylaxis for chemotherapy-induced nausea and vomiting. N. Engl. J. Med 374, 1356–1367 [DOI] [PubMed] [Google Scholar]
- 90.Busby J et al. (2018) Selective serotonin reuptake inhibitor use and breast cancer survival: a population-based cohort study. Breast Cancer Res. 20, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cowen P and Sargent P (1997) Changes in plasma prolactin during SSRI treatment: evidence for a delayed increase in 5-HT neurotransmission. J. Psychopharmacol 11, 345–348 [DOI] [PubMed] [Google Scholar]
- 92.Christensen DK et al. (2016) SSRI use and clinical outcomes in epithelial ovarian cancer. Oncotarget 7, 33179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mravec B et al. (2020) Stress and cancer. Part I: mechanisms mediating the effect of stressors on cancer. J. Neuroimmunol 346, 577311. [DOI] [PubMed] [Google Scholar]
- 94.Mravec B et al. (2020) Stress and cancer. Part II: therapeutic implications for oncology. J. Neuroimmunol 346, 577312. [DOI] [PubMed] [Google Scholar]
- 95.Elshayeb EI et al. (2016) Serum serotonin as a novel marker for hepatocellular carcinoma. Adv. Res. Gastroenterol. Hepatol 1, 555571 [Google Scholar]
- 96.Balasubramanian S and Paulose CS (1998) Induction of DNA synthesis in primary cultures of rat hepatocytes by serotonin: possible involvement of serotonin S2 receptor. Hepatology 27, 62–66 [DOI] [PubMed] [Google Scholar]
- 97.Lesurtel M et al. (2006) Platelet-derived serotonin mediates liver regeneration. Science 312, 104–107 [DOI] [PubMed] [Google Scholar]
- 98.Soll C et al. (2010) Serotonin promotes tumor growth in human hepatocellular cancer. Hepatology 51, 1244–1254 [DOI] [PubMed] [Google Scholar]
- 99.Jiang S-H et al. (2017) Increased serotonin signaling contributes to the Warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology 153, 277–291 [DOI] [PubMed] [Google Scholar]
- 100.Domínguez-Soto Á et al. (2017) Serotonin drives the acquisition of a profibrotic and anti-inflammatory gene profile through the 5-HT7R–PKA signaling axis. Sci. Rep 7, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nocito A et al. (2008) Serotonin regulates macrophage-mediated angiogenesis in a mouse model of colon cancer allografts. Cancer Res. 68, 5152–5158 [DOI] [PubMed] [Google Scholar]
- 102.Caraci F et al. (2011) Metabolic drug interactions between antidepressants and anticancer drugs: focus on selective serotonin reuptake inhibitors and hypericum extract. Curr. Drug Metab 12, 570–577 [DOI] [PubMed] [Google Scholar]
- 103.Sachlos E et al. (2012) Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell 149, 1284–1297 [DOI] [PubMed] [Google Scholar]
- 104.Cheng H et al. (2015) Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data. Cell Death Dis. 6, e1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ranjan A and Srivastava SK (2016) Penfluridol suppresses pancreatic tumor growth by autophagy-mediated apoptosis. Sci. Rep 6, 26165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang W et al. (2018) Antiproliferative activities of the second-generation antipsychotic drug sertindole against breast cancers with a potential application for treatment of breast-to-brain metastases. Sci. Rep 8, 15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bangalore S et al. (2008) Perioperative β blockers in patients having non-cardiac surgery: a meta-analysis. Lancet 372, 1962–1976 [DOI] [PubMed] [Google Scholar]
- 108.Hothersall JD et al. (2016) Can residence time offer a useful strategy to target agonist drugs for sustained GPCR responses? Drug Discov. Today 21, 90–96 [DOI] [PubMed] [Google Scholar]
- 109.O’Hayre M et al. (2014) Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol 27, 126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hauser AS et al. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Discov 16, 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Usman S et al. (2020) The current status of anti GPCR drugs against different cancers. J. Pharm. Anal Published online January 11, 2020 10.1016/j.jpha.2020.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bullard KM et al. (2018) Prevalence of diagnosed diabetes in adults by diabetes type – United States, 2016. Morb. Mortal. Wkly Rep 67, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shlomai G et al. (2016) Type 2 diabetes mellitus and cancer: the role of pharmacotherapy. J. Clin. Oncol 34, 4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou M et al. (2017) The anti-diabetic drug exenatide, a glucagon-like peptide-1 receptor agonist, counteracts hepatocarcinogenesis through cAMP–PKA–EGFR–STAT3 axis. Oncogene 36, 4135–4149 [DOI] [PubMed] [Google Scholar]
- 115.Nomiyama T et al. (2014) Exendin-4, a GLP-1 receptor agonist, attenuates prostate cancer growth. Diabetes 63, 3891–3905 [DOI] [PubMed] [Google Scholar]
- 116.He W et al. (2016) Exendin-4 inhibits growth and augments apoptosis of ovarian cancer cells. Mol. Cell. Endocrinol 436, 240–249 [DOI] [PubMed] [Google Scholar]
- 117.Koehler JA et al. (2015) GLP-1R agonists promote normal and neoplastic intestinal growth through mechanisms requiring Fgf7. Cell Metab. 21, 379–391 [DOI] [PubMed] [Google Scholar]
- 118.Elashoff M et al. (2011) Pancreatitis, pancreatic, and thyroid cancer with glucagon-like peptide-1-based therapies. Gastroenterology 141, 150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pinto LC et al. (2019) Glucagon-like peptide-1 receptor agonists and pancreatic cancer: a meta-analysis with trial sequential analysis. Sci. Rep 9, 2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nyborg NC et al. (2012) The human GLP-1 analog liraglutide and the pancreas: evidence for the absence of structural pancreatic changes in three species. Diabetes 61, 1243–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Körner M et al. (2007) GLP-1 receptor expression in human tumors and human normal tissues: potential for in vivo targeting. J. Nucl. Med 48, 736–743 [DOI] [PubMed] [Google Scholar]
- 122.Renz BW et al. (2018) Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discov. 8, 1458–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Goto Y et al. (2020) Muscarinic receptors promote castration-resistant growth of prostate cancer through a FAK–YAP signaling axis. Oncogene 39, 4014–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Magnon C et al. (2013) Autonomic nerve development contributes to prostate cancer progression. Science 341, 1236361. [DOI] [PubMed] [Google Scholar]
- 125.Hayakawa Y et al. (2017) Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell 31, 21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cheng K et al. (2003) Transactivation of the epidermal growth factor receptor mediates cholinergic agonist-induced proliferation of H508 human colon cancer cells. Cancer Res. 63, 6744–6750 [PubMed] [Google Scholar]