

Abstract
The careful development, validation, and implementation of serodiagnostic assays can provide reliable results that make them a valuable tool in microbial quality control for nonhuman primates. This article includes identification and description of the components of assay development, including formulas for calculating the number of positive serum samples needed for assay validation and methods for calculating their diagnostic sensitivity and specificity. To ensure that assays are performing within predetermined specifications, there must be a quality control system that includes appropriate system and sample suitability controls as well as mechanisms to track assay performance over time. The section on quality assurance includes definitions of precision and accuracy in assay performance, and how to interpret these two factors using the Levey-Jennings chart, Westgard's rules, and other monitoring methods. Because all serologic assays are prone to false positive and false negative results, it is essential to interpret all diagnostic test results using both the expected prevalence of disease in the population and the population-specific assay performance characteristics that are determined during assay validation. The discussion on interpreting diagnostic test results also includes guidelines for calculating the positive and negative predictive values of an assay and for interpreting results based on the disease prevalence of the test population. A glossary provides definitions of commonly used terms.
Keywords: assay, diagnostics, nonhuman primate, quality control, serology, validation
Nonhuman primates (NHPs) are our closest living animal relatives and, because of their phylogenetic similarity to humans, are critically important as animal models for human diseases. Unfortunately, adventitious infectious agents can be a significant cause of morbidity and mortality in NHPs and many of the viruses that infect them cause persistent or latent lifelong infections. In addition to being a potential source of confounding variables in biomedical research, some infectious agents of NHPs are zoonotic and present a potential health risk to humans. To limit research variability and to more fully understand the risks to biomedical research and personnel, it is important to monitor NHPs for adventitious infectious agents as part of a routine colony health monitoring program. Serology assays are sensitive, specific, and readily automated, and therefore form the foundation for most colony health screening programs.
Assay Development
The process of assay1 development is the foundation of high-quality diagnostics. The effectiveness of serodiagnostic assays for infectious disease monitoring in nonhuman primates begins with the development and validation1 of high-quality, reliable assays and includes the implementation of system and sample suitability controls as well as appropriate procedures and practices to ensure that the assays perform within specifications. But development does not end with the validation process—assays require continuous monitoring and refinement both to replenish consumed reagents and to address issues that arise in the course of normal assay use. A flowchart outlining a generalized process for serological assay development is shown in Figure 1.
Figure 1.

A process for serological assay development, validation, and quality control management. HAI, hemagglutination inhibition; IFA, indirect fluorescent antibody; PCR, polymerase chain reaction; WIB, western immunoblot.
Assay System
Assay development begins with identification of the assay system's intended use, which usually includes high-sensitivity, high-throughput screening assays and lower-throughput, high-specificity assays to confirm equivocal or positive results. For serological screening, most laboratories use assays that can be easily automated, such as the enzyme-linked immunosorbent assay (ELISA) or the Luminex xMAP®-based multiplexed fluorometric immunoassay (MFIA™). The ELISA is a singleplex system (one assay is performed on one serum sample in each assay well), whereas the MFIA is a multiplexed system (multiple assays and internal controls can be performed on a single serum sample in each assay well at the same time). For confirmation of equivocal or positive results, most laboratories use assays such as the indirect fluorescent antibody (IFA) test, western immunoblot (WIB), or even the polymerase chain reaction (PCR) when appropriate.
Antigens
After identification of the screening1 and confirmation1 assays that need to be developed, the next step is to identify the necessary antigens, which may include purified whole virus lysates or one or more immunodominant, recombinant proteins. Purified whole virus lysates typically contain a complete complement of virally expressed proteins; however, virus purification may result in the loss of some virus proteins, and virus adaptation to cell culture can change the expression profile of others. Additionally, virus lysates often include a complex mixture of copurifying proteins from the cell culture system, including eukaryotic cellular proteins and proteins from cell culture components such as fetal bovine serum, which may lead to artifactual reactivity with some serum samples (Levinson 1992; Pedersen et al. 1986).
For recombinant proteins, antigens can be produced in either prokaryotic or eukaryotic expression systems. Prokaryotic systems, such as Escherichia coli, produce large quantities of protein efficiently, but they are often expressed into insoluble inclusion bodies and lack eukaryotic post-translational processing. Eukaryotic systems, such as the insect cell-baculovirus recombinant expression systems, are often much more difficult to work with and produce lower quantities of protein, but the proteins they produce are more likely to be in a native conformation and they have glycosylation patterns similar to those of mammals.
Regardless of the expression system chosen, recombinant proteins can be expressed as chimeric proteins that have an affinity tag enabling antigen purification by metal or antibody affinity chromatography, thus yielding antigens with fewer copurifying contaminating proteins, a result that can boost the expected assay signal while at the same time decreasing background noise. Because of the inherent limitations and advantages of using whole virus lysates and recombinant proteins, laboratories often use one or both in combination to optimize assay results.
Serum Samples
The development of robust serological assays requires serum samples from a broad range of animals that represent the population(s) on which the assay is intended to be used. Thus an essential component of assay development—often just as critical as the identification of appropriate antigens—is the accumulation of positive and negative serum samples from animals of different sexes, ages, and a wide variety of health statuses.
Acquiring Positive and Negative Sera
Negative serum samples may come from specific pathogen-free (SPF) animals, if they exist, or from animals that have repeatedly tested negative by other tests or laboratories. Positive serum samples are frequently the most difficult to accumulate in large enough numbers for assay development. Ideally, they should come from natural infections that are confirmed by other diagnostic techniques and should represent a wide range of time points after infection, from incipient to convalescent. Positive sera can also be generated by the infection of naïve animals, when possible: sera from infection studies can be serially collected at predetermined time points, allowing for a more thorough assessment of how the assay performs during seroconversion. The least desirable positive serum samples are those from vaccinated animals, as they do not represent a normal process of seroconversion and may result in the production of interfering antibodies. However, if the assay is intended to monitor for seroconversion in vaccinated animals, these samples are quite appropriate and should be included (Barlough et al. 1984; Cook et al. 1989).
For assay development, control sera should be accumulated and pooled, when necessary, to achieve volumes of several milliliters for each sample, which should then be placed in single-use aliquots (e.g., 0.1 ml) and frozen at –70°C until use. Representative aliquots of frozen sera should be qualified1 by comparison to existing assays or by outside laboratories, where appropriate, and the results recorded in the serum lot documentation records. Known positive and negative serum samples for assay prevalidation and validation must also be accumulated and aliquoted in a similar manner.
The following formula is an effective way to calculate the number of positive serum samples required to validate an assay (Jacobson 1998):
| (1) |
where n is the number of known infected animals, DSn is the expected diagnostic sensitivity1 of the assay, e is the percentage error allowed in the estimate of diagnostic sensitivity (expressed as a decimal), and c is the confidence interval for the estimate (e.g., 1.96 for 95% confidence). Thus the number of positive serum samples required to validate an assay with an expected DSn of 95% ±5% with 95% confidence is 73. The number of negative serum samples required to validate an assay can be determined by replacing the expected diagnostic sensitivity in the above formula with the expected diagnostic specificity1 (DSp).
Prevalidation Testing and Retesting
In addition to developing suitable antigens, laboratories must also develop appropriate system and sample suitability controls to ensure that the assay and samples are performing within specifications to provide valid results during routine use. These controls are developed during the course of routine assay development along with the assays themselves. Because these controls are inextricably linked to a routine quality assurance1 program, there is a fuller description of them in the section below on Quality Assurance in Diagnostic Testing Laboratories.
Assay development is an intensive, iterative process that begins on a small scale by titrating antigen(s) and other critical reagents and then testing them against a panel of well-characterized antibody-positive and -negative sera. This process continues until antigens of the appropriate type, quality, and concentration are identified to produce the desired assay performance characteristics. Next, the assay is scaled up to a minimum production level, and the antigens and reagents are retitrated to ensure that assay performance characteristics have not changed with the scale-up.
After confirmation or reestablishment of the assay performance characteristics, a prevalidation study tests many (often hundreds) of positive, negative, and problematic serum samples to refine the assay and determines preliminary assay-specific performance characteristics, such as analytical sensitivity (ASn1) and analytical specificity (ASp1). Analytical sensitivity is the lowest amount of antibody that can be detected by the assay, also called the assay's limit of detection (LOD). ASn is determined by serially diluting one or more well-characterized positive control serum samples until the diluted sample becomes negative in the assay and, when possible, by comparing the new assay to similar results from preexisting assays (Figure 2). The result is often reported as a titer1 (e.g., 1:105,600), with a higher dilution indicating an increased ASn. ASp is an assessment of the selectivity of the antigen-antibody response and is often determined by performing the assay using a panel of heterologous, monotypic, positive control sera (Figure 3). The assay is considered analytically specific if it does not react when challenged with heterologous positive sera.
Figure 2.

Determination of the analytical sensitivity (ASn), or limit of detection (LOD), for four antigens that are used to detect simian type D retrovirus (SRV). Antigens include SRV-1, -2, and -5 whole virus lysates and SRV-2 recombinant gp20 antigen. The horizontal dashed line indicates the cutoff value for the assay. MFIA, multiplexed fluorescent immunoassay.
Figure 3.
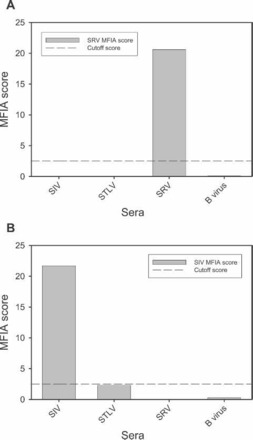
Determination of analytical specificity (ASp), or selectivity of a given antigen. ASp is determined by performing an assay for a given antigen versus a panel of known positive control sera. (A) illustrates the reactivity of SRV gp20 recombinant antigen and (B) shows the reactivity of SIV whole virus lysate antigen versus a panel of known positive control sera. MFIA, multiplexed fluorescent immunoassay; SIV, simian immunodeficiency virus; SRV, simian type D retrovirus; STLV, simian T lymphotropic virus.
Determination of Assay Cutoff Values
The goal of diagnostic testing is to accurately classify serum samples as positive or negative for a given infectious agent; unfortunately, for assays in the real world, there is an overlapping continuum of assay responses from negative to positive (Figure 4). It is possible to measure how well an assay classifies serum samples as positive or negative in a given population of animals by calculating the assay's diagnostic sensitivity (DSn) and diagnostic specificity (DSp): the DSn is the probability that an assay correctly identifies positive (infected or diseased) animals, and the DSp is the probability that an assay correctly identifies negative (normal1) animals.
Figure 4.
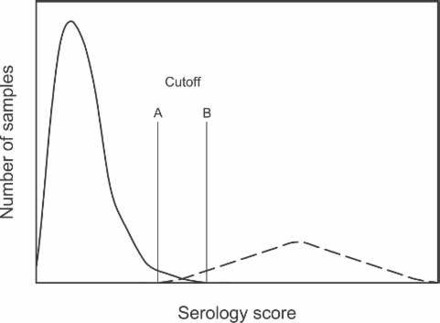
Frequency distribution for a hypothetical set of reference sera for serodiagnostic testing. The curve on the left represents known negative sera and the curve on the right, positive sera. An assay cutoff at line A would properly classify all positive serum samples (high diagnostic sensitivity, or DSn), and at line B would properly classify all negative serum samples (high diagnostic specificity, or DSp).
The DSn and DSp of an assay vary according to where the assay cutoff is set. Setting the assay cutoff at line A in Figure 4 results in an assay that correctly classifies all positive serum samples (higher DSn) but also gives a high number of false positive (FP)1 results (lower DSp); however, setting the assay cutoff at line B would correctly classify all true negative (TN)1 samples (higher DSp) but result in a high number of false negative (FN)1 classifications (lower DSn). For a given assay in a population of animals, changing the assay cutoff to increase DSn results in a similar decrease in DSp and, conversely, any increase in DSp is done at a similar expense to DSn. Assay cutoffs are commonly set at specific levels to accomplish the intended goal of the assay: for high-throughput screening assays the cutoff is often set to maximize DSn (and thus limit the number of false negative classifications), whereas for lower-throughput confirmation assays1, the cutoff is often set to maximize DSp (and thus limit the number of false positive classifications).
There are a variety of ways to determine assay cutoff values. Many laboratories perform a simple statistical analysis of the negative data and set the positive-negative assay cutoff at the mean value obtained from a large number of known negative serum samples plus two or three standard deviations (SD) (Barajas-Rojas et al. 1993). However, serological data do not typically follow a normal (Gaussian) distribution pattern. For example, data from negative sera (Figure 4) are typically skewed to the right (positive skew) and demonstrate a high degree of kurtosis (they are leptokurtic). Thus, if simple parametric statistics are the basis for determining assay cutoff values, the data should first be transformed to a normal distribution pattern verified either by a Kolmogorov-Smirnov or Lilliefors test or by another statistic that verifies their normal distribution. A simpler approach is to use nonparametric statistics, place the negative data in rank order from lowest to highest, and set the cutoff at the desired level of statistical significance for the assay (e.g., 95%, 97.5%, or 99%).
Another statistical method for determining assay cutoffs is to plot the data on a receiver operating characteristics (ROC) curve. On a traditional ROC curve, the true positive (TP)1 rate (DSn) is plotted on the ordinate of a graph versus the false positive rate (1-DSp) on the abscissa (Zweig and Campbell 1993)—i.e., the benefit of the true positive result is plotted versus the cost of a false positive result. Unfortunately, the results of the ROC curve are not intuitive and can be difficult for the unfamiliar to interpret. The development of a modified ROC curve, in which the assay DSn and DSp are both plotted on the ordinate of a graph as a function of assay cutoff value (plotted on the abscissa), has made the results of the curve more intuitive (Greiner et al. 1995, 2000). On a modified ROC curve it is easy to determine the assay cutoff(s) that maximize the intended goal of the assay. For example, if the goal is to maximize the assay's DSn and DSp, then the cutoff line is set at the intersection of the two curves.
A final method of assay cutoff determination is empirical: the data are plotted as a frequency distribution pattern as in Figure 4 and a line or lines are drawn to maximize the assay performance characteristics that are consistent with the goal of the assay in the population of animals under consideration. Many laboratories draw two cutoff lines, as in lines A and B in Figure 4: results to the left of A are classified as negative, results to the right of B are classified as positive, and results between the two lines are classified as intermediate. Intermediate results could represent a response that is either a "noisy" negative or an early positive result. For samples classified as intermediate, either an additional sample obtained one to several weeks later or additional diagnostic testing using other assays on the original serum sample may help to determine whether the sample should be classified as positive or negative.
Assay Validation
Assay validation is a formal process for determining the suitability of a given laboratory method for generating the data necessary to calculate assay performance characteristics and to develop an assay validation report (Jacobson 1998; Jacobson and Romatowski 1996). The data that result from assay validation are used to calculate population-specific assay performance characteristics such as DSn and DSp (where the "population" is the samples collected for assay validation).
For serodiagnostic assay validation, a predetermined number of known positive and known negative serum samples (see formula 1 above) are repeatedly tested by different technicians on different days, often under nonideal conditions (for example, with slight differences in incubation times, temperatures, or concentrations of certain critical reagents). Table 1 provides a list of variations in assay performance tests to demonstrate assay repeatability, robustness, and ruggedness.
Table 1.
Variations in assay erformance methods on a single set of serum samples to assess assay repeatability, robustness, and ruggedness
| Variation | Demonstrated trait |
|---|---|
| Multiple assay performances | Repeatability |
| Performance by different people or laboratories | Robustness |
| Minor alterations in assay conditions (e.g., temperature, incubation time) | Ruggedness |
Although the process of assay validation may sound daunting and should not be taken lightly, by the time an assay makes it to the formalized process of validation, its performance characteristics should be well known and the results of the validation process should be a foregone conclusion.
Calculation of Diagnostic Sensitivity and Specificity
With the completion of the validation study and the classification of serum samples as positive or negative, it is possible to calculate population-specific assay performance characteristics such as DSn and DSp. A useful way to accomplish this calculation is to enter the validation study data in a 2 x 2 contingency table (Figure 5) and compare them to the known (expected) infection status of the samples. Data from the validation study that agree with the known positive and negative status of the sera in question are classified as true positive (TP) and true negative (TN), respectively. Data from the validation study that do not agree with the known positive and known negative status of the sera in question are classified as false negative (FN) and false positive (FP), respectively. From this information the DSn and DSp can be calculated thus:
Figure 5.
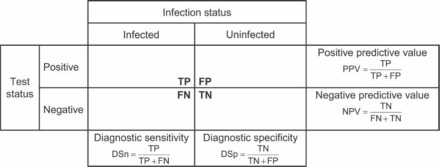
A 2 x 2 contingency table used to classify serum samples and to assess population-specific assay performance characteristics (DSn and DSp) as well as the predictive values for the assay in the test population. DSn, diagnostic sensitivity; DSp, diagnostic specificity; FN, false negative; FP, false positive; NPV, negative predictive value; PPV, positive predictive value; TN, true negative; TP, true positive.
| (2) |
| (3) |
The above formulas result in decimal equivalents, whereas DSn and DSp are most commonly reported as percentages. It is also important to note that these assay performance characteristics are population specific and vary according to the individual characteristics of the population being sampled (tested). Thus, validation studies performed on a large number of samples that broadly represent the characteristics of the population being tested will provide the most reliable and universally applicable estimates of DSn and DSp. Conversely, the use of a small number of carefully chosen positive and negative serum samples for the validation study can easily result in assays with a DSn and DSp of 100%, which is neither realistic nor representative of any real population of serum samples. So, while DSn and DSp are the most commonly used assay performance characteristics, they must be interpreted by understanding the population from which the serum samples were drawn. It is essential to thoroughly review unrealistically high values for DSn and DSp before using them to interpret assay results.
Quality Assurance in Diagnostic Testing Laboratories
The goal of a quality assurance program is to enhance the confidence of both the laboratory and the consumer in the reported diagnostic test results (MacWilliams and Thomas 1992). The reliability of these results depends on the constant monitoring of serodiagnostic assays during routine use to verify that they are performing within predetermined assay performance specifications. It is therefore useful to develop a quality assurance (QA) program that monitors as many steps as possible in reporting assay results, including sample processing, sample- and assay-specific components, and data analysis and reporting.
System and Sample Suitability Controls
Monitoring serodiagnostic assays during routine use requires the development of a complement of controls that assess as many components of the assay system as possible. Assay-specific controls typically fall into two categories: those that assess the system and those that assess the sample.
System suitability controls assess the fitness of the assay components and equipment for assay performance and thus ensure that the assay is performing within specifications. Common system suitability controls include high and low positive and negative controls as well as diluent controls, to ensure that the assay is providing reliable positive and negative results. In multiplexed assays it is also possible to include additional internal system suitability controls with each sample, such as a species-specific immunoglobulin G (IgG) control, which is an internal positive control that indicates whether all of the reagents necessary for a positive reaction (e.g., secondary antibody, reporter molecule) have been added to the sample assay well and whether the machinery is functioning properly when the sample is read (Martins 2002, 2003). Internal positive controls allow for more confidence in the validity of negative results when data are interpreted since they identify problems in an assay system that could produce false negative results.
Sample suitability controls assess the fitness of the sample itself for assay performance and often include tissue controls (lysates of one or more cell lines that were used to produce the antigens in the assay). The sample suitability controls indicate whether an animal has developed antibodies against cell culture proteins, insect cells, bovine serum albumin, or other contaminating cell culture components, any of which could result in a false positive assay response (Pedersen et al. 1986). In multiplexed assays it is also possible to include additional internal sample suitability controls in each sample well, such as an antispecies IgG control, which binds to a small amount of the IgG in the serum sample and ensures both that the serum has been added to the assay well and that it was not degraded in transport, handling, or storage (Martins 2002, 2003).
Both system and sample suitability controls are necessary with every assay and need to be reviewed with every assay run to ensure that they are performing within specifications before results of individual serum samples are interpreted or reported.
Accuracy and Precision
The goal of a quality assurance program is to provide results that are both accurate and precise. Accuracy1 is a measure of the agreement between a measured test value and the expected or "true" value for that sample (Figure 6). For serodiagnostic testing, correct classification of a sample as negative, intermediate, or positive determines accuracy. It is often quite difficult to determine the "true" status of any individual serum sample, so system suitability controls should be included with every assay run or plate to monitor serodiagnostic accuracy; positive and negative serum controls and a diluent control are most commonly used to accomplish this task. To challenge the system, the positive controls should include both a high and a low positive. The low positive control, which should be titrated so that it yields an assay result just above the assay positive cutoff value, assesses assay performance at the most critical point—where the distinction is made between positive and negative results.
Figure 6.

Diagram illustrating the difference between accuracy and precision: accuracy is how close the test value is to the "true" or known reference value, and precision refers to the assay result's reproducibility.
Precision1 is a measure of the reproducibility of a serodiagnostic test result and often is used as an indicator of the amount of random error in the system. Precision is different from and independent of accuracy: an assay can be extremely precise (i.e., it produces a result that is nearly identical with every run of the assay) but inaccurate (i.e., it incorrectly classifies the sample result), or vice versa. To assess assay precision, it is necessary to run aliquoted controls from the same control lot with every assay and compare the results to a predetermined standard for the control, by setting upper and lower values in the assay acceptance criteria for the high and low positive system suitability controls. Similar assay acceptance criteria can be set when anti-IgG controls are used in a multiplexed system, as an additional measure of assay precision. It is essential to perform these controls on every assay plate or in every run of the assay. The assay should not be accepted as valid if these controls fail.
Monitoring Serodiagnostic Assays for Variation over Time
Run-to-run variations in accuracy and precision lead to analytical error, which has two components: random error and systematic error. Random error is the result of erratic run-to-run variations in a diagnostic method that has a wide variety of sources and can be identified by a lack of precision (fluctuation) in assay controls over time. Systematic error is a sign of a consistent positive or negative bias in the assay results, and is often the result of effects such as deterioration of reagents or controls, drift in pipettor or instrument calibration, or deterioration in light sources or instrument readers over time.
How then does a diagnostic laboratory ensure the validity of serodiagnostic testing results over time? In the early 1930s Shewhart described a quality control process for manufacturing in which a single control value was tracked over time and the lot was rejected if it fell more than 3 SD from the expected mean value (Shewhart 1931). Levey and Jennings (1950) then applied Shewhart's quality control procedure to clinical diagnostic testing and introduced the L-J chart (Figure 7) as a simple mechanism to determine, by visual inspection, whether an individual assay run was in control or out of control (i.e., performing within predetermined specifications or not). Plotting control data from each run of an assay on an L-J chart allows a diagnostic laboratory to track control values and monitor them for trends that might develop. For example, on an L-J chart random error appears as increased dispersion about the expected mean (Figure 7A), and systematic error can be identified as a positive or negative trend in the data (Figure 7B). L-J charts are made by plotting the assay run number on the abscissa and the analyte value on the ordinate of a graph. The ordinate is typically labeled with the mean expected value of the analyte plus and minus 1, 2, and 3 standard deviations from the expected mean value.
Figure 7.
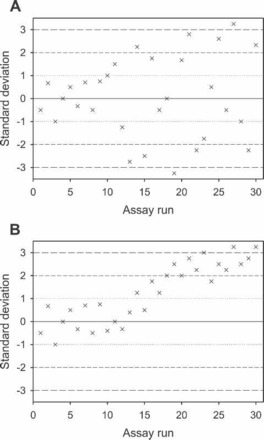
Levey-Jennings charts showing the mean value of a single control result (ordinate) versus the assay run number (abscissa). (A) shows increased dispersion (random error) in control results after assay run number 10, and (B) shows a bias or trend in the control data after assay run number 13 (systematic error).
Applying a single assay acceptance criterion to an individual control value does not, however, enable the laboratory to track an assay for signs of systematic error that might develop over multiple runs (like those shown in Figure 7). To address this concern, Westgard and colleagues (1981) proposed that four additional rules and one warning signal be added to Shewhart's original ±3 SD rule (Table 2). Westgard's rules include additional control limits at ±1 and ±2 SD that enable the laboratory to monitor control values for both dispersion and trends in the data that may develop over time. These control rules can be either applied to data plotted on an L-J chart and observed by visual inspection or programmed into a microcomputer for assessment (Eggert et al. 1987).
Table 2.
Westgard's rules for clinical reference standards. Clinical reference controls are considered to be out of control if any of thefollowing circumstances are observed, and the assay run should be rejected.a
| 12SD: | 1 control observation is more than ±3 SDb from the expected mean. |
| 22SD: | 2 consecutive control observations are more than 2 SD from and both control observations fall on the same side of the mean. |
| R4SD: | The difference between the largest and smallest control observation exceeds 4 SD. The two control observations must fall on opposite sides of the mean. |
| 41SD: | 4 consecutive control observations are greater than 1 SD from and fall on the same side of the mean. |
| 10 x: | 10 consecutive assay runs fall on the same side of the mean. |
| 12SD: | Warning rule that triggers more thorough inspection of control results if a single value exceeds the expected mean±2SD. |
Data from Westgard JO, Barry PL, Hunt MR, Groth T. 1981. A multi-rule Shewhart chart for quality control in clinical chemistry. Clin Chem 27:493-501.
SD, standard deviation from the mean value.
One caveat to bear in mind when applying control rules to clinical data is that random chance may result in the false rejection of data that are actually in control. For the ±3 SD rule, 99.73% of all normal data should be captured within ±3 SD of the mean. Thus there is a 1/370 chance of false assay rejection when the control value is greater than ±3 SD from the mean expected value; put another way, if a control value exceeds the expected mean ±3 SD 369 out of every 370 times, there is a problem with the assay system. So the likelihood of falsely rejecting a control run using a single control rule is fairly remote (0.27%), but there is a similar probability of false rejection with each control rule used (Carroll et al. 2003), and the overall probability of false rejection in a multirule quality control program is the sum of the individual probabilities of false rejection for each rule (Clifford 2001). Diagnostic laboratory personnel must therefore review routine quality control data that fall outside predetermined specifications to establish whether the data are valid or not. Interpretation of the quality control data will determine whether the assay run is in control and valid, or out of control and not valid.
Interpretation of Diagnostic Test Results
The goal of serodiagnostic testing is to use the resulting information to make management decisions about the infection status of animals from which the sera were taken. However, because of the inherent limitations of assay performance, both FN and FP classifications occur even with well-validated assays that have very good performance characteristics (Figure 4).
One common mistake in interpreting diagnostic testing results is the assumption that an assay with a DSn of 95% will result in 95 TP and 5 FN results for every 100 samples tested, and conversely that an assay with a DSp of 92% will result in 92 TN and 8 FP results for every 100 samples tested. In fact, it is not possible to calculate the number of TP, TN, FN, and FP results from the DSn and DSp without knowing (or being able to estimate) the prevalence1 of the disease in the test population.
Positive and Negative Predictive Values
Knowledge of the DSn, DSp, and disease prevalence in the target population makes it possible to calculate the positive and negative predictive values (PPV and NPV1) of the assay. The PPV is the probability that an animal that tests positive for an infectious agent or disease is truly infected with the agent. Conversely, the NPV is the probability that an animal that tests negative for an infectious agent is actually normal or not infected by the agent.
The PPV and NPV vary greatly with the prevalence of disease in the target population. For example, Figure 8 shows how to calculate the PPV and NPV if an infectious agent has an expected prevalence of 30% in a population of 10,000 animals and the assay used to detect the agent has a DSn of 98% and a DSp of 95%. From this calculation the resulting PPV is 89.4% and the NPV is 99.1%, both of which strongly support confidence in the assay's positive and negative results when the prevalence of disease in the population is 30%.
Figure 8.

Calculation of assay predictive values given an assay with a diagnostic sensitivity (DSn) of 98%, a diagnostic specificity (DSp) of 95%, and a disease prevalence of 30% in a target population of 10,000 animals.
Interpreting the Results for an SPF Population
Now, what will happen with the application of the same assay, which gave excellent results when the prevalence of disease in the population was 30%, to a specific pathogen-free (SPF) population? Because zeros often do not work well in mathematics and because it is rarely possible to have absolute confidence that a population is SPF, it is useful to approximate the SPF population by assuming that its prevalence of disease is near zero (0.1%; Figure 9). With the application of an assay with the same DSn and DSp to a population with a disease prevalence of only 0.1%, the PPV drops to 2% and the NPV increases to 100%. These results support very good confidence in the negative results from this population of animals, but indicate that only 2 out of every 100 animals that test positive are truly infected with the agent, even though the assay is identical to the one that gave outstanding results when the disease prevalence in the target population was 30%.
Figure 9.
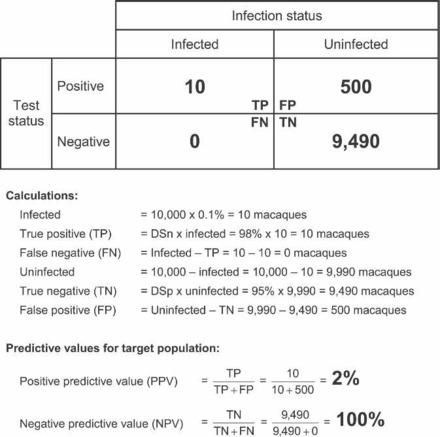
Calculation of assay predictive values given an assay with a diagnostic sensitivity (DSn) of 98%, a diagnostic specificity (DSp) of 95%, and a disease prevalence of 0.1% in a target population of 10,000 animals.
The Importance of Knowing Disease Prevalence
The example shown in Figure 9 illustrates the conundrum that confronts diagnostic laboratories and clinicians when interpreting unexpected positive test results in a population of animals that either have a low prevalence of disease or are believed to be SPF: the PPV of the assay indicates that the result is most likely an FP but there is a chance, albeit very small, that the result is correct. In this scenario, additional diagnostic testing using more specific diagnostic assays is advisable so that the laboratory can attempt to sort out the true diagnostic test result for the sample. Additional serodiagnostic tests that often have greater DSp, and thus a lower rate of FP results, include IFA, WIB, hemagglutination inhibition, and PCR.
Other information should be considered as well in the interpretation of an unexpected serodiagnostic test result. For example, was there a possible risk of exposure to the agent? How long has the colony been SPF and was the agent in question previously present in the population? Does a sporadic positive result fit with the expected biology of the virus or with the epidemiology of the agent in a naïve population?
The two examples described above illustrate several critical issues in the interpretation of serodiagnostic test results. First, it is important to know the population-specific assay performance characteristics (DSn and DSp) and whether the population from which these values were determined is similar to the population of animals being tested. Second, it is very important to either know the expected prevalence of the agent in the population being tested or be able to estimate the prevalence from previous experience, clinical impression, or potential risk of exposure analysis. Knowing the DSn, DSp, and disease prevalence one can then calculate the positive and negative predictive values for the assay as in Figures 8 and 9.
Interestingly, as long as the DSn and DSp are above 50%, the predictive values for an assay follow the same trends: as the prevalence of an agent in a population falls to zero, so does the PPV (Figure 10A); the NPV, however, increases to 100% (Figure 10B). The converse is also true: as the prevalence of an agent in the population approaches 100% so does the PPV for that population (Figure 10A), while the NPV falls to zero (Figure 10B). Because of these trends in predictive value curves, if the DSn, DSp, and disease prevalence can be estimated with reasonable accuracy, then the PPV and NPV for a given diagnostic test result can be interpolated as shown in Figures 10A and B, respectively.
Figure 10.
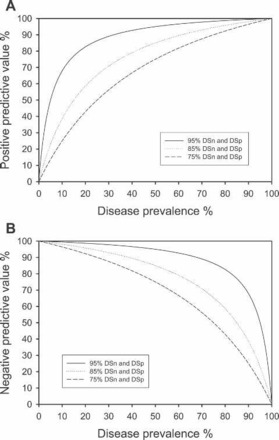
Calculated curves for positive predictive value (A) and negative predictive value (B) versus disease prevalence for assays with a diagnostic sensitivity (DSn) and specificity (DSp) of 95%, 85%, and 75%. These curves can be used to interpolate the predictive values for an assay if the DSn, DSp, and disease prevalence in the population either are known or can be estimated with reasonable accuracy.
Conclusion
Serodiagnostic assay development begins with the identification and accumulation of high-quality antigens and serum samples that represent a wide range of animals known to be positive and negative for the agent in question. Assay development and validation determine both assay-specific (ASn and ASp) and population-specific (DSn and DSp) assay performance characteristics. Assay validation does not end with implementation of a serodiagnostic test but rather is an ongoing process that requires the replacement and requalification of consumed reagents and the refinement of the assay's performance characteristics to address changing populations and diagnostic testing requirements.
When diagnostic assays are implemented, they must also include a complement of system and sample suitability controls. A rigorous quality control (QC) program often includes as many as eight control assays to determine whether the assay is performing within specifications and the sample is appropriate for the assay. System and sample suitability controls may indicate that the results are valid but not necessarily that they are correct, as all assays are prone to both false positive and false negative results. To limit day-to-day variation in reported assay results, the diagnostic laboratory should implement a process for tracking and monitoring QC results over time; L-J charts, for example, are an effective way to track control data.
Given the risk of false positive and false negative results, the clinician must consider the possibility that diagnostic test results do not reflect the true infection status of the animal. It is therefore important to carefully interpret all such results before making management decisions about the animal in question. When determining an animal's infection status (i.e., interpreting the result), a clinician can make an informed decision about how much weight to assign to the reported diagnostic result by using the assay's DSn and DSp and by having a reasonable estimate of the expected prevalence of the disease in the population of animals being studied.
The assay's predictive values are useful tools for the interpretation of diagnostic test results; however, if this information is unknown or if it is unavailable, the predictive values can also be estimated from the curves of the predictive value versus the population's disease prevalence. All unexpected results should be confirmed by additional diagnostic testing before management decisions are made about the animals in question.
Glossary
- accuracy:
how close an assay result is to the true value
- assay:
a procedure used to approximate infection or disease status
- assay-specific performance characteristics:
- analytical sensitivity (ASn):
smallest detectable amount of the analyte in question, or the limit of detection of the assay
- analytical specificity (ASp):
the degree to which the analyte in question cross reacts with other analytes, or the selectivity of the assay
- confirmation assay:
generally a lower throughput assay, such as an indirect fluorescent antibody (IFA) test or western immunoblot (WIB), that is often used to confirm the results of a screening assay. Confirmation assays are often biased to have a high diagnostic specificity and thus fewer false positive responses
- false positive (FP):
identification of a normal animal as test positive
- false negative (FN):
identification of an infected or diseased animal as test negative
- normal:
absence of the disease or infection of interest
- population-specific performance characteristics:
- diagnostic sensitivity (DSn):
- probability of correctly identifying true positive (infected or diseased) animals
- diagnostic specificity (DSp):
- probability of identifying normal or true negative animals
- precision:
degree of variability in an assay result
- predictive values:
- negative predictive value (NPV):
- probability that an animal that tests negative for an infectious agent or disease is truly normal
- positive predictive value (PPV):
- probability that an animal that tests positive for an infectious agent or disease is truly positive
- prevalence:
the proportion of a population that has the infection or disease of interest
- qualification:
a process of ensuring the quality of a given reagent, antigen, assay, etc. by comparison to previous versions of the same reagent
- quality assurance:
a program to create, monitor, and enhance confidence in diagnostic testing results
- screening assay:
generally a high-throughput assay, such as an enzyme-linked immunosorbent assay (ELISA) or multiplexed fluorescent immunoassay (MFIA), that is biased to have a high diagnostic sensitivity and thus fewer false negative responses
- titer:
highest dilution of a sample that is capable of producing a positive test result (limit of detection)
- true positive (TP):
identification of a diseased or infected animal as test positive
- true negative (TN):
identification of a normal animal as test negative
- validation:
a process of determining the suitability of a given laboratory method for providing useful analytical data
Footnotes
The definitions of this and other terms used in this article are included in the Glossary on pages 168-169.
References
- Barajas-Rojas JA, Riemann HP, Franti CE. 1993. Notes about determining the cutoff value in enzyme linked immunosorbent assay (ELISA). Prev Vet Med 15:231–233. [Google Scholar]
- Barlough JE, Jacobson RH, Pepper CE, Scott FW. 1984. Role of recent vaccination in production of false-positive coronavirus antibody titers in cats. J Clin Microbiol 19:442–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TA, Pinnick HA, Carroll WE. 2003. Probability and the Westgard rules. Ann Clin Lab Sci 33:113–114. [PubMed] [Google Scholar]
- Clifford CB. 2001. Samples, sample selection and statistics: Living with uncertainty. Lab Anim (NY) 30:26–31. [DOI] [PubMed] [Google Scholar]
- Cook RF, Gann SJ, Mumford JA. 1989. The effects of vaccination with tissue culture-derived viral vaccines on detection of antibodies to equine arteritis virus by enzyme-linked immunosorbent assay (ELISA). Vet Microbiol 20:181–189. [DOI] [PubMed] [Google Scholar]
- Eggert AA, Westgard JO, Barry PL, Emmerich KA. 1987. Implementation of a multirule multistage quality control program in a clinical laboratory computer system. J Med Syst 11:391–411. [DOI] [PubMed] [Google Scholar]
- Greiner M, Sohr D, Gobel P. 1995. A modified ROC analysis for the selection of cutoff values and the definition of intermediate results of serodiagnostic tests. J Immunol Methods 185:123–132. [DOI] [PubMed] [Google Scholar]
- Greiner M, Pfeiffer D, Smith RD. 2000. Principles and practical application of the receiver-operating characteristic analysis for diagnostic tests. Prev Vet Med 45:23–41. [DOI] [PubMed] [Google Scholar]
- Jacobson RH. 1998. Validation of serological assays for diagnosis of infectious diseases. Rev Sci Tech 17:469–526. [DOI] [PubMed] [Google Scholar]
- Jacobson RH, Romatowski J. 1996. Assessing the validity of serodiagnostic test results. Semin Vet Med Surg (Small Anim) 11:135–143. [DOI] [PubMed] [Google Scholar]
- Levey S, Jennings ER. 1950. The use of control charts in the clinical laboratory. Am J Clin Pathol 20:1059–1066. [DOI] [PubMed] [Google Scholar]
- Levinson SS. 1992. Antibody multispecificity in immunoassay interference. Clin Biochem 25:77–87. [DOI] [PubMed] [Google Scholar]
- MacWilliams PS, Thomas CB. 1992. Basic principles of laboratory medicine. Semin Vet Med Surg (Small Anim) 7:253–261. [PubMed] [Google Scholar]
- Martins TB. 2002. Development of internal controls for the luminex instrument as part of a multiplex seven-analyte viral respiratory antibody profile. Clin Diagn Lab Immunol 9:41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins TB. 2003. The application of true internal controls to multiplexed fluorescent immunoassays. J Clin Ligand Assay 26:93–97. [Google Scholar]
- Pedersen NC, Lowenstine L, Marx P, Higgins J, Baulu J, McGuire M, Gardner MB. 1986. The causes of false-positives encountered during the screening of old-world primates for antibodies to human and simian retroviruses by ELISA. J Virol Methods 14:213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewhart WA. 1931. Economic Control of Quality of the Manufactured Product. New York: Van Nostrand. [Google Scholar]
- Westgard JO, Barry PL, Hunt MR, Groth T. 1981. A multi-rule Shewhart chart for quality control in clinical chemistry. Clin Chem 27:493–501. [PubMed] [Google Scholar]
- Zweig MH, Campbell G. 1993. Receiver-operating characteristic (ROC) plots: A fundamental evaluation tool in clinical medicine. Clin Chem 39:561–577. [PubMed] [Google Scholar]