


Abstract
The AID/APOBEC enzymes deaminate cytosines in single-stranded DNA (ssDNA) and play key roles in innate and adaptive immunity. The resulting uracils cause mutations and strand breaks that inactivate viruses and diversify antibody repertoire. Mutational evidence suggests that two members of this family, APOBEC3A (A3A) and APOBEC3B, deaminate cytosines in the lagging-strand template during replication. To obtain direct evidence for the presence of these uracils, we engineered a protein that covalently links to DNA at uracils, UdgX, for mammalian expression and immunohistochemistry. We show that UdgX strongly prefers uracils in ssDNA over those in U•G or U:A pairs, and localizes to nuclei in a dispersed form. When A3A is expressed in these cells, UdgX tends to form foci. The treatment of cells with cisplatin, which blocks replication, causes a significant increase in UdgX foci. Furthermore, this protein- and hence the uracils created by A3A- colocalize with replication protein A (RPA), but not with A3A. Using purified proteins, we confirm that RPA inhibits A3A by binding ssDNA, but despite its overexpression following cisplatin treatment, RPA is unable to fully protect ssDNA created by cisplatin adducts. This suggests that cisplatin treatment of cells expressing APOBEC3A should cause accumulation of APOBEC signature mutations.
INTRODUCTION
Uracil is a rare base in DNA but plays an important role in a number of different organisms and biological processes. As dUTP is a precursor for TTP, some level of dUTP exists in most cells. The bacterial and eukaryotic DNA polymerases do not discriminate against this nucleotide and they occasionally incorporate dU instead of dT across an adenine in DNA (1). The second source of uracils in DNA is through the deamination of cytosines in DNA (2). This may be caused by water within cells, exposure to chemicals such as bisulfite (3) or by the vertebrate AID/APOBEC family enzymes (4). Of these proteins, the APOBEC3 enzymes (APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D, APOBEC3F, APOBEC3G and APOBEC3H) play an important role in innate immunity, where their expression in response to virus-induced cytokines creates mutations in viral genomes (5–7). Another member of this family, AID, is required for two processes essential for antibody diversification, somatic hypermutation and class-switch recombination, and targets cytosines in the immunoglobulin genes (4,8).
Although the amount of uracil in most genomes is small, about 1 uracil per 106 bp, its level can be as high as 2000–3000 uracils per 106 bp depending on the organism, cell type and the genetic background (9,10). Such quantitation of uracils in DNA has been accomplished using a number of different methods. The quantification methods include ELISA-like assays (e.g. (11)), a PCR-based assay (12) and LC/MS/MS analysis (e.g. (13)). More recently, a number of groups have succeeded in mapping uracils in whole genomes. This is generally accomplished using selective whole-genome re-sequencing. These uracil-mapping techniques require the excision of uracils by a uracil–DNA glycosylase followed by either conversion of the resulting abasic site to a strand break (14) or a biotinylated chemical species (15,16). These tagged DNA fragments are then sequenced on Next-Gen sequencing platforms and the obtained sequences are mapped to the known genome sequences. While useful, none of these methods are able to identify uracils in intact cells and nuclei. Consequently, they cannot answer many biological and biochemical questions about the creation of uracils. For example, they cannot easily determine what proteins were present near uracils when they were created or what cellular processes may inhibit their creation.
The AID/APOBEC enzymes strongly prefer single-stranded DNA (ssDNA) and such DNA occurs in the lagging strand template during replication, non-template strand during transcription, telomeres, recombination intermediates and non-B DNA structures such as G-quadruplexes (17–21). However, whether these enzymes act upon cytosines in all these potential targets is unknown. AID, causes mutations within a few kilobases in the immunoglobulin genes at a very high frequency (22), but it also causes mutations in many non-immunoglobulin genes at a lower frequency (23,24). Because of these off-target effects, AID plays a key role in promoting B cell cancers (25,26) and uracil load is very high in the genomes of many B lymphocyte-derived tumors (27,28). Additionally, analysis of cancer genome sequences has revealed that two members of the AID/APOBEC family, APOBEC3A and APOBEC3B, play a major role in creating mutations during the growth of different types of tumors strongly suggesting that under some physiological conditions these enzymes can target cytosines in cellular genes (29–31). Consequently, there is a need for a technique that can localize uracils created by the AID/APOBEC enzymes at a cellular level and which does not require complex biochemical manipulations or Next-Gen sequencing.
We describe below the development of a method by which uracils in DNA can be detected in situ in mammalian cells using an unusual protein from Mycobacterium smegmatis, UdgX (32). This family 4 uracil-DNA glycosylase (UDG), excises uracils from DNA and covalently links to the resulting abasic site through a histidine (Supplementary Figure S1). It does not excise normal DNA bases and its covalent link with abasic sites is stable under harsh conditions used for denaturing gels-boiling of samples in the presence of formamide under strong alkaline conditions and electrophoresis in 8 M urea gels (32). This protein has been expressed in Escherichia coli (32,33), purified to homogeneity and the structure of its covalent complex with ssDNA substrate has been reported (34,35). We have now expressed this protein in mammalian cells and used it to detect uracils in the cellular genome using immunohistochemistry. We demonstrate usefulness of this approach by confirming the known preference of the APOBEC enzymes for single-stranded DNA in replication forks (36–39) under normal growth conditions and when replication is thwarted by cisplatin adducts.
MATERIALS AND METHODS
Construction of UdgX expression plasmids
The pET-14b-polyHis-mCherry-UdgX was generously provided by Dr. Umesh Varshney (Indian Institute of Science, Bangalore, India). This mCherry-UdgX protein was purified from E. coli to near homogeneity and was found to have poor activity towards uracil-containing single-stranded DNA (ssU; Supplementary Table S1A; data not shown). To improve the activity of this protein, the mCherry gene was deleted from this plasmid as a NdeI fragment to create pHis-UdgX (Supplementary Figure S2A). This new construct contained 10 amino acids between the polyHis-tag and the N-terminus of UdgX and had higher activity than the mCherry-UdgX protein. However, it did not form a covalent complex with ssU at 1:1 molar ratio. To improve the activity of UdgX, its gene was recloned into FLAG-HA-pcDNA3.1 plasmid (Addgene, Watertown, MA) to fuse it with the FLAG and HA tags separated by flexible polyGly linkers (Supplementary Figure S2B). The FLAG-HA-pcDNA3.1 vector has both CMV and T7 promoters but lacks a ribosome-binding site for E. coli expression. Using site-directed mutagenesis (SDM) a Shine-Delgarno sequence was introduced 5 bp upstream of the start codon (Supplementary Figure S2B) using the primers listed in the Supplementary Table S1B. The UdgX gene was amplified from pHis-UdgX using primers listed in Supplementary Table S1B to create pFLAG-UdgX (Supplementary Figure S2B). The H109S mutation was engineered into the UdgX gene in pFLAG-UdgX using primers listed in Supplementary Table S1B. The pEGFP-N3-A3A plasmid was obtained from Dr Reuben Harris (University of Minnesota) and the generation of its catalytically inactive pEGFP-N3-A3A(E72A) mutant has been described previously (40). The pET28a+-based A3A expression plasmid (pA3A-His) has also been described previously (41). All the new plasmid constructs were subjected to Sanger sequencing (University of Michigan sequencing core) to confirm their accuracy.
Expression and purification of proteins
pHis-UdgX was introduced into E. coli strain BL21(DE3) and grown to an OD600 of 0.8. The expression of UdgX was induced using 0.5 mM Isopropyl ß-d-1-thiogalactopyranoside (IPTG) and 0.01%FeCl3 and grown at 18°C for 18 h. The cells were pelleted and resuspended in 30 mL of lysis buffer (20 mM Tris–Cl pH 7.5, 500 mM NaCl, 2 mM beta-mercaptoethanol (BME), 0.01% FeCl3, 10% glycerol, 10 mM imidazole) supplemented with cOmplete™ protease inhibitor cocktail (Thermofisher). Using a French Press, cells were broken at 1500 psi and the lysate clarified using centrifugation of 16 000g for 30 min. Ni-NTA agarose (Thermofisher) was washed in lysis buffer and was added to the clarified lysate. The suspension was rotated at 4°C overnight. The Ni-NTA agarose was successively washed with Buffer A (20 mM Tris–Cl pH 7.5, 500 mM NaCl, 2 mM BME, 0.01% FeCl3, 10% glycerol) containing 20 mM imidazole and 50 mM imidazole. The protein was eluted using 500 μL aliquots of Buffer A containing 250 mM imidazole. The elutions found to have the protein in an SDS-PAGE gel were combined and dialyzed using Buffer A. The protein was further purified using an AKTA pure FPLC equipped with a sephadex G-50 gel filtration resin. The peaks of UdgX fractions were concentrated to 4 mg/mL using a 10 kDa MWCO centrifugal filter (ThermoFisher) and the buffer was exchanged for UdgX storage buffer (20 mM Tris–Cl pH 7.5, 500 mM NaCl, 2 mM BME, 0.01% FeCl3, 50% glycerol) for storage at –80°C.
pFLAG-UdgX and the pFLAG-UdgX(H109S) were transformed into the same strain of E. coli and the proteins expressed using the same conditions as described above for the polyHis-UdgX. The cell pellets were resuspended in 30 mL of FLAG Buffer A (20 mM Tris–Cl pH 7.5, 500 mM NaCl, 10% glycerol) and broken using a French Press. Anti-FLAG M2 magnetic beads (Sigma) were washed in 1× TBS, then added to the cleared cell lysate and the mixture was rotated at 4°C overnight. The beads were separated from the supernatant using a DynaMag-2 magnet (Thermofisher) and washed three times with 1× TBS. Protein was eluted with 500 μL of FLAG Elution Buffer (3× FLAG peptide at 150 ng/μL in 1× TBS). The eluted FLAG-UdgX fractions were concentrated to 2 mg/mL using a 10 kDa MWCO centrifugal filter (ThermoFisher) and the buffer was exchanged for UdgX storage buffer for storage at –80°C. The purity of all the preparation was assessed using SDS-PAGE (Supplementary Figure S3).
The purification of A3A was performed as described previously, with small modifications (41). The plasmid pA3A-His was transformed into BL21(DE3), A3A expression was induced using 0.5 mM IPTG when the cells reached OD600 of 0.6 and the cells were grown at 18°C for additional 18 h. The cells were pelleted and resuspended in 30 mL of lysis buffer (20 mM Tris–Cl pH 8.0, 500 mM NaCl, 50 mM NaCl, 10 mM imidazole) supplemented with cOmplete™ protease inhibitor cocktail (Thermofisher). Cells were broken using a French Press, the lysate clarified, using centrifugation and the supernatant was incubated with the Ni-NTA agarose as described above. The agarose was successively washed with Buffer B (20 mM Tris–Cl pH 8, 50 mM imidazole) containing 50, 250 or 500 mM NaCl. The A3A protein was eluted from the agarose using 500 μL aliquots of elution buffer (20 mM Tris–Cl pH 8.0, 50 mM NaCl, 250 mM imidazole). The elution fractions found to have the protein when analyzed on an SDS-PAGE gel were combined and dialyzed using Buffer C (20 mM Tris–Cl pH 8.0, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, 10% glycerol). The protein was concentrated to 10 mg/mL using 10kDa MWCO centrifugal filter and stored at –80°C.
RPA was purified by co-expressing RFA3-pCDF (His-RFA3) and RFA1-2-pET (RFA1 and RFA2) in BL21 (DE3) pLyS competent cells under ampicillin and kanamycin selection (42). Cells were grown at 25°C to an OD600 of 0.8 and induced with 0.5 mM IPTG for 20 h at 16°C. The cells were lysed in a French Press and pelleted in a Beckman JA-20 rotor at 20 000 RPM for 1 h, and the supernatant was brought to 20 mM Tris–HCl pH 7.9, 200 mM NaCl, 1mM PMSF, 1mM TCEP and 20 mM Imidazole. The clarified lysate was added to a 5 mL Ni-NTA column (Cytiva) and washed with 40 column volumes of buffer containing 20 mM Tris pH 7.9, 600 mM NaCl, 1 mM TCEP, and 5% glycerol. RPA was eluted with a gradient of 20–500 mM Imidazole in a buffer containing 200 mM NaCl, 20 mM Tris–HCl pH 7.9, 1 mM TCEP and 5% glycerol. Peak fractions were diluted 2-fold with 20 mM Tris–HCl and 5% glycerol to bring the final solution to 100 mM NaCl. The dilution was quickly loaded onto a 1 ml Mono Q anion exchange column (Cytiva) and eluted with a gradient of 100–500 mM NaCl in a buffer containing 20 mM Tris–HCl pH 7.9, 5% glycerol and 1 mM TCEP. Peak fractions were aliquoted and stored at –80°C. The purity of A3A and RPA preparations were assessed using SDS-PAGE (Supplementary Figure S9).
Comparison of FLAG-UdgX and polyHis-UdgX Activities
The activities of polyHis-UdgX and FLAG-UdgX were compared using a 6-FAM labeled ssU24 substrate (Supplementary Table S1A). These reaction mixtures contained either UDG buffer (50 mM Tris–HCl pH 8.0, 1 mM Na2EDTA, 1 mM DTT) or UdgX buffer (350 mM Tris–HCl pH 8.0, 5 mM Na2EDTA, 5 mM DTT, 250 μg/ml BSA). These reactions were incubated at 37°C for 1 h and then stopped by adding 0.1 M NaOH and boiling at 95°C for 10 min. Formamide dye was added to 50% (v/v) and the reaction products were separated on a 15% denaturing polyacrylamide gel and visualized by scanning for Cy2 fluorescence and the band intensities were quantified using ImageJ software.
Inactivation of FLAG-UdgX by Imidazole
Imidazole was added to a preparation of FLAG-UdgX to a final concentration ranging from 1 to 1000 mM in the UdgX buffer. These mixtures were incubated at 4°C for 1 h. The ssU24 substrate (2 pmol) was added to the reaction and incubated at 37°C for 1 h. The reactions were stopped by adding 0.1 M NaOH and boiling at 95°C for 10 min. The reaction buffer was exchanged with 1× TBS three times using 3 kDa MWCO centrifugal filters to get rid of the imidazole. Formamide dye was added and the reaction products were separated and analyzed as described above.
Comparison of FLAG-UdgX for ssDNA and dsDNA substrates
The dsDNA substrates were created by annealing 6-FAM labeled ssU11 (Supplementary Table S1A) to complementary unlabeled oligomers G-complement or A-complement (Supplementary Table S1A) to generate respectively the double-stranded U·G or U:A containing substrates. The FLAG-UdgX protein was added to reactions containing ssU11, dsU·G, or dsU:A substrates in the UdgX buffer. After 1 h at 37°C, the reactions were terminated, and the products analyzed as described above.
FLAG-UdgX binding to ssU in the presence of genomic uracils
Escherochia coli strains GM31 (K-12 dcm6 thr1 hisG4 leuB6 rpsl ara14 supE44 lacY1 tonA31 tsx78 galK2 galE2 xyl5 thi1 mtl1) and CJ236 (F’CmR, dut- ung-1 thi- RelA1) were grown in LB media and their genomic DNA was extracted. The DNAs were digested with the restriction endonuclease HaeIII and different amounts of these DNAs were incubated with UdgX at 37°C for 10 min. The ssU11 substrate (2 pmol) was added to these reactions and incubation was continued for an additional one hour.
Cell growth and viability analysis
Human HEK293T and HeLa cells were obtained from ATCC (Manassas, VA, USA). The construction of the HeLa Tet-On A3A-EGFP (HTO-A3A-EGFP) cell line has been described previously (40). All the cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) of penicillin/streptomycin. To determine cell counts and viability, the cells were harvested using trypsinization, mixed at a 1:1 ratio with 0.4% Trypan blue solution (Sigma) and analyzed using a Bio-Rad TC20 cell counter. Counts for every sample were obtained using biological and technical replicates.
Manipulations of HEK293T cells
HEK293T cells were grown to 60% confluence in 8-well chamber slides and the cells were transfected with pFLAG-UdgX WT, pFLAG-UdgX H109S, pA3A-EGFP WT, or A3A-EGFP (E72A) using Lipofectamine 3000. In some experiments, the cells were treated with cisplatin for a duration of 18 h prior to plasmid transfections. All cells were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton-X100 and blocked for 1 h at 25°C with 10% goat serum. The FLAG-UdgX protein was detected using rabbit anti-DYKDDDDK primary antibody (Cell Signaling; 1:500 dilution in 1.5% goat serum) and incubated with the cells for 2 h at 25°C. The cells were further labeled with goat anti-rabbit IgG (H + L) Cy3-conjugated secondary antibody (Thermofisher; 1:1000 dilution) for 1 h at 25°C. Replication protein A (RPA) was detected using mouse anti-RPA antibody, clone RPA34–19 (Millipore Sigma; 1:500 dilution) and goat anti-mouse IgG (H+L) labeled with AlexaFluor 647. The slides were mounted with ProLong™ Gold Antifade Mountant with DAPI (Thermofisher). Cell images were collected using an Axiovert.A1 inverted microscope (Carl Zeiss) and high-resolution images were obtained using a Zeiss LSM 800 confocal microscope with an Airyscan detector, both equipped with oil immersion 63× objective lens (NA 1.4). Airyscan deconvolution and image processing was done using Zen Blue and FIJI (ImageJ) software. In some cases, image colors were inverted in FIJI to see UdgX foci more clearly.
Imaging of HTO-A3A-EGFP cells transfected with UdgX
HTO-A3A-EGFP cells were grown to 80% confluence and A3A expression was induced for 12 h using Doxycycline (Dox; 2 μg/ml). These cells were transfected with pFLAG-UdgX using Lipofectamine 3000 (Thermofisher) for 18 h with or without cisplatin (5 μM) treatment. Cells were fixed and stained for UdgX as described above. Images were acquired using an Axiovert.A1 inverted microscope (Carl Zeiss) with 40× and oil immersion 63X objective lens. Image processing was done using Zen lite software (Carl Zeiss) and in some cases, the colors were inverted in FIJI to clearly show the concentrated foci of UdgX.
Quantification of genomic uracils
HTO-A3A-EGFP and HeLa cells were grown to 60–80% confluence and the HTO-A3A-EGFP cells were induced with 2 μg/ml Dox for 24 h. The cells were trypsinized and the genomic DNA was extracted and digested with HaeIII. The quantification of uracils in the DNA samples was carried out using the alkoxyamine AA6 as previously described with minor changes (16,43). Briefly, digested DNA abasic sites were blocked using AA7 (10 mM; (11)). This DNA was sequentially treated with uracil-DNA glycosylase and AA6 (2 mM). The AA6-labeled DNA was purified by phenol:chloroform extraction and ethanol precipitation then dissolved in ddH2O. A strain-promoted azide-alkyne cyclization between the azide within AA6 and DBCO-Cy5 (1.6 μM) was carried out at 37°C for 2 h. This DNA was subsequently purified using a DNA Clean & Concentrator kit (ZYMO Research) then spotted on a Nylon membrane in technical replicates with a standard (Cy5-labeled CJ236 DNA; (43)). The membrane was scanned using a Typhoon FLA 9500 phosphorimager for Cy5 fluorescence. The fluorescence intensity of the standard was measured using FIJI then plotted to generate a standard curve. The sample intensities were measured in FIJI and were interpolated in the standard curve to determine the uracils per million base pair. These values were analyzed in GraphPad Prism 8 and the significance of the data values was determined using the Mann-Whitney U test with a 95% confidence interval.
Quantification of UdgX Foci
The UdgX signal in HTO-A3A-EGFP cells was detected using a Cy3 emission filter and the images were collected for each experimental condition. Each image field contained ∼10–15 cells positive for UdgX signal. Cells with a punctate appearance with more than five UdgX aggregates were identified as containing foci. Only those cells with detectable UdgX signal were used to calculate the percentage of cells with foci (i.e. percentage of cells with foci = 100 × (number of cells with foci/total number of cells with UdgX signal)). In samples with co-expression of A3A-EGFP and UdgX proteins, only cells with both A3A and UdgX signals were used to determine the foci ratio. The ratio of cells with foci in a single field was determined for 10 images in each experimental condition. Violin plots were generated in GraphPad Prism 8 and statistical significance was determined using Mann-Whitney U test with a 95% confidence interval.
Quantification of RPA signal in cisplatin treated cells
HEK293T cells were grown to 60% confluence with different concentrations of cisplatin for 24 h. The cells were immunostained using an anti-RPA antibody and DAPI to identify individual nuclei. Cell images were collected using an Axiovert.A1 inverted microscope equipped with a 20× objective. RPA images were obtained using the Cy5 emission filter and 400 ms exposure. Images obtained with the DAPI channel were converted to an 8-bit image then to binary masks using FIJI software. These masks were overlaid to the RPA images and each cell was analyzed for fluorescence intensity. For each concentration of cisplatin, ∼100 cells were analyzed and plotted using GraphPad Prism 8.
1D analysis of fluorescence signals in cisplatin treated HEK293T cells
A Zeiss LSM 800 confocal microscope with Airyscan was used to acquire images with four channels in one field: DAPI, GFP, Cy3 and AlexaFluor 647 (AF647). The images were deconvoluted using the Zen Blue software, the individual channels were merged, and an area of interest was defined by drawing an arrow across one cell nucleus. This cell was selected due to the presence of both UdgX and A3A-EGFP signals. The signal intensities across the defined area were measured in 0.285-micron increments and deposited into a table by the software. The intensities were plotted using GraphPad Prism 8 for each individual merged image.
2D analysis and quantitative colocalization analysis for HEK293T cells
HEK293T cells treated with cisplatin and transfected for expression of FLAG-UdgX and A3A-EGFP were visualized at 63× magnification (NA = 1.4) and deconvoluted as described previously. The images were obtained for multiple cells (n = 14) and used for quantitative colocalization analysis using Coloc2 plugin in FIJI. Image background was subtracted with a rolling ball radius of 50 pixels and the image was converted to 8-bit images. Coloc2 analysis of fluorescence signals was done for two channels (e.g. UdgX and RPA signal) at a time for a single cell. The nucleus of the cell was identified using the DAPI channel, this signal was converted to a binary mask and overlaid with the other channels. In the Coloc2 plugin, two channels of interest were defined, and the mask was selected as the region of interest for colocalization analysis. In addition, the Costes threshold was selected with the Costes’ significance test, the size of the point spread function was set at 3.0 and 100 Costes randomizations were requested. The Coloc2 software generates a Pearson correlation coefficient (PCC) with and without the threshold. Pearson values above the threshold define the colocalization of the signals analyzed. These values were accepted for the analysis if they met the following criteria: the below threshold Pearson value was equal to or close to zero and the Costes significance P-value was 1.0. This P-value means that none of the randomized images had a better correlation. This analysis was carried out for each sample and treatment type, and the PCCs were plotted in a violin plot generated in GraphPad Prism 8. Significance was determined using Mann–Whitney U test with a 95% confidence interval.
Z-stack intensity acquisition and colocalization analysis
Using the same field and cell analyzed for the 1D intensity analysis, the Z-stack was defined. There were 26 slices of 0.15 μM thickness in the Z-stack, and fluorescence intensity was determined in all four channels. The nucleus of the cell of interest was defined and the stack was deconvoluted using the built-in colocalization processing tool for the RPA and UdgX signals. A Costes threshold was applied and a PCC was generated for each slice. These coefficients were plotted across the Z-stacks and the strength of correlation (positive PCC) at each slice was represented as a heatmap. In parallel, DAPI signal at each slice was measured and plotted across the Z-stack to define the volume occupied by the nucleus. All the graphs were generated using GraphPad Prism 8 software.
RPA binding and A3A deamination assay
RPA (4 pmol) was incubated with a 6-FAM labeled 30-mer DNA oligomer containing a single A3A substrate site, 5′-TC (2 pmol; Supplementary Table S1C), in UDG buffer containing 5 mM MgCl2 at 25°C for 30 min. One picomole A3A was added to the reaction the reaction was continued at 37°C for 10 min. The deaminase reaction was stopped by boiling at 95°C for 10 min. To completely release the RPA from DNA, 200 pmol of the unlabeled form of the same 30-mer was added to the mixture and the reaction was continued at 25°C for an additional 15 min. This was followed by the addition of E. coli Ung protein (0.2 pmol; NEB) and the reactions were incubated at 37°C for 10 min. The reaction products were separated on a 15% denaturing polyacrylamide gel and visualized by scanning for Cy2 fluorescence and the band intensities were quantified using ImageJ software. In control experiments, following the addition of excess unlabeled oligomer, the DNAs were electrophoresed on an 8% native polyacrylamide gel and visualized as before to demostrate that the RPA was no longer bound to the 6-FAM labeled oligomer.
RESULTS
UdgX is inhibited by imidazole
In the previous studies, this protein was purified with a polyHis-tag, bound to a Ni-based affinity column and eluted using 500 mM (35) or 1000 mM imidazole (32–34). When we expressed polyHis-UdgX in E. coli (Supplementary Figure S2A), purified it to apparent homogeneity (Supplementary Figure S3), and tested it for binding to a uracil-containing DNA oligomer (ssU) using a buffer utilized by these investigators, the protein functioned poorly. Although the protein is a monomer and binds the DNA covalently, about four-fold molar excess of the protein was needed to quantitatively convert the DNA oligomer to its denaturation resistant form (Figure 1A). We were able to improve the binding somewhat by optimizing the buffer, but still a 2-fold excess of the protein was required for near 100% binding (Figure 1B). Several efforts to improve the activity of this protein were unsuccessful (data not shown).
Figure 1.

Binding of poly-His-tagged UdgX to uracils in single-stranded DNA. Homogeneous polyHis-tagged UdgX protein was reacted with a 6FAM-labeled oligomer containing a single uracil in E. coli uracil-DNA glycosylase reaction buffer (New England Biolabs, A) or a buffer optimized for UdgX (B). E. coli Ung was used in one reaction to confirm that the oligomer contained a uracil.
To address this problem, we recloned the UdgX gene into a different vector fusing it to an N-terminal FLAG tag (Supplementary Figure S2B) and then purified the protein over an anti-FLAG monoclonal antibody column. This preparation was also nearly homogeneous (Supplementary Figure S3) and, unlike the polyHis-tagged protein, gave quantitative conversion of the ssU oligomer to its covalent complex at a 1:1 molar ratio (Figure 2A). This discrepancy between the activities of polyHis- and FLAG-tagged proteins made us wonder whether the manner of purification of the polyHis-UdgX creates a partially inactive protein. In particular, we wondered whether the imidazole used during its purification replaces one or more histidine residues involved in catalysis or the binding of the Fe–S cluster (34,35). To test this, the FLAG-tagged UdgX was preincubated with increasing concentrations of imidazole and then used in the covalent binding assay. At 250 mM or higher concentration, imidazole clearly inhibited the enzyme eliminating its activity at 1 M (Figure 2B). Therefore, we suspect that some of the imidazole used to elute the polyHis-tagged protein from Ni-based affinity columns remains bound to the protein, replaces important histidine side-chains in the protein and inhibits its activity. For these reasons, we used only the FLAG-tagged UdgX in subsequent experiments.
Figure 2.

FLAG-UdgX is inhibited by imidazole. (A) Homogeneous FLAG-UdgX protein was reacted with a 6FAM-labeled oligomer containing a single uracil. The reactions were performed in UdgX optimized buffer and the products were separated on a denaturing gel. (B) FLAG-UdgX protein was pre-incubated with imidazole at the concentration shown and then reacted with the 6FAM-labeled oligomer in UdgX buffer. The imidazole was removed by buffer exchange and the products were analyzed on a denaturing gel.
UdgX prefers uracils in single-stranded DNA
We wanted to obtain the protein in a more active form because we wanted to compare the activity of the protein for ssDNA with that for U:A pairs in double-stranded DNA (dsDNA). U:A pairs are directly created in DNA during replication through the utilization of dUTP. Alternatively, cytosines may deaminate to uracil within ssDNA in the replication forks and then be copied over by the replicative polymerases again creating U:A pairs. If UdgX binds as well or better to uracils in U:A pairs compared to those in ssDNA, then visualization of UdgX–DNA complexes would show complexes in post-replicative DNA in addition to those at replication forks, and this would complicate data analysis.
Fortuitously, UdgX prefers strongly uracils in ssDNA. Under conditions that UdgX quantitatively converted the ssU oligomer to its covalent complex, only 8% or 29% of respectively the U:A or U•G containing duplexes formed complexes (Figure 3A). Furthermore, preincubation of the protein with DNA from a strain with about 2 uracils/106 bp (GM31; (43)) did not significantly decrease complex formation even at a 100-fold molar excess of uracil-containing dsDNA over ssDNA (Figure 3B). The complex formation was significantly inhibited only when >1500-fold excess of uracils in dsDNA was preincubated with UdgX prior to its incubation with ssU oligomer and was not completely eliminated even in the presence of 150 000-fold molar excess of uracils in dsDNA (Figure 3B). Together, these results suggest that UdgX reacts with uracils in dsDNA very slowly and hence in mammalian cells with normal dUTP levels, UdgX should overwhelmingly bind to uracils in the single-stranded regions of the genome.
Figure 3.
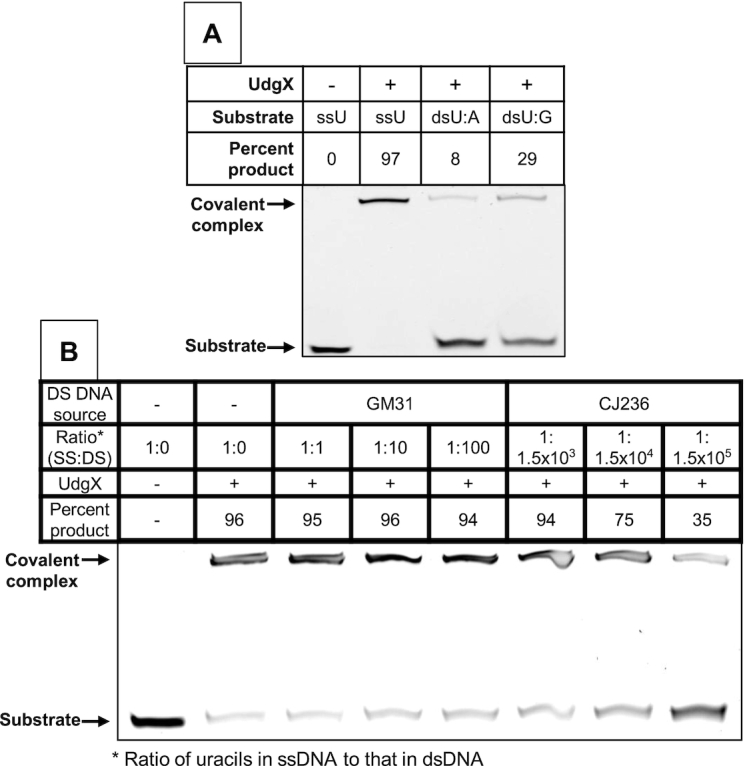
UdgX preferentially binds uracils in ssDNA. (A) The 6FAM-labeled oligomer with a uracil (ssU) was annealed to unlabeled complement strands with either a guanine (dsU:G) or adenine (dsU:A) base across from the uracil. FLAG-UdgX was reacted with these substrates in UdgX buffer. (B) Two pmol of the ssU substrate was mixed with different amounts of E. coli strain GM31 or CJ236 DNA and reacted with FLAG-UdgX.
UdgX forms nuclear foci in the presence of A3A
When a plasmid in which FLAG-UdgX gene is transcribed from the CMV promoter (Supplementary Figure S2B) was transfected into HEK293T cells, the protein was found in the nucleus within 24 h post-transfection (Figure 4A). While it was dispersed in most nuclei, a small fraction of cells showed granule-like aggregation (see below). As no nuclear localization signal had been added to the protein, the reasons for its transport into the nucleus are unknown. The H109S mutant of UdgX, which does not form a stable covalent complex with DNA (Supplementary Figure S4), also migrated to the nuclei (Figure 4A), eliminating the possibility that UdgX is found in the nuclei because the protein becomes covalently bound to DNA backbone at uracils. Consistent with this conclusion, transfection of UdgX plasmid into HEK293T cells did not affect cell growth or decrease viability for at least the first 24 h (Supplementary Figure S5A and B). After that time, UdgX appeared to slow the growth of the cells and slightly reduce their viability. However, this deleterious effect on growth and viability was also seen for the H109S mutant. These results suggest that UdgX is not strongly associated with the genomic DNA in these cells and hence does not cause major toxic effects for at least 24 h. For these reasons, the subsequent experiments were terminated at 24 or fewer hours post-transfection of the UdgX plasmid.
Figure 4.

UdgX forms foci in nuclei in the presence of active A3A. (A) HEK293 cells were transfected with a plasmid expressing WT or H109S mutant of FLAG-UdgX and visualized using anti-FLAG antibodies. (B) HeLa Tet-ON A3A-EGFP (HTO-A3A-EGFP) cells were either uninduced (-Dox) or induced for A3A expression (+Dox). The genomic uracils were quantified as described previously. HeLa cells served as a control. (C) Uninduced and induced HTO-A3A EGFP cells were transfected with FLAG-UdgX plasmid. The cells were stained appropriately, photographed using a fluorescence microscope and the colors in the images were inverted to allow clearer visualization of the foci. (D) The number of cells with UdgX foci of fluorescence images of HTO-A3A-EGFP cells that were induced or not induced for A3A expression was quantified. The percentage of cells with foci for these two conditions were compared using violin plots. The median values are shown using dashed lines (- - - -), while the upper and lower quartiles are shown using light lines (___). (E) HEK293T cells were co-transfected with plasmids expressing FLAG-UdgX and A3A-EGFP and their fluorescence images are shown. In different experiments WT or H109S mutant of UdgX, and WT or E72A mutant of A3A were used.
We previously reported construction of a HeLa-derived cell line called HTO in which A3A-EGFP protein is expressed from a doxycycline-regulated (Dox-regulated) Tet promoter (HTO-A3A-EGFP; (40)). When these cells were transfected with the FLAG-UdgX plasmid, the genomic DNA acquired about two-fold higher levels of uracils even without doxycycline treatment, presumably because of the leakiness of the promoter. The induction of expression A3A from the Tet promoter caused a further >60% increase in genomic uracils (Figure 4B). In the HTO-A3A-EGFP cells UdgX appeared in aggregates or foci in a small fraction of the cells even without Dox treatment, and this fraction increased 2.5-fold after doxycycline treatment (Figure 4C and D). To demonstrate that UdgX foci formation required the cytosine deamination activity of A3A and the ability of UdgX to link to DNA, HEK293T cells were transfected with plasmids expressing WT or mutant versions of FLAG-UdgX and A3A-EGFP. The UdgX foci were seen only when both the enzymes were catalytically active (Figure 4E). It should be noted that, in contrast with the distribution of UdgX, A3A was largely dispersed in the nuclei (Figure 4C and Supplementary Figure S6). Together these results are consistent with a model in which A3A deaminates multiple cytosines in some single-stranded regions in the genome and FLAG-UdgX covalently links at the resulting uracils creating foci that are visualized using anti-FLAG antibody.
Cisplatin treatment increases ssDNA and UdgX foci
It is well-established that cisplatin treatment of cells blocks DNA and RNA polymerases (44,45) and causes replication forks to stall (46,47). One of the consequences of such replication stress is the persistence of single-stranded DNA at the stalled replication forks. Although the single-strand DNA-binding protein, replication protein A (RPA), is recruited to protect this ssDNA and initiate DNA damage response (48), we hypothesized that cytosines at replication forks may be more accessible to AID/APOBEC enzymes following cisplatin treatment than during normal replication.
As expected, cisplatin treatment of HEK293T cells stopped growth within 24 h but did not decrease cell viability (Supplementary Figure S7). This cessation in cell growth was associated with an increase in RPA signal that was largely concentrated in the nuclei (Figure 5A) and increasing the dose of cisplatin resulted in a stronger RPA signal in the treated cells (Figure 5B). The cells treated with 10 μM cisplatin showed a six-fold higher RPA signal than untreated cells (Figure 5B) suggesting an upregulation of RPA expression. Such overproduction of RPA following treatment of cells with DNA damaging agents has been reported previously (49,50). These data confirm that the cisplatin treatment created regions of ssDNA in the genome-presumably at stalled replication forks.
Figure 5.

Cisplatin treatment increases cellular RPA and UdgX foci. (A) HEK293T cells were treated with cisplatin at the different concentrations and RPA was visualized using antibodies. (B) The RPA signal intensity of cells in part A was quantified at each concentration. (C) HTO-A3A-EGFP cells were induced for A3A-EGFP expression using doxycycline and then treated with cisplatin and transfected with pFLAG-UdgX. The fluorescence image colors are inverted for clarity. (D) The cells from part C were used to quantify the number of cells with UdgX foci. The median values are shown using dashed lines (- - - -), while the upper and lower quartiles are shown using light lines (___).
When the HTO-A3A-EGFP cells were treated with cisplatin and transfected with the UdgX plasmid, a number of cells contained UdgX foci even without doxycycline treatment (Figure 5C). It is likely that once again the leaky expression of the A3A gene in the absence of doxycycline (Figure 4B), are the cause of these UdgX foci. When the cells were induced for A3A expression using doxycycline and then treated with cisplatin treatment and transfected with the UdgX plasmid, the number of cells with UdgX foci increased ∼2-fold (Figure 5D). We interpret these data to mean that a combination of cisplatin treatment and A3A expression creates uracils at the stalled replication forks and these regions appear as UdgX foci.
UdgX and RPA colocalize in the presence of A3A
To study the distribution of RPA, A3A and UdgX in cells more quantitatively, HEK293T cells were treated with cisplatin and transfected with FLAG-UdgX and A3A-EGFP expression plasmids. When the cells were stained with appropriate antibodies and visualized using a confocal microscope, RPA and UdgX appeared largely as foci in the nuclei, while A3A was dispersed throughout the cells (Figure 6A and Supplementary Figure S8). When the fluorescence signals for UdgX and RPA were overlaid, many light brown spots were seen suggesting colocalization of the two fluorophores (Figure 6B, left). Similar colocalization was not apparent when UdgX and A3A or RPA and A3A signals were overlaid (Figure 6C, left and Figure 6D, left). This suggests that many of the RPA and UdgX molecules were near each other, but neither protein congregated near A3A.
Figure 6.

Distribution of UdgX, RPA and A3A in cisplatin treated HEK293 cells. (A) The cells were treated with cisplatin and transfected with pA3A-EGFP and pFLAG-UdgX, fixed and stained with DAPI and appropriate antibodies. Fluorescence image shows distribution of DAPI (blue), A3A (green), RPA (pink) and UdgX protein (yellow). (B) through (D) Individual channels were overlaid two at a time and an intensity profile was defined over 10 μm (panels on the left; white arrow). The values of fluorescence intensity are plotted UdgX and RPA (B), UdgX and A3A (C) and RPA and A3A (D). Regions in this scan where the intensities of a pair of fluorophores change in opposite directions are indicated by red bars below each plot (C and D, graphs on the right).
To determine more rigorously whether the fluorescence signals of the three proteins colocalize, we performed three types of analysis on these images. In the first approach, the fluorescence intensities were determined across a nucleus in a line within a single focal plane and fluorescence intensities of two different fluorophores were plotted in a graph (1D scan). These graphs show that while the RPA and UdgX fluorescence signals increased and decreased in rough unison (Figure 6B, right); the intensity distributions of the other two pairs of fluorophores did not show much similarities (Figure 6C and D, right). Thus, based on this semi-quantitative measure, UdgX and RPA show colocalization, but UdgX and A3A or RPA and A3A do not appear to colocalize.
To quantify the colocalization of these proteins, cells were transfected with pFLAG-UdgX alone or both pFLAG-UdgX and pA3A-EGFP and many cells were visualized in a one focal plane. The fluorescence intensities were analyzed using the ImageJ Coloc2 software to calculate Pearson correlation coefficient (‘R’). This was done for cells with and without cisplatin treatment. In the absence of cisplatin treatment and without A3A transfection, the median R for RPA and UdgX correlation was nearly zero (0.08) and it increased to 0.21 when cells were pre-treated with cisplatin, but this change was not statistically significant (P-value = 0.56; Figure 7A). However, when A3A-EGFP plasmid was co-transfected with the FLAG-UdgX plasmid, the R-value increased to 0.42 even in the absence of cisplatin treatment. Furthermore, when this was combined with pre-treatment of cells with cisplatin, the correlation became stronger (median R = 0.70) and this change was statistically highly significant (P-value < 0.0001; Figure 7A). In contrast, A3A showed poor correlation with either UdgX or RPA and this was true regardless of whether the cells had been treated with cisplatin (Figure 7B and C). These 2D analyses of correlations strongly argue that RPA and UdgX are frequently close to each other when A3A is present in the cells and the closeness is enhanced by treatment of cells with cisplatin. However, neither of these proteins is physically aggregated with or near A3A under these conditions.
Figure 7.

Correlation between localization of RPA and UdgX. HEK293T cells were transfected with pFLAG-UdgX and/or pFLAG-A3A-EGFP and the cells were either treated with cisplatin or were untreated. Fluorescence intensities were determined for UdgX, A3A and RPA across one focal plane for multiple cells and the Pearson correlation coefficient was determined for each pair of fluorophores using FIJI Colc2 software. The median values of correlations are shown by dashed lines while the upper and lower quartiles are marked by dotted lines. Transfected plasmid(s) and cisplatin treatment condition are shown below each plot. The different correlation analyses are- UdgX and RPA (A); UdgX and A3A (B) and A3A and RPA (C). (D) The cell profiled in Figure 6 was divided into 26 × 0.15 micron z-stacks, and the fluorescence intensities were determined across each z-stack. The Pearson coefficients were calculated for UdgX and RPA signals at each stack and is shown as a heatmap. DAPI signal intensity at each slice is also shown as a heatmap to identify the position of the nucleus in the cell.
Finally, to demonstrate that UdgX and RPA are together in the nucleus and not in the cytoplasm, fluorescence intensities were determined across the Z-axis for one cell that had been treated with cisplatin and transfected with UdgX and A3A-EGFP plasmids. The Pearson correlation coefficient was determined for each of the 26 Z-stacks and this was compared with the intensities of the DAPI fluorescence in these stacks (Figure 7D). The DAPI signal was most intense in the top half of the cell suggesting that the nucleus was near the top of the cell. In the same half of the molecule the mean R-values for the Z-stacks ranged from ∼0.60 to ∼0.75 and these were much higher than those in the bottom half of the cell (Figure 7D). This 3D analysis of the cell shows that UdgX and RPA have a good correlation within the nucleus, but not in the cytoplasm. Together these different analyses demonstrate that treatment of cells with cisplatin creates single-stranded regions in the cellular genome, and despite the upregulation of RPA, A3A converts cytosines in this ssDNA to uracil. Furthermore, UdgX is able to access these uracils and covalently link to their deoxyribose sugar while RPA binds to nearby ssDNA.
DISCUSSION
We have engineered the Mycobacterial protein UdgX for greater activity towards uracils in DNA and developed it as an in vivo uracil labeling reagent. The optimization of UdgX activity allowed us to compare its activity for uracils in ssDNA with that towards two double-stranded substrates, U:A and U•G. This comparison shows that UdgX strongly prefers ssDNA. The FLAG-tagged UdgX is transported into human cell nuclei without the need to add nuclear localization signal and is not very toxic to cells. This property allowed us to study the distribution of the protein following expression of the DNA-cytosine deaminase A3A and treatment of cells with cisplatin. In the absence of A3A expression and cisplatin treatment, the UdgX protein was largely dispersed in the nuclei of most cells but became more localized forming foci in the presence of A3A and this redistribution was aided by the pre-treatment of cells with cisplatin. In these experiments, UdgX served as the proxy for uracils in DNA and allowed us to show that uracils were being introduced by A3A in the ssDNA created by the cisplatin treatment even as RPA bound to the same regions of DNA. These results confirm the known preference of A3A for cytosines in the ssDNA at replication forks, enhance our understanding of the consequences of cisplatin treatment when APOBEC3 family proteins are present in the nuclei, while demonstrating the utility and simplicity of the use of UdgX for visualizing uracils in DNA at the cellular level.
Although we have not isolated DNA from the replication forks to directly demonstrate that UdgX links to uracils in the ssDNA within the forks, there are strong reasons to draw such a conclusion. First, it is well-established that A3A and A3B mostly create uracils in the lagging-strand template during replication (36–39,43,51). Second, induction of A3A expression in HeLa cells causes about a three-fold increase in genomic uracil levels (Figure 4B) and hence the overwhelming majority of uracils in the genome are expected to be due to cytosine deamination by A3A. Third, the median percentage of cells with UdgX foci doubles upon A3A induction (Figure 4D) and this is dependent on catalytic activity of A3A (Figure 4E). This shows a correlation between foci formation by UdgX and the ability of A3A to cause deamination. Four, in cisplatin treated cells, the percentage of cells with UdgX foci doubles when A3A is induced (Figure 5D). As cisplatin is known to create blocked replication forks- which contain ssDNA- this observation further confirms the conclusion that UdgX links to uracils created by A3A at replication forks.
Another observation from this study is that A3A does not colocalize with RPA or the uracils it creates. The lack of its colocalization with uracils (i.e. UdgX) is not surprising, because A3A is an enzyme and presumably releases its reaction product. But, the lack of colocalization between A3A and RPA suggests that A3A does not stay loosely bound to the single-stranded regions of the genome. This is consistent with the high KD of A3A for ssDNA and its frequent jumping and short scanning mechanism of search for a TC dinucleotide substrate (52). Moreover, these data argue that unlike replication and recombination during which RPA interacts with proteins such as RAD52 and PRIMPOL involved in these processes (53), RPA does not interact with A3A. It is likely that RPA and A3A compete, rather than cooperate, for binding to ssDNA. This makes biological sense because of the hazardous nature of the A3A catalytic activity.
To directly test the inference that RPA may compete with A3A for binding DNA at replication forks, we purified the two proteins (Supplementary Figure S9) and tested the ability of RPA to inhibit A3A activity in vitro. A fluorescently tagged 30-mer was used in these experiments and it forms a stable complex with RPA (Supplementary Figure S10, lane 2). This oligomer contains a single 5′-TC dinucleotide (Supplementary Table S1C) and A3A deaminates it efficiently (Figure 8, lane 2). However, when this DNA is first bound with RPA and then challenged with A3A followed by the inactivation of A3A, the ability of A3A to deaminate the cytosine in DNA was eliminated (Figure 8, lane 3). This shows that when RPA is bound to ssDNA, it can inhibit A3A activity. Similar inhibition of the APOBEC3G activity by RPA was reported previously (54).
Figure 8.

RPA protects cytosines against deamination by A3A. A 30 nucleotide DNA oligomer was either untreated (lane 1) or successively treated with A3A and Ung, and then heated in the presence of NaOH (lane 2). Alternately, the DNA was bound with excess RPA and then subjected to A3A. The A3A was inactivated by heating and RPA was released from DNA by the addition of excess unlabeled oligomer and then treated with Ung and electrophoresed under denaturation conditions (lane 3).
Before we embarked on the cellular studies, we wanted to determine the preferences of UdgX for the secondary structure of DNA within which uracils can be found. This is because different family 4 UDGs show different substrate preferences. For example, while the Sulfolobus solfataricus UDG has the following order of substrate preference- U•G>ssU>U:A (55), the Sulfolobus acidocaldarius UDG has a small preference for dsDNA over ssDNA regardless of the base opposite the uracil (56). We were concerned that if M. smegmatis UdgX reacted with uracils in dsDNA as well or better than those in ssDNA, it would not localize to ssDNA regions in the genome such as the replication forks.
To study the structural preference of UdgX, we had to improve the purification of UdgX and optimize its activity. This was accomplished by introducing polyGly linkers between the protein and its amino-terminal tags (Supplementary Figure S2B), use of the FLAG-, instead of a polyHis-, tag for its purification and improving the reaction buffer. The protein purified in this manner quantitatively formed covalent complexes with ssU DNA at 1:1 molar ratio of protein to DNA. This was not achieved in previous reports about this protein (32–35). Under these same reaction conditions, UdgX reacted poorly with U•G and U:A in duplex DNA (Figure 3A) and was not strongly inhibited by genomic DNA containing uracils (Figure 3B). These results show that M. smegmatis UdgX strongly prefers uracils in ssDNA in vitro. This preference is consistent with the increased foci formation by UdgX following cisplatin treatment and its high colocalization with RPA when the cisplatin treatment is combined with A3A expression (Figure 7A). These results demonstrate that UdgX can be used as a tool to understand where the AID/APOBEC enzymes can cause cytosine deaminations when cellular physiology is altered to create ssDNA.
Cisplatin is the standard of care treatment for a wide variety of cancers including malignancies of breast, lung and head-and-neck (57,58). It is one member of a large family of anticancer agents that react with DNA and create blocks in DNA replication (47). This family includes other DNA cross-linking agents like melphalan and chlorambucil (59), and also includes topoisomerase poisons such as doxorubicin and camptothecin (60). In addition, many endogenous and exogenous chemical and physical agents create DNA adducts that cannot be copied by replicative DNA polymerases creating replication stress (61). The data presented here suggests that in these situations if A3A or one of the other members of the AID/APOBEC family enzymes is present in the nucleus, it may deaminate cytosines in the stalled replication forks creating mutations. The uracil lesions may also be converted to strand breaks through the action of UDG and AP endonuclease causing genome instability.
The mutations created by APOBEC3A and APOBEC3B in human cancer genomes are collectively referred to as APOBEC signature mutations (29). Although the number of mutations attributable to APOBECs varies considerably from tumor to tumor and among the different cancer types, in some tumors a majority of mutations have the APOBEC signature (29–31). They are prevalent in a number of different cancer types, including malignancies such as breast and head-and-neck cancers that are routinely treated with cisplatin and other replication blocking drugs. As yet, few whole-genome sequencing (WGS) studies have been done on tumors that become resistant to cisplatin treatment or newly arise after a chemotherapy regimen. The results presented here point to a need for studies that systematically explore APOBEC signature mutations that arise in human cells following DNA damaging anti-cancer therapy in the context of expression of the AID/APOBEC genes.
UdgX could be developed further to make it even more useful as a tool to investigate uracils in DNA. For example, purified UdgX may be used to detect uracils in the genomes of fixed cells. Such an approach alleviates the need to express the protein in mammalian cells and may be adequate for some applications. An attractive potential in vivo application of UdgX is to detect uracils in live cells so that the creation or insertion of uracils in DNA and their repair could be monitored in real-time. To accomplish this, we expressed an mCherry-polyGly-UdgX fusion in mammalian cells but found it to be too toxic to the cells (data not shown). It is possible that other fluorescent proteins may be less toxic to the cells. Other possible uses of UdgX include pull-down of DNA fragments containing uracils in a single step for WGS. Such an approach would be much simpler than the existing methods (15,16) which require multiple biochemical steps. In summary, we have demonstrated here the potential of UdgX as a beacon for uracils, but many of its applications remain untapped.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr Umesh Varshney and Madhurima Datta (Indian Institute of Science, Bangalore, India) for providing the plasmid pET-14b-polyHis-mCherry-UdgX and help with the purification of the polyHis-mCherry-UdgX protein.
Contributor Information
Jessica A Stewart, Department of Chemistry, Wayne State University, Detroit, MI 48202, USA.
Grant Schauer, Department of Biochemistry & Molecular Biology, Colorado State University, Fort Collins, CO 80523, USA.
Ashok S Bhagwat, Department of Chemistry, Wayne State University, Detroit, MI 48202, USA; Department of Biochemistry, Microbiology and Immunology, Wayne State University School of Medicine, Detroit, MI 48201, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
A competitive graduate research assistantship and an Initiative for Maximizing Student Development (IMSD) fellowship from the Wayne State University (to J.A.S.); National Institutes of Health [1R21AI144708]; Bridge Funding grant from Wayne State University (to A.S.B.). Funding for open access charge: NIH [R21AI144708].
Conflict of interest statement. None declared.
REFERENCES
- 1. Berger S.H., Pittman D.L., Wyatt M.D.. Uracil in DNA: consequences for carcinogenesis and chemotherapy. Biochem. Pharmacol. 2008; 76:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lindahl T., Nyberg B.. Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry. 1974; 13:3405–3410. [DOI] [PubMed] [Google Scholar]
- 3. Hayatsu H. Bisulfite modification of nucleic acids and their constituents. Prog. Nucleic Acid Res. Mol. Biol. 1976; 16:75–124. [DOI] [PubMed] [Google Scholar]
- 4. Siriwardena S.U., Chen K., Bhagwat A.S.. Functions and malfunctions of mammalian DNA-cytosine deaminases. Chem. Rev. 2016; 116:12688–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Green A.M., Weitzman M.D.. The spectrum of APOBEC3 activity: from anti-viral agents to anti-cancer opportunities. DNA Repair (Amst.). 2019; 83:102700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salter J.D., Polevoda B., Bennett R.P., Smith H.C.. Regulation of antiviral innate immunity through APOBEC ribonucleoprotein complexes. Subcell. Biochem. 2019; 93:193–219. [DOI] [PubMed] [Google Scholar]
- 7. Stavrou S., Ross S.R.. APOBEC3 proteins in viral immunity. J. Immunol. 2015; 195:4565–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T.. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000; 102:553–563. [DOI] [PubMed] [Google Scholar]
- 9. Lari S.U., Chen C.Y., Vertessy B.G., Morre J., Bennett S.E.. Quantitative determination of uracil residues in Escherichia coli DNA: Contribution of ung, dug, and dut genes to uracil avoidance. DNA Repair (Amst.). 2006; 5:1407–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muha V., Horváth A., Békési A., Pukáncsik M., Hodoscsek B., Merényi G., Róna G., Batki J., Kiss I., Jankovics F. et al.. Uracil-containing DNA in Drosophila: stability, stage-specific accumulation, and developmental involvement. PLoS Genet. 2012; 8:e1002738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wei S., Shalhout S., Ahn Y.H., Bhagwat A.S.. A versatile new tool to quantify abasic sites in DNA and inhibit base excision repair. DNA Repair (Amst.). 2015; 27:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horváth A., Vértessy B.G.. A one-step method for quantitative determination of uracil in DNA by real-time PCR. Nucleic Acids Res. 2010; 38:e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Galashevskaya A., Sarno A., Vagbo C.B., Aas P.A., Hagen L., Slupphaug G., Krokan H.E.. A robust, sensitive assay for genomic uracil determination by LC/MS/MS reveals lower levels than previously reported. DNA Repair (Amst.). 2013; 12:699–706. [DOI] [PubMed] [Google Scholar]
- 14. Bryan D.S., Ransom M., Adane B., York K., Hesselberth J.R.. High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res. 2014; 24:1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shu X., Liu M., Lu Z., Zhu C., Meng H., Huang S., Zhang X., Yi C.. Genome-wide mapping reveals that deoxyuridine is enriched in the human centromeric DNA. Nat. Chem. Biol. 2018; 14:680–687. [DOI] [PubMed] [Google Scholar]
- 16. Sakhtemani R., Senevirathne V., Stewart J., Perera M.L.W., Pique-Regi R., Lawrence M.S., Bhagwat A.S.. Genome-wide mapping of regions preferentially targeted by the human DNA-cytosine deaminase APOBEC3A using uracil-DNA pulldown and sequencing. J. Biol. Chem. 2019; 294:15037–15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anindya R. Single-stranded DNA damage: Protecting the single-stranded DNA from chemical attack. DNA Repair (Amst.). 2020; 87:102804. [DOI] [PubMed] [Google Scholar]
- 18. Jinks-Robertson S., Bhagwat A.S.. Transcription-associated mutagenesis. Annu. Rev. Genet. 2014; 48:341–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pannunzio N.R., Lieber M.R.. Concept of DNA lesion longevity and chromosomal translocations. Trends Biochem. Sci. 2018; 43:490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith E.M., Pendlebury D.F., Nandakumar J.. Structural biology of telomeres and telomerase. Cell. Mol. Life Sci. 2020; 77:61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang D., O’Donnell M.. The eukaryotic replication machine. Enzymes. 2016; 39:191–229. [DOI] [PubMed] [Google Scholar]
- 22. Methot S.P., Di Noia J.M.. Molecular mechanisms of somatic hypermutation and class switch recombination. Adv. Immunol. 2017; 133:37–87. [DOI] [PubMed] [Google Scholar]
- 23. Alvarez-Prado A.F., Perez-Duran P., Perez-Garcia A., Benguria A., Torroja C., de Yebenes V.G., Ramiro A.R.. A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J. Exp. Med. 2018; 215:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu M., Duke J.L., Richter D.J., Vinuesa C.G., Goodnow C.C., Kleinstein S.H., Schatz D.G.. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008; 451:841–845. [DOI] [PubMed] [Google Scholar]
- 25. Gazumyan A., Bothmer A., Klein I.A., Nussenzweig M.C., McBride K.M.. Activation-induced cytidine deaminase in antibody diversification and chromosome translocation. Adv. Cancer. Res. 2012; 113:167–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robbiani D.F., Nussenzweig M.C.. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol. 2013; 8:79–103. [DOI] [PubMed] [Google Scholar]
- 27. Shalhout S., Haddad D., Sosin A., Holland T.C., Al-Katib A., Martin A., Bhagwat A.S.. Genomic uracil homeostasis during normal B cell maturation and loss of this balance during B cell cancer development. Mol. Cell. Biol. 2014; 34:4019–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pettersen H.S., Galashevskaya A., Doseth B., Sousa M.M., Sarno A., Visnes T., Aas P.A., Liabakk N.B., Slupphaug G., Saetrom P. et al.. AID expression in B-cell lymphomas causes accumulation of genomic uracil and a distinct AID mutational signature. DNA Repair (Amst.). 2015; 25:60–71. [DOI] [PubMed] [Google Scholar]
- 29. Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. et al.. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A. et al.. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012; 149:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roberts S.A., Lawrence M.S., Klimczak L.J., Grimm S.A., Fargo D., Stojanov P., Kiezun A., Kryukov G.V., Carter S.L., Saksena G. et al.. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013; 45:970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sang P.B., Srinath T., Patil A.G., Woo E.J., Varshney U.. A unique uracil-DNA binding protein of the uracil DNA glycosylase superfamily. Nucleic Acids Res. 2015; 43:8452–8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Datta M., Aroli S., Karmakar K., Dutta S., Chakravortty D., Varshney U.. Development of mCherry tagged UdgX as a highly sensitive molecular probe for specific detection of uracils in DNA. Biochem. Biophys. Res. Commun. 2019; 518:38–43. [DOI] [PubMed] [Google Scholar]
- 34. Ahn W.C., Aroli S., Kim J.H., Moon J.H., Lee G.S., Lee M.H., Sang P.B., Oh B.H., Varshney U., Woo E.J.. Covalent binding of uracil DNA glycosylase UdgX to abasic DNA upon uracil excision. Nat. Chem. Biol. 2019; 15:607–614. [DOI] [PubMed] [Google Scholar]
- 35. Tu J., Chen R., Yang Y., Cao W., Xie W.. Suicide inactivation of the uracil DNA glycosylase UdgX by covalent complex formation. Nat. Chem. Biol. 2019; 15:615–622. [DOI] [PubMed] [Google Scholar]
- 36. Bhagwat A.S., Hao W., Townes J.P., Lee H., Tang H., Foster P.L.. Strand-biased cytosine deamination at the replication fork causes cytosine to thymine mutations in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:2176–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haradhvala N.J., Polak P., Stojanov P., Covington K.R., Shinbrot E., Hess J.M., Rheinbay E., Kim J., Maruvka Y.E., Braunstein L.Z. et al.. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell. 2016; 164:538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hoopes J.I., Cortez L.M., Mertz T.M., Malc E.P., Mieczkowski P.A., Roberts S.A.. APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging Strand Template during DNA Replication. Cell Rep. 2016; 14:1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seplyarskiy V.B., Soldatov R.A., Popadin K.Y., Antonarakis S.E., Bazykin G.A., Nikolaev S.I.. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016; 26:174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stewart J.A., Holland T.C., Bhagwat A.S.. Human Herpes Simplex Virus-1 depletes APOBEC3A from nuclei. Virology. 2019; 537:104–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wijesinghe P., Bhagwat A.S.. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 2012; 40:9206–9217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schauer G., Finkelstein J., O’Donnell M.. In vitro assays for eukaryotic leading/lagging strand DNA replication. Bio Protoc. 2017; 7:2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Siriwardena S.U., Perera M.L.W., Senevirathne V., Stewart J., Bhagwat A.S.. A tumor-promoting phorbol ester causes a large increase in APOBEC3A expression and a moderate increase in APOBEC3B expression in a normal human keratinocyte cell line without increasing genomic uracils. Mol. Cell. Biol. 2019; 39:e00238-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Donaldson K.L., Goolsby G.L., Wahl A.F.. Cytotoxicity of the anticancer agents cisplatin and taxol during cell proliferation and the cell cycle. Int. J. Cancer. 1994; 57:847–855. [DOI] [PubMed] [Google Scholar]
- 45. Jamieson E.R., Lippard S.J.. Structure, recognition, and processing of cisplatin-DNA adducts. Chem. Rev. 1999; 99:2467–2498. [DOI] [PubMed] [Google Scholar]
- 46. McHugh P.J., Sones W.R., Hartley J.A.. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol. Cell. Biol. 2000; 20:3425–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wagner J.M., Karnitz L.M.. Cisplatin-induced DNA damage activates replication checkpoint signaling components that differentially affect tumor cell survival. Mol. Pharmacol. 2009; 76:208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marechal A., Zou L.. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015; 25:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cruet-Hennequart S., Villalan S., Kaczmarczyk A., O’Meara E., Sokol A.M., Carty M.P.. Characterization of the effects of cisplatin and carboplatin on cell cycle progression and DNA damage response activation in DNA polymerase eta-deficient human cells. Cell Cycle. 2009; 8:3039–3050. [PubMed] [Google Scholar]
- 50. Liu J.S., Kuo S.R., Beerman T.A., Melendy T.. Induction of DNA damage responses by adozelesin is S phase-specific and dependent on active replication forks. Mol. Cancer Ther. 2003; 2:41–47. [PubMed] [Google Scholar]
- 51. Green A.M., Landry S., Budagyan K., Avgousti D.C., Shalhout S., Bhagwat A.S., Weitzman M.D.. APOBEC3A damages the cellular genome during DNA replication. Cell Cycle. 2016; 15:998–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Love R.P., Xu H., Chelico L.. Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J. Biol. Chem. 2012; 287:30812–30822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bhat K.P., Cortez D.. RPA and RAD51: fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 2018; 25:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lada A.G., Waisertreiger I.S., Grabow C.E., Prakash A., Borgstahl G.E., Rogozin I.B., Pavlov Y.I.. Replication protein A (RPA) hampers the processive action of APOBEC3G cytosine deaminase on single-stranded DNA. PLoS One. 2011; 6:e24848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dionne I., Bell S.D.. Characterization of an archaeal family 4 uracil DNA glycosylase and its interaction with PCNA and chromatin proteins. Biochem. J. 2005; 387:859–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yi G.S., Wang W.W., Cao W.G., Wang F.P., Liu X.P.. Sulfolobus acidocaldarius UDG can remove dU from the RNA backbone: insight into the specific recognition of uracil linked with deoxyribose. Genes (Basel). 2017; 8:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dasari S., Tchounwou P.B.. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 2014; 740:364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer. 2007; 7:573–584. [DOI] [PubMed] [Google Scholar]
- 59. Rycenga H.B., Long D.T.. The evolving role of DNA inter-strand crosslinks in chemotherapy. Curr. Opin. Pharmacol. 2018; 41:20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 2013; 8:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kaufmann W.K. Initiating the uninitiated: replication of damaged DNA and carcinogenesis. Cell Cycle. 2007; 6:1460–1467. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.