


Abstract
Specific elements of viral genomes regulate interactions within host cells. Here, we calculated the secondary structure content of >2000 coronaviruses and computed >100 000 human protein interactions with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The genomic regions display different degrees of conservation. SARS-CoV-2 domain encompassing nucleotides 22 500–23 000 is conserved both at the sequence and structural level. The regions upstream and downstream, however, vary significantly. This part of the viral sequence codes for the Spike S protein that interacts with the human receptor angiotensin-converting enzyme 2 (ACE2). Thus, variability of Spike S is connected to different levels of viral entry in human cells within the population. Our predictions indicate that the 5′ end of SARS-CoV-2 is highly structured and interacts with several human proteins. The binding proteins are involved in viral RNA processing, include double-stranded RNA specific editases and ATP-dependent RNA-helicases and have strong propensity to form stress granules and phase-separated assemblies. We propose that these proteins, also implicated in viral infections such as HIV, are selectively recruited by SARS-CoV-2 genome to alter transcriptional and post-transcriptional regulation of host cells and to promote viral replication.
INTRODUCTION
A disease named Covid-19 by the World Health Organization and caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been recognized as responsible for the pneumonia outbreak that started in December 2019 in Wuhan City, Hubei, China (1) and spread in February to Milan, Lombardy, Italy (2) becoming pandemic.
SARS-CoV-2 is a positive-sense single-stranded RNA virus that shares similarities with other beta-coronavirus such as severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) (3). Bats have been identified as the primary host for SARS-CoV and SARS-CoV-2 (4,5) but the intermediate host linking SARS-CoV-2 to humans is still unknown, although a recent report indicates that pangolins could be involved (6).
Coronaviruses use species-specific proteins to mediate the entry in the host cell and the spike S protein activates the infection in human respiratory epithelial cells in SARS-CoV, MERS-CoV and SARS-CoV-2 (7). Spike S is assembled as a trimer and contains around 1300 amino acids within each unit (8,9). The receptor binding domain (RBD) of Spike S, which contains around 300 amino acids, mediates the binding with angiotensin-converting enzyme (ACE2), attacking respiratory cells. A region upstream of the RBD, present in MERS-CoV but not in SARS-CoV, is involved in the adhesion to sialic acid-containing oligosaccharides and plays a key role in regulating viral infection (7,10).
At present, few molecular details are available on SARS-CoV-2 and its interactions with the human host, which are mediated by specific RNA elements (11). To study the RNA structural content, we used CROSS (12) that was previously developed to investigate large transcripts such as the human immunodeficiency virus HIV-1 (13). CROSS predicts the structural profile of RNA molecules (single- and double-stranded state) at single-nucleotide resolution using sequence information only. Here, we performed sequence and structural alignments among SARS-CoV-2 strains available and identified the conservation of specific elements in the spike S region, which provides clues on the evolution of domains involved in the binding to ACE2 and sialic acid.
As highly structured RNAs have strong propensity to form stable contacts with different proteins (14) and promote specific assembly of complexes (15,16), SARS-CoV-2 domains enriched in double-stranded content are expected to establish interactions within host cells that are important to replicate the virus (17). To investigate the interactions of SARS-CoV-2 RNA with human proteins, we employed catRAPID (18,19). catRAPID (20) estimates the binding potential of a specific protein for an RNA molecule through van der Waals, hydrogen bonding and secondary structure propensities allowing identification of interaction partners with high confidence (21). The computational analysis of more than 100 000 interactions with SARS-CoV-2 RNA reveals that the 5′ end of SARS-CoV-2 has strong propensity to bind to human proteins involved in viral infection and reported to be associated with HIV infection. A comparison between SARS-CoV and HIV reveals similarities (22) that are still unexplored. Interestingly, HIV and SARS-CoV-2, but not SARS-CoV nor MERS-CoV, have a furin-cleavage site occurring in the spike S protein, which could explain the high velocity spread of SARS-CoV-2 compared to SARS-CoV and MERS-CoV (23,24).
We hope that our large-scale calculations of structural properties and binding partners of SARS-CoV-2 will be useful to identify the mechanisms of virus replication within the human host.
MATERIALS AND METHODS
Structure prediction
We computed the secondary structure of transcripts using CROSS (Computational Recognition of Secondary Structure) (12,13). The algorithm predicts the structural profile (single- and double-stranded state) at single-nucleotide resolution using sequence information only and without sequence length restrictions (scores > 0 indicate double stranded regions). We used the Vienna RNA Package (25) to further investigate the RNA secondary structure of minima and maxima identified with CROSS (13).
CROSS alive was employed to predict SARS-CoV-2 secondary structure in vivo (26). CROSS alive (m6A+ fast option) predicts long range interactions and can identify pseudoknots of 50–100 nucleotides. The RF-Fold algorithm of the RNAFramework suite (26) was used to identify pseudoknots in SARS-CoV-2. In this analysis, the partition function was calculated using CROSS calculations as soft-constraints. RNA was then folded employing Vienna RNA Package (25) and pseudo-knotted bases were hard-constrained to be single-stranded.
MN908947 predictions are available at http://crg-webservice.s3.amazonaws.com/submissions/2020-05/270257/output/index.html?unlock=fd65439e7b (CROSS) and also http://crg-webservice.s3.amazonaws.com/submissions/2020-05/271372/output/index.html?unlock=1de1d3a54a (CROSS alive).
Structural conservation
We used CROSSalign (12,13), an algorithm based on Dynamic Time Warping (DTW), to check and evaluate the structural conservation between different viral genomes (13). CROSSalign was previously employed to study the structural conservation of ∼5000 HIV genomes. SARS-CoV-2 fragments (1000 nt, not overlapping) were searched inside other complete genomes using the OBE (open begin and end) module, in order to search a small profile inside a larger one. The lower the structural distance, the higher the structural similarities (with a minimum of 0 for almost identical secondary structure profiles). The significance is assessed as in the original publication (13).
The Infernal package (version 1.1.3) was employed to build covariance models (CMs) for fragments 22, 23 and 24 (27). The package was then used to search for sequence and structural similarities among RNAs in our database (267 representative sequences), which allows to identify a series of matches below a specific E-value threshold (0.1, 1 and 10). The analysis shows agreement with CROSSalign (12,13) results. The minimum and maximum number of identified motifs were 224 and 4878 (E-value of 10), 136 and 3093 (E-value of 1) and 94 and 1060 (E-value of 0.1). The motifs in Spike S region were counted for annotated coronaviruses (239 genomes out of 246, of which 161 within E-value of 0.1).
Sequence collection
The FASTA sequences of the complete genomes of SARS-CoV-2 were downloaded in March 2020 from Virus Pathogen Resource (VIPR; www.viprbrc.org), for a total of 62 strains. An additional non-redundant set was downloaded in August 2020 for further analyses (462 sequences). Regarding the other coronaviruses, the sequences were downloaded in March 2020 from NCBI selecting only complete genomes, for a total of 2040 genomes. The reference Wuhan sequence with available annotation (EPI_ISL_402119) was downloaded from Global Initiative on Sharing All Influenza Data in March 2020 (GISAID https://www.gisaid.org/).
Protein-RNA interaction prediction
Interactions between each fragment of target sequence and the human proteome were predicted using catRAPID omics (18,19), an algorithm that estimates the binding propensity of protein–RNA pairs by combining secondary structure, hydrogen bonding and van der Waals contributions. As reported in a recent analysis of about half a million of experimentally validated interactions (21), the algorithm is able to separate interacting vs non-interacting pairs with an area under the ROC curve of 0.78. The complete list of interactions between the 30 fragments and the human proteome is available at http://crg-webservice.s3.amazonaws.com/submissions/2020-03/252523/output/index.html?unlock=f6ca306af0. The output then is filtered according to the Z-score column, which is the interaction propensity normalised by the mean and standard deviation calculated over the reference RBP set (http://s.tartaglialab.com/static_files/shared/faqs.html#4). We used three different thresholds in ascending order of stringency: Z greater or equal than 1.50, 1.75 and 2 respectively and for each threshold we then selected the proteins that were unique for each fragment for each threshold. omiXscore calculations of ADAR and ADARB1 are interactions are respectively at http://crg-webservice.s3.amazonaws.com/submissions/2020-04/263420/output/index.html?unlock=f9375fdbf9 and http://crg-webservice.s3.amazonaws.com/submissions/2020-04/263140/output/index.html?unlock=bb28d715ea.
GO terms analysis
cleverGO (28), an algorithm for the analysis of Gene Ontology annotations, was used to determine which fragments present enrichment in GO terms related to viral processes. Analysis of functional annotations was performed in parallel with GeneMania (29). The link to cleverGO analyses for fragment 1 is at http://www.tartaglialab.com/GO_analyser/render_GO_universal/3073/0d66e887c3/ (Z≥2).
RNA and protein alignments
We used Clustal W (30) for 62 SARS-CoV-2 strains alignments and T-Coffee (31) for spike S proteins alignments. The variability in the spike S region was measured by computing Shannon entropy on translated RNA sequences. The Shannon entropy is computed as follows:
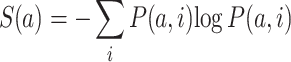 |
where a correspond to the amino acid at the position i and P(a,i) is the frequency of a certain amino-acid a at position i of the sequence. Low entropy indicates poorly variability: if P(a,x) = 1 for one a and 0 for the rest, then S(x) = 0. By contrast, if the frequencies of all amino acids are equally distributed, the entropy reaches its maximum possible value.
Predictions of phase separation
catGRANULE (32) was employed to identify proteins assembling into biological condensates. Scores >0 indicate that a protein is prone to phase separate. Structural disorder, nucleic acid binding propensity and amino acid patterns such as arginine–glycine and phenylalanine–glycine are key features combined in this computational approach (32).
RESULTS
SARS-CoV-2 contains highly structured elements
Structured elements within RNA molecules attract proteins (14) and reveal regions important for interactions with the host (33). Indeed, each gene expressed from SARS-CoV-2 is preceded by conserved transcription-regulating sequences that act as signal for the transcription complex during the synthesis of the RNA minus strand to promote a strand transfer to the leader region to resume the synthesis. This process is named discontinuous extension of the minus strand and is a variant of similarity-assisted template switching that operates during viral RNA recombination (17).
To analyze SARS-CoV-2 structure (reference Wuhan strain MN908947.3), we employed CROSS (12) that was previously developed to predict the double- and single-stranded content of RNA genomes such as HIV-1 (13). We found the highest density of double-stranded regions in the 5′ end (nucleotides 1–253), membrane M protein (nucleotides 26 523–27 191), spike S protein (nucleotides 21 563–25 384), and nucleocapsid N protein (nucleotides 28 274–29 533; Figure 1A) (34). The lowest density of double-stranded regions were observed at nucleotides 6 000–6 250 and 20 000–21 500 and correspond to the regions between the non-structural proteins nsp14 and nsp15 and the upstream region of the spike surface protein S (Figure 1) (34). In addition to the maximum corresponding to nucleotides 22 500–23 000, the structural content of Spike S protein shows minima at around nucleotides 21 500–22 000 and 23 500–24 000 (Figure 1). We used the Vienna method (25) to further investigate the RNA secondary structure of specific regions identified with CROSS (13). Employing a 100-nucleotide window centered around CROSS maxima and minima, we found good match between CROSS scores and Vienna free energies (Figure 1).
Figure 1.
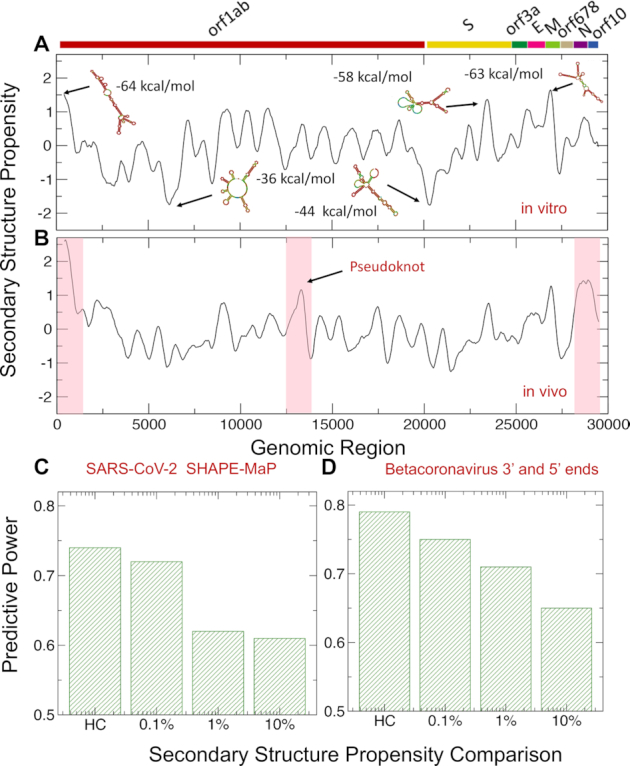
Predictions of SARS-CoV-2 structure. (A) Using the CROSS approach (12,13), (A) we predicted the structural content of SARS-CoV-2 in vitro. We found the highest density of double-stranded regions in the 5′ end (nucleotides 1–250) and within membrane M protein (nucleotides 26 500–27 000), and spike S protein (nucleotides 22 500–23 000) regions. Regions with the highest structural content are predicted by Vienna to have the lowest free energies. (B) Using CROSSalive (26), we studied the structural content of SARS-CoV-2 in vivo. The 5′ and 3′ ends (indicated by red boxes) are predicted to be highly structured. In addition, nucleotides 22 500–23 000 in Spike S region and nucleotides 13 400–13 600 (indicated by a red box) forming a pseudoknot (35) show high density of contacts. (C) Comparison of CROSS predictions with the secondary structure landscape of SARS-CoV-2 revealed by SHAPEMaP (38). From low (10%) to high (0.1%) confidence scores, the predictive power, measured as the Area Under the Curve (AUC) of Receiver Operating Characteristics (ROC), increases monotonically (HC corresponds to 10 nucleotides with highest/lowest scores). (D) CROSS performances on betacoronavirus 5′ and 3′ ends (39–42). Using different confidence scores, we show that CROSS is able to identify double and single stranded regions with great predictive power.
RNA structure in vitro and in vivo could be significantly different due to interactions with proteins and other molecules (26). Using CROSS alive to predict the double- and single-stranded content of SARS-CoV-2 in the cellular context, we found that both the 5′ and 3′ ends are the most structured regions followed by nucleotides 22 500–23 000 in the Spike S region, while nucleotides 6 000–6 250 and 20 000–21 500 have the lowest density of double-stranded regions (Figure 1B). The region corresponding to nucleotides 13 400–13 600 shows high density of contacts. This part of SARS-CoV-2 sequence has been proposed to form a pseudoknot (35) that is also visible in CROSS profile (Figure 1A), but CROSS alive is able to identify long range interactions and better identifies the region. Additionally, we used the RF-Fold algorithm of the RNAFramework suite (36) (Material and Methods) to search for pseudoknots. Employing CROSS as a soft-constraint for RF-Fold, we predicted 6 pseudoknots (nucleotides 3 394–3 404, 13 723–13 732, 14 677–14 711, 16 867–16 905, 24 844–24 884, 27 969–27 990). The pseudoknot at nucleotides 13 723–13 732 is in close proximity to the one proposed for SARS-CoV-2 (35) and the one at nucleotides 27 969–27 990 is at the 3′ end, where pseudonoknots have been shown to occur in coronaviruses (37).
To validate our results, we compared CROSS predictions of double- and single-stranded content (as released in March 2020) with the secondary structure landscape of SARS-CoV-2 revealed by SHAPE mutational profiling (SHAPEMaP) (38). In their experimental work, Manfredonia et al. carried out in vitro refolding of RNA followed by probing with 2-methylnicotinic acid imidazolide. In our comparison, balanced lists of single and double stranded regions were used for the calculations: A confidence score of 10% indicates that we compared the SHAPE reactivity values of 3000 nucleotides associated with the highest CROSS scores (i.e. double stranded) and 3000 nucleotides associated with the lowest CROSS scores (i.e. single stranded). From low (10%) to high (0.1%) confidence scores, we observed that the predictive power, measured as the Area Under the Curve (AUC) of Receiver Operating Characteristics (ROC), increases monotonically reaching the value of 0.73 (the AUC is 0.74 for the 10 highest/lowest scores; Figure 1C), which indicates that CROSS reproduces SHAPEMaP in great detail.
We also assessed CROSS performances on structures of betacoronavirus 5′ and 3′ ends (39–42) (Figure 1D). In this analysis, we used RFAM multiple sequence alignments of betacoronavirus 5′ and 3′ ends and relative consensus structures (RF03117 and RF03122) (39–42). We generated the 2D representation of nucleotide chains of consensus structures. We extracted the ‘secondary structure occupancy’, as defined in a previous work (20), and counted the contacts present around each nucleotide. Following the procedure used for the comparison with SHAPEMaP, different progressive cut-offs were used for ranking all the structures using balanced lists of single and double stranded regions: 10% indicates that we compared 600 nucleotides associated with the highest amount of contacts and 600 nucleotides associated with the lowest amount of contacts. From low (10%) to high (0.1%) confidence scores we observed that the AUC of ROC increases monotonically reaching the value of 0.75 (10 highest/lowest scores have an AUC of 0.78; Figure 1D), which indicates that CROSS is able to identify known double and single stranded regions reported in great detail. We also tested the ability of CROSS to recognize specific secondary structures in representative cases for which we studied both the 3′ and 5′ ends: NC_006213 or Human coronavirus OC43 strain ATCC VR-759, NC_019843 or Middle East respiratory syndrome coronavirus, NC_026011 or Betacoronavirus HKU24 strain HKU24-R05005I, NC_001846 or Mouse hepatitis virus strain MHV-A59 C12 and NC_012936 or Rat coronavirus Parker (Supplementary Figure S1).
In summary, our analysis identifies several structural elements in SARS-CoV-2 genome (11). Different lines of experimental and computational evidence indicate that transcripts containing a large amount of double-stranded regions have a strong propensity to recruit proteins (14,43) and can act as scaffolds for protein assembly (15,16). We therefore expected that the 5′ end attracts several host proteins because of the enrichment in secondary structure elements. The binding would not just involve proteins interacting with double-stranded regions. If a specific protein contact occurs in a loop at the end of a long RNA stem, the overall region is enriched in double-stranded nucleotides but the specific interaction takes place in a single-stranded element.
Structural comparisons reveal that a spike S region of SARS-CoV-2 is conserved among coronaviruses
We employed CROSSalign (13) to study the structural conservation of SARS-CoV-2 in different strains (Materials and Methods).
In our analysis, we compared the Wuhan strain MN908947.3 with 2040 coronaviruses (reduced to 267 sequences upon redundancy removal at 95% sequence similarity (44); Figure 2; full data shown in Supplementary Figure S2).
Figure 2.
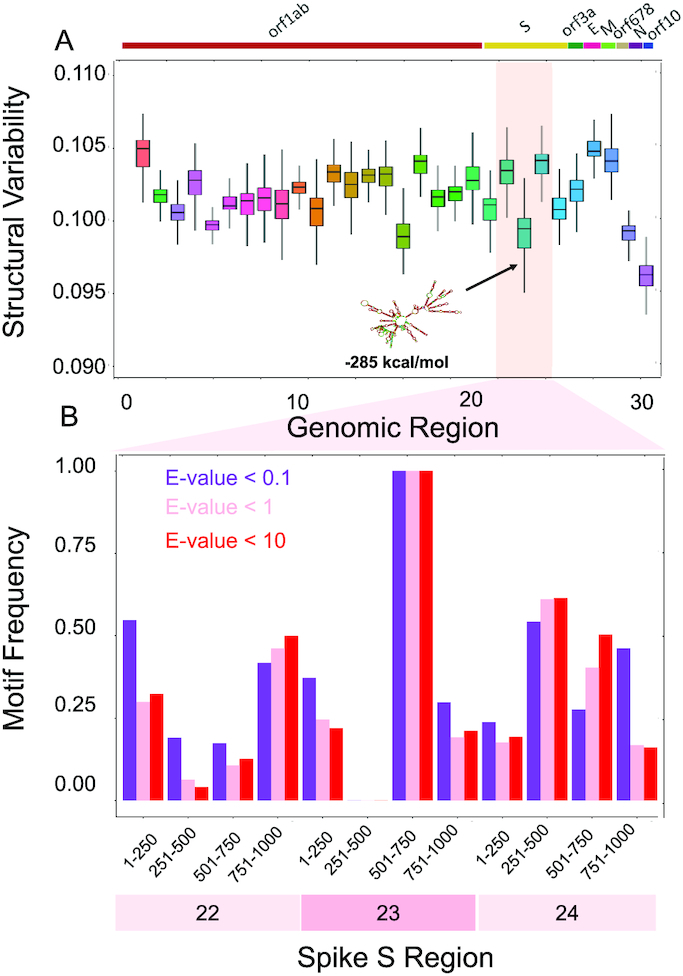
Structural comparisons of coronaviruses. (A) We employed the CROSSalign approach (12,13) to compare Wuhan strain MN908947.3 with other coronaviruses. One of the regions with the lowest structural variability encompasses nucleotides 22 000–23 000. The centroid structure and free energy computed with the Vienna method (25) are displayed. (B) We studied the conservation of nucleotides 22 000–23 000 (fragment 23) and the adjacent regions using structural motives identified with RF-Fold algorithm of the RNAFramework suite (36) with CROSS as soft-constraint. We found that nucleotides 501–750 within fragment 23 are the ones with the highest number of matches at confidence thresholds (E-values).
We note that the regulatory regions located at the 3′ end are slightly longer (about 250–500 nts containing a bulged stem loop, a pseudoknot plus a poly-A tail) than the ones at the 5′ end (the 1–4 stem loops are within the first 200 nucleotides) and their structural elements are therefore better recognized within the 1000 nucleotides window that we use for our analysis (45). Although the 5′ end is variable, it is more structured in SARS-CoV-2 than other coronaviruses (average structural content of 0.56, indicating that 56% of the CROSS signal is >0). The 3′ end is less variable and slightly less structured (average structural content of 0.49). By contrast, the other coronaviruses have lower average structural content of 0.49 in the 5′ end and 0.42 in the 3′ end.
One conserved region falls inside the Spike S genomic locus between nucleotides 22 000 and 23 000 and exhibits an intricate and stable secondary structure (RNAfold minimum free energy = −285 kcal/mol) (25). High conservation of a structured region suggests a functional activity that is relevant for host infection.
To demonstrate the conservation of nucleotides 22 000–23 000 (fragment 23), we divided this region and the adjacent ones (nucleotides 21 000–22 000 and 23 000–24 000) into sub-fragments. We then used the RF-Fold algorithm of the RNAFramework suite (36) to fold the different sub-regions using CROSS predictions as soft-constraints. The structural motives identified with this procedure were employed to build covariance models (CMs) that were then searched in our set of coronaviruses using the ‘Infernal’ package (27). We found that nucleotides 501–750 within fragment 23 have the highest number of matches for different confidence thresholds, implying a higher chance of sequence and structure conservation across coronaviruses (E-values of 10,1, 0.1; Figure 2B). We specifically counted the matches falling in the Spike S region (±1000 nucleotides to take into account the division of the genome into fragments; Supplementary Table S1). For the large majority of annotated sequences, we found a match falling in the Spike S region (239 genomes out of 246, of which 161 with E-value below 0.1) This further emphasizes the conservation of the region in exam.
Sequence and structural comparisons among SARS-CoV-2 strains
To better investigate the sequence conservation of SARS-CoV-2, we compared 62 strains isolated from different countries during the pandemic (including China, USA, Japan, Taiwan, India, Brazil, Sweden and Australia; data from NCBI and in VIPR www.viprbrc.org; Materials and Methods). Our analysis aims to determine the relationship between structural content and sequence conservation.
Using ClustalW for multiple sequence alignments (30), we observed general conservation of the coding regions (Figure 3A). The 5′ and 3′ ends show high variability due to practical aspects of RNA sequencing and are discarded in this analysis (46). Indeed, their sequences are less well characterized (47), and their variation results higher than other parta of the viral sequence. One highly conserved region is between nucleotides 22 000 and 23 000 in the Spike S genomic locus, while sequences up- and downstream are variable (purple bars in Figure 3A). We then used CROSSalign (13) to compare the structural content (Material and Methods). High variability of structure is observed for both the 5′ and 3′ ends and for nucleotides 21 000–22 000 as well as 24 000–25 000, associated with the Spike S region (purple bars in Figure 3A). The rest of the regions are significantly conserved at a structural level (P-value < 0.0001; Fisher's test).
Figure 3.
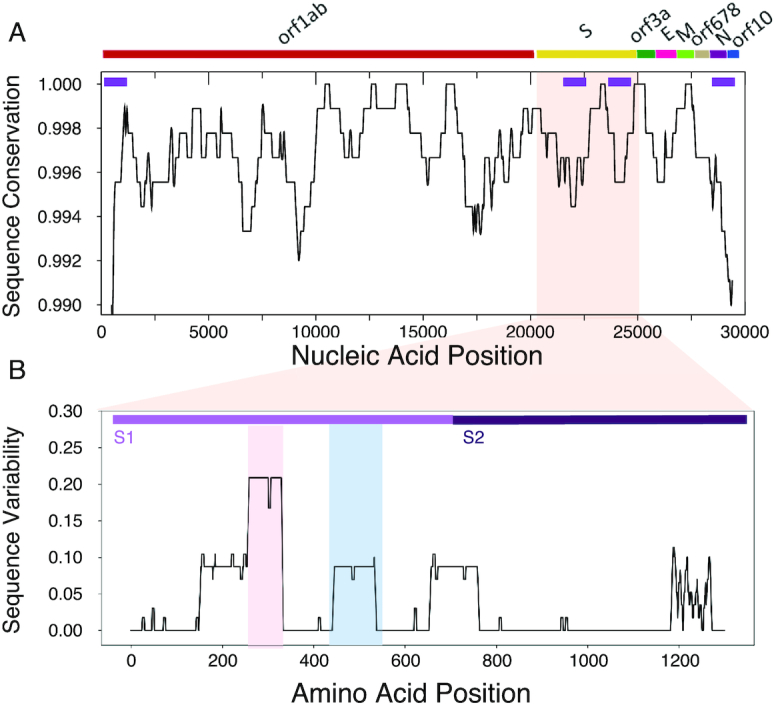
Sequence and structural comparison of human SARS-CoV-2 strains. (A) Strong sequence conservation (ClustalW multiple sequence alignments (28)) is observed in coding regions, including the region between nucleotides 22 000 and 23 000 of spike S protein. High structural variability (purple bars on top) is observed for both the UTRs and for nucleotides between 21 000 and 22 000 as well as 24 000 and 25 000, associated with the S region. The rest of the regions are significantly conserved at a structural level. (B) The sequence variability (Shannon entropy computed on T-Coffee multiple sequence alignments (31)) in the spike S protein indicate conservation between amino-acids 460 and 520 (blue box) binding to the host receptor angiotensin-converting enzyme 2 ACE2. The region encompassing amino-acids 243 and 302 is highly variable and is implicated in sialic acids in MERS-CoV (red box). The S1 and S2 domains of Spike S protein are displayed.
We note that sequence conservation (Figure 3A) and secondary structure profiles (Figure 1A) are statistically related. Following the analysis to compare CROSS and SHAPE scores, we selected balanced groups of nucleotides with the highest and lowest sequence conservation and measured their single and double stranded content: a conservation score of 1% indicates that we compared 300 nucleotides with the highest sequence similarity and 300 nucleotides with the lowest sequence similarity. At conservation score of 1% (or less stringent threshold of 10%), the match between similarity and structure, measured as the AUC of ROC is 0.76 (or 0.60, respectively). The association is statistically significant: shuffling the sequence conservation profiles, the empirical P-values are <0.02 (at both 10% and 1% conservation scores).
We also compared protein sequences coded by the Spike S genomic locus (NCBI reference QHD43416) and found that both sequence (Figure 3A) and structure (Figure 2) of nucleotides 22 000–23 000 are highly conserved. The region corresponds to amino acids 460–520 that contact the host receptor angiotensin-converting enzyme 2 (ACE2) (48) promoting infection and provoking lung injury (24,49). By contrast, the region upstream of the binding site receptor ACE2 and located in correspondence to the minimum of the structural profile at around nucleotides 22 500–23 000 (Figure 1) is highly variable (31), as indicated by T-coffee multiple sequence alignments (31) (Figure 3A). This part of the Spike S region corresponds to amino acids 243–302 that in MERS-CoV binds to sialic acids regulating infection through cell–cell membrane fusion (Figure 3B; see related manuscript by E. Milanetti et al.) (10,50,51).
Our analysis suggests that the structural region between nucleotides 22 000 and 23 000 of Spike S region is conserved among coronaviruses (Figure 2) and that the binding site for ACE2 has poor variation in human SARS-CoV-2 strains (Figure 3B). By contrast, the region upstream of the ACE2 binding site, which has also propensity to bind sialic acids (10,50,51), showed poor structural content and high variability (Figure 3B). The region downstream of the ACE2 binding site and located at the beginning of S2 domain shows high variability (Figure 3B). The results are confirmed by analysing a pool of 462 genomes having a ±5 nucleotides length difference with respect to MN908947.3 (August 2020; Supplementary Figure S3).
Analysis of human interactions with SARS-CoV-2 identifies proteins involved in viral replication
In order to obtain insights on how the virus replicates in human cells, we predicted SARS-CoV-2 interactions with the whole RNA-binding human proteome. Following a protocol to study structural conservation in viruses (13), we first divided the Wuhan sequence in 30 fragments of 1000 nucleotides each moving from the 5′ to 3′ end and then calculated the protein–RNA interactions of each fragment with catRAPID omics (3 340 canonical and putative RNA-binding proteins, or RBPs, for a total 102 000 interactions) (18). Proteins such as Polypyrimidine tract-binding protein 1 PTBP1 (Uniprot P26599) showed the highest interaction propensity (or Z-score; Materials and Methods) at the 5′ end while others such as heterogeneous nuclear ribonucleoprotein Q HNRNPQ (O60506) showed the highest interaction propensity at the 3′end, in agreement with previous studies on coronaviruses (Figure 4A) (52).
Figure 4.

Predictions of protein interactions with SARS-CoV-2 RNA. (A) In agreement with studies on coronaviruses (52), PTBP1 shows the highest interaction propensity at the 5′ and HNRNPQ at 3′ (indicated by red bars). (B) Number of RBP interactions for different SARS-CoV-2 regions (colours indicate catRAPID (18,19) confidence levels: Z = 1.5 or low Z = 1.75 or medium and Z = 2.0 or high; regions with scores lower than Z = 1.5 are omitted); (C) enrichment of viral processes in the 5′ of SARS-CoV-2 (precision = term precision calculated from the GO graph structure lvl = depth of the term; go_term = GO term identifier, with link to term description at AmiGO website; description = label for the term; e/d = enrichment / depletion compared to the population; %_set = coverage on the provided set; %_pop = coverage of the same term on the population; p_bonf = P-value of the enrichment. To correct for multiple testing bias, use Bonferroni correction) (28); (D) viral processes are the third largest cluster identified in our analysis;
For each fragment, we predicted the most significant interactions by filtering according to the Z score. We used three different thresholds in ascending order of stringency: Z ≥ 1.50, 1.75 and 2 respectively and we removed from the list the proteins that were predicted to interact promiscuously with more than one fragment. Fragment 1 corresponds to the 5′ end and is the most contacted by RBPs (∼120 with Z ≥ 2 high-confidence interactions; Figure 4B), which is in agreement with the observation that highly structured regions attract a large number of proteins (14). Indeed, the 5′ end contains multiple stem loop structures that control RNA replication and transcription (53,54). By contrast, the 3′ end and fragment 23 (Spike S), which are still structured but to a lesser extent, attract fewer proteins (10 and 5, respectively) and fragment 20 (between Orf1ab and Spike S) that is predicted to be unstructured, does not have predicted binding partners. Fragments 1 and 29 together with the adjacent regions are also predicted to be the most structured in vivo and show the highest amount of contacts for different Z scores (Figure 1B).
The interactome of each fragment was analysed using cleverGO, a tool for Gene Ontology (GO) enrichment analysis (28). Proteins interacting with fragments 1, 2 and 29 were associated with annotations related to viral processes (Figure 4C; Supplementary Table S2). Considering the three thresholds applied (Material and Methods), we found 23 viral proteins (including 2 pseudogenes), for fragment 1, 2 proteins for fragment 2 and 11 proteins for fragment 29 (Figure 4D). Among the high-confidence interactors of fragment 1, we discovered RBPs involved in positive regulation of viral processes and viral genome replication, such as double-stranded RNA-specific editase 1 ADARB1 (Uniprot P78563), 2–5A-dependent ribonuclease RNASEL (Q05823) and 2–5-oligoadenylate synthase 2 OAS2 (P29728; Figure 5A). Interestingly, 2–5-oligoadenylate synthase 2 OAS2 has been reported to be upregulated in human alveolar adenocarcinoma (A549) cells infected with SARS-CoV-2 (log fold change of 4.2; P-value of 10−9 and q-value of 10−6) (55). While double-stranded RNA-specific adenosine deaminase ADAR (P55265) is absent in our library due to its length that does not meet catRAPID omics requirements (18), the omiXcore extension of the algorithm specifically developed for large molecules (56) attributes the same binding propensity to both ADARB1 and ADAR, thus indicating that the interactions with SARS-CoV-2 are likely to occur (Materials and Methods). Moreover, experimental works indicate that the family of ADAR deaminases is active in bronchoalveolar lavage fluids derived from SARS-CoV-2 patients (57) and is upregulated in A549 cells infected with SARS-CoV-2 (log fold change of 0.58; P-value of 10−8 and q-value of 10−5) (55).
Figure 5.

Characterization of protein interactions with SARS-CoV-2 RNA. (A) Protein interaction network of SARS-CoV-2 5′ end (inner circle) and associations with other human genes retrieved from literature (blue: genetic associations; purple: physical associations); (B) number of RBP interactions identified by Gordon et al. (71) and Schmidt et al. (76) for different SARS-CoV-2 regions. Representative cases are shown in black (Gordon et al. (71)) and gray (Schmidt et al. (76)). (C) Proteins binding to the 5′ with Z score ≥1.5 show high propensity to accumulate in stress-granules (same number of proteins with Z score <−1.5 are used in the comparison; *** P-value <0.0001; Kolmogorov−Smirnoff).
We also identified proteins related to the establishment of integrated proviral latency, including X-ray repair cross-complementing protein 5 XRCC5 (P13010) and X-ray repair cross-complementing protein 6 XRCC6 (P12956; Figure 5A). In accordance with our calculations, comparison of A549 cells responses to SARS-CoV-2 and respiratory syncytial virus, indicates upregulation of XRCC6 in SARS-CoV-2 (log fold-change of 0.92; P-value of 0.006 and q-value of 0.23) (55). Moreover, previous evidence suggests that the binding of XRCC6 takes places at the 5′ end of SARS-CoV-2, thus giving further support to our predictions (58). Nucleolin NCL (P19338), a protein known to be involved in coronavirus processing, was also predicted to bind tightly to the 5′ end (Supplementary Table S2) (59).
Importantly, we found proteins related to defence response to viruses, such as ATP-dependent RNA helicase DDX1 (Q92499), that are involved in negative regulation of viral genome replication. Some DNA-binding proteins such as Cyclin-T1 CCNT1 (O60563), Zinc finger protein 175 ZNF175 (Q9Y473) and Prospero homeobox protein 1 PROX1 (Q92786) were included because they could have potential RNA-binding ability (Figure 5A) (60). As for fragment 2, we found two canonical RBPs: E3 ubiquitin-protein ligase TRIM32 (Q13049) and E3 ubiquitin−protein ligase TRIM21 (P19474), which are listed as negative regulators of viral release from host cell, negative regulators of viral transcription and positive regulators of viral entry into host cells. Among these genes, DDX1 (log fold change of 0.36; P-value of 0.007 and q-value of 0.23) and TRIM21 (log fold change of 0.44; P-value of 0.003 and q-value of 0.18) are also upregulated in A549 cells infected with SARS-CoV-2 (55). Ten of the 11 viral proteins detected for fragment 29 are members of the Gag polyprotein family, that perform different tasks during HIV assembly, budding, and maturation. More than just scaffold elements, these proteins are elements that accompany viral and host proteins as they traffic to the cell membrane (Supplementary Table S2) (61). Finally, among the RBPs with the highest interaction propensity for fragment 23, we found nucleosome assembly protein 1-like 1 NAP1L1 and E3 ubiquitin-protein ligase makorin-1 MKRN1, which could have an effect on the regulation of cell proliferation.
Analysis of functional annotations carried out with GeneMania (29) revealed that proteins interacting with the 5′ of SARS-CoV-2 RNA are associated with regulatory pathways involving NOTCH2, MYC and MAX that have been previously connected to viral infection processes (Figure 5A) (62,63). Interestingly, some proteins, including DDX1, CCNT1 and ZNF175 for fragment 1 and TRIM32 for fragment 2, have been shown to be necessary for HIV functions and replication inside the cell, as well as SARS-CoV-1. DDX1 has been shown to enable the switch from discontinuous to continuous transcription in SARS-CoV-1 infection and its knockdown reduced the number of longer sub-genomic mRNA (sgmRNA) through interaction with the SARS-CoV-1 nucleocapsid protein N (64) and Nsp14 (65). It functions as a bidirectional helicase, which distinguishes it from the coronaviruses helicases, which can only unwind RNA in the 5′ to 3′ direction (66), a very important function in highly structured RNA such SARS-CoV-2. DDX1 is also required for HIV-1 Rev as well as for avian coronavirus IBV replication and it binds to the RRE sequence of HIV-1 RNAs (67,68), while CCNT1 binds to 7SK snRNA and regulates transactivation domain of the viral nuclear transcriptional activator, Tat (69,70).
Analyses of SARS-CoV-2 proteins interactomes reveal common protein targets
Recently, Gordon et al. reported a list of human proteins binding to Open Reading Frames (ORFs) translated from SARS-CoV-2 (71). Identified through affinity purification followed by mass spectrometry quantification, 332 proteins from HEK-293T cells interact with viral ORF peptides. By selecting 274 proteins binding at the 5′ with Z score ≥1.5 (Supplementary Table S2), of which 140 are exclusively interacting with fragment 1 (Figure 4B), we found that 8 are also reported in the list by Gordon et al. (71), which indicates significant enrichment (representation factor of 2.5; P-value of 0.02; hypergeometric test with human proteome in background). The fact that our list of protein-RNA binding partners contains elements identified also in the protein-protein network analysis is not surprising, as ribonucleoprotein complexes evolve together (14) and their components sustain each other through different types of interactions (16).
We note that out of 332 interactions, 60 are RBPs (as reported in Uniprot), which represents a considerable fraction (i.e. 20%), considering that there are around 1500 RBPs in the human proteome (i.e. 6%). Comparing the RBPs present in Gordon et al. (71) and those present in our list (79 RBP annotated in Uniprot), we found an overlap of six proteins (representation factor = 26.5; P-value < 10−8; hypergeometric test), including: Janus kinase and microtubule-interacting protein 1 JAKMIP1 (Q96N16), A-kinase anchor protein 8 AKAP8 (O43823) and A-kinase anchor protein 8-like AKAP8L (Q9ULX6), which in case of HIV-1 infection is involved as a DEAD/H-box RNA helicase binding (72), signal recognition particle subunit SRP72 (O76094), binding to the 7S RNA in presence of SRP68, La-related protein 7, LARP7 (Q4G0J3) and La-related protein 4B LARP4B (Q92615), which are part of a system for transcriptional regulation acting by means of the 7SK RNP system (73) (Figure 5B; Supplementary Table S3). We speculate that sequestration of these elements is orchestrated by a viral program aiming to recruit host genes (74). LARP7 is also upregulated in A549 cells infected with SARS-CoV-2 (log fold change of 0.48; P-value of 0.006 and q-value of 0.23) (55).
Moreover, by directly analysing the RNA interaction potential of all the 332 proteins by Gordon et al. (71), catRAPID identified 38 putative binders at the 5′ end (Z score ≥ 1.5; 27 occurring exclusively in the 5′ end and not in other regions of the RNA) (18), including Serine/threonine-protein kinase TBK1 (Q9UHD2), among which 10 RBPs (as reported in Uniprot) such as: Splicing elements U3 small nucleolar ribonucleoprotein protein MPP10 (O00566) and Pre-mRNA-splicing factor SLU7 (O95391), snRNA methylphosphate capping enzyme MEPCE involved in negative regulation of transcription by RNA polymerase II 7SK (Q7L2J0) (75), Nucleolar protein 10 NOL10 (Q9BSC4) and protein kinase A Radixin RDX (P35241; in addition to those mentioned above; Supplementary Table S3).
Using the liver cell line HuH7 a recent experimental study by Schmidt et al. (76). identified SARS-CoV-2 RNA associations within the human host (76). Through the RAP-MS approach, 571 interactions were detected, of which 250 are RBPs (as reported in Uniprot) (76).
In common with our library we found an overlap of 148 proteins. We compared predicted (as released in March 2020) and experimentally-validated interactions employing balanced lists of high-affinity (high fold-change with respect to RNA Mitochondrial RNA Processing Endoribonuclease RMRP) and low-affinity (low fold-change with respect to RNA Mitochondrial RNA Processing Endoribonuclease RMRP) associations: a confidence score of 25% indicates that we compared the interaction scores of 35 proteins with the highest fold-change values and 35 interactions associated with the lowest fold-change values. From low (25%) to high (5%) confidence scores, we observed that the predictive power, measured as the AUC of ROC, increases monotonically reaching the remarkable value of 0.99 (the AUC is 0.72 for 25% confidence score; Supplementary Figure S4), which indicates strong agreement between predictions and experiments. In addition to DDX1 and DDX3X (O00571), other interactions corresponding to catRAPID scores >1.5 and fold-change >1 include Insulin-like growth factor 2 mRNA-binding protein 1 IGF2BP1 (Q9Y6M1), Insulin-like growth factor 2 mRNA-binding protein 2 IGF2BP2 2 (Q9Y6M1) and La-related protein 4 LARP4 (Q71RC2; also in Gordon et al. (71)).
By directly analysing RNA interactions of all the 571 proteins by Schmidt et al. (76), catRAPID identified 18 strong RBP binders at the 5′ end (Z score ≥ 1.5; fold-change >1; P-value of 0.008 computed with respect to all the interactions; Fisher exact test; Supplementary Table S4), including Helicase MOV-10 (Q9HCE1), Cold shock domain-containing protein E1 CSDE1 (O75534), Staphylococcal nuclease domain-containing protein 1 SND1 (Q7KZF4), Pumilio homolog 1 PUM1 (Q14671), and La-related protein 1 LARP1 (Q6PKG0), among other interactions (Supplementary Table S4).
The 5′ end is enriched in host interactions implicated in other viral infections
In the list of 274 proteins binding to the 5′ end (fragment 1) with Z score ≥1.5, we found 10 hits associated with HIV (Supplementary Table S5), which represents a significant enrichment (P-value = 0.0004; Fisher's exact test), considering that the total number of HIV-related proteins is 35 in the whole catRAPID library (3340 elements). The complete list of proteins includes ATP-dependent RNA helicase DDX1 (Q92499), ATP-dependent RNA helicase DDX3X (O00571 also involved in Dengue and Zika Viruses), Tyrosine-protein kinase HCK (P08631, nucleotide binding), Arf-GAP domain and FG repeat-containing protein 1 (P52594), Double-stranded RNA-specific editase 1 ADARB1 (P78563), Insulin-like growth factor 2 mRNA-binding protein 1 IGF2BP1 (Q9NZI8), A-kinase anchor protein 8-like AKAP8L (Q9ULX6; its partner AKAP8 is also found in Gordon et al. (71)) Cyclin-T1 CCNT1 (O60563; DNA-binding) and Forkhead box protein K2 FOXK2 (Q01167; DNA-binding; Figures 4B and 5A; Supplementary Table S5).
Smaller enrichments were found for proteins related to Hepatitis B virus (HBV; P-value = 0.01; three hits out of seven in the whole catRAPID library; Fisher's exact test), including Nuclear receptor subfamily 5 group A member 2 NR5A2 (DNA-binding; O00482), Interferon-induced, double-stranded RNA-activated protein kinase EIF2AK2 (P19525), and SRSF protein kinase 1 SRPK1 (Q96SB4) as well as Influenza A (P-value = 0.03; two hits out of four; Fisher's exact test), including Synaptic functional regulator FMR1 (Q06787) and RNA polymerase-associated protein RTF1 homologue (Q92541; Supplementary Table S5). By contrast, no significant enrichments were found for other viruses such as for instance Ebola.
Very importantly, specific chemical compounds have been developed to interact with HIV- and HVB-related proteins. The list of HIV-related targets reported in ChEMBL (77) includes ATP-dependent RNA helicase DDX1 (CHEMBL2011807), ATP-dependent RNA helicase DDX3X (CHEMBL2011808), Cyclin-T1 CCNT1 (CHEMBL2348842) and Tyrosine-protein kinase HCK (CHEMBL2408778), among other targets. In addition, HVB-related targets are Nuclear receptor subfamily 5 group A member 2 NR5A2 (CHEMBL3544), Interferon-induced, double-stranded RNA-activated protein kinase EIF2AK2 (CHEMBL5785) and SRSF protein kinase 1 SRPK1 (CHEMBL4375). We hope that this list can be the starting point for further pharmaceutical studies.
Phase-separating proteins are enriched in the 5′ end interactions
As SARS-CoV-2 represses host gene expression through a number of unknown mechanisms, sequestration of cell transcription machinery elements could be exploited to alter biological pathways in the host cell. A number of proteins identified in our catRAPID calculations have been previously reported to coalesce in large ribonucleoprotein assemblies similar to stress granules. Among these proteins, we found double-stranded RNA-activated protein kinase EIF2AK2 (P19525), Nucleolin NCL (P19338), ATP-dependent RNA helicase DDX1 (Q92499), Cyclin-T1 CCNT1 (O60563), signal recognition particle subunit SRP72 (O76094), LARP7 (Q4G0J3) and La-related protein 4B LARP4B (Q92615) as well as Polypyrimidine tract-binding protein 1 PTBP1 (P26599) and Heterogeneous nuclear ribonucleoprotein Q HNRNPQ (O60506) (78). To further investigate the propensity of these proteins to phase separate, we used the catGRANULE algorithm (Material and Methods) (32). Differently from other methods to predict solid-like aggregation (79,80), catGRANULE estimates the propensity of proteins to form liquid-like assemblies such as stress granules (81). We found that the 274 proteins binding to the 5′ end (fragment 1) with Z score ≥1.5 are highly prone to accumulate in assemblies similar to stress-granules (274 proteins with the lowest Z score are used in the comparison; P-value <0.0001; Kolmogorov−Smirnoff; Figure 5C; Supplementary Table S6). We note that there is not a direct correlation between RNA-binding scores (catRAPID) and phase-separation propensities (catGRANULE; Supplementary Figure S5).
Supporting this hypothesis, DDX1 and CCNT1 have been shown to condense in membrane-less organelles such as stress granules (82–84) that are the direct target of RNA viruses (85). DDX1 is also the primary component of distinct nuclear foci (86), together with factors associated with pre-mRNA processing and polyadenylation. Similarly, SRP72, LARP7 and LARP4B proteins have been found to assemble in stress granules (78,87,88). A recent work also suggests that the binding of LARP4 and XRCC6 takes places at the 5′ end of SARS-CoV-2 and contributes to SARS-CoV-2 phase separation (58). Moreover, emerging evidence indicates that the SARS-CoV-2 nucleocapsid protein N has a strong phase separation propensity that is modulated by the viral genome (58,89,90) and can enter into host cell protein condensates (89), suggesting a possible mechanism of cell protein sequestration. Notably, catGRANULE does predict that nucleocapsid protein N is the viral protein with highest propensity to phase separate (91).
As is the case with molecular chaperones (92), RNAs can influence the liquid-like or solid-like state of proteins (93). This observation is particularly relevant because RNA viruses are known to antagonize stress granules formation (85). Stress granules and other phase-separated assemblies such as processing bodies regulate translation suppression and RNA decay, which could have a strong impact on virus replication (94).
DISCUSSION
Our study is motivated by the need to identify molecular mechanisms involved in Covid-19 spreading. Using advanced computational approaches, we investigated the structural content of SARS-CoV-2 genome and predicted human proteins that bind to it.
We employed CROSS (12,13) to compare the structural properties of ∼2000 coronaviruses and identified elements conserved in SARS-CoV-2 strains. The regions containing the highest amount of structure are the 5′ end as well as glycoproteins spike S and membrane M.
We found that the Spike S protein domain encompassing amino acids 460–520 is conserved across SARS-CoV-2 strains. This result suggests that Spike S must have evolved to specifically interact with its host partner ACE2 (48) and mutations increasing the binding affinity should be highly infrequent. As nucleic acids encoding for this region are enriched in double-stranded content, we speculate that the structure might attract host regulatory elements, such as nucleosome assembly protein 1-like 1 NAP1L1 and E3 ubiquitin-protein ligase makorin-1 MKRN1, further constraining its variability. The fact that this region of the Spike S region is highly conserved among all the analysed SARS-CoV-2 strains suggests that a specific drug could be designed to prevent interactions within the host.
The highly variable region at amino acids 243–302 in spike S protein corresponds to the binding site of sialic acids in MERS-CoV (7,10,51) and could play a role in infection (50). The fact that the binding region is highly variable suggests different affinities for sialic acid-containing oligosaccharides and polysaccharides such as heparan sulfate, which provides clues on the specific responses in the human population. At present, a glycan microarray technology indicated that SARS-CoV-2 Spike S binds more tightly to heparan sulfate than sialic acids (95).
Using catRAPID (18,19) we computed >100 000 protein interactions with SARS-CoV-2 and found previously reported interactions such as Heterogeneous nuclear ribonucleoprotein Q HNRNPQ and Nucleolin NCL (59), among others. We discovered that the highly structured region at the 5′ end has the largest number of protein partners including ATP-dependent RNA helicase DDX1, which was previously reported to be essential for HIV-1 and coronavirus IBV replication (96,97), and the double-stranded RNA-specific editases ADAR and ADARB1, which catalyse the hydrolytic deamination of adenosine to inosine. Other predicted interactions are XRCC5 and XRCC6 members of the HDP-RNP complex associating with ATP-dependent RNA helicase DHX9 (98) as well as and 2–5A-dependent ribonuclease RNASEL and 2–5-oligoadenylate synthase 2 OAS2 that control viral RNA degradation (99,100). Interestingly, DDX1, XRCC6 and OAS2 were found upregulated in human alveolar adenocarcinoma cells infected with SARS-CoV-2 (55) and DDX1 knockdown has been shown to reduce the number of sgmRNA in SARS-CoV-1 infected cells (64). In agreement with our predictions, recent experimental work indicates that the family of ADAR deaminases is active in bronchoalveolar lavage fluids derived from SARS-CoV-2 patients (57).
Comparison with protein-RNA interactions detected in the liver cell line HuH7 (76) shows agreement with our predictions. We note that the experiments have been carried out 24 h after infection (76) and the protein interaction landscape might have changed with respect to the early events of replication. Yet, the accordance with our calculations indicates participation of elements involved in controlling RNA processing and editing (DDX1, DDX3X) and translation (IGF2BP1 and IGF2BP2), although proteins such as ADAR and XRCC5 were reported to have poorer binding capacity (76).
A significant overlap exists with the list of protein interactions reported by Gordon et al. (71) and among the candidate partners we identified AKAP8L, involved as a DEAD/H-box RNA helicase binding protein involved in HIV infection (72). In general, proteins associated with retroviral replication are expected to play different roles in SARS-CoV-2. As SARS-CoV-2 massively represses host gene expression (74), we hypothesize that the virus hijacks host pathways by recruiting transcriptional and post-transcriptional elements interacting with polymerase II genes and splicing factors such as for instance A-kinase anchor protein 8-like AKAP8L and La-related protein 7 LARP7. In concordance with our predictions LARP7 has been reported to be upregulated in human alveolar adenocarcinoma cells infected with SARS-CoV-2 (55). The link to proteins previously studied in the context of HIV and other viruses, if further confirmed, is particularly relevant for the repurposing of existing drugs (77).
The idea that SARS-CoV-2 sequesters different elements of the transcriptional machinery is particularly intriguing and is supported by the fact that a large number of proteins identified in our screening are found in stress granules (78). Indeed, stress granules protect the host innate immunity and are hijacked by viruses to favour their own replication (94). As coronaviruses transcription uses discontinuous RNA synthesis that involves high-frequency recombination (59), it is possible that pieces of the viruses resulting from a mechanism called defective interfering RNAs (101) could act as scaffold to attract host proteins (14,15). In agreement with our hypothesis, it has been very recently shown that the coronavirus nucleocapsid protein N can form protein condensates based on viral RNA scaffold and can merge with the human cell protein condensates (89), which provides a potential mechanism of host protein sequestration.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr Mattia Miotto, Dr Lorenzo Di Rienzo, Dr Alexandros Armaos, Dr Alessandro Dasti, Dr Claudia Giambartolomei for discussions. We are particularly grateful to Prof. Annalisa Pastore for critical reading, Dr Gilles Mirambeau for the RT versus RdRP analysis, Dr Andrea Cerase for the discussing on stress granules and Dr Roberto Giambruno for pointing to PTBP1 and HNRNPQ experiments. We are very much in debt with Dr Tommaso Muto, Giampaolo Fiore for their advice and friendship.
Authors contributions: G.G.T. and R.D.P. conceived the study. A.V. and A.A. carried out catRAPID analysis of protein interactions, R.D.P. calculated CROSS structures of coronaviruses, G.G.T., M.M. and E.M. performed and analysed sequence alignments, J.R., E.Z. and E.B. analysed the prediction results. A.V., R.D.P. and G.G.T. wrote the paper.
Contributor Information
Andrea Vandelli, Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Dr. Aiguader 88, 08003 Barcelona, Spain and Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain; Systems Biology of Infection Lab, Department of Biochemistry and Molecular Biology, Biosciences Faculty, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Spain.
Michele Monti, Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Dr. Aiguader 88, 08003 Barcelona, Spain and Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain; Center for Human Technologies, Istituto Italiano di Tecnologia, Via Enrico Melen 83, 16152 Genoa, Italy.
Edoardo Milanetti, Department of Physics, Sapienza University, Piazzale Aldo Moro 5, 00185 Rome, Italy; Center for Life Nanoscience, Istituto Italiano di Tecnologia, Viale Regina Elena 291, 00161 Rome, Italy.
Alexandros Armaos, Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Dr. Aiguader 88, 08003 Barcelona, Spain and Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain; Center for Human Technologies, Istituto Italiano di Tecnologia, Via Enrico Melen 83, 16152 Genoa, Italy.
Jakob Rupert, Center for Human Technologies, Istituto Italiano di Tecnologia, Via Enrico Melen 83, 16152 Genoa, Italy; Department of Biology ‘Charles Darwin’, Sapienza University of Rome, P.le A. Moro 5, Rome 00185, Italy.
Elsa Zacco, Center for Human Technologies, Istituto Italiano di Tecnologia, Via Enrico Melen 83, 16152 Genoa, Italy.
Elias Bechara, Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Dr. Aiguader 88, 08003 Barcelona, Spain and Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain; Center for Human Technologies, Istituto Italiano di Tecnologia, Via Enrico Melen 83, 16152 Genoa, Italy.
Riccardo Delli Ponti, School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore.
Gian Gaetano Tartaglia, Centre for Genomic Regulation (CRG), The Barcelona Institute for Science and Technology, Dr. Aiguader 88, 08003 Barcelona, Spain and Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain; Center for Human Technologies, Istituto Italiano di Tecnologia, Via Enrico Melen 83, 16152 Genoa, Italy; Department of Biology ‘Charles Darwin’, Sapienza University of Rome, P.le A. Moro 5, Rome 00185, Italy; Institucio Catalana de Recerca i Estudis Avançats (ICREA), 23 Passeig Lluis Companys, 08010 Barcelona, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council [RIBOMYLOME_309545, ASTRA_855923]; H2020 projects [IASIS_727658 and INFORE_825080]; Spanish Ministry of Economy and Competitiveness [BFU2017-86970-P]; collaboration with Peter St. George-Hyslop financed by the Wellcome Trust. Funding for open access charge: ERC [ASTRA 855923].
Conflict of interest statement. None declared.
REFERENCES
- 1. Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R. et al.. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020; 382:727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. D’Antiga L. Coronaviruses and immunosuppressed patients. The facts during the third epidemic. Liver Transpl. 2020; 26:832–834. [DOI] [PubMed] [Google Scholar]
- 3. Cascella M., Rajnik M., Cuomo A., Dulebohn S.C., Di Napoli R.. Features, evaluation and treatment coronavirus (COVID-19). StatPearls. 2020; Treasure Island: StatPearls Publishing. [PubMed] [Google Scholar]
- 4. Ge X.-Y., Li J.-L., Yang X.-L., Chmura A.A., Zhu G., Epstein J.H., Mazet J.K., Hu B., Zhang W., Peng C. et al.. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013; 503:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Follis K.E., York J., Nunberg J.H.. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell-cell fusion but does not affect virion entry. Virology. 2006; 350:358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xiao K., Zhai J., Feng Y., Zhou N., Zhang X., Zou J.-J., Li N., Guo Y., Li X., Shen X. et al.. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature. 2020; 583:286–289. [DOI] [PubMed] [Google Scholar]
- 7. Park Y.-J., Walls A.C., Wang Z., Sauer M.M., Li W., Tortorici M.A., Bosch B.-J., DiMaio F., Veesler D.. Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat. Struct. Mol. Biol. 2019; 26:1151–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Walls A.C., Tortorici M.A., Bosch B.-J., Frenz B., Rottier P.J.M., DiMaio F., Rey F.A., Veesler D.. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature. 2016; 531:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J. et al.. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020; 11:1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li W., Hulswit R.J.G., Widjaja I., Raj V.S., McBride R., Peng W., Widagdo W., Tortorici M.A., van Dieren B., Lang Y. et al.. Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E8508–E8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang D., Leibowitz J.L.. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015; 206:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Delli Ponti R., Marti S., Armaos A., Tartaglia G.G.. A high-throughput approach to profile RNA structure. Nucleic Acids Res. 2017; 45:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Delli Ponti R., Armaos A., Marti S., Tartaglia G.G.. A method for RNA structure prediction shows evidence for structure in lncRNAs. Front. Mol. Biosci. 2018; 5:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sanchez de Groot N., Armaos A., Graña-Montes R., Alriquet M., Calloni G., Vabulas R.M., Tartaglia G.G.. RNA structure drives interaction with proteins. Nat. Commun. 2019; 10:3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cid-Samper F., Gelabert-Baldrich M., Lang B., Lorenzo-Gotor N., Ponti R.D., Severijnen L.-A.W.F.M., Bolognesi B., Gelpi E., Hukema R.K., Botta-Orfila T. et al.. An integrative study of protein-RNA condensates identifies scaffolding RNAs and reveals players in fragile X-Associated Tremor/Ataxia syndrome. Cell Rep. 2018; 25:3422–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cerase A., Armaos A., Neumayer C., Avner P., Guttman M., Tartaglia G.G.. Phase separation drives X-chromosome inactivation: a hypothesis. Nat. Struct. Mol. Biol. 2019; 26:331. [DOI] [PubMed] [Google Scholar]
- 17. Moreno J.L., Zúñiga S., Enjuanes L., Sola I.. Identification of a coronavirus transcription enhancer. J. Virol. 2008; 82:3882–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Agostini F., Zanzoni A., Klus P., Marchese D., Cirillo D., Tartaglia G.G.. catRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics. 2013; 29:2928–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cirillo D., Blanco M., Armaos A., Buness A., Avner P., Guttman M., Cerase A., Tartaglia G.G.. Quantitative predictions of protein interactions with long noncoding RNAs. Nat Meth. 2017; 14:5–6. [DOI] [PubMed] [Google Scholar]
- 20. Bellucci M., Agostini F., Masin M., Tartaglia G.G.. Predicting protein associations with long noncoding RNAs. Nat. Methods. 2011; 8:444–445. [DOI] [PubMed] [Google Scholar]
- 21. Lang B., Armaos A., Tartaglia G.G.. RNAct: Protein–RNA interaction predictions for model organisms with supporting experimental data. Nucleic Acids Res. 2019; 47:D601–D606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kliger Y., Levanon E.Y.. Cloaked similarity between HIV-1 and SARS-CoV suggests an anti-SARS strategy. BMC Microbiol. 2003; 3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hallenberger S., Bosch V., Angliker H., Shaw E., Klenk H.D., Garten W.. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature. 1992; 360:358–361. [DOI] [PubMed] [Google Scholar]
- 24. Glowacka I., Bertram S., Herzog P., Pfefferle S., Steffen I., Muench M.O., Simmons G., Hofmann H., Kuri T., Weber F. et al.. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010; 84:1198–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lorenz R., Bernhart S.H., Höner zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorith. Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ponti R.D., Armaos A., Vandelli A., Tartaglia G.G.. CROSSalive: a web server for predicting the in vivo structure of RNA molecules. Bioinformatics. 2020; 36:940–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nawrocki E.P., Eddy S.R.. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013; 29:2933–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Klus P., Ponti R.D., Livi C.M., Tartaglia G.G.. Protein aggregation, structural disorder and RNA-binding ability: a new approach for physico-chemical and gene ontology classification of multiple datasets. BMC Genomics. 2015; 16:1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Warde-Farley D., Donaldson S.L., Comes O., Zuberi K., Badrawi R., Chao P., Franz M., Grouios C., Kazi F., Lopes C.T. et al.. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010; 38:W214–W220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Madeira F., Park Y. mi, Lee J., Buso N., Gur T., Madhusoodanan N., Basutkar P., Tivey A.R.N., Potter S.C., Finn R.D. et al.. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019; 47:W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J.-M., Taly J.-F., Notredame C.. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011; 39:W13–W17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bolognesi B., Lorenzo Gotor N., Dhar R., Cirillo D., Baldrighi M., Tartaglia G.G., Lehner B.. A Concentration-Dependent liquid phase separation can cause toxicity upon increased protein expression. Cell Rep. 2016; 16:222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gultyaev A.P., Richard M., Spronken M.I., Olsthoorn R.C.L., Fouchier R.A.M.. Conserved structural RNA domains in regions coding for cleavage site motifs in hemagglutinin genes of influenza viruses. Virus Evol. 2019; 5:vez034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu A., Peng Y., Huang B., Ding X., Wang X., Niu P., Meng J., Zhu Z., Zhang Z., Wang J. et al.. Genome composition and divergence of the novel coronavirus (2019-nCoV) Originating in China. Cell Host Microbe. 2020; 27:325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kelly J.A., Olson A.N., Neupane K., Munshi S., San Emeterio J., Pollack L., Woodside M.T., Dinman J.D.. Structural and functional conservation of the programmed -1 ribosomal frameshift signal of SARS coronavirus 2 (SARS-CoV-2). J. Biol. Chem. 2020; 295:10741–10748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Incarnato D., Morandi E., Simon L.M., Oliviero S.. RNA Framework: an all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Res. 2018; 46:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Williams G.D., Chang R.-Y., Brian D.A.. A phylogenetically conserved hairpin-type 3′ untranslated region pseudoknot functions in coronavirus RNA replication. J Virol. 1999; 73:8349–8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manfredonia I., Nithin C., Ponce-Salvatierra A., Ghosh P., Wirecki T.K., Marinus T., Ogando N.S., Snider E.J., van Hemert M.J., Bujnicki J.M., Incarnato D.. Genome-wide mapping of therapeutically-relevant SARS-CoV-2 RNA structures. 2020; bioRxiv doi:15 June 2020, preprint: not peer reviewed 10.1101/2020.06.15.151647. [DOI] [PMC free article] [PubMed]
- 39. Goebel S.J., Taylor J., Masters P.S.. The 3′ cis-acting genomic replication element of the severe acute respiratory syndrome coronavirus can function in the murine coronavirus genome. J. Virol. 2004; 78:7846–7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang D., Leibowitz J.L.. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015; 206:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sola I., Mateos-Gomez P.A., Almazan F., Zuñiga S., Enjuanes L.. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol. 2011; 8:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Madhugiri R., Fricke M., Marz M., Ziebuhr J.. Coronavirus cis-Acting RNA elements. Adv. Virus Res. 2016; 96:127–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Agostini F., Cirillo D., Bolognesi B., Tartaglia G.G.. X-inactivation: quantitative predictions of protein interactions in the Xist networkitle. Nucleic Acids Res. 2013; 41:e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li W., Godzik A.. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- 45. Yang D., Leibowitz J.L.. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015; 206:120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hrdlickova R., Toloue M., Tian B.. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA. 2017; 8:e1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ozsolak F., Milos P.M.. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011; 12:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Andersen K.G., Rambaut A., Lipkin W.I., Holmes E.C., Garry R.F.. The proximal origin of SARS-CoV-2. Nat. Med. 2020; 26:450–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. et al.. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Qing E., Hantak M., Perlman S., Gallagher T.. Distinct roles for sialoside and protein receptors in coronavirus infection. mBio. 2020; 11:e02764–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Milanetti E., Miotto M., Di Rienzo L., Monti M., Gosti G., Ruocco G.. In-Silico evidence for two receptors based strategy of SARS-CoV-2. 2020; bioRxiv doi:27 March 2020, preprint: not peer reviewed 10.1101/2020.03.24.006197. [DOI] [PMC free article] [PubMed]
- 52. Galán C., Sola I., Nogales A., Thomas B., Akoulitchev A., Enjuanes L., Almazán F.. Host cell proteins interacting with the 3′ end of TGEV coronavirus genome influence virus replication. Virology. 2009; 391:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu K., Heng X., Summers M.F.. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 2011; 410:609–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fehr A.R., Perlman S.. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015; 1282:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D. et al.. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020; 181:1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Armaos A., Cirillo D., Tartaglia G.G.. omiXcore: a web server for prediction of protein interactions with large RNA. Bioinformatics. 2017; 33:3104–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giorgio S.D., Martignano F., Torcia M.G., Mattiuz G., Conticello S.G.. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Science Advances. 2020; 6:eabb5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iserman C., Roden C., Boerneke M., Sealfon R., McLaughlin G., Jungreis I., Park C., Boppana A., Fritch E., Hou Y.J. et al.. Specific viral RNA drives the SARS CoV-2 nucleocapsid to phase separate. 2020; bioRxiv doi:12 June 2020, preprint: not peer reviewed 10.1101/2020.06.11.147199. [DOI]
- 59. Sola I., Mateos-Gomez P.A., Almazan F., Zuñiga S., Enjuanes L.. RNA-RNA and RNA-protein interactions in coronavirus replication and transcription. RNA Biol. 2011; 8:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M. et al.. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012; 149:1393–1406. [DOI] [PubMed] [Google Scholar]
- 61. Bell N.M., Lever A.M.L.. HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol. 2013; 21:136–144. [DOI] [PubMed] [Google Scholar]
- 62. Hayward S.D. Viral interactions with the Notch pathway. Semin. Cancer Biol. 2004; 14:387–396. [DOI] [PubMed] [Google Scholar]
- 63. Dudley J.P., Mertz J.A., Rajan L., Lozano M., Broussard D.R.. What retroviruses teach us about the involvement of c- Myc in leukemias and lymphomas. Leukemia. 2002; 16:1086–1098. [DOI] [PubMed] [Google Scholar]
- 64. Wu C.-H., Chen P.-J., Yeh S.-H.. Nucleocapsid phosphorylation and RNA helicase DDX1 recruitment enables coronavirus transition from discontinuous to continuous transcription. Cell Host Microbe. 2014; 16:462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu L., Khadijah S., Fang S., Wang L., Tay F.P.L., Liu D.X.. The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. J. Virol. 2010; 84:8571–8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shu T., Huang M., Wu D., Ren Y., Zhang X., Han Y., Mu J., Wang R., Qiu Y., Zhang D.-Y. et al.. SARS-Coronavirus-2 Nsp13 possesses NTPase and RNA helicase activities that can be inhibited by bismuth salts. Virol. Sin. 2020; 35:321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Edgcomb S.P., Carmel A.B., Naji S., Ambrus-Aikelin G., Reyes J.R., Saphire A.C.S., Gerace L., Williamson J.R.. DDX1 is an RNA-dependent ATPase involved in HIV-1 rev function and virus replication. J Mol Biol. 2012; 415:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu L., Khadijah S., Fang S., Wang L., Tay F.P.L., Liu D.X.. The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. J. Virol. 2010; 84:8571–8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ivanov D., Kwak Y.T., Nee E., Guo J., García-Martínez L.F., Gaynor R.B.. Cyclin T1 domains involved in complex formation with Tat and TAR RNA are critical for tat-activation. J. Mol. Biol. 1999; 288:41–56. [DOI] [PubMed] [Google Scholar]
- 70. Kwak Y.T., Ivanov D., Guo J., Nee E., Gaynor R.B.. Role of the human and murine cyclin T proteins in regulating HIV-1 tat-activation. J. Mol. Biol. 1999; 288:57–69. [DOI] [PubMed] [Google Scholar]
- 71. Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L. et al.. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020; 583:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xing L., Zhao X., Guo F., Kleiman L.. The role of A-kinase anchoring protein 95-like protein in annealing of tRNALys3 to HIV-1 RNA. Retrovirology. 2014; 11:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Markert A., Grimm M., Martinez J., Wiesner J., Meyerhans A., Meyuhas O., Sickmann A., Fischer U.. The La-related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes. EMBO Rep. 2008; 9:569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim D., Lee J.-Y., Yang J.-S., Kim J.W., Kim V.N., Chang H.. The architecture of SARS-CoV-2 transcriptome. Cell. 2020; 181:914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jeronimo C., Forget D., Bouchard A., Li Q., Chua G., Poitras C., Thérien C., Bergeron D., Bourassa S., Greenblatt J. et al.. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol. Cell. 2007; 27:262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schmidt N., Lareau C.A., Keshishian H., Melanson R., Zimmer M., Kirschner L., Ade J., Werner S., Caliskan N., Lander E.S. et al.. A direct RNA-protein interaction atlas of the SARS-CoV-2 RNA in infected human cells. 2020; bioRxiv doi:15 July 2020, preprint: not peer reviewed 10.1101/2020.07.15.204404. [DOI]
- 77. Mendez D., Gaulton A., Bento A.P., Chambers J., De Veij M., Félix E., Magariños M.P., Mosquera J.F., Mutowo P., Nowotka M. et al.. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019; 47:D930–D940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Markmiller S., Soltanieh S., Server K.L., Mak R., Jin W., Fang M.Y., Luo E.-C., Krach F., Yang D., Sen A. et al.. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell. 2018; 172:590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lee Y., Zhou T., Tartaglia G.G., Vendruscolo M., Wilke C.O.. Translationally optimal codons associate with aggregation-prone sites in proteins. Proteomics. 2010; 10:4163–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Agostini F., Cirillo D., Livi C.M., Ponti R.D., Tartaglia G.G.. ccSOL omics: a webserver for large-scale prediction of endogenous and heterologous solubility in E. coli. Bioinformatics. 2014; 30:2975–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gotor N.L., Armaos A., Calloni G., Torrent Burgas M., Vabulas R.M., De Groot N.S., Tartaglia G.G.. RNA-binding and prion domains: the Yin and Yang of phase separation. Nucleic Acids Res. 2020; 48:9491–9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ning W., Guo Y., Lin S., Mei B., Wu Y., Jiang P., Tan X., Zhang W., Chen G., Peng D. et al.. DrLLPS: a data resource of liquid–liquid phase separation in eukaryotes. Nucleic Acids Res. 2020; 48:D288–D295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shin Y., Chang Y.-C., Lee D.S.W., Berry J., Sanders D.W., Ronceray P., Wingreen N.S., Haataja M., Brangwynne C.P.. Liquid nuclear condensates mechanically sense and restructure the genome. Cell. 2018; 175:1481–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Su X., Ditlev J.A., Hui E., Xing W., Banjade S., Okrut J., King D.S., Taunton J., Rosen M.K., Vale R.D.. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science. 2016; 352:595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. White J.P., Lloyd R.E.. Regulation of stress granules in virus systems. Trends Microbiol. 2012; 20:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Uversky V.N. Intrinsically disordered proteins in overcrowded milieu: membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017; 44:18–30. [DOI] [PubMed] [Google Scholar]
- 87. Schäffler K., Schulz K., Hirmer A., Wiesner J., Grimm M., Sickmann A., Fischer U.. A stimulatory role for the La-related protein 4B in translation. RNA. 2010; 16:1488–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Küspert M., Murakawa Y., Schäffler K., Vanselow J.T., Wolf E., Juranek S., Schlosser A., Landthaler M., Fischer U.. LARP4B is an AU-rich sequence associated factor that promotes mRNA accumulation and translation. RNA. 2015; 21:1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Perdikari T.M., Murthy A.C., Ryan V.H., Watters S., Naik M.T., Fawzi N.L.. SARS-CoV-2 nucleocapsid protein undergoes liquid-liquid phase separation stimulated by RNA and partitions into phases of human ribonucleoproteins. 2020; bioRxiv doi:10 June 2020, preprint: not peer reviewed 10.1101/2020.06.09.141101. [DOI]
- 90. Cubuk J., Alston J.J., Incicco J.J., Singh S., Stuchell-Brereton M.D., Ward M.D., Zimmerman M.I., Vithani N., Griffith D., Wagoner J.A. et al.. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. 2020; bioRxiv doi:18 June 2020, preprint: not peer reviewed 10.1101/2020.06.17.158121. [DOI] [PMC free article] [PubMed]
- 91. Cascarina S.M., Ross E.D.. A proposed role for the SARS-CoV-2 nucleocapsid protein in the formation and regulation of biomolecular condensates. FASEB J. 2020; doi:10.1096/fj.202001351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tartaglia G.G., Dobson C.M., Hartl F.U., Vendruscolo M.. Physicochemical determinants of chaperone requirements. J. Mol. Biol. 2010; 400:579–588. [DOI] [PubMed] [Google Scholar]
- 93. Zacco E., Graña-Montes R., Martin S.R., de Groot N.S., Alfano C., Tartaglia G.G., Pastore A.. RNA as a key factor in driving or preventing self-assembly of the TAR DNA-binding protein 43. J. Mol. Biol. 2019; 431:1671–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhang Q., Sharma N.R., Zheng Z.-M., Chen M.. Viral regulation of RNA granules in infected cells. Virol. Sin. 2019; 34:175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hao W., Ma B., Li Z., Wang X., Gao X., Li Y., Qin B., Shang S., Cui S., Tan Z.. Binding of the SARS-CoV-2 Spike Protein to Glycans. 2020; bioRxiv doi:17 May 2020, preprint: not peer reviewed 10.1101/2020.05.17.100537. [DOI] [PMC free article] [PubMed]
- 96. Fang J., Kubota S., Yang B., Zhou N., Zhang H., Godbout R., Pomerantz R.J.. A DEAD box protein facilitates HIV-1 replication as a cellular co-factor of Rev. Virology. 2004; 330:471–480. [DOI] [PubMed] [Google Scholar]
- 97. Xu L., Khadijah S., Fang S., Wang L., Tay F.P.L., Liu D.X.. The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. J. Virol. 2010; 84:8571–8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhang S., Schlott B., Görlach M., Grosse F.. DNA-dependent protein kinase (DNA-PK) phosphorylates nuclear DNA helicase II/RNA helicase A and hnRNP proteins in an RNA-dependent manner. Nucleic Acids Res. 2004; 32:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sarkar S.N., Ghosh A., Wang H.W., Sung S.S., Sen G.C.. The nature of the catalytic domain of 2′-5′-oligoadenylate synthetases. J. Biol. Chem. 1999; 274:25535–25542. [DOI] [PubMed] [Google Scholar]
- 100. Sarkar S.N., Bandyopadhyay S., Ghosh A., Sen G.C.. Enzymatic characteristics of recombinant medium isozyme of 2′-5′ oligoadenylate synthetase. J. Biol. Chem. 1999; 274:1848–1855. [DOI] [PubMed] [Google Scholar]
- 101. Pathak K.B., Nagy P.D.. Defective interfering RNAs: foes of viruses and friends of virologists. Viruses. 2009; 1:895–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.