


Abstract
Type I restriction-modification (R-M) systems consist of a DNA endonuclease (HsdR, HsdM and HsdS subunits) and methyltransferase (HsdM and HsdS subunits). The hsdS sequences flanked by inverted repeats (referred to as epigenetic invertons) in certain Type I R-M systems undergo invertase-catalyzed inversions. Previous studies in Streptococcus pneumoniae have shown that hsdS inversions within clonal populations produce subpopulations with profound differences in the methylome, cellular physiology and virulence. In this study, we bioinformatically identified six major clades of the tyrosine and serine family invertases homologs from 16 bacterial phyla, which potentially catalyze hsdS inversions in the epigenetic invertons. In particular, the epigenetic invertons are highly enriched in host-associated bacteria. We further verified hsdS inversions in the Type I R-M systems of four representative host-associated bacteria and found that each of the resultant hsdS allelic variants specifies methylation of a unique DNA sequence. In addition, transcriptome analysis revealed that hsdS allelic variations in Enterococcus faecalis exert significant impact on gene expression. These findings indicate that epigenetic switches driven by invertases in the epigenetic invertons broadly operate in the host-associated bacteria, which may broadly contribute to bacterial host adaptation and virulence beyond the role of the Type I R-M systems against phage infection.
INTRODUCTION
Bacterial restriction-modification (R-M) systems were originally discovered in enterobacteria as a defense mechanism against bacteriophage invasion (1–3), and later found in the vast majority of prokaryotic organisms (4,5). At present, more than 20 000 R-M systems are listed in the REBASE restriction enzyme database (6). The R-M systems have been divided into four Types based on their constituents and modes of the action. All of the R-M systems possess restriction endonuclease (R) and nucleotide modification (M) enzyme activities except for Type IV R-Ms, which encode only endonucleases for digestion of methylated DNA and do not have a modification function. Type II R-M systems are the best-known type, in which both the R and M subunits function independently as an endonuclease (R) and modification (M) enzyme, respectively. The Type I systems represent the most complex R-M type, possessing three subunits encoded by host specificity determinant (hsd) genes: hsdR (restriction), hsdM (modification) and hsdS (specificity). The Type I R-M enzyme is a pentameric complex with two copies of the HsdR (R) and HsdM (M) subunits together with one HsdS (S) subunit (7). Unlike the counterparts in Type II R-M systems, the Type I R and M subunits are incapable of recognizing target DNA sequences and thus depend on the S subunit to do so. In the absence of the R subunit, the M and S subunits can still retain DNA methyltransferase (MTase) activity. Each HsdS protein consists of two target recognition domains (TRD); each TRD recognizes a half of the target sequence (8). Two TRDs in a given HsdS protein are functionally separated from each other and form a functional S subunit of the Type I R-M MTase by combining with another TRD from an unrelated HsdS protein (9); the resulting MTase methylates a new sequence.
Recent studies have uncovered many special Type I and III R-M systems or phasevarions, which enable the host bacteria to undergo reversible switches or phase variations in genome methylation pattern and virulence-associated traits (10). At least three mechanisms have been described to drive reversible sequence changes in phasevarions: (i) natural frame-shifting mutations in the hsdM genes in Type III R-M systems, (ii) reversible excisions-reintegration in the hsdS genes of Type I R-M systems and (iii) inversions in the hsdS genes of Type I R-M systems. Changes in polynucleotide repeat tract of R-M genes are shown to result in ON-or-OFF phase variations in the expression of functional R-M proteins by the slipped-strand mispairing mechanism (11–13). Recombinase-mediated excision-reintegration between two hsdS genes in the SpnVI Type I R-M system of Streptococcus pneumoniae leads to phase variations in pneumococcal methylome (14). Programmed inversions in the hsdS genes of Type I R-M systems have been reported in many bacteria, such as Bacteroides fragilis (15), Lactobacillus salivarius (16), Listeria monocytogenes (17), Mycoplasma pulmonis (18,19), S. pneumoniae (20–24) and Streptococcus suis (25). More importantly, the resultant phase variations in bacterial methylome are associated with profound changes in transcription of bacterial genes, particularly those encoding virulence factors (5,10,23).
The biochemical mechanisms and epigenetic impacts of the hsdS inversions are most extensively studied in the SpnIII Type I R-M system or colony opacity determinant (cod) locus of S. pneumoniae. The inversions among the three hsdS genes are predominantly catalyzed by a tyrosine recombinase family invertase PsrA or CreX through recognizing pairs of inverted repeats flanking the invertible sequences (21,23,26–27). The hsdS inversions in S. pneumoniae allow a single cell to generate multiple allelic variants of the HsdS subunits, each of which forms a DNA MTase that methylates a unique sequence (21,23), resulting in extensive differences in methylome. More importantly, the methylomes defined by hsdS allelic variants determine the transcriptome, colonies opacity phases and pathogenic traits of S. pneumoniae (21,23,28).
Previous studies have revealed that many archaeal and bacterial species possess Type I R-M systems with multiple hsdS genes (5,21,29). DNA inversions catalyzed by serine recombinases and tyrosine recombinases have been well documented to drive ON/OFF orientations of the promoter sequences flanked by inverted repeats or invertons, and thereby cause phase variations in the expression of flagella, pilus, capsule and surface proteins in many bacteria (30–32). Because the well-characterized inversions in Type I R-M systems share the same biochemical principles with the reactions in the phase variable promoter invertons: invertase and invertible sequences flanked by a set of inverted repeats (33,34), we will use the term epigenetic inverton to define the invertible hsdS sequences bound by pairs of inverted repeats.
This study bioinformatically characterized the general features of potentially invertible Type I R-M systems, or epigenetic invertons, in phylogenetically diverse bacteria and experimentally analyzed inversions in the epigenetic invertons of B. fragilis, Enterococcus faecalis, Streptococcus agalactiae and Treponema denticola. By determining the DNA methylomes in these bacteria, we establish that the allelic variants of the Type I R-M DNA MTases generated by the hsdS inversions result in distinct DNA methylation patterns in these bacteria. Finally, the epigenetic impact of the recombinations on gene expression was assessed in E. faecalis. The broad significance of the epigenetic invertons is discussed.
MATERIALS AND METHODS
Bacterial strains and cultivation, and chemical reagents
All bacterial strains used in this study are listed in Supplementary Table S1. B. fragilis NCTC9343 (ATCC25285) and T. denticola ATCC35405 were obtained from the American Type Culture Collection (ATCC); S. agalactiae strain 515 from Michael R. Wessels (35). E. faecalis strain TH4125 was originally isolated from a urine culture of a patient with liver cirrhosis in the 302 People's Liberation Army (PLA) Hospital (Beijing, China); strain H25 was a clinical isolate from Hospital Universitario Ramón y Cajal (Madrid, Spain) (36). S. pneumoniae TH5160 was constructed as described below and used as a host strain to determine DNA methylation activities of the hsdS allelic variants generated by hsdS inversions. S. pneumoniae and S. agalactiae strains were cultured in Todd-Hewitt broth supplemented with 0.5% yeast extract (THY) or on tryptic soy agar plates containing 3% sheep blood as previously described (21). E. faecalis was grown in brain-heart infusion (BHI) broth or on BHI agar plates. B. fragilis and T. denticola were grown in tryptone-yeast extract-gelatin-volatile fatty acids-serum (TYGVS) medium at 37°C in an anaerobic chamber with 85% nitrogen, 5% hydrogen and 10% CO2 as previously described (37,38). Escherichia coli DH5α was used for molecular cloning of hsdS sequences and grown in Luria–Bertani (39) broth or on LB agar plates as described (21). All chemicals and molecular biology reagents were obtained from Sigma (Shanghai, China) and New England Biolabs (Beijing, China), unless stated otherwise. All primers were supplied by Ruibio Biotech (Beijing, China) and listed in Supplementary Table S2.
Bioinformatic analysis of epigenetic invertons
Putative epigenetic invertons in the REBASE database were identified by downloading all available Type I R-M systems (on 10 July 2020) and filtered for those with at least two hsdS genes. The sequences generated from metagenome or mixed sample sequencing were excluded from further analysis due to potential hsdS gene mis-assembly during the sequencing process. The sequences from redundant genomes of the same bacteria (the same genome with different accession numbers) were also excluded. The genomic information of the resulting 3292 Type I R-M loci from 2829 strains was obtained from NCBI using Entrez Direct (EDirect, https://www.ncbi.nlm.nih.gov/books/NBK179288). The protein sequences of hsdS genes were downloaded from REBASE (http://rebase.neb.com/cgi-bin/seqsget?L+gd), and used to determine the coding regions and orientations in genomes. The Type I R-M loci with hsdS genes with opposite orientations were probed for the presence of adjacent invertase homologs within 10 kilo-bases (kb) from either the 5′ or 3′ coding boundary of hsdS based on their sequence annotations. Amino acid sequences of the resulting invertase homologs were compared by Molecular Evolutionary Genetics Analysis (MEGA) software (version 10.1.7 for Mac). Phylogenetic placement of the target bacteria was carried out by searching the NCBI databases and LPSN website (https://www.bacterio.net). Habitat placement of the invertase-positive species was carried out in the ProGenomes site (progenomes.embl.de) as described (40).
The invertase-positive epigenetic invertons in the NCBI databases were identified by a combination of multiple searches. A single representative invertase sequence was first chosen from each of the five major clades identified in the REBASE search based on the available information from the literature and this work: PsrA of S. pneumoniae (21,23) for clade 1, TvrR of S. pneumoniae (41) for clade 2, BfiA (B. fragilisinvertase A) for clade 3 (15), EfiA (E. faecalisinvertase A) for clade 4 and MfiA (Mycoplasma fermentansinvertase A) for clade 5. These five proteins, along with the HvsR invertase of M. pulmonis (19) (referred to as clade 6), were used as queries to identify homologs in the non-redundant database by Protein BLAST as previously described (5). Because the queries of clades 1–4 yielded much large numbers of hits with higher homology levels than those of clades 5 and 6, subsequent analyses were focused on the hits with a 10−100 or lower probability (e) value for clades 1–4 and the hits representing the top 100 species for clades 5–6. Due to overrepresentation of certain bacterial species by too many strains, the original hits were filtered to leave only one representative hit from each species in the subsequent analyses (with highest homology with the query). The simplified six lists were then used to trace the corresponding DNA sequences in NCBI and identify Type I R-M genes based on their annotations. A physical distance of 2 kb was arbitrarily set as a cutoff between an invertase homolog and its nearest R-M genes (hsdR, hsdM or hsdS). In case more than one invertase homolog was identified within this range, only the one proximal to the R-M gene was considered. The resulting R-M systems were analyzed for multiple hsdS genes by the existing annotations in the accessions or BLAST analysis of unannotated ORFs in the absence of functional annotations for the ORFs surrounding the target invertase genes. The phylogenetic placement of the inverton-positive bacteria was identified as described above.
Detection of hsdS inversions
DNA inversions in Type I R-M loci were detected by polymerase chain reaction (PCR) and DNA sequencing as described in our previous work (21). Briefly, potentially invertible hsdS sequences were amplified from genomic DNA with combinations of a forward primer in the non-invertible hsdM gene and another primer in the downstream hsdS sequences. Both the forward and reverse orientations of the downstream primers were used to detect DNA inversions. The genome sequences of T. denticola ATCC35405 (accession AE017226), B. fragilis NCTC9343 (CR626927), S. agalactiae 515 (AAJP01000001-AAJP01000255) and E. faecalis H25 (GCA_002289045.2) were used to map the R-M loci and design the primers. Initial sequencing analysis of the Efa4125I locus in E. faecalis TH4125 was carried out using primers Pr12967 and Pr12979, which were designed with the sequence of E. faecalis strain L12 (CP018102). Successful amplification in the reactions with the same-orientation primer sets was an initial indicator of DNA inversion(s) for the target sequences, which was verified by DNA sequencing of the amplicons. The sizes of the secondary hsdS genes are defined by the nucleotides between the 5′ ends of their relevant IR sequences and termination codons.
In E. faecalis H25, the orientation of the hsdS alleles in the EfaH25I locus was quantified as described (23). In brief, using the primer pair ZJ41F 5′-TTAGCTATGCTTGATGACCTAGTGGTAACGGAAG-3′ (FAM labeled) and ZJ41R 5′-TTTTCTGTTACGCATACTTCCTCCAAATGATAGTTGTT-3′, the segment containing both hsdS alleles was amplified and then digested with BciVI and Hpy99I. This restriction digestion yielded allele specific labeled fragments (allele A 878 bp, B 996 bp, C 963 bp and D 843 bp), which were quantified by capillary electrophoresis and analyzed using Peak Scanner v1.0 software (Thermo Fisher, UK). To generate wild type stocks expressing the same hsdS allele, H25 was plated and single colonies were selected and passaged. Three independent stocks expressing the same hsdS allele in over 70% of cells were generated for each of the four variants (allele A stocks MRO768, MRO769, MRO770; allele B stocks MRO771, MRO772, MRO773; allele C stocks MRO774, MRO775, MRO776; allele D stocks MRO777, MRO778, MRO779).
DNA amplification and analysis
Target DNA fragments were amplified by PCR with a high fidelity PrimeSTAR DNA polymerase (TaKaRa, Dalian, China) as described (20). Sequences of PCR products and plasmids were determined using the Sanger sequencing method by Ruibio Biotech (Beijing, China) (42). Rho-independent transcription terminators were characterized by the internet-based ARNold program (http://rna.igmors.u-psud.fr/toolbox/arnold/index.php#Results); promoters were predicted using Neural Network Promoter Prediction (http://www.fruitfly.org/seq_tools/promoter.html). Routine DNA sequence analyses for inverted repeats, sequence homology, coding, restriction features and primer design were carried out with the Lasergene package (version 15.0.0 for Mac).
Detection of DNA methylation
Methylated DNA sequence motifs in bacterial genomes were identified by single molecule real-time (SMRT) sequencing essentially as described (20). The methylomes of B. fragilis NCTC9343, E. faecalis TH4125, S. agalactiae 515 and T. denticola ATCC35405 were determined by the PacBio RS II system in the W. M. Keck Foundation Biotechnology Resource Laboratory at Yale University, and deposited in the SRA database under the PRJNA593863 project. The genomes and methylation motifs of E. faecalis H25 and its hsdS variants were determined by PacBio Sequel II system in the Wellcome Sanger Institute (Cambridge, UK). The genomic DNA of hsdS variants EfaH25-A, EfaH25-B, EfaH25-C and EfaH25-D was extracted from exponential bacterial cultures grown in BHI broth by the Zymo clean and concentrator kit (Zymo, USA) after the pellet was treated with Mutanolysin (Sigma, UK). The SMRT sequencing data for E. faecalis H25 and its hsdS variants were deposited in the SRA database under the PRJNA400682 project.
Expression of hsdS allelic variants in S. pneumoniae
The allele-specific MTase activities of the hsdSA variants in the four bacteria were determined in S. pneumoniae strain TH5160 by SMRT sequencing (20). TH5160 was chosen because it carried unmarked deletions of both the Type I R-M loci (Spn556II and Spn556III) and our preliminary study did not detect any Type I R-M methylation motifs in the genome. In addition, high transformation frequency of this strain allowed us to directly compare the MTase activities of all 21 hsdSA variants in a single genetic background. TH5160 (ΔSpn556II/ΔSpn556III) was constructed in strain ST5444 (ΔSpn556II), which lacked the Spn556II Type I R-M (cod) system (21). First, the Spn556III Type I R-M system was replaced with the Janus cassette JC1 to generate strain TH6760 (ΔSpn556II/ΔSpn556III::JC1). The up- and downstream sequences of the Spn556III locus were separately amplified by PCR from genomic DNA of ST606 using primer pairs Pr9405/Pr9406 and Pr9407/Pr9408, respectively; JC1 from genomic DNA of strain TH7187 (21) with primers Pr9840 and Pr1098. The three amplicons were digested with XbaI and XhoI, and linked with T4 DNA ligase before being transformed into TH5444 and selected for kanamycin-resistant (400 μg/ml) colonies. Second, JC1 was removed by transformation with a fusion DNA of the Spn556III flanking sequence amplicons. The up- and down-stream sequences of the Spn556III locus were PCR amplified from genomic DNA of TH5444 using primer pairs Pr6940/Pr7958 and Pr6934/Pr7957, respectively, and linked by fusion PCR with primers Pr6940 and Pr6934.
The native sequences spanning the coding regions of the hsdSA1 allele and upstream hsdM gene were PCR amplified from the genomic DNA of each target bacterium and cloned into pIB166, a shuttle vector of E. coli–S. pneumoniae (43). The plasmid containing hsdM and hsdSA1 for each of the four target bacteria was subsequently used as a backbone to construct the DNA inserts of additional hsdSAalleles from the same species by replacing the hsdSA1-specific sequence with the counterparts of other alleles using allele-specific primers. The primers and DNA templates used for cloning hsdSA variants are specified in Supplementary Table S3. The recombinant derivatives of pIB166 were subsequently transformed in S. pneumoniae TH5160. Genomic DNA samples of the resultant chloramphenicol resistant pneumococcal derivatives (4 μg/ml) were used to determine the methylated DNA sequences by SMRT sequencing. The methylomes specified by Ef-hsdSA1, Sa-hsdSA1 and Td-hsdSA1 were detected at the Yale University platform as described above; the activities of the remaining hsdSA alleles by the PacBio Sequel system at the Novogene Bioinformatics Technology (Beijing, China). The SMRT sequencing data for all 21 hsdSA variants are available under the PRJNA593863 project in the SRA database.
RNA sequencing (RNA-seq)
RNA-seq analysis in E. faecalis H25 variants was carried out as described previously (23). Bacteria were grown to mid-log phase in BHI broth, harvested by centrifugation and lysed with 10 μg/ml lysozyme (Sigma, UK) and 10 U/ml Mutanolysin (Sigma, UK). RNA was extracted using the Maxwell® 16 LEV SimplyRNA Cells Kit (Promega, USA) following the manufacturer's instructions. After depletion of rRNAs with Ribo-Zero from Illumina, the RNA samples were sequenced as 75-bp paired-end reads on an Illumina HiSeq 4000 and trimmed with trimmomatic/0.32 (44). Trimmed FASTq reads were aligned to the E. faecalis H25 genome generated by SMRT sequencing (accession GCA_002289045.2). RNA-seq was performed in triplicates on one clone for each variant and then a single RNA-seq for the second clone of each variant. The FASTq reads have been deposited in the SRA (accession PRJNA400682).
RESULTS
Identification of epigenetic invertons in a wide range of bacteria
To better understand the phylogenetic spectrum of the epigenetic invertons, Type I R-M systems with multiple hsdS genes and invertase homologs, in the genomes of prokaryotic organisms, we first sought to identify the Type I R-M loci with at least two hsdS genes by taking advantage of the R-M annotations in the restriction enzyme database, REBASE (6). Epigenetic inverton is here defined as an invertible hsdS sequence of Type I R-M system that is flanked by a pair of inverted repeats. As exemplified by the invertons of four representative bacteria in the below sections, a Type I R-M system can possess multiple invertons. This search identified a total of 3292 Type I R-M systems with at least two hsdS genes (referred to as S+ systems) (Supplementary Table S4). We further searched for the S+ systems that encode invertase homologs as described in ‘Materials and Methods’ section.
This analysis identified 827 Type I R-M loci with putative invertons and at least one invertase homolog from 235 species (Supplementary Table S5). Based on sequence homology of the invertases, the host bacteria are placed into 10 of the 41 currently established bacterial phyla and one candidate phylum (Candidatus Saccharibacteria). As summarized in Table 1, the vast majority of the hits are concentrated in Firmicutes, Bacteroidetes, Proteobacteria, Tenericutes, Verrucomicrobia and Actinobacteria. Further characterization of environmental niches of the composite bacteria based on the information from ProGenomes (40) divided these bacteria into the residents of host-associated (216/235, 91.9%), terrestrial (11/235, 4.7%) and aquatic (8/235, 3.4%) habitats (Supplementary Table S5). The overwhelming representation of host-associated genomes in the inverton-positive Type I R-M suggests functional association between the epigenetic invertons and host adaptation. Additional sequence comparison further divided the majority of the 827 invertases (98.3%) into five major invertase clades (Figure 1A). All of the invertases in clades 1–4 belong to the tyrosine recombinase family. The sequences in clade 5 included the members of the serine recombinase family (six loci), the tyrosine recombinase family (three loci) and completely unknown proteins (without conserved tyrosine and serine residues, four loci). The additional 14 invertase homologs beyond the five major clades have < 30% amino acid sequence homology to the members of the major clades and possessed either the conserved tyrosine (13 loci) or serine (one loci) residue (45) (Figure 1A and Supplementary Table S5).
Table 1.
Phylogenetic distribution of the invertase homologs identified in REBASE
| Phylum | No. of loci clade 1 | No. of loci clade 2 | No. of loci clade 3 | No. of loci clade 4 | No. of loci clade 5 | No. of loci others | Total loci | % of loci |
|---|---|---|---|---|---|---|---|---|
| Actinobacteria | 2 | 9 | 0 | 9 | 1 | 0 | 21 | 2.5 |
| Bacteroidetes | 46 | 19 | 65 | 0 | 0 | 7 | 137 | 16.6 |
| Candidatus Saccharibacteria | 0 | 2 | 0 | 0 | 0 | 0 | 2 | 0.2 |
| Chloroflexi | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0.1 |
| Euryarchaeota | 0 | 1 | 0 | 0 | 3 | 0 | 4 | 0.5 |
| Firmicutes | 91 | 74 | 4 | 273 | 0 | 2 | 444 | 53.7 |
| Fusobacteria | 0 | 3 | 0 | 0 | 0 | 0 | 3 | 0.4 |
| Proteobacteria | 95 | 2 | 0 | 20 | 1 | 0 | 118 | 14.3 |
| Spirochaetes | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0.1 |
| Tenericutes | 16 | 17 | 1 | 22 | 8 | 5 | 69 | 8.3 |
| Verrucomicrobia | 16 | 11 | 0 | 0 | 0 | 0 | 27 | 3.3 |
| Total loci | 267 | 139 | 70 | 324 | 13 | 14 | 827 | 100.0 |
Figure 1.

Sequence diversity and phylogenetic distribution of the inverton-associated invertases. (A) Sequence diversity of the invertase encoded by Type I R-M systems. The invertase homologs encoded by the representative S+ Type I R-M systems initially identified from REBASE (as summarized in Table 1) were used to construct a cladogram by the Neighbor Joining method. The five major clades are colored and labeled with the names of phyla, in which bacteria housing the composite sequences were identified. (B) Phylogenetic distribution of the six invertase clades. The inverton-associated invertase homologs were identified from the NCBI database using the representative proteins of the six invertase clades. Percentage representations of the 16 phyla in the final hits of six clades (as summarized in Table 2) were proportionally presented as colored sections in concentric donuts by Microsoft Excel.
As represented by the pneumococcal PsrA/CreX (21,23), the 267 clade-1 invertases are encoded by the S+ Type I R-M systems in seven bacterial phyla. Arrangement of the hsdS genes in these systems also resembles that of the pneumococcal cod locus (21,23) and the S+ Type I R-M systems of the four bacteria characterized later in this study. The S+ Type I R-M loci encoding the clade-2 invertases are found in 10 bacterial phyla, the widest phylogenetic distribution among the major invertase clades. The hsdS genes in the 139 Type I R-M loci encoding clade-2 invertases are mostly arranged in peculiar manners. As illustrated in Supplementary Figure S1, the first and downstream hsdS genes of these loci are arranged in mixed coding orientations; however, inversion reactions between these hsdS genes would mostly yield truncated HsdS proteins due to the positions of the inverted repeats, and thereby lead to ON/OFF phase switches in the methylome. This clade also include the TvrR recombinase, which catalyzes reversible hsdS excision/reintegration of SpnIV or Spn556III Type I R-M system of S. pneumoniae (41). In this context, the clade-2 invertases may catalyze both inversion and excision/reintegration reactions between hsdS genes depending on the orientations of target repeat sequences as demonstrated for the Cre recombinase (46). The clade-3, −4 and −5 invertases were found only in a few phyla.
Because the REBASE databases only collect the sequences of complete genomes or whole genome contigs, we further examined the phylogenetic spectrum of the invertases encoded by the S+ Type I R-M systems by sampling the NCBI non-redundant protein database, which has a much larger collection of sequences. A protein sequence from each of the five invertase clades identified in our initial search was used to perform individual Protein BLAST search as described in ‘Materials and Methods’ section. The HvsR invertase was also included as a query for clade 6, because it catalyzes hsdS inversions in M. pulmonis although it is not encoded by its target Type I R-M loci (19). After a series of filtrations specified in ‘Materials and Methods’ section, the combined results of six separate searches identified a total of 1140 invertases encoded by the S+ Type I R-M systems (Supplementary Tables S6–11). In addition to the 11 invertase-positive invertons identified from the REBASE search (Table 1), the NCBI search identified five new bacterial phyla harboring this type of invertible structures, including four established bacterial phyla (Chlamydiae, Fibrobacteres, Lentisphaerae and Synergistetes) and one candidate phyla (Candidatus Melainabacteria) (Table 2 and Figure 1B). Consistent with the REBASE search result (Figure 1A), the hits were predominantly found in the same five phyla: Firmicutes (52.7%), Bacteroidetes (23.9%), Actinobacteria (8.6%), Proteobacteria (5.5%) and Tenericutes (4.7%) (Table 2). Likewise, the invertases of clades 1–4 were more abundantly and broadly identified in the S+ Type I R-M systems, but clades 5 and 6 were limited to Mycoplasma species in the phylum of Tenericutes. Taken together, the REBASE and NCBI searches indicate that Type I R-M systems employ highly divergent tyrosine and serine site-specific recombinases to catalyze hsdS inversions and/or excisions in a vast diversity of bacteria, in particular in those host-associated pathogens.
Table 2.
Phylogenetic distribution of the six invertase clades identified in NCBI
| Phylum | No. of loci clade 1 PsrA | No. of loci clade 2 TvrR | No. of loci clade 3 BfiA | No. of loci clade 4 EfiA | No. of loci clade 5 MfiA | No. of loci clade 6 HvsR | Total loci | % of loci |
|---|---|---|---|---|---|---|---|---|
| Actinobacteria | 1 | 43 | 0 | 54 | 0 | 0 | 98 | 8.6 |
| Bacteroidetes | 72 | 88 | 111 | 2 | 0 | 0 | 273 | 23.9 |
| Candidatus Saccharibacteria | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0.1 |
| Chlamydiae | 1 | 0 | 0 | 1 | 0 | 0 | 2 | 0.2 |
| Chloroflexi | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0.1 |
| Candidatus Melainabacteria | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0.1 |
| Euryarchaeota | 0 | 11 | 0 | 0 | 0 | 0 | 11 | 1.0 |
| Fibrobacteres | 1 | 2 | 0 | 0 | 0 | 0 | 3 | 0.3 |
| Firmicutes | 56 | 267 | 4 | 274 | 0 | 0 | 601 | 52.7 |
| Fusobacteria | 1 | 7 | 0 | 0 | 0 | 0 | 8 | 0.7 |
| Lentisphaerae | 0 | 2 | 0 | 0 | 0 | 0 | 2 | 0.2 |
| Proteobacteria | 19 | 11 | 0 | 33 | 0 | 0 | 63 | 5.5 |
| Spirochaetes | 7 | 8 | 0 | 1 | 0 | 0 | 16 | 1.4 |
| Synergistetes | 0 | 1 | 0 | 1 | 0 | 0 | 2 | 0.2 |
| Tenericutes | 13 | 14 | 1 | 16 | 3 | 8 | 54 | 4.7 |
| Verrucomicrobia | 1 | 2 | 1 | 0 | 0 | 0 | 4 | 0.4 |
| Total loci | 173 | 458 | 117 | 382 | 3 | 8 | 1140 | 100.0 |
General features of the epigenetic invertons
Sequence comparisons revealed several interesting features among the epigenetic invertons identified thus far. There is a remarkable sequence diversity among the invertase-positive invertons. As exemplified in Figure 2A, the invertases, HsdM, HsdR and HsdS proteins in the pneumococcal cod locus, are highly homologous to their counterparts in S. agalactiae NCTC8182; however, the same proteins showed marginal sequence homology with the counterparts from clades 2 (Bifidobacterium longum F8), 3 (B. fragilis NCTC9343), 4 (E. faecalis 4125), 5 (M. fermentans M64) and 6 (M. pulmonis UAB-CTIP). In particular, there is a remarkable sequence heterogeneity among the invertases from different clades. For instance, the clade-1 invertase PsrA does not share significant sequence homology with any of the representative invertases from the other clades (Figure 2A). Even among the tyrosine family invertases of clades 1, 2, 3, 4 and 6, there is a marginal homology, except for the conserved catalytic tyrosine residue (34). Identification of serine recombinase family invertases in S+ Type I R-M loci has significantly broadened the enzymatic spectrum of the epigenetic invertons because this recombinase family has not yet been reported to catalyze hsdS inversions. This result strongly suggests that the invertases in different clades represent the products of convergent evolution, which are derived from unrelated proteins.
Figure 2.

Genetic features of the epigenetic invertons. (A) Schematic illustration of the invertase and R-M genes in seven representative invertase-positive Type I R-M loci from the six invertase clades. The sequences and coding orientations of hsdR (red), hsdM (green), hsdS (blue) and invertase (yellow) genes in each locus are indicated with colored arrows. Percent amino acid sequence homology between each pneumococcal gene and the counterpart of other bacterium is marked on the right side of the gene cluster. (B) Presence of multiple invertons in the same bacteria. Two Type I R-M loci in M. bovis PG45 (accession CP002188) homologous to the invertons from Streptococcus pneumoniae and Enterococcus faecalis are schematically displayed as in (A). Percent sequence similarity between the two related sequences (indicated with a dashed line) is provided between each gene pair. (C) Duplication of the invertons. Two Type I R-M loci with identical genetic organization and similar sequences in Jeotgalibaca sp. H21T32 and Mycoplasma pulmonis UAB-CTIP are schematically presented as in (A). The sequences encoding each TRD of the hsdS genes are marked with various patterns of striped rectangles to reflect similarity between the related sequences.
Certain bacteria possess multiple epigenetic invertons. As an example, Mycoplasma bovis PG4 (accession CP002188) carries two invertons of clades 1 and 4 (Figure 2B). The clade 1 inverton is homologous to the pneumococcal cod locus, whereas the clade 4 is similar to a Type I R-M locus in E. faecalis TH4125, suggesting that these invertons are separately acquired by horizontal gene transfer. In contrast, some bacteria can harbor similar invertons as exemplified by the two Type I R-M loci in Jeotgalibaca sp. H21T32 and M. pulmonis UAB-CTIP (Figure 2C), which appear to be generated by gene duplication.
Interestingly, epigenetic invertons are occasionally found in plasmids. Certain S+ Type I R-M loci identified in REBASE and NCBI databases are annotated as the elements of plasmids. As an example, 61 out of the 3292 S+ Type I R-M loci identified from the REBASE search are plasmid-based, such as those in the Ruminococcus albus 7 plasmid pRUMAL02 (accession CP002405), L. salivarius ZLS006 plasmid (CP020859) and Lactococcus lactis JM1 plasmid pMPJM1 (CP016746). We also noticed that the allelic variant sequences generated by hsdS inversions in Type I R-M loci were often incorrectly annotated as plasmids in the genome sequences because they could not be properly assembled in the genomes.
Because invertases perform DNA cut/ligation reactions by interacting with enzyme-specific inverted repeats in target sequences (34), we sampled inverted repeats in 46 Type I R-M loci representing all of the 16 phyla with invertase-positive invertons identified in the NCBI search. This identified inverted repeat sequences in the hsdS coding regions of 45 tested Type I R-M loci (Supplementary Table S12). The only exception is a Type I R-M locus of Sphaerochaeta halotolerans (NZ_WUJG01000003), in which direct repeat sequences, instead of inverted repeats, were found in the hsdS genes. This feature resembles the direct sequence-bound hsdS sequences in the SpnIV phase-variable Type I R-M locus of S. pneumoniae, which has been shown to undergo recombinase-catalyzed reversible excisions/reintegrations (41). Collectively, these findings strongly suggest that a small proportion of the S+ Type I R-M systems identified above in the NCBI search undergo hsdS excisions/reintegrations instead of inversions (Table 2).
Programmed DNA rearrangements in the inverton of B. fragilis NCTC9343
Based on the sequence and phylogenetic features of the invertase homologs in the epigenetic invertons (Figure 1), we assessed hsdS inversions in the four phylogenetically distinct bacterial species that represent three major clades of tyrosine family invertases encoded by the S+ Type I R-M systems: S. agalactiae 515 (clade 1), T. denticola ATCC35405 (clade 1), B. fragilis NCTC9343 (clade 3) and E. faecalis TH4125 (clade 4). Our clades/species selection was based on medical importance of the target bacteria and significant epigenetic role of the hsdS inversions catalyzed by PsrA (clade-1 tyrosine invertase) in pneumococcal pathobiology (5,47). The clade-2 invertase was not included for further methylome analysis because of the complex configurations of the associated hsdS genes as described above (Supplementary Figure S1).
Bacteroides fragilis is a Gram-negative anaerobe normally residing in gastrointestinal tract and an opportunistic pathogen of abscesses and bacteremia (48). It is well known for phase and antigenic variation of surface structures, but the underlying molecular mechanisms are not fully characterized (15). The NCTC9343 genome contains nine putative R-M systems as predicted in REBASE. Bfa9343I, one of the three Type I R-M systems, harbors an inverton with four hsdS genes and a putative invertase gene (BF1843 or bfiA) (Figure 3A). For clarity, these hsdS genes are denoted Bf-hsdSA (BF1839, 1119 bp), Bf-hsdSB (BF1840, 644 bp), Bf-hsdSC (BF1841, 628 bp) and Bf-hsdSD (BF1842, 1143 bp). While Bf-hsdSA and Bf-hsdSD consist of two TRDs, Bf-hsdSB and Bf-hsdSC harbor only one TRD.
Figure 3.
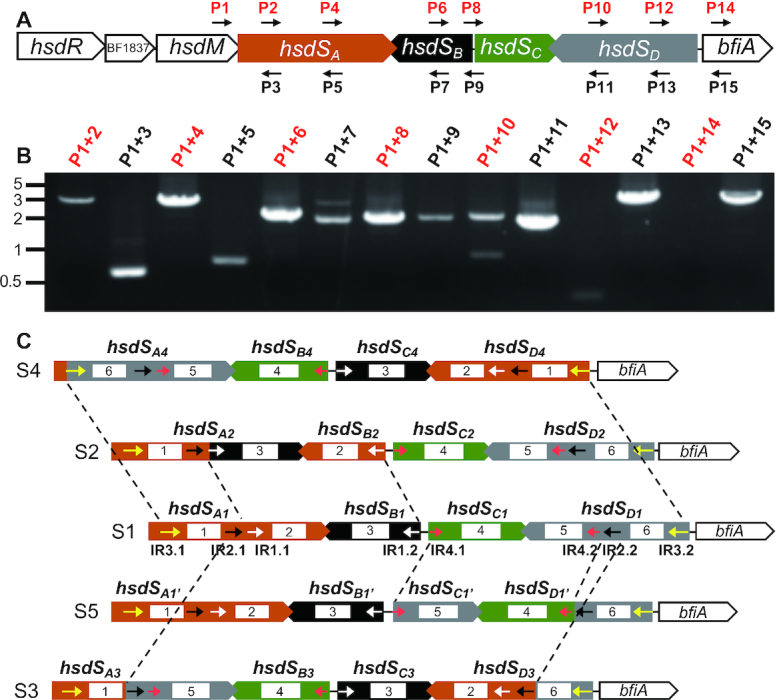
Genetic arrangements of four epigenetic invertons in Bacteroides fragilis NCTC9343. (A) Genetic arrangement of the Bfa9343I Type I R-M locus. The genes are marked with either their predicted functions or tags annotated in the genome. The primers used for detecting DNA inversions in (B) are indicated with small arrowheads. (B) Detection of hsdS inversions by PCR. The primers used for each reaction are marked at the top of each lane. The reactions with the same-orientation primer pairs are marked with red characters. (C) Schematic representation of variable configurations in the Bfa9343I locus. The DNA configurations generated by hsdS inversions are marked as S1–5. The TRDs encoded by hsdS genes are indicated by white rectangles and numbered. The position and orientation of inverted repeats IR1 (white), IR2 (black), IR3 (yellow) and IR4 (red) are indicated with small arrowheads. The invertible sequences are marked with dashed lines.
Although a previous sequencing study indicated that inversions occur among the hsdS genes in the Bfa9343I locus (15), there is no experimental proof that these inversions occur. We thus tested inversions among the four hsdS genes by PCR using a combination of the same- and opposite-orientation primer sets because the reactions with the two same-orientation primers could not amplify the intervening sequence unless the orientation for one of the primers is switched to the opposite direction by inversion (21) (Figure 3A). This experiment yielded multiple amplicons with combinations of primer P1 in the coding region of hsdM and one of the downstream primers with the same sequence orientation (Figure 3B). Amplicons were generated with the primer sets targeting Bf-hsdSA (P1-P2/P4), Bf-hsdSB (P1-P6), Bf-hsdSC (P1-P8) and Bf-hsdSD (P1-P10/P12). This result confirmed that the Bfa9343I locus undergoes extensive inversions. Sequencing analysis of the PCR products identified four invertible sequences in the coding regions of the hsdS genes. Each of the invertible sequence is bound by one of the four pairs of inverted repeats: 20-bp IR1 (5′-AATCTCTAATTAAAGGGCTT-3′, white arrowheads), 29-bp IR2 (5′-GAGCGTATCGCAACCCAAAACAAAATAAT-3′, black arrowheads), 47-bp IR3 (5′-AATTTCCCAACTTTGCGATTTCCAGAGTTCTCGGGTGAGTGGAAAAA-3′, yellow arrowheads) and 16-bp IR4 (5′-GAATGCACTTGCGGAA-3′, red arrowheads) (Figure 3C). IR1, IR2 and IR3 control inversions of Bf-hsdSA with Bf-hsdSB and Bf-hsdSD, whereas IR4 defines the boundary of the inversion between Bf-hsdSC and Bf-hsdSD.
The sequencing data indicated that inversions of the four invertible sequences could potentially generate 16 different DNA configurations in this locus, which were designated as Bf-S1 to S16 (Figure 3C and Supplementary Figure S2). The invertible sequences in Bf-S1 can be converted into four new DNA forms (S2–S5) (Figure 3C). Bf-S2 is generated by inversion of a 1175-bp IR1-bound sequence between Bf-hsdSA1 and Bf-hsdSB1; Bf-S3 and Bf-S4 by the inversions of IR2- (2466 bp) and IR3-bound (3492 bp) sequences between Bf-hsdSA and Bf-hsdSD, respectively. Inversion of a 1188-bp IR4-flanked region between Bf-hsdSC and Bf-hsdSD yields Bf-S5, which does not alter the sequence of the hsdSA gene, but the new TRD in Bf-hsdSD1’ makes it possible to move the TRD encoded by Bf-hsdSD1’ into hsdSA in subsequent reactions (Supplementary Figure S2). Further inversions in Bf-S2, Bf-S3, Bf-S4, Bf-S6, Bf-S7, Bf-S9 and Bf-S12 generate additional 11 new configurations (S6–S16) (Supplementary Figure S2) and four new hsdSA alleles (Bf-hsdSA5, Bf-hsdSA6, Bf-hsdSA7 and Bf-hsdSA8). This result has demonstrated that extensive DNA inversions occur among the four hsdS genes in the Bfa9343I locus, resulting in genetic heterogeneity in a clonal population.
Impact of hsdS inversions on the B. fragilis methylome
To define the epigenetic impact of hsdS inversions in the Bfa9343I locus, we first determined the NCTC9343 methylome using SMRT sequencing, which identified a total of seven credible methylation motifs, each with an m6A nucleotide (Figure 4A). Four of these motifs fully matched the predicted methylation sequences for three R-M loci in REBASE: 5′-Gm6ACN5GRTY-3′/5′-Rm6AYCN5GTC-3′ for Bfa9343I, 5′-GANGGm6AG-3′ for Bfa9343II and 5′-ATGCm6AT-3′ for Bfa9343III. Three remaining motifs could not be assigned to any specific R-M MTases because there are at least six putative R-M loci with unknown recognition sequences in the genome. Two of the unassigned motifs (5′-CNm6AGN6TGA-3′ and 5′-TCm6AN6 CTNG-3′) should be the target sequence for one of the two Type I R-M systems (Bfa9343IV and Bfa9343V). Motif 5′-CYC m6AT-3′ appears to be methylated by one of the two Type III R-M systems (Bfa9343VI and Bfa9343VII) (Figure 4A).
Figure 4.
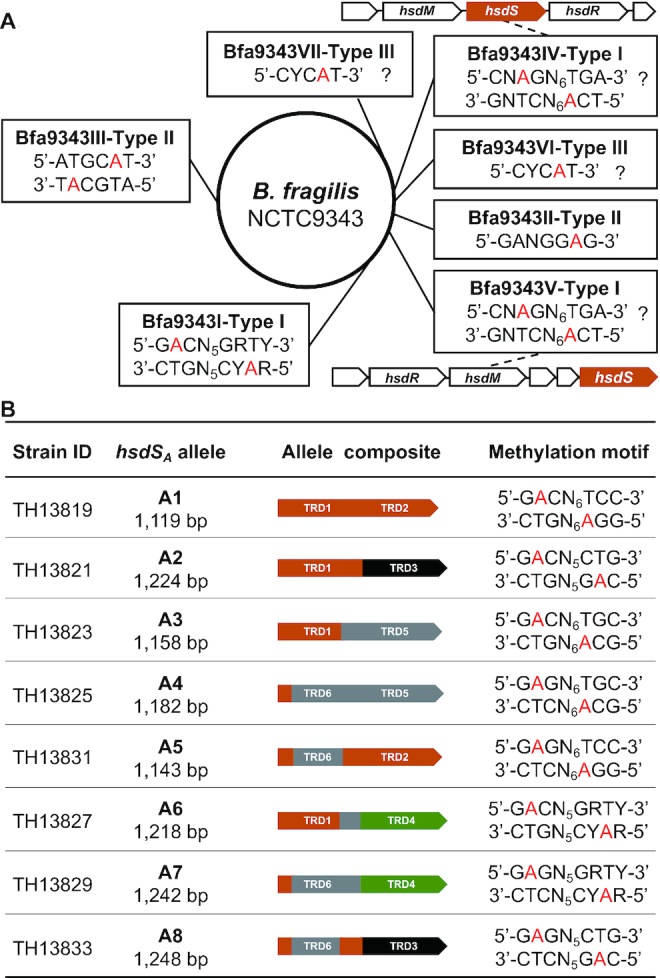
The DNA sequences methylated by the Bacteroides fragilis MTases. (A) The methylated sequences in the genome of B. fragilis NCTC9343. The methylated motifs identified by SMRT sequencing and corresponding R-M systems are placed together in single rectangles. The modified nucleotides are indicated with red characters. The genetic arrangements of the Bfa9343IV and Bfa9343V loci, the inverton-negative Type I R-M systems, are depicted; the sequences with uncertain MTase assignments are indicated with question marks. (B) The DNA motifs methylated by the hsdSA alleles. The sequence recognized by each of the eight hsdSA alleles is presented along with the identification of the pneumococcal strain used for the sequencing. R = A or G, Y = T or C, N = any four nucleotides.
Since the Bf-hsdSA gene likely encodes a functional S subunit of the Type I R-M MTase (explained in ‘Discussion’ section), we selectively assessed the methylation activities of the hsdSA alleles produced by the hsdS inversions in the Bfa9343I locus and the Type I R-M loci of T. denticola, E. faecalis and S. agalactiae by expressing these alleles in S. pneumoniae TH5160, a strain lacking the two endogenous Type I R-M systems (Figure 4B). Each of the eight hsdSA alleles and cognate Bf-hsdM were placed after the P23 promoter in the E. coli–S. pneumoniae shuttle vector pIB166. SMRT sequencing analysis revealed a unique Type I R-M MTase motif for each of the eight Bf-hsdSA alleles: Bf-hsdSA1 (5′-Gm6ACN6TCC-3′), Bf-hsdSA2 (5′-Gm6ACN5CTG-3′), Bf-hsdSA3 (5′-Gm6ACN6TGC-3′), Bf-hsdSA4 (5′-Gm6AGN6TGC-3′), Bf-hsdSA5 (5′-Gm6AGN6TCC-3′), Bf-hsdSA6 (5′-Gm6ACN5GRTY-3′), Bf-hsdSA7 (5′-Gm6AGN5GRTY-3′) and Bf-hsdSA8 (5′-Gm6AGN5CTG-3′) (Figure 4B). This result demonstrates that each of the eight hsdSA alleles generated by inversions in the Bfa9343I locus has a unique methylation specificity, and further indicates that a clonal population of B. fragilis NCTC9343 is capable of generating progeny cells acquiring one of the eight hsdSA alleles and thereby one of the eight methylomes through reversible hsdS inversions in the Bfa9343I locus.
Programmed DNA rearrangements in the inverton of T. denticola ATCC35405
Because spirochetes represent a phylogenetically distinct group of bacteria, we determined potential DNA inversions in the Tde35405VII Type I R-M locus of strain ATCC35405 in T. denticola, an anaerobic oral spirochete associated with human periodontal disease (49), The Tde35405VII locus possesses an inverton with three hsdS genes and a putative invertase gene (referred to as T. denticolainvertase A or TdiA), in addition to hsdR (TDE2747) and hsdM (TDE2746) (Figure 5A) (50). The three hsdS genes were designated as Td-hsdSA (TDE2744), Td-hsdSB (TDE2743) and Td-hsdSC (TDE2740). Td-hsdSA (1599 base pair, bp) encodes two TRDs, whereas Td-hsdSB (520 bp) and Td-hsdSC (572 bp) code for only one TRD.
Figure 5.

Epigenetic variations caused by hsdS inversions of two epigenetic invertons in Treponema denticola ATCC35405. Genetic composition of the Tde35405VII Type I R-M locus (A) is depicted as in Figure 3A; detection of DNA inversions (B) as in Figure 3B; the DNA configurations directly derived from S1 (C) by inversions as in Figure 3C. Inverted repeats IR1 (white) and IR2 (yellow) are marked with colored arrowheads. The methylated DNA motifs detected by SMRT sequencing in T. denticola ATCC35405 (D) are presented as in Figure 4A. The inverton-negative Type I R-M locus is illustrated at the bottom left corner. The DNA motifs methylated by the three hsdSA allelic variants (E) are presented as in Figure 4B. D = A or G or T, H = A or C or T, R = A or G, Y = T or C, N = any four nucleotides.
The reaction with the same-orientation primers in hsdM (P1) and 5′ region of hsdSA (P2) did not yield a detectable product, but two amplicons of ∼2 and 4.5 kb in size were obtained with the same-orientation primers P1 and P4 (Figure 5B), indicating that Td-hsdSB is invertible with at least two DNA configurations. Likewise, the reaction of primers P1 and P6 also generated an approximately 2-kb amplicon, which suggests that Td-hsdSC is subjective to inversion. Sequencing analysis of the P1/P4 amplicons identified the small segment as the product of inversion reaction between Td-hsdSA and Td-hsdSB, which was flanked by two 18-bp inverted repeats (5′-TTGTTATTATGAGAAGTT-3′, IR1) (Figure 5C). The IR1-mediated inversion replaced the 3′ region (586 bp) of Td-hsdSA with the entire coding sequence of Td-hsdSB (520 bp), which generated two new alleles: Td-hsdSA2 (1533 bp) and Td-hsdSB2 (586 bp). Sequencing analysis of the P1/P6 product revealed the inversion between Td-hsdSA and Td-hsdSC and identified two 14-bp inverted repeats (5′-ATAATTGTTATTAT-3′, IR2) at the 5′ and 3′ ends of the invertible region (Figure 5C). The IR2-mediated inversion placed Td-hsdSC (572 bp) in the original position of the 3′ Td-hsdSA, yielding allelic variants Td-hsdSA3 (1581 bp) and Td-hsdSC3 (590 bp). This result demonstrates that the IR1 and IR2 sequences mediate two separate inversions between Td-hsdSA and the downstream Td-hsdSB and Td-hsdSC genes in the Tde35405VII Type I R-M locus. These reactions generate three distinct configurations of this locus (S1–S3) (Figure 5C).
To define the epigenetic impact of the hsdS inversions, we characterized the methylomes of T. denticola ATCC35405 and pneumococcal derivatives each expressing one of the three Td-hsdSA alleles generated by the inversions in the Tde35405VII locus. The analysis revealed a total of eight methylated DNA sequences, including seven with N6-methyladenine (m6A) and one with N4-methylcytosine (m4C) (Figure 5D). All motifs completely matched the predicted recognition sequences of the MTases for five R-M loci in REBASE: 5′-Cm6AGN7TDCC-3′/5′-GGHm6AN7CTG-3′ (Tde35405VII, Type I), 5′-CNm6ACN6TTC-3′/5′-GA m6AN6CTNG-3′ (Tde35405VI, Type I), 5′-m4CTCTTC-3′/5′-GAAGm6AG-3′ (Tde35405II, Type II), 5′-RAm6ATTY-3′ (Tde35405V, Type II) and 5′-CTAm6AT-3′ (Tde35405IV, Type III). Motifs 5′-GAAGm6AG-3′ and 5′-m4CTCTTC-3′ possess different methylation types, but they appear to be methylated by the same M.TdeII enzyme encoded by the Tde35405II Type II R-M locus because the two sequences are reverse complemented (Figure 5D). This result suggests that M.TdeII as a novel MTase is able to carry out two different reactions by a single enzyme molecule, which is elaborated in the ‘Discussion’ section.
We further identified the DNA motifs that are methylated by the three Td-hsdSA alleles derived from the inversions in the Tde35405VII locus (Figure 5C). Each of the three hsdSA alleles along with the upstream sequence encoding TED2745 and Td-hsdM was placed on pIB166 and transformed into TH5160. SMRT sequencing revealed a unique Type I R-M MTase recognition motif for Td-hsdSA1 (5′-Cm6AGN7TDCC-3′), Td-hsdSA2 (5′-Cm6AGN7TCC-3′) and Td-hsdSA3 (5′-Cm6AGN7TGC-3′) (Figure 5E). Since only the motif methylated by Td-hsdSA1 was detected in wildtype T. denticola, this observation indicates that Td-hsdSA1 is the dominant allele under our culture conditions. Consistent with the common sequence for the first TRDs of the three hsdSA alleles, the first halves of the three methylation motifs share the same sequence (5′-Cm6AG-3′). This result indicates that all three Td-hsdSA alleles generated by inversions in the Tde35405VI locus are capable of generating completely different methylomes in T. denticola.
Programmed DNA rearrangements in the inverton of S. agalactiae 515
Streptococcus agalactiae or Group B Streptococcus is a leading Gram-positive pathogen of new-borne meningitis (51). The Type I R-M locus of S. agalactiae strain 515 (Sag515I) harbors an inverton with three hsdS genes (e.g. Sa-hsdSA, 1548 bp; Sa-hsdSB, 580 bp; and Sa-hsdSC, 1342 bp) and a putative 798-bp invertase gene (tentatively named S. agalactiaeinvertase A or saiA) (Figure 6A). Consistent with the sequence feature of the Sag515I locus, PCR amplifications revealed inversions in Sa-hsdSA (P1/P2), Sa-hsdSB (P1/P4) and Sa-hsdSC (P1/P8) (Figure 6B). DNA sequencing analysis of the amplicons identified eight different inversion reactions in three invertible sequences, which are flanked by three pairs of inverted repeats: 15-bp IR1 (5′-CTCTCCTTATGGGAA-3′, white arrowheads), 27-bp IR2 (5′-ACAAAAGGAGATGATAACTCTCCTTAT-3′, yellow arrowheads) and 18-bp IR3 (5′-GAAACCTTATGAGAAGTT-3′, black arrowheads) (Figure 6C). These reactions drive the reversible formation of eight DNA configurations (S1-S8) (Figure 6C and Supplementary Figure S3). Sa-S1 can be directly converted to configurations S2, S3 or S4 after inversions of the sequences flanked by IR1 (1095 bp), IR2 (2566 bp) and IR3 (4128 bp), respectively (Figure 6C). Additional four inversions switch DNA from Sa-S3 to S5 (IR1-bound sequence) and S6 (IR3-bound sequence) (Supplementary Figure S3A), Sa-S4 to S7 (IR1-bound sequence) (Supplementary Figure S3B) and Sa-S7 to S8 (IR2-bound sequence) (Supplementary Figure S3B). These inversions generate five new alleles of hsdSA with the replacement of one (Sa-hsdSA2, Sa-hsdSA3, Sa-hsdSA6, and Sa-hsdSA8) or two TRDs (Sa-hsdSA4) in a clonal population of S. agalactiae 515.
Figure 6.

Epigenetic variations caused by hsdS inversions of three epigenetic invertons in Streptococcus agalactiae 515. Inversions among the three hsdS genes in the Sag515I Type I R-M locus (A) were tested by PCR (B) and are presented as in Figure 3A; the three DNA configurations generated directly from S1 (C) are illustrated as in Figure 3C; inverted repeats IR1 (white), IR2 (yellow) and IR3 (black) are marked as colored arrowheads. The two methylation motifs identified by SMRT sequencing in the genome of S. agalactiae 515 (D) are assigned to the Sag515I and Sag515II systems and presented as in Figure 4A. The sequences methylated by the six hsdSA alleles (E) are shown as in Figure 4B. N = any four nucleotides.
Because the genome of S. agalactiae 515 was partially sequenced without any information of genome methylation (52), we further determined the complete genome and methylome of S. agalactiae 515 by SMRT sequencing. The result revealed that the genome consists of 2 032 743 bp with two putative R-M systems: a Type I system (Sag515I) and a Type II system (Sag515II) (Figure 6D). Consistently, SMRT sequencing identified methylated sequences for both the Sag515I (5′-Gm6ACN7CTT-3′/5′-Am6AGN7GTC-3′) and Sag515II (5′-Gm6ATC-3′).
To define the specific activity of the six hsdSA allelic variants generated by the inversions in the Sag515I locus, we expressed each of these hsdSA alleles together with the upstream hsdM genes (Figure 6D) in TH5160 as described above. SMRT sequencing identified a unique Type I R-M MTase recognition motif for each of the six hsdSA alleles: Sa-hsdSA1 (5′-Gm6ACN7CTT-3′), Sa-hsdSA2 (5′-Gm6ACN8TTC-3′), Sa-hsdSA3 (5′-Gm6ACN7GTC-3′), Sa-hsdSA4 (5′-CCm6AN8GTC-3′), Sa-hsdSA5 (5′-CCm6AN8CTT-3′) and Sa-hsdSA6 (5′-CCm6AN9TTC-3′) (Figure 6D). This result indicates that the hsdSA alleles generated by inversions in the Sag515I locus are capable of generating six different methylomes.
Programmed DNA rearrangements in two unique invertons of E. faecalis
Enterococcus faecalis is a Gram-positive pathogen of nosocomial infections (e.g. endocarditis, sepsis and meningitis) and one of several pathogens with the widest spectrum of drug resistance (43). In our preliminary screen of enterococcal genomes in the NCBI database, we identified two types of inverton loci with unique genes and organizations. While the most prevalent type had the gene organization of hsdR, hsdM, hsdSA, efiA and hsdSB as exemplified in strain L12 (locus tag BSG25_11645–11665), the genes in the second type are ordered as hsdM, hsdSA, efiA, hsdSB and hsdR represented by isolate T8 (Genbank accession ACOC00000000.1; M.EfaT8ORFDP in REBASE). E. faecalis strain TH4125 (sequence type ST16; accession CP051005) and H25 (ST9; accession GCF_002289045.2) in our collection were found to carry the L12-like locus, which were designated as Efa4125I and EfaH25I, respectively, in the context of the complete genomes (see below). Efa4125I carries two hsdS genes (referred to as Ef-hsdSA and Ef-hsdSB) and a putative invertase gene (GRB94_13125 or efiA) (Figure 7A). The loci of EfaH25I and Efa4125I share the same structure and show a high level of nucleotide identity in the hsdR (2976/2988, 99%), hsdM (1584/1593, 99%) and efiA (911/930, 98%) genes. To the contrary only two of the four hsdS target recognition domains were highly conserved between the strains (Figure 7A) with a nucleotide identity between the 5′ part of S.Efa4125ORF13135P and S1.EfaH25ORF14845P 539/539 (100%) and the 3′ part of S.Efa4125ORF13120P and S1.EfaH25ORF14845P 638/638 (100%) (TRD1 and TRD3 domains in Figure 7A).
Figure 7.

Epigenetic variations resulted from hsdS inversions of two epigenetic invertons in Enterococcus faecalis. (A) Genetic arrangement of the Efa4125I and EfaH25I Type I R-M loci. The hsdS genes are marked with different colors, and the same color and shape between hsdS of the Efa4125I and EfaH25I indicate similar amino acid sequences. The TRDs encoded by hsdS genes are indicated by white rectangles and numbered. (B) The methylated sequences in the genome of E. faecalis TH4125 (left panel) and H25 (right panel). The methylated motifs and corresponding R-M systems are placed together in single rectangles. The modified nucleotides are indicated with red characters. The EfaH25II Type II R-M locus could not be assigned to any of the 4 Type II methyltransferases. (C) The DNA motifs methylated by the hsdSA alleles derived from inversions in Efa4125I (left panel) and H25 (right panel) are presented as in Figure 4A. Different hsdS alleles of TH4125 are named hsdSA1-A4, and the hsdS alleles of H25 are named A–D. R = A or G, Y = T or C, W = A or T, N = any four nucleotides.
Our PCR amplifications using the same-orientation primer sets yielded multiple amplicons, indicating that multiple inversions occurred between Ef-hsdSA and Ef-hsdSB (Supplementary Figure S4A). Subsequent sequencing analyses of the PCR products uncovered four DNA configurations (S1–S4) in the invertible region of the Efa4125I locus, which are bound by two inverted repeats: 60-bp IR1 (5′-ATCACTCTTCATCAGCGTAAGTTAGAACAGCTAAAAGAGTT-GAAGAAGGCTTATTTACAG-3′, white arrowheads) and 15-bp IR2 (5′-TGGGAACAGTGTAAG-3′, yellow arrowheads) (Supplementary Figure S4B). Ef-S1 contains two full-length hsdS genes: Ef-hsdSA1 (1218 bp) and Ef-hsdSB1 (1077 bp). It can be converted to Ef-S2 by an inversion of the IR1-flanked sequence in the middle of Ef-hsdSA and Ef-hsdSB. This reaction generates two new alleles, Ef-hsdSA (hsdSA2, 1164 bp) and Ef-hsdSB (hsdSB2, 1131 bp) after the Ef-hsdSA1 and Ef-hsdSB1 swapped their second TRDs to each other. Ef-S1 also produces Ef-S3 through inversion of the IR2-bound region, resulting in new alleles of Ef-hsdSA (Ef-hsdSA3, 1134 bp) and Ef-hsdSB (Ef-hsdSB3, 1161 bp). Ef-S4 is derived from Ef-S3 through inversion of the IR1-flanked sequence. This reaction generates new allelic variants Ef-hsdSA4 (1188 bp) and Ef-hsdSB4 (1107 bp). Likewise, analysis of the EfaH25I locus in E. faecalis H25 revealed three inversion reactions, generating four hsdSA alleles (Ef-hsdSA1, 1206 bp; Ef-hsdSA2, 1197 bp; Ef-hsdSA3, 1164 bp; Ef-hsdSA4, 1173 bp). These reactions are mediated by two pairs of inverted repeats (15-bp IR1: 5′-GTGTAAGTTGGGGGA-3′ and 30-bp IR2: 5′-ATCACTCTTCATCAGCGTAAGTTAGAACAG-3′). In summary, the three identified inversion events in both the Efa4125II and EfaH25I loci lead to reversible switch of one or both of the TRDs in the two hsdS genes.
We further determined the complete genome of E. faecalis TH4125 and H25 by SMRT sequencing. The result revealed that the TH4125 genome consists of 2 993 403 bp with three uncharacterized R-M systems: two Type I systems (Efa4125I and the Efa4125III) and a Type II system (Efa4125II) (Figure 7B). The sequencing analysis identified five sequences each with an m6A nucleotide: 5′-CRTm6AN7TTG-3′/5′-CAm6AN7TAYG-3′, 5′-TTTm6AN6TTAC-3′/5′-GTAm6AN6TAAA-3′, 5′-ATGCm6AT-3′. The last sequence was assigned to the Efa4125II Type II R-M system based on the characteristics of the gene locus and methylated sequence. To differentiate the methylation activities of the two Type I R-M loci, we expressed the hsdM and hsdSA1 genes of the Efa4125I locus with the cognate hsdM gene in TH5160 and determined the methylome. The resulting strain TH11346 gained two complementary Type I R-M sequences: 5′-CRTm6AN7TTG- 3′/5′-CAm6AN7TAYG-3′ (Figure 7C). This result allowed us to assign 5′-TTTm6AN6TTAC-3′/5′-GTAm6AN6TAAA-3′ as the target sequence of the Efa4125III system.
The H25 genome (accession GCF_002289045.2) was found to be 3 132 715 bp in size and confirmed to be a ST9 CC9 strain. REBASE lists six complete R-M system for H25 which include the phase variable EfaH25I Type I R-M system (CNQ40_14845 AWQ41023.1 efaH25ORF14845MP), four Type II N6-adenine DNA methyltransferases (CNQ40_04255 AWQ39088.1 efaH25ORF4255RMP, CNQ40_10430 AWQ40212.1 efaH25ORF10430MP, CNQ40_10475 AWQ40220.1 efaH25ORF10475MP, CNQ40_13780 AWQ40833.1 efaH25ORF13780MP) and one C5-cytosine methyltransferase (CNQ40_11255 AWQ40364.1 efaH25ORF11255MP). Except for EfaH25I, none of the other R-M systems shows any nucleotide identity to the R-M systems of TH4125. Methylome analysis identified one Type I R-M system motif in each of the four EfaH25 variants (Figure 7C), and one Type II R-M system motif 5′-GTTGGm6A-3′ (EfaH25II) that could not be assigned to any of the four Type II methyltransferases. We assigned a C-5 cytosine methylation motif 5′-m5CCGGA-3′ EfaH25III which we assign to the only cytosine methyltransferase (CNQ40_11255).
Impact of the programmed inversions on gene expression in E. faecalis
To define the epigenetic impact of hsdS shuffling, we performed RNA-seq on triplicate samples of independent stocks expressing prevalently a single hsdS variant. Pairwise comparisons of gene expression were made between each of the hsdS variants and filtered for significance. Using a 2-fold cutoff, we identified a pool of differentially regulated genes and operons. In order to confirm that differences did not arise from random mutations in passaged strains, an additional biological replicate was also analyzed in the same RNAseq run. Only significant, 2-fold differences observed in both biological replicates for each pairwise comparison were investigated further (Supplementary Table S13). Multicomponent analysis of the triplicate samples and the single independent control sample showed that expression profiles of stocks with a specific prevalent hsdS grouped together (Supplementary Table S13).
To investigate how methylation could impact gene expression, we mapped the EfaH25I sites onto the H25 genome and analyzed the DNA sequences immediately upstream of the differentially regulated genes. The monocistronic CNQ40_06785 gene encoding for the sex pheromone cAM373 precursor lipoprotein (E. faecalis V583 EF_1340 AAO81131.1) shows a reduced level of expression in EfaH25I-D with respect to variants A, B and C. Analysis of the upstream region of cAM373 reveals the presence of an EfaH25I-D methylation target site in the −10 promoter element (53) (Figure 8). Similarly, operon CNQ40_05565/05570/05575, (downregulated in the C variant) encodes three hypothetical proteins present only in two published genomes. As with cAM373, analysis of the upstream region identified an EfaH25I-C site mapping to the predicted −10 promoter site (Figure 8). In strains expressing EfaH25I-A, the operon CNQ40_00420/00425, encoding an MFS transporter and hypothetical protein, was upregulated in variants B, C and D. Despite that there were no sites could be detected in the putative promoter region, two EfaH25I-A target sites at 416- and 581-bp were found near the transcriptional start. In the case of the PTS operon CNQ40_00130/00135/00140, it was upregulated in EfaH25I-C and no EfaH25I sites were detected in the 1.5 kb upstream of the operon.
Figure 8.

Sequence features of phase-regulated genes in Enterococcus faecalis H25. (A) Genetic arrangement of two phase-regulated gene loci. The ORFs encoding pheromone cAM373 precursor lipoprotein (CNQ40_06785, upper panel) and uncharacterized operon (CNQ40_05565, CNQ40_05570 and CNQ40_05575, bottom panel) are marked with their genome tags and sizes of the coding and intergenic regions. The predicted promoters of the two loci are illustrated with arrows. (B) Features of the CNQ40_06785 and CNQ40_05565 promoters. Putative −10 and −35 elements of the CNQ40_06785 (upper panel) and CNQ40_05565 (bottom panel) promoters are underlined; the EfaH25I-D (upper panel) and EfaH25I-D (bottom panel) recognition sequences are boxed; the translational start codon is in bolded.
DISCUSSION
Sequence heterogeneity of the inverton-associated invertases
Programmed inversions in Type I R-M systems have recently been shown to exert profound epigenetic impact on phenotypic diversity of the human pathogen S. pneumoniae (21,23). Although similar inverton systems broadly exist in Type I R-M loci of many phylogenetically distant bacteria (5,29), the invertases that catalyze the inversions are only characterized in M. pulmonis (19) and S. pneumoniae (21,23,26–27,54). The work reported here has uncovered a large number of representative invertase homologs encoded by the S+ Type I R-M loci and their mediated epigenetic modifications by using a comprehensive approach of bioinformatics, genetics, SMRT sequencing and RNA-seq. These putative recombinases are predominantly clustered in six major invertase clades on the basis of their sequence heterogeneity. The low sequence homology levels among the invertase clades and among associated R-M proteins strongly suggest that the epigenetic invertons are derived from multiple molecular origins under selection pressure. Identification of serine recombinase family invertases in S+ Type I R-M loci is consistent with this projection.
Biological implications of the epigenetic variability
The prevalence of epigenetic invertons in phylogenetically divergent bacteria suggests that this type of DNA recombination systems provides broad and important functions. Epigenetic invertons should broaden the spectrum of phage resistance in the host bacteria on the basis of the well-known anti-phage function of the Type I R-M systems (55). For instance, hsdS inversions have been demonstrated to alter the specificity in bacterial resistance to phages in M. pulmonis (56) and S. pneumoniae (21,23). Thus, the levels of epigenetic sophistication brought about by the invertons may impact the anti-phage specificity. In this case, a bacterium with a relatively large secondary hsdS reservoir should be resistant to more variety of phages (e.g. B. fragilis).
Epigenetic invertons should fulfill important roles in enhancing environmental adaptation beyond the anti-phage function. Our previous studies have shown that the hsdS inversions in the Type I R-M cod locus have profound impact on pneumococcal physiology and virulence (21,23). The similarities in the genetic arrangement between the pneumococcal invertons and those of other bacteria, as well as dramatic impact of hsdS inversions on the methylomes of B. fragilis, T. denticola, E. faecalis and S. agalactiae, argue for functional similarity among different epigenetic invertons. The overwhelming enrichment of the epigenetic invertons in host-associated bacteria also suggest that they are involved in bacterial responses to uncharacterized host conditions. This hypothesis agrees with our recent finding that the hsdS inversions in the pneumococcal inverton are regulated by multiple two-component signal transduction systems (57). A recent study also uncovered many non-epigenetic inverton systems that are also enriched in host-associated bacteria, which catalyze inversions of bacterial promoters and thereby drive phase variations in cell surface structures and antibiotic resistance (31). In this context, it appears that host-associated bacteria utilize the same basic principles of DNA inversion between the epigenetic and non-epigenetic invertons to promote bacterial responses to host conditions. As exemplified by the presence of both the epigenetic and non-epigenetic invertons in B. fragilis (15), the two types of phase variation mechanisms can operate in the same bacterial cells.
Significant impact of the E. faecalis epigenetic invertons on the transcriptome strongly suggests that epigenetic invertons can promote the fitness of host-associated bacteria by phase-dependent modulation of gene expression. Our transcriptional analysis of four E. faecalis H25 inverton variants has revealed a number of phase-regulated genes. This finding is consistent with the observations that phase variations in the MTase activities of Type I and III R-M systems modulate gene transcription in many host-adapted bacteria (10,23). The transcription for some of the phase-regulated genes appears to be modulated by the ON-or-OFF methylation of the promoters. The allele-specific methylation sites are found to be embedded in the predicted −10 promoter motifs of two phase-regulated gene loci (Figure 8), which may affect interactions between the promoters and RNA polymerase and/or transcriptional regulators. Methylation of the −35 promoter motif has been shown to effectively regulate expression of the CfrBI Type II R-M genes (58,59). Moreover, alternative methylation of promoter sequences by solitary DNA methyltransferase Dam has been well documented for driving phase variations of pyelonephritis-associated pili and antigen 43 in E. coli by affecting transcriptional factors and target DNA motifs (60,61).
Characteristics of the epigenetic invertons
The epigenetic inverton shares the same molecular characteristics with non-epigenetic invertons in the non-Type I R-M loci (33). It represents an invertible sequence flanked by a pair of inverted repeats, which serve as the sites for precise recognition and cleavage/ligation activities of a sequence-specific invertase. Moreover, the invertible hsdS sequences typically encode two or more TRDs, which enable reversible exchange of one (e.g. from Bf-S1 to Bf-S2 in B. fragilis) or two TRDs (e.g. from Bf-S1 to Bf-S4 in B. fragilis) between two hsdS genes in a single reaction. The inversion products always maintain the translational reading frames of the two hsdS genes involved in the reactions. Functionally, each of the resulting MTases methylates a unique sequence. Lastly, the inverton-positive Type I R-M systems should possess at least two hsdS genes, a criterion we used to identify new epigenetic invertons in this study.
The epigenetic invertons and invertase genes are mostly co-localized in the same Type I R-M loci. In the cod locus of S. pneumoniae and the epigenetic invertons of four bacteria investigated in this work, invertase genes are placed either within (e.g. E. faecalis, S. pneumoniae, S. agalactiae and T. denticola) or outside (e.g. B. fragilis) the invertons. A deviation from this genetic arrangement is the lack of the invertase homologs in certain Type I R-M systems with multiple hsdS genes. Our REBASE search identified many S+ Type I R-M loci that don’t encode any identifiable invertase homologs. These invertase-negative epigenetic invertons may still undergo hsdS inversions with the help of invertases encoded elsewhere in the genomes. For instance, the HvsR invertase of M. pulmonis catalyzes the inversions of three loci distantly located in the genome: two S+ Type I R-M loci and the V-1 lipoprotein genes (19). It is also possible that certain invertons are subjective to ‘spontaneous’ inversions as shown for other invertible sequences (26,62–63).
There are great variations in the number of functionally unique MTases generated by various epigenetic invertons. The four invertons investigated in this report are able to yield variable numbers of hsdSA allelic variants, ranging from three in T. denticola to eight in B. fragilis. This diversity is mainly defined by the number of the secondary hsdS genes in the invertons, which is reflected by the difference between E. faecalis (one) and B. fragilis (three). Another contributing factor is the number of the TRDs encoded by the secondary hsdS genes. Although the T. denticola inverton possesses one more secondary hsdS genes than the E. faecalis counterpart, the latter is capable of producing one more hsdSA allele than the former because the hsdSB gene of E. faecalis encodes two TRDs as compared with one TRD encoded by the hsdSB and hsdSC genes of T. denticola. Finally, the genetic organization of the TRDs encoded by the secondary hsdS gene also affects the epigenetic output of the inversions. Specifically, a full hsdS gene encoding two TRDs is more productive in generating functionally unique MTases. This point is also exemplified by the generation of four hsdSA alleles with a full hsdSBgene in E. faecalis, as compared with three hsdSA alleles from two ‘half’ secondary hsdS genes (hsdSB and hsdSC) in T. denticola.
Functional partition of the hsdS genes in the same Type I R-M systems
The existing data strongly suggest that only the first hsdS genes in the epigenetic invertons encode the intact S subunits of the Type I R-M MTases. Our recent work has revealed that the first hsdS gene (hsdSA) in the pneumococcal cod locus is co-transcribed from the upstream hsdR promoter but the two secondary hsdS genes are not expressed due to the lack of promoters (26). Sequence analysis suggested that the first hsdS genes in the four selected bacteria are co-transcribed with the upstream neighbors on the basis of short intergenic regions and lack of Rho-independent transcription terminators. Similar to the hsdS genes in the cod locus of S. pneumoniae (26), no promising promoters were identified for the secondary hsdS genes in the invertons of B. fragilis, E. faecalis, T. denticola and S. agalactiae. Functionally, all of the sequences methylated by the Type I R-M systems in this study are completely matched to the sequence specificities of the first hsdS genes in the invertons. These observations support our conclusion that the hsdSA genes in the epigenetic invertons are expressed and encode the enzymatically active S subunit, whereas the downstream homologs function as DNA templates for inversion-driven sequence changes in the hsdSA genes. Combining the transcriptional active and silent hsdS genes for phase variation in bacterial methylomes is reminiscent of the similar genetic organizations governing the antigenic variations in other pathogenic microorganisms (64). Programmed site-specific recombinations between the transcriptionally active and silent copies of homologous genes are responsible for antigenic variations in the surface-exposed proteins in pathogenic Borrelia (65), Neisseria (66) and African Trypanosome (67).
M.TdeII - a novel methyltransferase with dual activities
The M.TdeII MTase encoded by the Tde3505II Type II R-M locus of T. denticola represents a unique enzyme that catalyzes dual m4C/m6A modifications in two DNA strands of the same sequence. Our SMRT sequencing analysis has indicated that M.TdeII carries out an m4C modification in one strand (5′-m4CTCTTCNG-3′) and another m6A modification in the opposite strand (5′-Gm6AAGAG-3′). A single Type II R-M system in Bacillus coagulans has been shown to recognize the sequence 5′-CTCTTC-3′/5′-GAAGAG-3′ and converts the first cytosine to N4-methylcytosine in one strand and the second adenine to N6-methyladenine in the opposite strand (68). However, in that case, the two strands are separately methylated by two MTases (M1.BcoKI and M2.BcoKI) encoded by different hsdM genes. The Type II R-M systems of Croceibacter atlanticus and Enterobacter aerogenes are also predicted to possess the dual methylations of the same 5′-CTCTTC-3′/5′-GAAGAG-3′ sequence in REBASE, apparently by the two different MTases. Certain Type I R-M MTases are also able to carry out similar m4C/m6A modifications with two different HsdM proteins in a single MTase complex (69). Our preliminary sequence analysis revealed that the amino (∼500 amino acids, aa) and carboxyl (∼370 aa) regions of M.TdeII (877 aa in total) are homologous to the M1.CatHI (412 aa) and M2.CatHI (374 aa) MTases of C. atlanticus HTCC2559 (REBASE), respectively. This information indicates that the single M.TdeII protein of T. denticola consists of two different MTases, each of which is responsible for modifying one of the two DNA strands of the target sequence. Our BLAST search revealed the homologs of the full-length M.TdeII in many Gram-negative bacteria (e.g. Acinetobacter, Citrobacter and Klebsiella), in which they are annotated as adenine-specific DNA methyltransferases or hypothetical proteins. This work provides insightful information for future characterization of the M.TdeII homologs in other bacteria.
DATA AVAILABILITY
Rho-independent transcription terminators were characterized by the internet-based ARNold program at http://rna.igmors.u-psud.fr/toolbox/arnold/index.php#Results; promoters by http://www.fruitfly.org/seq_tools/promoter.html.
The complete genomes of E. faecalis TH4125 and S. agalactiae 515 were uploaded to Genbank, under accession numbers CP051005 and CP051004, respectively. The raw data of the SMRT sequencing runs are clustered under the project PRJNA593863 and PRJNA400682 at the SRA database (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA593863 and https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA400682).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Teresa Coque (Ramón y Cajal University Hospital, Madrid, Spain) for providing E. faecalis strain H25.
Contributor Information
Xueting Huang, Department of Basic Medical Science, School of Medicine, Tsinghua University, Beijing 100084, China; Tsinghua-Peking Center for Life Sciences, Tsinghua University, Beijing 100084, China.
Juanjuan Wang, Department of Basic Medical Science, School of Medicine, Tsinghua University, Beijing 100084, China.
Jing Li, Department of Basic Medical Science, School of Medicine, Tsinghua University, Beijing 100084, China.
Yanni Liu, Department of Basic Medical Science, School of Medicine, Tsinghua University, Beijing 100084, China.
Xue Liu, Department of Fundamental Microbiology, Faculty of Biology and Medicine, University of Lausanne, Lausanne, CH 1015, Switzerland.
Zeyao Li, Tsinghua-Peking Center for Life Sciences, Tsinghua University, Beijing 100084, China.
Kurni Kurniyati, Department of Oral and Craniofacial Molecular Biology, School of Dentistry, Virginia Commonwealth University, Richmond, VA 23298, USA.
Yijie Deng, Department of Oral and Craniofacial Molecular Biology, School of Dentistry, Virginia Commonwealth University, Richmond, VA 23298, USA.
Guilin Wang, W. M. Keck Foundation Biotechnology Resource Laboratory, Yale University, New Haven, CT 06520, USA.
Joseph D Ralph, Department of Genetics and Genome Biology, University of Leicester, Leicester, LE1 7RH, UK.
Megan De Ste Croix, Department of Genetics and Genome Biology, University of Leicester, Leicester, LE1 7RH, UK.
Sara Escobar-Gonzalez, Department of Genetics and Genome Biology, University of Leicester, Leicester, LE1 7RH, UK.
Richard J Roberts, New England Biolabs, 240 County Road, Ipswich, MA 01938, USA.
Jan-Willem Veening, Department of Fundamental Microbiology, Faculty of Biology and Medicine, University of Lausanne, Lausanne, CH 1015, Switzerland.
Xun Lan, Department of Basic Medical Science, School of Medicine, Tsinghua University, Beijing 100084, China; Tsinghua-Peking Center for Life Sciences, Tsinghua University, Beijing 100084, China.
Marco R Oggioni, Department of Genetics and Genome Biology, University of Leicester, Leicester, LE1 7RH, UK.
Chunhao Li, Department of Oral and Craniofacial Molecular Biology, School of Dentistry, Virginia Commonwealth University, Richmond, VA 23298, USA.
Jing-Ren Zhang, Department of Basic Medical Science, School of Medicine, Tsinghua University, Beijing 100084, China; Tsinghua-Peking Center for Life Sciences, Tsinghua University, Beijing 100084, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31820103001, 31530082, 81671972, 31728002]; United States Public Health Service Grants [AI078958, DE023080]; BBSRC [BB/N002903/1, BB/P504737/1 to M.R.O., in part]. Funding for open access charge: National Natural Science Foundation of China [31530082].
Conflict of interest statement. R.J.R. is an employee of New England Biolabs, a company that sells research reagents, including restriction enzymes and DNA methyltransferases, to the scientific community.
REFERENCES
- 1. Anderson E.S., Felix A.. Variation in Vi-phage II of Salmonella typhi. Nature. 1952; 170:492–494. [DOI] [PubMed] [Google Scholar]
- 2. Luria S.E., Human M.L.. A nonhereditary, host-induced variation of bacterial viruses. J. Bacteriol. 1952; 64:557–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bertani G., Weigle J.J.. Host controlled variation in bacterial viruses. J. Bacteriol. 1953; 65:113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vasu K., Nagaraja V.. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. R. 2013; 77:53–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Ste Croix M., Vacca I., Kwun M.J., Ralph J.D., Bentley S.D., Haigh R., Croucher N.J., Oggioni M.R.. Phase-variable methylation and epigenetic regulation by type I restriction-modification systems. FEMS Microbiol. Rev. 2017; 41:S3–S15. [DOI] [PubMed] [Google Scholar]
- 6. Roberts R.J., Vincze T., Posfai J., Macelis D.. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015; 43:D298–D299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kennaway C.K., Taylor J.E., Song C.F., Potrzebowski W., Nicholson W., White J.H., Swiderska A., Obarska-Kosinska A., Callow P., Cooper L.P. et al.. Structure and operation of the DNA-translocating type I DNA restriction enzymes. Genes Dev. 2012; 26:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loenen W.A.M., Dryden D.T.F., Raleigh E.A., Wilson G.G.. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014; 42:20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gann A.A., Campbell A.J., Collins J.F., Coulson A.F., Murray N.E.. Reassortment of DNA recognition domains and the evolution of new specificities. Mol. Microbiol. 1987; 1:13–22. [DOI] [PubMed] [Google Scholar]
- 10. Seib K.L., Srikhanta Y.N., Atack J.M., Jennings M.P.. Epigenetic regulation of virulence and immunoevasion by phase-variable restriction-modification systems in bacterial pathogens. Annu. Rev. Microbiol. 2020; 74:655–671. [DOI] [PubMed] [Google Scholar]
- 11. Adamczyk-Poplawska M., Lower M., Piekarowicz A.. Deletion of one nucleotide within the homonucleotide tract present in the hsdS gene alters the DNA sequence specificity of type I restriction-modification system NgoAV. J. Bacteriol. 2011; 193:6750–6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anjum A., Brathwaite K.J., Aidley J., Connerton P.L., Cummings N.J., Parkhill J., Connerton I., Bayliss C.D.. Phase variation of a Type IIG restriction-modification enzyme alters site-specific methylation patterns and gene expression in Campylobacter jejuni strain NCTC11168. Nucleic Acids Res. 2016; 44:4581–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Srikhanta Y.N., Fox K.L., Jennings M.P.. The phasevarion: phase variation of type III DNA methyltransferases controls coordinated switching in multiple genes. Nat. Rev. Microbiol. 2010; 8:196–206. [DOI] [PubMed] [Google Scholar]
- 14. Kwun M.J., Oggioni M.R., De Ste Croix M., Bentley S.D., Croucher N.J.. Excision-reintegration at a pneumococcal phase-variable restriction-modification locus drives within- and between-strain epigenetic differentiation and inhibits gene acquisition. Nucleic Acids Res. 2018; 46:11438–11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cerdeno-Tarraga A.M., Patrick S., Crossman L.C., Blakely G., Abratt V., Lennard N., Poxton I., Duerden B., Harris B., Quail M.A. et al.. Extensive DNA inversions in the B. fragilis genome control variable gene expression. Science. 2005; 307:1463–1465. [DOI] [PubMed] [Google Scholar]
- 16. Claesson M.J., Li Y., Leahy S., Canchaya C., van Pijkeren J.P., Cerdeno-Tarraga A.M., Parkhill J., Flynn S., O'Sullivan G.C., Collins J.K. et al.. Multireplicon genome architecture of Lactobacillus salivarius. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:6718–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fagerlund A., Langsrud S., Schirmer B.C., Moretro T., Heir E.. Genome analysis of Listeria monocytogenes sequence type 8 strains persisting in salmon and poultry processing environments and comparison with related strains. PLoS One. 2016; 11:e0151117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dybvig K., Yu H.. Regulation of a restriction and modification system via DNA inversion in Mycoplasma pulmonis. Mol. Microbiol. 1994; 12:547–560. [DOI] [PubMed] [Google Scholar]
- 19. Sitaraman R., Denison A.M., Dybvig K.. A unique, bifunctional site-specific DNA recombinase from Mycoplasma pulmonis. Mol. Microbiol. 2002; 46:1033–1040. [DOI] [PubMed] [Google Scholar]
- 20. Feng Z., Li J., Zhang J.R., Zhang X.. qDNAmod: a statistical model-based tool to reveal intercellular heterogeneity of DNA modification from SMRT sequencing data. Nucleic Acids Res. 2014; 42:13488–13499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li J., Li J.W., Feng Z., Wang J., An H., Liu Y., Wang Y., Wang K., Zhang X., Miao Z. et al.. Epigenetic switch driven by DNA inversions dictates phase variation in Streptococcus pneumoniae. PLoS Pathog. 2016; 12:e1005762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Croucher N.J., Coupland P.G., Stevenson A.E., Callendrello A., Bentley S.D., Hanage W.P.. Diversification of bacterial genome content through distinct mechanisms over different timescales. Nat. Commun. 2014; 5:5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manso A.S., Chai M.H., Atack J.M., Furi L., De Ste Croix M., Haigh R., Trappetti C., Ogunniyi A.D., Shewell L.K., Boitano M. et al.. A random six-phase switch regulates pneumococcal virulence via global epigenetic changes. Nat. Commun. 2014; 5:5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tettelin H., Nelson K.E., Paulsen I.T., Eisen J.A., Read T.D., Peterson S., Heidelberg J., DeBoy R.T., Haft D.H., Dodson R.J. et al.. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. 2001; 293:498–506. [DOI] [PubMed] [Google Scholar]
- 25. Atack J.M., Weinert L.A., Tucker A.W., Husna A.U., Wileman T.M., N F.H., Hoa N.T., Parkhill J., Maskell D.J., Blackall P.J. et al.. Streptococcus suis contains multiple phase-variable methyltransferases that show a discrete lineage distribution. Nucleic Acids Res. 2018; 46:11466–11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li J.W., Li J., Wang J., Li C., Zhang J.R.. Molecular mechanisms of hsdS inversions in the cod locus of Streptococcus pneumoniae. J. Bacteriol. 2019; 201:e00581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Ste Croix M., Chen K.Y., Vacca I., Manso A.S., Johnston C., Polard P., Kwun M.J., Bentley S.D., Croucher N.J., Bayliss C.D. et al.. Recombination of the phase-variable spnIII locus is independent of all known pneumococcal site-specific recombinases. J. Bacteriol. 2019; 201:e00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oliver M.B., Basu Roy A., Kumar R., Lefkowitz E.J., Swords W.E.. Streptococcus pneumoniae TIGR4 phase-locked opacity variants differ in virulence phenotypes. mSphere. 2017; 2:e00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Atack J.M., Guo C., Litfin T., Yang L., Blackall P.J., Zhou Y., Jennings M.P.. Systematic analysis of REBASE identifies numerous type I restriction-modification systems with duplicated, distinct hsdS specificity genes that can switch system specificity by recombination. mSystems. 2020; 5:e00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van der Woude M.W., Baumler A.J.. Phase and antigenic variation in bacteria. Clin. Microbiol. Rev. 2004; 17:581–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jiang X., Hall A.B., Arthur T.D., Plichta D.R., Covington C.T., Poyet M., Crothers J., Moses P.L., Tolonen A.C., Vlamakis H. et al.. Invertible promoters mediate bacterial phase variation, antibiotic resistance, and host adaptation in the gut. Science. 2019; 363:181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Henderson I.R., Owen P., Nataro J.P.. Molecular switches–the ON and OFF of bacterial phase variation. Mol. Microbiol. 1999; 33:919–932. [DOI] [PubMed] [Google Scholar]
- 33. Johnson R.C. Site-specific DNA inversion by serine recombinases. Microbiol. Spectr. 2015; 3:MDNA3–0047–2014. [DOI] [PubMed] [Google Scholar]
- 34. Grindley N.D.F., Whiteson K.L., Rice P.A.. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 2006; 75:567–605. [DOI] [PubMed] [Google Scholar]
- 35. Wessels M.R., Paoletti L.C., Rodewald A.K., Michon F., DiFabio J., Jennings H.J., Kasper D.L.. Stimulation of protective antibodies against type Ia and Ib group B streptococci by a type Ia polysaccharide-tetanus toxoid conjugate vaccine. Infect. Immun. 1993; 61:4760–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ruiz-Garbajosa P., Bonten M.J., Robinson D.A., Top J., Nallapareddy S.R., Torres C., Coque T.M., Canton R., Baquero F., Murray B.E. et al.. Multilocus sequence typing scheme for Enterococcus faecalis reveals hospital-adapted genetic complexes in a background of high rates of recombination. J. Clin. Microbiol. 2006; 44:2220–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bacic M.K., Smith C.J.. Laboratory maintenance and cultivation of bacteroides species. Curr. Protoc. Microbiol. 2008; 9:13C.11.11–13C.11.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kurniyati K., Liu J., Zhang J.R., Min Y., Li C.. A pleiotropic role of FlaG in regulating the cell morphogenesis and flagellar homeostasis at the cell poles of Treponema denticola. Cell. Microbiol. 2019; 21:e12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sartingen S., Rozdzinski E., Muscholl-Silberhorn A., Marre R.. Aggregation substance increases adherence and internalization, but not translocation, of Enterococcus faecalis through different intestinal epithelial cells in vitro. Infect. Immun. 2000; 68:6044–6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mende D.R., Letunic I., Huerta-Cepas J., Li S.S., Forslund K., Sunagawa S., Bork P.. proGenomes: a resource for consistent functional and taxonomic annotations of prokaryotic genomes. Nucleic Acids Res. 2017; 45:D529–D534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kwun M.J., Oggioni M.R., De Ste Croix M., Bentley S.D., Croucher N.J.. Excision-reintegration at a pneumococcal phase-variable restriction-modification locus drives within- and between-strain epigenetic differentiation and inhibits gene acquisition. Nucleic Acids Res. 2018; 46:11438–11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sanger F., Nicklen S., Coulson A.R.. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 1977; 74:5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Biswas I., Jha J.K., Fromm N.. Shuttle expression plasmids for genetic studies in Streptococcus mutans. Microbiology. 2008; 154:2275–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tjaden B. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol. 2015; 16:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grindley N.D.F., Whiteson K.L., Rice P.A.. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 2006; 75:567–605. [DOI] [PubMed] [Google Scholar]
- 46. Meinke G., Bohm A., Hauber J., Pisabarro M.T., Buchholz F.. Cre recombinase and other tyrosine recombinases. Chem. Rev. 2016; 116:12785–12820. [DOI] [PubMed] [Google Scholar]
- 47. Li J., Zhang J.R.. Phase variation of Streptococcus pneumoniae. Microbiol. Spectr. 2019; 7:GPP3–0005–2018. [DOI] [PubMed] [Google Scholar]
- 48. Patrick S. Tang Y-.W., Sussman M., Liu D., Poxton I., Schwartzman J.. Bacteroides: Molecular Medical Microbiology. 2015; 2:Amsterdam: Academic Press; 917–944. [Google Scholar]
- 49. Loesche W.J., Grossman N.S.. Periodontal disease as a specific, albeit chronic, infection: diagnosis and treatment. Clin. Microbiol. Rev. 2001; 14:727–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Seshadri R., Myers G.S., Tettelin H., Eisen J.A., Heidelberg J.F., Dodson R.J., Davidsen T.M., DeBoy R.T., Fouts D.E., Haft D.H. et al.. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:5646–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Raabe V.N., Shane A.L.. Group B Streptococcus (Streptococcus agalactiae). Microbiol. Spectr. 2019; 7:GPP3–0007–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tettelin H., Masignani V., Cieslewicz M.J., Donati C., Medini D., Ward N.L., Angiuoli S.V., Crabtree J., Jones A.L., Durkin A.S. et al.. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:13950–13955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Flannagan S.E., Clewell D.B.. Identification and characterization of genes encoding sex pheromone cAM373 activity in Enterococcus faecalis and Staphylococcus aureus. Mol. Microbiol. 2002; 44:803–817. [DOI] [PubMed] [Google Scholar]
- 54. Li J.-W., Wang J., Ruiz-Cruz S., Espinosa M., Zhang J.-R., Bravo A.. In vitro DNA inversions mediated by the PsrA site-specific tyrosine recombinase of Streptococcus pneumoniae. Front. Mol. Biosci. 2020; 7:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Murray N.E. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol. Mol. Biol. R. 2000; 64:412–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dybvig K., Sitaraman R., French C.T.. A family of phase-variable restriction enzymes with differing specificities generated by high-frequency gene rearrangements. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:13923–13928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang J., Li J.W., Li J., Huang Y., Wang S., Zhang J.R.. Regulation of pneumococcal epigenetic and colony phases by multiple two-component regulatory systems. PLoS Pathog. 2020; 16:e1008417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beletskaya I.V., Zakharova M.V., Shlyapnikov M.G., Semenova L.M., Solonin A.S.. DNA methylation at the CfrBI site is involved in expression control in the CfrBI restriction-modification system. Nucleic Acids Res. 2000; 28:3817–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zakharova M., Minakhin L., Solonin A., Severinov K.. Regulation of RNA polymerase promoter selectivity by covalent modification of DNA. J. Mol. Biol. 2004; 335:103–111. [DOI] [PubMed] [Google Scholar]
- 60. Sanchez-Romero M.A., Cota I., Casadesus J.. DNA methylation in bacteria: from the methyl group to the methylome. Curr. Opin. Microbiol. 2015; 25:9–16. [DOI] [PubMed] [Google Scholar]
- 61. Casadesus J., Low D.A.. Programmed heterogeneity: epigenetic mechanisms in bacteria. J. Biol. Chem. 2013; 288:13929–13935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bi X., Liu L.F.. DNA rearrangement mediated by inverted repeats. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schofield M.A., Agbunag R., Miller J.H.. DNA inversions between short inverted repeats in Escherichia coli. Genetics. 1992; 132:295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vink C., Rudenko G., Seifert H.S.. Microbial antigenic variation mediated by homologous DNA recombination. FEMS Microbiol. Rev. 2012; 36:917–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Norris S.J. vls antigenic variation systems of Lyme disease Borrelia: eluding host immunity through both random, segmental gene conversion and framework heterogeneity. Microbiol. Spectr. 2014; 2:MDNA3–0038–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Obergfell K.P., Seifert H.S.. Mobile DNA in the pathogenic Neisseria. Microbiol. Spectr. 2015; 3:MDNA3–0015–2014. [DOI] [PubMed] [Google Scholar]
- 67. McCulloch R., Morrison L.J., Hall J.P.J.. DNA recombination strategies during antigenic variation in the African Trypanosome. Microbiol. Spectr. 2015; 3:MDNA3–0016–2014. [DOI] [PubMed] [Google Scholar]
- 68. Svadbina I.V., Matvienko N.N., Zheleznaya L.A., Matvienko N.I.. Location of the bases modified by M.BcoKIA and M.BcoKIB methylases in the sequence 5 -CTCTTC-3 /5 -GAAGAG-3. Biochemistry (Mosc). 2005; 70:1126–1128. [DOI] [PubMed] [Google Scholar]
- 69. O'Sullivan D., Twomey D.P., Coffey A., Hill C., Fitzgerald G.F., Ross R.P.. Novel type I restriction specificities through domain shuffling of HsdS subunits in Lactococcus lactis. Mol. Microbiol. 2000; 36:866–875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Rho-independent transcription terminators were characterized by the internet-based ARNold program at http://rna.igmors.u-psud.fr/toolbox/arnold/index.php#Results; promoters by http://www.fruitfly.org/seq_tools/promoter.html.
The complete genomes of E. faecalis TH4125 and S. agalactiae 515 were uploaded to Genbank, under accession numbers CP051005 and CP051004, respectively. The raw data of the SMRT sequencing runs are clustered under the project PRJNA593863 and PRJNA400682 at the SRA database (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA593863 and https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA400682).