

Summary
Engineered cardiac tissues hold tremendous promise for in vitro drug discovery, studies of heart development and disease, and therapeutic applications. Here, we describe a versatile “frame-hydrogel” methodology to generate engineered cardiac tissues with highly mature functional properties. This methodology has been successfully utilized with a variety of cell sources (neonatal rat ventricular myocytes, human and mouse pluripotent stem cell-derived cardiomyocytes) to generate tissues with diverse 3D geometries (patch, bundle, network) and levels of structural and functional anisotropy. Maturation of such engineered cardiac tissues is rapidly achieved without the need for exogenous electrical or mechanical stimulation or use of complex bioreactors, with tissues routinely reaching conduction velocities and specific forces of 25 cm/sec and 20 mN/mm2, respectively, and forces per input cardiomyocyte of up to 12 nN. This method is reproducible and readily scalable to generate small tissues ideal for in vitro testing as well as tissues with large, clinically-relevant dimensions.
Keywords: Engineered cardiac tissues, Human pluripotent stem cells, Tissue engineering, Cardiac patch, Cardiac bundle, Cardiomyocytes, Hydrogel
1. Introduction
Ischemic heart disease is a leading cause of death worldwide, resulting in approximately 17.7 million deaths per year [1]. In order to facilitate studies of cardiovascular disease, drug discovery, and regenerative therapies, extensive work has gone into mimicking myocardium in vitro [2–7]. Two-dimensional (2D) cultures of cardiomyocytes can provide valuable electrophysiological and structural information, as well as drug responses, but do not fully recapitulate the in vivo environment. Furthermore, cardiomyocytes cultured in 2D fail to exhibit mature structural characteristics and electromechanical function [8]. To combat these limitations, various methodologies for 3D cardiomyocyte culture have been developed. Early work relied on the use of neonatal rat ventricular myocytes (NRVMs) to generate 3D cardiac tissues due to their wide availability and ease of culture [9–13]. More recent advances in cardiac differentiation protocols for human induced pluripotent and embryonic stem cells (iPSCs and ESCs) have enabled generation of functional human cardiac tissues [8,14,15].
Here, we describe a “frame-hydrogel” methodology, whereby cardiomyocytes are cast in a fibrin hydrogel and anchored to a flexible nylon frame to form highly functional 3D engineered cardiac tissues [2,16–23]. Such formed cardiac tissues are subsequently cultured in a low-shear, dynamic culture environment to rapidly mature heart cells without the need for external electrical or mechanical stimulation or use of complex bioreactors [18]. After two to three weeks of culture, cardiac tissues generated using this methodology exhibit physiologically high cell density and electromechanical properties approximating those of adult myocardium. We have successfully utilized this method with both rodent (neonatal rat ventricular and mouse ESC-derived) and human (iPSC- and ESC-derived) cardiomyocytes (CMs), as well as mouse ESC- and iPSC-derived cardiovascular progenitors (CVPs) [2,17–22]. The frame-hydrogel casting method also allows generation of a wide variety of tissue architectures and sizes while consistently yielding highly advanced levels of CM maturity. Specifically, we have applied this methodology to generate isotropic tissue patches with random CM orientations, network patches with staggered elliptical pores to guide local CM alignment and allow control of tissue anisotropy, epicardial-mimetic network patches with locally varying CM alignment replicating realistic fiber orientations of human myocardium determined with cardiac MRI, and cylindrical bundles with highly aligned CMs. The aforementioned tissues have dimensions that range from 7 × 7 mm to 36 × 36 mm in surface area.
2. Materials
2.1. Manufacturing of tissue molds
Polytetrafluoroethylene (PTFE)
Computer numerical control (CNC) router
Laser cutter
SYLGARD™ 184 Silicone Elastomer Kit (Polydimethylsiloxane, PDMS)
Petri dishes, non-treated
Oven set to 60 °C
Cerex® nylon material (Cerex® Advanced Fabrics Inc) (see Note 1)
70% Ethanol solution
Ultrasonic bath
Razor blades
2.2. 3D engineered tissue culture media and cell sources
A list of cell types, sources, and 3D engineered tissue culture media is provided in Table 1.
Table 1 –
Cardiogenic cell types, sources, and culture media used for maintenance of engineered cardiac tissues generated by the frame-hydrogel method.
| Cell Type | Cell Source | Culture Media |
|---|---|---|
| Human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) | Differentiated from monolayers of human iPSCs using small molecule modulation of the Wnt pathway [24]. Purified using metabolic selection [25]. |
Days 0–7 days of culture: RPMI-1640, 2% B-27 supplement, 2 mg/mL aminocaproic acid, 50 ug/mL ascorbic acid 2-phosphate, 1% penicillin-streptomycin, 1% non-essential amino acids, 1% sodium pyruvate, 0.45μM 1-thioglycerol Days 7–21 of culture: Low-glucose DMEM, 5% fetal bovine serum, 2 mg/mL aminocaproic acid, 50 ug/mL ascorbic acid 2-phosphate, 1% penicillin-streptomycin, 1% non-essential amino acids, 0.45μM 1-thioglycerol [2] |
| Human embryonic stem cell-derived cardiomyocytes (hESC-CMs) | Differentiated from embryoid bodies of human ESCs using previously published protocols [26]. Purified using magnetic-activated cell sorting for SIRPA-positive cells [26]. | DMEM, 5% fetal bovine serum, 1 mg/mL aminocaproic acid, 50 μg/mL ascorbic acid 2-phosphate, 1 mM sodium pyruvate, 2 mM glutamine, 0.1 mM non-essential amino acids, 0.45 mM 1-thioglycerol [20] |
| Neonatal rat ventricular myocytes (NRVMs) | Enzymatically digested from the ventricles of 2-day-old Sprague Dawley rats. Purified using two pre-plating steps [11]. | Low-glucose DMEM, 10% horse serum, 1% chick embryo extract, 1 mg/mL aminocaproic acid, 50 μg/mL ascorbic acid 2-phosphate, 5 U/mL penicillin, 2 μg/mL vitamin B-12 [19] |
| Mouse induced pluripotent stem cell-derived cardiovascular progenitors (miPSC-CVPs) | Differentiated from embryoid bodies of mouse iPSCs using published methods [22]. miPSCs stably transfected with puromycin N-acetyl transferase under the control of a Nkx2–5 cardiac-specific enhancer element to allow for purification of progenitor cells by puromycin selection [22]. | 1:1 ratio of DMEM/F12 and Neurobasal base medium, 1x N2 supplement, 1x B-27 supplement, Glutamax-I, non-essential amino acids, β-Mercaptoethanol, 5 mg/mL BSA, Gentamicin, 1 mg/mL aminocaproic acid [22] |
| Mouse embryonic stem cell-derived cardiomyocytes and cardiovascular progenitors (mESC-CMs and mESC-CVPs) | Differentiated from embryoid bodies of mouse ESCs using published protocols [21,27]. mESCs stably transfected with puromycin aminotransferase under the control of a mouse Myh6 promotor or a Nkx2.5 enhancer element to allow for purification by puromycin selection [21]. | 1:1 ratio of DMEM/F12 and Neurobasal base medium, 1% L-glutamine, 0.1% gentamicin reagent solution, 1% B-27 supplement, 0.0125 mg/mL insulin, 0.05 mg/mL apo-transferrin, 2.5 ng/mL sodium selenite, 3 ng/mL progesterone, 8 ug/mL putrescine, 0.0334% BSA fraction V, 1 mg/mL aminocaproic acid [21] |
2.3. 3D tissue generation
All components should be prepared in a sterile fashion and filtered using a 0.22 μm filter before use.
Pluronic F-127: 0.2% (w/v) in deionized H2O
Molecular biology grade water
Dulbecco’s phosphate buffered saline (DPBS) without CaCl2 and MgCl2
Bovine Thrombin: 50 U/mL in 0.1% (w/v) BSA solution
Human or Bovine Fibrinogen: 10 mg/mL in DPBS, dissolved at 37°C and stored on ice (see Note 2)
2X Media: 2X concentrated low glucose DMEM (reconstituted from powder at half recommended volume), 20% heat inactivated horse serum, 10 U/mL penicillin, 4 μg/mL B-12, 2 mg/mL aminocaproic acid
Matrigel matrix
Incubator set to 37 °C, 5 % CO2
Water bath set to 37 °C
High angle rocker set to ± 30° tilt and 0.4 Hz rocking frequency
Hemocytometer
Trypan blue 0.4%
Standard forceps, curved
Fine point forceps, straight
10 cm culture dishes, non-treated
6 cm culture dishes, non-treated
12-well or 6-well culture plates, non-treated
3. Methods
3.1. Manufacturing of PDMS tissue molds
Machine polytetrafluoroethylene (PTFE, Teflon) into negative master shape using a CNC router (see Note 4).
Prepare PDMS by thoroughly mixing SYLGARD™ 184 Silicone Elastomer base and SYLGARD™ 184 Silicone Elastomer curing solution in a 10:1 ratio (w:w).
Transfer the PDMS to a vacuum desiccator and degas for 30 minutes, until all bubbles are removed.
Place the PTFE master(s) face up into a petri dish and pour PDMS over the master until it is completely submerged. There should be approximately 2 mm of PDMS above the highest point of the master.
Transfer the dish containing the PDMS and master to a vacuum desiccator and degas for 30 minutes, ensuring all bubbles are evacuated. Remaining bubbles may be removed with a needle or pipette tip.
Cure PDMS in a 60°C oven for at least 4 hours, ensuring the dish is level.
After curing is complete, remove the PDMS from the dish and carefully extract the PTFE master (see Note 5).
Remove excess PDMS that surrounds the mold(s) using a sharp razor blade, producing a final PDMS mold. (Figure 1C–D)
Submerge PDMS molds in 70% ethanol and sonicate in an ultrasonic bath for 60 minutes at 60°C.
Remove PDMS molds from 70% ethanol and allow them to dry at room temperature, then steam autoclave to sterilize (see Note 6).
Figure 1 –

(A) PTFE master negatives used to manufacture PDMS molds for generating a 7 × 2 mm bundle, a 7 × 7 mm patch, a 10 × 10 mm patch with border pores, and a 36 × 36 mm patch. Scale bar = 10 mm. (B) Left: laser cutter path to generate frames within a nylon sheet; Middle: laser-cut frames attached to sheet with thin bridges (dashed lines). Right: nylon frames cut from the main sheet at the bridges. Scale bar = 5 mm. (C) Resulting PDMS molds generated from the masters shown in (A) with nylon frames pressed in. Scale bar = 10 mm. (D) PDMS molds for fabrication of a 7 × 7 mm and a 14 × 14 mm network patch and a 25 × 25 mm epicardial mimetic patch. These molds were generated using high aspect ratio soft lithography. Scale bar = 10 mm. Appearance of 7 × 7mm (E) bundle, (F) patch, and (G) network engineered cardiac tissues after 2–3 weeks of culture. Scale bar = 1 mm. (Figure contains elements reproduced from references [2,16,18] with permission)
3.2. Frame manufacturing
Design a frame template using 2D drawing software compatible with the laser cutter. The frame should surround the entire perimeter of the final tissue (Figure 1B, E–G). Frames typically have edges that are, at minimum, 1 mm thick and dimensions slightly smaller than the mold interior in which they will be press-fitted. Frames are connected to the main sheet of Cerex® nylon material by a thin bridge of uncut material (Figure 1B).
Laser cut frames out of nylon material, ensuring that the laser settings do not burn the Cerex® nylon material.
Carefully separate individual frames by cutting the thin bridges holding the frames to the larger sheet of nylon material (Figure 1B).
Sterilize frames using UV or gas sterilization.
3.3. Initial mold preparation for tissue culture
All steps should be performed in a BSL-2 biosafety cabinet.
- Using sterile curved forceps, place autoclaved PDMS molds into a sterile 10 cm petri dish. Leave at least 3 mm of space between molds to facilitate eventual mold removal.
- A 10 cm petri dish can accommodate up to twelve 10 × 10 mm (length × width) molds, three 18 × 18 mm molds, or one 41 × 41 mm mold.
To decrease cell attachment, increase the hydrophilicity of the PDMS molds by filling each mold with Pluronic F-127 solution until the internal surface is fully covered, approximately 250 μL for a 10 × 10 × 1.5 mm mold (length × width × depth). Remove any air bubbles using a sterile pipette tip.
Allow Pluronic F-127 filled molds to sit undisturbed at room temperature for 1 to 2 hours. Proceed to cell preparation after the 1–2 hours have passed.
3.4. Cell preparation
Prepare cardiac cells in suspension using previously published methods (see Table 1 and Note 7).
Resuspend cells in recommended culture media (see Table 1) and count the number of live cells using a hemocytometer and trypan blue.
Centrifuge cells at 300xG for 5 minutes.
- Determine the required live cell concentration using the following formula, where V is equal to the volume of cell/hydrogel mixture required for one engineered tissue (in mL).
- For a 7 × 7 mm patch, V is equal to 120 μL and the number of cells per tissue is 500,000. Therefore, the required cell concentration is 8.68×106 cells/mL.
- For a 7 × 2 mm bundle, V is equal to 37.5 μL and the number of cells per tissue is 375,000. Therefore, the required cell concentration is 20.8×106 cells/mL.
Resuspend cells in culture media at the calculated concentration and store on ice.
Immediately proceed to final mold preparation and hydrogel preparation. Cells should be kept as a single cell suspension on ice for a minimal duration of time.
3.5. Final mold preparation
Immediately after the cardiomyocytes are prepared, aspirate the Pluronic F-127 from the molds, taking care to not touch the molds with the aspirating tip.
Rinse the molds by filling each mold with molecular biology grade water until the internal surface is covered.
Aspirate the water from the molds, drying the molds as much as possible while ensuring the aspirating tip does not touch or scrape the mold surface.
Allow the molds to air-dry in the BSL-2 biosafety cabinet with the petri dish lids removed to improve airflow and evaporation. This step can occur while the hydrogel is prepared and aliquoted. Molds with more complex architecture should be allowed a longer period of time to dry.
Proceed immediately to hydrogel preparation.
3.6. Hydrogel preparation
All solutions should be prepared and kept on ice in a BSL-2 biosafety cabinet.
Place stock solutions of fibrinogen, matrigel, thrombin, and 2X media on ice (see Note 8).
-
Use Table 2 to calculate volumes required for each component (see Note 9).
Define V as the volume of cell/hydrogel mixture required for one engineered tissue.-
aFor a patch with dimensions of 7 × 7 mm (length × width), V is equal to 120 μL. V can be scaled in proportion to the area of the tissue.
Define Ntissues as the number of tissues to make consecutively.-
bThe number of tissues that can be cast consecutively (Ntissues) is limited by the rapid cross-linking of the fibrinogen by thrombin. Therefore, tissues must be generated in batches. We have found that the maximum Ntissues is 4. This value should be reduced for larger or more complex molds.
-
a
Using the volumes calculated in step 2, combine fibrinogen, matrigel, and cell solutions in an Eppendorf tube on ice, mixing thoroughly but carefully.
In a separate Eppendorf tube on ice, combine the 2X media and thrombin solutions using the volumes calculated in step 2.
- If number of needed engineered tissues is larger than Ntissues, repeat steps 3 and 4 until there are a sufficient number of Eppendorf tubes to generate the desired number of tissues.
- For example, if 12 total engineered cardiac tissues are to be generated, and Ntissues is 4, prepare 3 Eppendorf tubes containing the fibrinogen, matrigel and cell solutions and 3 Eppendorf tubes containing the 2X media and thrombin solutions.
Proceed to casting of engineered tissues in PDMS molds, limiting the amount of time between hydrogel preparation and tissue casting.
Table 2 –
Hydrogel composition for generation of engineered cardiac tissues using the frame-hydrogel method. V is defined as the volume of cell/hydrogel mixture required for one engineered tissue (in mL) and Ntissues is defined as the number of tissues to make consecutively.
| Starting Concentration | Final Concentration | Percent of Final Hydrogel Volume | Volume to Prepare | |
|---|---|---|---|---|
| Fibrinogen | 10 mg/mL | 2 mg/mL | 20% | 0.2×V×Ntissues |
| Matrigel | 100% | 10% (v/v) | 10% | 0.1×V×Ntissues |
| Thrombin | 50 U/mL | 1 U/mL | 2% | 0.02×V×Ntissues |
| 2X Media | 100% | 20% | 20% | 0.2×V×Ntissues |
| Cells suspended in culture media | 48% | 0.48×V×Ntissues |
3.7. Casting of engineered tissues in PDMS molds
Using sterile fine point forceps, place one sterile nylon frame into each PDMS mold, ensuring that the mold is entirely dry and that no water is wicking into the frame (see Note 10) (Figure 1C). The frame should be in contact with the base of the PDMS mold along the entire perimeter of the frame.
To initiate polymerization and form the final hydrogel/cell mixture, add the contents of the 2X media/thrombin Eppendorf tube to the fibrinogen/matrigel/cell Eppendorf tube and pipette up and down to mix, taking care to not introduce any bubbles.
Quickly and carefully pipette one volume’s worth (V) of the hydrogel/cell mixture into one PDMS mold containing a frame. Do not touch or scrape the PDMS mold with the pipette tip and avoid introducing bubbles into the hydrogel. The hydrogel should completely cover the bottom surface of the mold and wick into the nylon frame (see Note 11).
Repeat step 3 without changing pipette tips until hydrogel mixture is fully distributed into Ntissues molds (see Note 12).
Repeat steps 2–4, making tissues in batches, until the desired total number of molds are filled.
Place the 10 cm petri dish containing the hydrogel-filled molds into a 37°C 5% CO2 incubator for 30 – 45 minutes to allow for complete gelling of the hydrogel.
After 30 – 45 minutes, return the 10 cm petri dish containing the hydrogel-filled molds to the biosafety cabinet. Place each hydrogel-filled mold into a well of a tissue culture plate. 10 × 10 mm molds fit in the wells of a 12-well plate and 18 × 18 mm molds fit in the wells of a 6-well plate. To prevent the molds from floating, press the PDMS mold firmly to the bottom of dry plates.
Add culture media to each well, ensuring that the mold is fully submerged. Two mL of media is sufficient to cover the 10 mm × 10 mm molds in a well of a 12-well culture plate and five mL of media is sufficient to cover the 18 mm × 18 mm molds in a well of a 6-well culture plate.
Place the tissue culture plate onto a high angle rocker set to ± 30° tilt and 0.4 Hz rocking frequency in a 37°C 5% CO2 incubator for 24 hours (see Note 13). The molds should not be free-floating at this stage.
24 hours after the tissues were cast, proceed to removing the engineered tissues from molds.
3.8. Removing engineered tissues from molds
All steps should be performed in a BSL-2 biosafety cabinet.
Fill a 6-cm culture dish with 20mL DPBS, or to a depth that will fully submerge the molds.
Remove the tissues from the high angle rocker and transfer them to the biosafety cabinet.
Using a pair of sterile curved forceps, remove one hydrogel-filled mold from the culture plate and submerge it in the DPBS-filled dish.
Hold and stabilize the sides of the mold using a pair of curved forceps. Use another pair of fine point forceps to remove the engineered tissue from the mold by gently sliding the fine point forceps under the nylon frame and lifting the frame upward. Continue gently lifting the frame at several points along one edge of the mold. When the center of the hydrogel (the area not supported by the frame) has separated from the PDMS mold, repeat the lifting technique on each of the remaining three sides until entire tissue is separated from the PDMS mold and is free-floating in the DPBS bath. Only touch the frame with the forceps--touching the hydrogel with forceps will damage the tissue (see Note 14).
Return the engineered tissue to the cell culture plate, holding the frame with fine point forceps. The tissue should sink to the bottom of the well.
Repeat steps 3–5 until all tissues are removed from molds and placed in culture plates (see Note 15).
Return culture dish to a high angle rocker in a 37°C 5% CO2 incubator. Tissues should be free-floating in the culture media.
3.9. Tissue Culture
- Replace culture media (Table 1) every other day as follows:
- Remove 2/3 of the culture media with an aspirating pipette while keeping the culture plate flat. Take care to not place the aspirating pipette too close to the tissue.
- Replace removed cell culture media with new media, adding the new media away from the engineered tissue to avoid damaging it.
Culture the tissues on the high angle rocker in a 37°C 5% CO2 incubator for 2–3 weeks.
Analyze the tissues for force generation, electrical propagation, and structure 2–3 weeks after initial tissue formation. Expected functional values for tissues generated from various cell sources and with different geometries are provided in Table 2 and Figure 2.
Figure 2 –
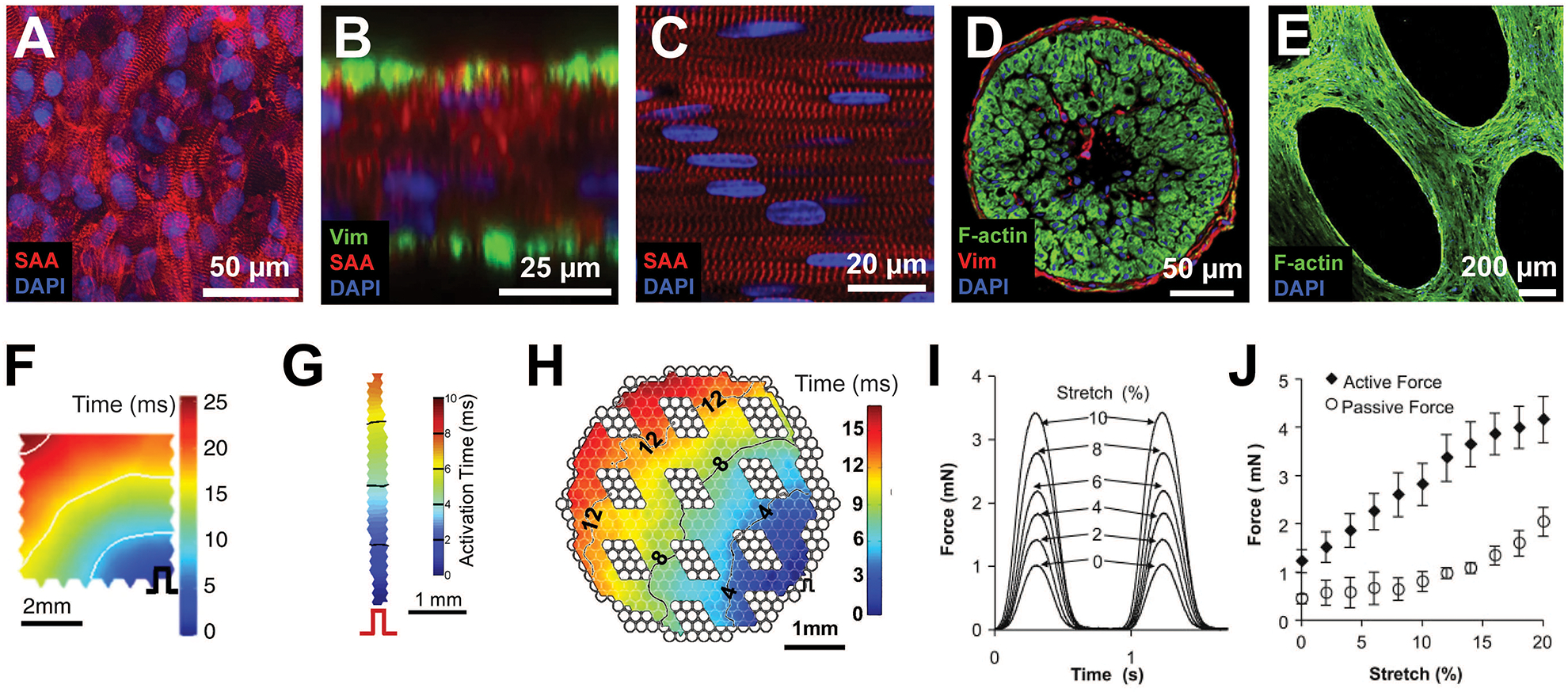
Functional properties of engineered cardiac tissues. Representative immunohistochemistry images of (A) a 7 × 7 mm hiPSC-CM patch, and (B) its cross section, (C) a 7 × 2 mm NRVM bundle and (D) its cross section, and (E) a 7 × 7 mm hESC-CM network patch. Representative isochrone activation maps from (F) a 7 × 7 mm hiPSC-CM patch, (G) a 7 × 2 mm NRVM bundle, and (H) a 7 × 7 mm mESC-CM network patch. (I) Representative force traces during stretch of a 1 Hz electrically stimulated 7×7 mm hESC-CM cardiac network patch. (J) Active and passive force-length relationships for 7×7 mm hESC-CM cardiac network patches. (Figure contains elements reproduced from references [18,2,21,20] with permission) (SAA = sarcomeric alpha actinin; Vim = vimentin)
4. Notes
We have also utilized standard Velcro® as a frame material.
Fibrinogen should be prepared less than four hours before use and kept on ice. It is recommended to limit the amount of time the fibrinogen is in the 37°C water bath to 10 minutes or until solids have just dissolved. Sterile filtration should occur when the solution is still warm, then placed on ice. Ensure the solution is fully cooled to 4°C before use.
It is recommended to first determine the desired final shape and size of tissues, subsequently design a tissue mold using 3D software, and then fabricate the master for tissue mold generation.
Some geometries, for example small network patches, may have features that are smaller than can be achieved with a CNC router. In order to form these intricate molds we recommend using high aspect ratio soft lithography, as has been previously described in detail [16]. Soft lithography will allow generation of a PDMS master that can be used in the same manner as the PFTE master described here.
If the PDMS is difficult to remove from the petri dish, use a pair of cutting pliers to fracture the side of the petri dish. Carefully remove fragments of the petri dish until the PDMS can be extracted.
PDMS molds can be generated weeks to months in advance and stored in sterile autoclaving pouches until needed.
It is recommended to begin with a cell population that contains approximately 10% fibroblasts. If necessary, fibroblasts can be separately cultured and added to highly pure cardiomyocyte populations.
Thrombin, matrigel, and 2X media stock solutions may be prepared in advance, aliquoted, and stored at −20°C. These components should be thawed on ice the day of tissue generation, ensuring that each solution is fully thawed before beginning cell collection.
There is some volume loss during the hydrogel generation procedure. Therefore, it is recommended to prepare a minimum of 105% of the total calculated volumes of each solution.
The plate lid will attract the frames through static electricity. Avoid placing the lid back on the plate once frames are press-fitted into the molds.
Due to the surface tension of the hydrogel, it can be difficult to fully spread the small volume of hydrogel over the entire surface of the mold. We recommend pipetting from the walls of the mold towards the center, instead of ejecting the hydrogel into the middle of the mold. This will help the hydrogel wick up the side walls of the mold.
If the hydrogel begins to take on a viscous appearance while being injected into the molds, the hydrogel has begun to polymerize. The number of tissues made at one time (Ntissues) should be decreased.
If the media spills out of the culture plate or splashes onto the lid while the culture plate is on the high angle rocker, reduce the media volume. If the mold cannot be completely submerged with a smaller volume of media, a shorter mold is recommended. If the mold exceeds the dimensions of a standard tissue culture dish (for example, the 41 × 41 mm patch molds), a custom-built chamber may be required [2].
If the hydrogel is adhering the to the PDMS mold, the Pluronic F-127 solution can be used at higher concentration or applied to the molds for a longer period of time. Alternatively, tissues can be removed from the PDMS molds earlier than 24 hours after formation.
PDMS molds can be reused after tissues are removed. Follow steps 10–11 in the Manufacturing of PDMS tissue molds section to clean and prepare the molds for reuse.
Table 3 –
Average functional outputs achieved by 2–3 week old engineered cardiac tissues generated using the frame-hydrogel method. Results of functional measurements are presented for various cell types and tissue geometries achieved during the last decade.
| Cell Type | Geometry | Max Active Force (mN) | Specific Force (mN/mm2) | Force per Input CM (nN) | Conduction Velocity (cm/sec) | Citation |
|---|---|---|---|---|---|---|
| hiPSC-CMs | 7×7 mm patch | 5.2 ± 0.2 | 22.4 ± 0.9 | 11.9 ± 0.5 | 28.5 ± 1.0 | Shadrin, IY. et al. 2017 [2] |
| hiPSC-CMs | 15×15 mm patch (megapatch) | 9.4 ± 1.0 | 19.4 ± 2.1 | 4.7 ± 0.5 | 27.2 ± 1.1 | Shadrin, IY. et al. 2017 [2] |
| hiPSC-CMs | 36×36 mm patch (gigapatch) | 17.5 ± 1.1 | 17.0 ± 0.8 | 1.75 ± 0.1 | 28.9 ± 1.8 | Shadrin, IY. et al. 2017 [2] |
| NRVMs | 10×10 mm patch with border pores | 18.0 ± 1.4 | 18.0 ± 1.4 | 3.0 ± 0.2 | 32.3 ± 1.8 | Jackman, CP. et al. 2018 [19] |
| NRVMs | 7×2 mm bundle | 2.10 ± 0.10 | 59.7 ± 4.3 | 5.6 ± 0.27 | 52.5 ± 0.9 | Jackman, CP. et al. 2016 [18] |
| hiPSC-CMs | 7×2 mm bundle | 1.3 ± 0.051 | 23.2 ± 1.6 | 3.47 ± 0.14 | 25.8 ± 1.2 | Jackman, CP. et al. 2016 [18] |
| NRVMs | 7×7 mm network patch, 1.2 mm pore length | 2.39 ± 0.25 | 8.9 ± 1.1 | 4.35 ± 0.45 | 26.8 ± 0.8 | Bian, W. et al. 2014 [17] |
| hESC-CMs | 7×7 mm network patch, 1.2 mm pore length | 3.0 ± 1.1 | 11.8 ± 4.5 | 5.7 ± 1.1 | 25.1 | Zhang, D. et al. 2013 [20] |
| mESC-CMs | 7×7 mm network patch, 1.2 mm pore length | 1.96 ± 0.54 | Not reported | 2.51 ± 0.69 | 24.1 ± 1.4 | Liau, B. et al. 2011 [21] |
| mESC-CVPs | 7×7 mm network patch, 1.2 mm pore length | 1.28 ± 0.11 | Not reported | 1.64 ± 0.14 | 19.2 ± 0.4 | Liau, B. et al. 2011 [21] |
Acknowledgements
This work was supported by NIH grants U01HL134764, UG3TR002142, HL132389, HL126524, and a grant from Foundation Leducq to NB and an NSF Graduate Research Fellowship (2017–2020) to AH.
5. References
- 1.Thomas H, Diamond J, Vieco A, Chaudhuri S, Shinnar E, Cromer S, Perel P, Mensah GA, Narula J, Johnson CO, Roth GA, Moran AE (2018) Global Atlas of Cardiovascular Disease 2000–2016: The Path to Prevention and Control. Glob Heart 13 (3):143–163. doi: 10.1016/j.gheart.2018.09.511 [DOI] [PubMed] [Google Scholar]
- 2.Shadrin IY, Allen BW, Qian Y, Jackman CP, Carlson AL, Juhas ME, Bursac N (2017) Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nature communications 8 (1):1825. doi: 10.1038/s41467-017-01946-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lundy SD, Zhu WZ, Regnier M, Laflamme MA (2013) Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem cells and development 22 (14):1991–2002. doi: 10.1089/scd.2012.0490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, Jiang J, Masse S, Gagliardi M, Hsieh A, Thavandiran N, Laflamme MA, Nanthakumar K, Gross GJ, Backx PH, Keller G, Radisic M (2013) Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods 10 (8):781–787. doi: 10.1038/nmeth.2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mannhardt I, Breckwoldt K, Letuffe-Breniere D, Schaaf S, Schulz H, Neuber C, Benzin A, Werner T, Eder A, Schulze T, Klampe B, Christ T, Hirt MN, Huebner N, Moretti A, Eschenhagen T, Hansen A (2016) Human Engineered Heart Tissue: Analysis of Contractile Force. Stem cell reports 7 (1):29–42. doi: 10.1016/j.stemcr.2016.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C, Drowley L, Plowright AT, Needham EJ, Wang QD, Gregorevic P, Xin M, Thomas WG, Parton RG, Nielsen LK, Launikonis BS, James DE, Elliott DA, Porrello ER, Hudson JE (2017) Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proceedings of the National Academy of Sciences of the United States of America. doi: 10.1073/pnas.1707316114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M, Vunjak-Novakovic G (2018) Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556 (7700):239–243. doi: 10.1038/s41586-018-0016-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pomeroy JE, Helfer A, Bursac N (2019) Biomaterializing the promise of cardiac tissue engineering. Biotechnol Adv. doi: 10.1016/j.biotechadv.2019.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimmermann WH, Fink C, Kralisch D, Remmers U, Weil J, Eschenhagen T (2000) Three-dimensional engineered heart tissue from neonatal rat cardiac myocytes. Biotechnology and bioengineering 68 (1):106–114 [PubMed] [Google Scholar]
- 10.Zimmermann WH, Schneiderbanger K, Schubert P, Didie M, Munzel F, Heubach JF, Kostin S, Neuhuber WL, Eschenhagen T (2002) Tissue engineering of a differentiated cardiac muscle construct. Circulation research 90 (2):223–230 [DOI] [PubMed] [Google Scholar]
- 11.Bursac N, Papadaki M, Cohen RJ, Schoen FJ, Eisenberg SR, Carrier R, Vunjak-Novakovic G, Freed LE (1999) Cardiac muscle tissue engineering: toward an in vitro model for electrophysiological studies. Am J Physiol 277 (2):H433–444. doi: 10.1152/ajpheart.1999.277.2.H433 [DOI] [PubMed] [Google Scholar]
- 12.Papadaki M, Bursac N, Langer R, Merok J, Vunjak-Novakovic G, Freed LE (2001) Tissue engineering of functional cardiac muscle: molecular, structural, and electrophysiological studies. American journal of physiology Heart and circulatory physiology 280 (1):H168–178. doi: 10.1152/ajpheart.2001.280.1.H168 [DOI] [PubMed] [Google Scholar]
- 13.Bursac N, Papadaki M, White JA, Eisenberg SR, Vunjak-Novakovic G, Freed LE (2003) Cultivation in rotating bioreactors promotes maintenance of cardiac myocyte electrophysiology and molecular properties. Tissue engineering 9 (6):1243–1253. doi: 10.1089/10763270360728152 [DOI] [PubMed] [Google Scholar]
- 14.Jackman CP, Shadrin IY, Carlson AL, Bursac N (2015) Human Cardiac Tissue Engineering: From Pluripotent Stem Cells to Heart Repair. Curr Opin Chem Eng 7:57–64. doi: 10.1016/j.coche.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shadrin IY, Khodabukus A, Bursac N (2016) Striated muscle function, regeneration, and repair. Cell Mol Life Sci 73 (22):4175–4202. doi: 10.1007/s00018-016-2285-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bian W, Liau B, Badie N, Bursac N (2009) Mesoscopic hydrogel molding to control the 3D geometry of bioartificial muscle tissues. Nature protocols 4 (10):1522–1534. doi: 10.1038/nprot.2009.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bian W, Jackman CP, Bursac N (2014) Controlling the structural and functional anisotropy of engineered cardiac tissues. Biofabrication 6 (2):024109–024109. doi: 10.1088/1758-5082/6/2/024109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackman CP, Carlson AL, Bursac N (2016) Dynamic culture yields engineered myocardium with near-adult functional output. Biomaterials 111:66–79. doi: 10.1016/j.biomaterials.2016.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackman CP, Ganapathi AM, Asfour H, Qian Y, Allen BW, Li Y, Bursac N (2018) Engineered cardiac tissue patch maintains structural and electrical properties after epicardial implantation. Biomaterials 159:48–58. doi: 10.1016/j.biomaterials.2018.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang D, Shadrin IY, Lam J, Xian HQ, Snodgrass HR, Bursac N (2013) Tissue-engineered cardiac patch for advanced functional maturation of human ESC-derived cardiomyocytes. Biomaterials 34 (23):5813–5820. doi: 10.1016/j.biomaterials.2013.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liau B, Christoforou N, Leong KW, Bursac N (2011) Pluripotent stem cell-derived cardiac tissue patch with advanced structure and function. Biomaterials 32 (35):9180–9187. doi: 10.1016/j.biomaterials.2011.08.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christoforou N, Liau B, Chakraborty S, Chellapan M, Bursac N, Leong KW (2013) Induced pluripotent stem cell-derived cardiac progenitors differentiate to cardiomyocytes and form biosynthetic tissues. PloS one 8 (6):e65963. doi: 10.1371/journal.pone.0065963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bian W, Badie N, Himel HDt, Bursac N (2014) Robust T-tubulation and maturation of cardiomyocytes using tissue-engineered epicardial mimetics. Biomaterials 35 (12):3819–3828. doi: 10.1016/j.biomaterials.2014.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP (2013) Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nature protocols 8 (1):162–175. doi: 10.1038/nprot.2012.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M, Fukuda K (2013) Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell stem cell 12 (1):127–137. doi: 10.1016/j.stem.2012.09.013 [DOI] [PubMed] [Google Scholar]
- 26.Dubois NC, Craft AM, Sharma P, Elliott DA, Stanley EG, Elefanty AG, Gramolini A, Keller G (2011) SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat Biotechnol 29 (11):1011–1018. doi: 10.1038/nbt.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christoforou N, Miller RA, Hill CM, Jie CC, McCallion AS, Gearhart JD (2008) Mouse ES cell-derived cardiac precursor cells are multipotent and facilitate identification of novel cardiac genes. J Clin Invest 118 (3):894–903. doi: 10.1172/jci33942 [DOI] [PMC free article] [PubMed] [Google Scholar]