

Abstract
Purpose of review
Recent advances in genetic evaluation improved the identification of several variants in the NOTCH3 gene causing Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL). Despite improved diagnosis, the disease mechanism remains an elusive target and an increasing number of scientific/clinical groups are investigating CADASIL to better understand it. The purpose of this review is to summarize the current knowledge in CADASIL.
Recent findings
CADASIL is a genotypically and phenotypically diverse condition involving multiple molecular systems affecting small blood vessels. Cerebral white matter changes observed by MRI are a key CADASIL characteristic in young adult patients often before severe symptoms and trigger NOTCH3 genetic testing. NOTCH3 mutation locations are highly variable, correlate to disease severity and consistently affect the cysteine balance within extracellular Notch3. Granular osmiophilic material deposits around blood vessels are also a unique CADASIL feature and appear to have a role in sequestering proteins that are essential for blood vessel homeostasis. As potential biomarkers and therapeutic targets are being actively investigated, neurofilament light chain can be detected in patient serum and may be a promising circulating biomarker.
Summary
CADASIL is a complex, devastating disease with unknown mechanism and no treatment options. As we increase our understanding of CADASIL, translational research bridging basic science and clinical findings needs to drive biomarker and therapeutic target discovery.
Keywords: Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, dementia, Notch3, small vessel disease, stroke
INTRODUCTION
Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) is a monogenic hereditary small vessel disease (SVD) and currently considered the most common genetic cause of stroke and dementia in adults. It is caused by mutations, commonly heterozygous missense (95%), in the NOTCH3 gene on Chromosome 19p13.2-p13.1 that are inherited in an autosomal dominant pattern. The disease is of slow onset, with initial clinical manifestations in the third and fourth decade of life. Predominant clinical features include migraines with aura, recurrent strokes of ischemic nature, transient ischemic attacks, progressive white matter degeneration, memory loss, debilitating dementia and disability and multiple psychiatric symptoms [1–3].
The path to diagnosis for most CADASIL patients is complex and often lengthy because of lack of awareness about the disease in the general clinical community. Adding complexity is the symptomatic heterogeneity of the disease, frequently even between family members that carry the same mutation but develop different clinical features. Diagnosis generally occurs through a combination of family history of migraines/strokes/dementia, MRI findings of white matter changes characteristic of CADASIL and NOTCH3 genetic testing. Prior to the availability of a genetic test and sometimes still used for diagnostic confirmation, electron microscopy of skin biopsies from CADASIL patients were the gold standard for the identification of deposits near blood vessels called granular osmiophilic material (GOM), which are unique to CADASIL and, the composition and function of which has not yet been fully characterized.
Notch3 is a transmembrane receptor expressed by vascular smooth muscle cells (VSMCs) and pericytes in vasculature throughout the body. The majority of CADASIL NOTCH3 mutations affect one of the 34 extracellular domain epidermal growth factor repeats (EGFr) of the Notch3 protein by either adding or removing a cysteine residue, resulting in accumulation within the extracellular space/within GOMs and, ultimately, leading to degradation of the VSMC layer in blood vessels. Through this process, subjects with CADASIL become susceptible to strokes but the exact mechanism has not yet been elucidated. The disease is progressive and fatal. There are currently no treatment options for patients with CADASIL as a therapeutic target has not been identified. This review focuses on recent research and clinical advances that increase our understanding of CADASIL as well as recent efforts to identify biomarkers for the disease.
BASIC AND CLINICAL ADVANCES IN CEREBRAL AUTOSOMAL DOMINANT ARTERIOPATHY WITH SUBCORTICAL INFARCTS AND LEUKOENCEPHALOPATHY
As more CADASIL patients are identified because of advances in genetic testing and whole exome/genome sequencing becomes more available, increasing efforts are being made to better understand the pathophysiology of the disease and to increase awareness throughout the clinical community. As of 2014, there were more than 230 unique CADASIL mutations already reported [4], a number that has likely increased in the last several years, and it is estimated that two to five people in 100 000 carry a CADASIL mutation, although this number varies depending on population nationality [5,6■]. Approximately 98% of these mutations occur within exons 2–23 of NOTCH3 and result in a gain or loss of a cysteine residue within one of the 34 EGF repeats of the Notch3 Receptor [7]. Even though mutations have been reported throughout all 33 NOTCH3 exons, high clustering is documented for exons 1 through 6, particularly within exons 3–4 [8]. This high genotypic variability is likely responsible, at least partially, for the high phenotypic variability of the disease, although progress is still being made to better characterize known mutations. Most recently, Rutten et al. [9■■] assessed a cohort of 664 CADASIL patients with pathogenic variants in EGFr domains 1 through 34 and found that individuals with EGFr 1–6 variants have an earlier stroke onset, higher brain lesion load and lower survival rates than those with variants in EGFr 7–34 that had a much milder version of the disease. This points to the location of the mutation potentially being critical for how CADASIL develops and progresses, although an effect might also be present through modifier genes, environmental effects or other concomitant risk factors to explain the phenotypic variability observed in CADASIL subjects, even when carrying the same mutation.
Upon ligand binding, the Notch3 receptor is activated by proteolytic cleavages that lead to release of the extracellular domain (Notch3ECD or N3ECD) into the interstitial space between cells. This also frees the intracellular domain (Notch3ICD or N3ICD), which translocates to the nucleus to regulate gene expression in a process that helps maintain VSMC homeostasis [10,11]. In CADASIL, mutant Notch3ECD accumulates in the extracellular space and eventually leads to VSMC degeneration in a mechanism that is poorly understood. However, work by several investigators suggests that Notch3ECD has binding affinity for a variety of molecules in a way that sequesters them from physiological pathway signaling to cause pathological dysfunction in blood vessel homeostasis [12].
In support of this hypothesis, GOMs have been identified near the surface of VSMCs and pericytes of brain, skeletal muscle, retina, kidney, pericardium and skin of CADASIL patients [13■]. It remains unclear whether GOMs are merely accumulated deposits that result from smooth muscle cell degradation or if they also impart toxicity to the cells further causing vessel degeneration by sequestering molecules that are important for normal physiological regulation. It has been established that extracellular Notch3, tissue inhibitor of metalloproteinases 3 (TIMP-3), vitronectin (VTN) and latent TGF-β binding protein 1 (LTBP-1) co-localize within these deposits by immunohistochemistry and mass spectroscopy of CADASIL brains and arteries obtained by microvessel isolation [12,14,15]. TIMP-3, VTN and TGF-β, regulated by LTBP-1, are proteins that have important roles in blood vessel formation and maintenance. Using a combination of laser microdissection, liquid chromatography-tandem spectrometry and immunohistochemistry, Nagatoshi et al. [16■■] recently found co-localization of serum amyloid P component (SAP) with Notch3 in GOM-enriched blood vessels as well as annexin 2 and periostin within GOMs. SAP has been implicated in cognitive diseases such as Alzheimer’s as having a role in the stabilization of amyloid plaques and may have a similar role in the formation and stabilization of GOMs in CADASIL [16■■,17]. All combined, these findings point to a possibility that a variety of molecules having key roles in blood vessel homeostasis become sequestered by Notch3ECD into GOMs, leading to a dysregulation of critical pathways.
In an effort to better understand the process leading to vessel degradation in CADASIL, several groups are focusing on how the disease affects blood vessels on a molecular level. In a recent study, Dziewulska et al. [18] analyzed blood vessels isolated from CADASIL brain, skeletal muscle and skin tissue with immunohistochemistry and electron microscopy. They found that VSMCs within the tissues often had nuclei with very condensed chromatin, in irregular shapes and sizes and that were also multinucleated. In combination with positive staining for proliferative markers, this group suggested that there may be a mitotic instability in CADASIL VSMCs because of poor regulation by mutant Notch3 that potentially leads to degeneration. Also focusing on VSMC, Hanemaaijer et al. [19■■] investigated whether Notch3ECD accumulation is a result of impaired lysosomal autophagy in CADASIL patient derived cerebral VSMCs. Confocal imaging with labeling for Notch3 and lysosomal markers pointed to a defect in the autophagosome-lysosome fusion step, resulting in reduced clearance of Notch3ECD by CADASIL VSMCs as compared with healthy controls.
Endothelial dysfunction also appears to play a role in CADASIL as plasma levels of endothelial damage/repair markers such as vonWillebrand factor, endothelial progenitor cells and circulating progenitor cells appear to be affected in CADASIL. While vonWillebrand factor levels were significantly elevated in patient plasma samples, endothelial progenitor cells and circulating progenitor cells levels were lower but the later appeared to correlate with worsened outcomes as measured by neuropsychometric testing and MRI [20■]. These results point to impaired endothelial homeostasis playing a role in the CADASIL pathophysiology.
Also implicating the gliovascular unit, astrocyte damage has been observed within the white matter of CADASIL patients at different stages of the disease and CADASIL brain tissue had a higher percentage of astrocytopathy and astrocyte turnover with evidence of a dysregulation in autophagy particularly within the anterior temporal lobe [21■■]. Using a combination of electroencephalograms and arterial spin labeling MRI, Huneau et al. [22■] focused on the coupling of neural activity and hemodynamic changes (neurovascular coupling or NVC) in response to visual and motor stimuli in CADASIL patients as compared with healthy controls. They found that, even at early stages of the disease, there was a significant difference in the dynamic of blood flow responses in CADASIL patients but not in neural activity, hypothesizing that this may be because of an early dysfunction in the cerebral vasculature. These results have been since confirmed in an additional cohort of 21 CADASIL patients by using functional MRI while performing go/No-go tasks and implicate an observed reduction of blood oxygen levels in several areas of the brain, possibly because of impaired vasoreactivity in CADASIL [23].
Biomarker discovery is particularly important in CADASIL as a tool to diagnose, track progression and measure therapeutic effectiveness in CADASIL. In the recent years, neurofilament light chain (NfL) has emerged as a promising circulating biomarker. NfL is a structural protein in the axons of neurons and appears in blood and cerebrospinal fluid upon neuronal damage in diseases such as multiple sclerosis and Alzheimer’s [24,25]. In 2018, Tiedt et al. [26] investigated serum NfL as a marker of ischemic stroke in a cohort of patients recruited within 24 h of ischemic stroke symptoms and followed over a period of 6 months. They found that serum NfL levels were not only elevated and peaked immediately after ischemic stroke but that they remained elevated at 3 and 6 months after ischemic stroke. Additionally, NfL levels correlated with infarct volumes as well as recurrent ischemic lesions and secondary neurodegeneration on MRI at 6 months and they independently predicted clinical outcomes at 3 months post-ischemic stroke. In a comparison study between CADASIL individuals, sporadic SVD patients and healthy controls, serum NfL was confirmed to correlate with SVD MRI markers for both patient groups but also with clinical neurological features such as impaired processing speed performance, focal neurological deficits and disability for the CADASIL patient cohort only [27■■]. Gravesteijn et al. [28■■] obtained similar results with serum NfL and found a strong correlation not only in measured MRI neuroimaging but also with cognitive function and long-term disease outcomes. These studies show a strong association between serum NfL and several clinical characteristics of CADASIL and is a good marker in patients with ischemic stroke during the acute phase but also several months after ischemic stroke. However, serum NfL levels can become elevated in aging as well as any situations leading to axonal damage because of other diseases, lacking specificity for CADASIL and lead to confounding conclusions. Further studies are needed to completely characterize how NfL serum levels can be used in combination with other clinical measures and biomarkers of the disease rather than on its own.
One of the most distinctive characteristics of CADASIL are the white matter hyperintensities (WMH) in the brain tissue that can be visualized by MRI and are often present as an early sign of the disease before a patient, usually in their 30s, develops many severe symptoms. However, CADASIL patients are often misdiagnosed as having multiple sclerosis or other types of SVDs despite similar but unique neuroimaging differences. Characteristics of CADASIL are symmetry, morphology and location of WMH with involvement of the anterior temporal lobe, the external capsule and the superior frontal gyrus [6■]. Figure 1 shows the typical CADASIL MRI findings for an acute ischemic event and the longer-term white matter changes. Other MRI features often observed in CADASIL are incident lacunes, cerebral microbleeds, enlarged perivascular space and brain atrophy [5], although these features can be observed in a variety of other SVDs. In an MRI neuroimaging comparison between CADASIL and myotonic dystrophy type 1 (DM1) patients, which are sometimes confused with one another, the CADASIL cohort appeared to have more involvement of the temporal lobes, external capsule and basal ganglia as well as a significantly higher incidence of microbleeds than DM1 [29]. Similarly, in an effort to differentiate MS from CADASIL patients, Vinciguerra et al. [30] have shown that a novel automated MRI marker, peak width skeletonized mean diffusivity (PSMD), correlates with the white matter lesion load in both conditions but is higher in MS than in CADASIL.
FIGURE 1.
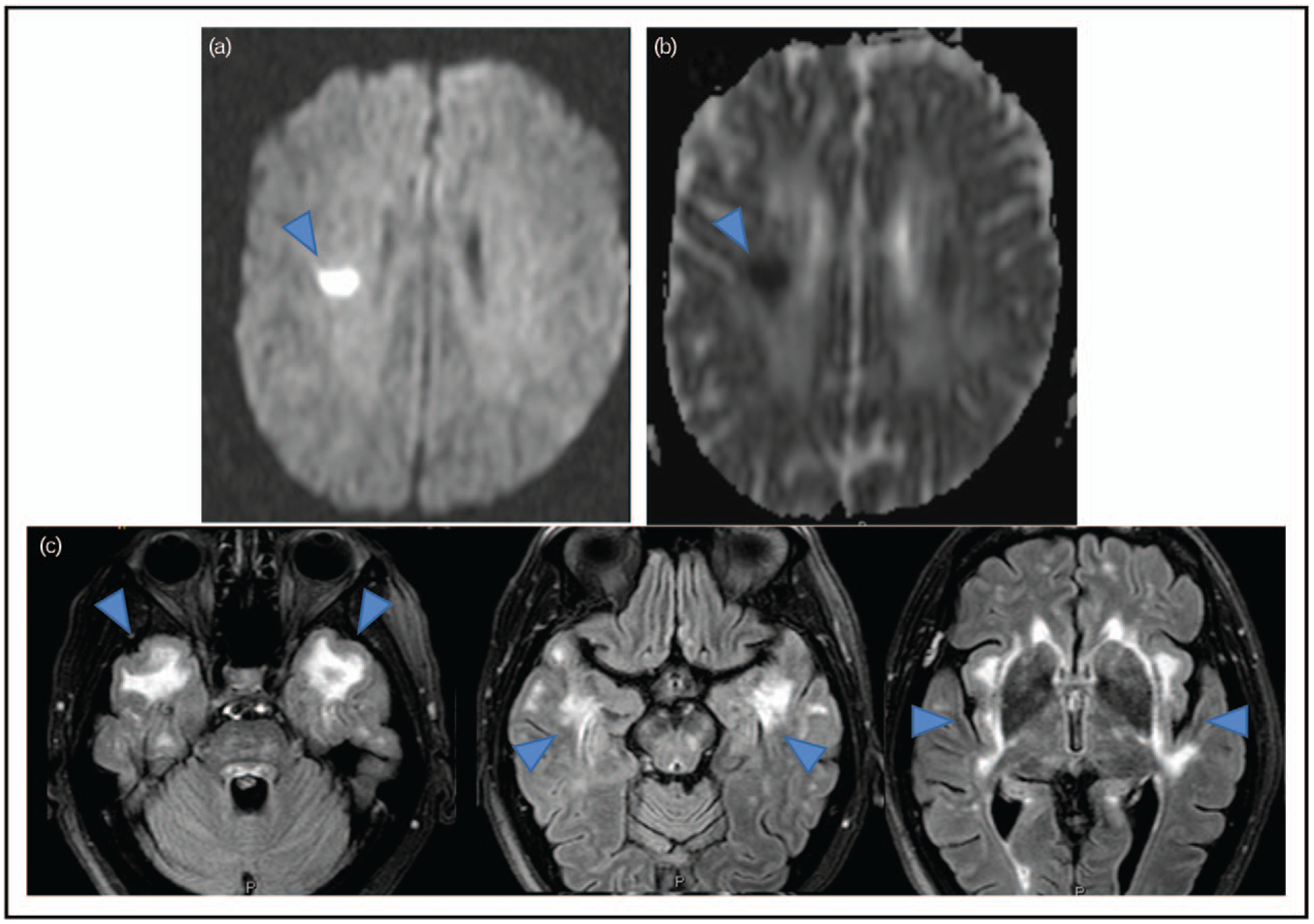
Typical Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) MRI findings: panels a and b show typical acute ischemic changes in a CADASIL patient after stroke. Panel c consists of three cross-sectional MRI images to show typical white matter changes in a CADASIL patient. Blue arrows point to the typical changes.
Aiming to determine clinical correlations with conventional MRI markers, a study following 160 CADASIL individuals with confirmed genetic diagnosis over 3 years, established that brain atrophy and the number of incident lacunes independently and sensitively correlated with clinical changes (strokes, dementia, disability) in the patient cohort and can be used as markers of disease progression on conventional MRI [31]. The extent of WMH changes in patients, however, has consistently been found to not have strong correlations with disease severity in CADASIL. Hypothesizing that regional rather global WMH changes in CADASIL contribute to differences clinical outcomes, Duchesnay et al. [32■] divided the brain in different spatial sections and found that higher WMH associated with the temporal lobes and frontal gyri correlated with a milder version of the disease as compared with WMH in the pyramidal tracts in 301 CADASIL patients.
CONCLUSION
While CADASIL has been extensively studied over the last several decades, our understanding increases as research and clinical techniques evolve. With whole exome and genome testing, we can now sequence entire families and identify individuals that are affected by the disease before development of symptoms. With proteomics, we can investigate how molecules and signaling pathways are dysregulated as a result of CADASIL, but we can also see how they interact to modulate its pathophysiology. Genomics and proteomics have opened the door to the critical and much needed possibilities of identifying biomarkers and potential therapeutic targets for this disease. Yet, this remains a complex condition, the mechanism of which still has to be elucidated.
It has become clear that this is a genotypically and phenotypically diverse disorder as more case studies appear in the scientific literature. Notch signaling is a highly conserved pathway that regulates blood vessel formation during development and vessel homeostasis in adulthood [33]. In CADASIL, NOTCH3 mutations and their effect on the Notch3 receptor appear to set off a cascade of imbalances with a profound effect at multiple levels as extracellular accumulation of Notch3ECD can sequester other molecules from their normal physiological functions. CADASIL vessel disorder, shown in Fig. 2, similarly involves multiple molecular systems on the endothelial, smooth muscle and neural cell levels related to maintenance of cellular health, delivery of important nutrients and vasoreactivity. Advances in MRI neuroimaging techniques and postprocessing are also enabling us to better discern regional differences in white matter changes within areas of the brain making MRI a better marker to track disease progression.
FIGURE 2.
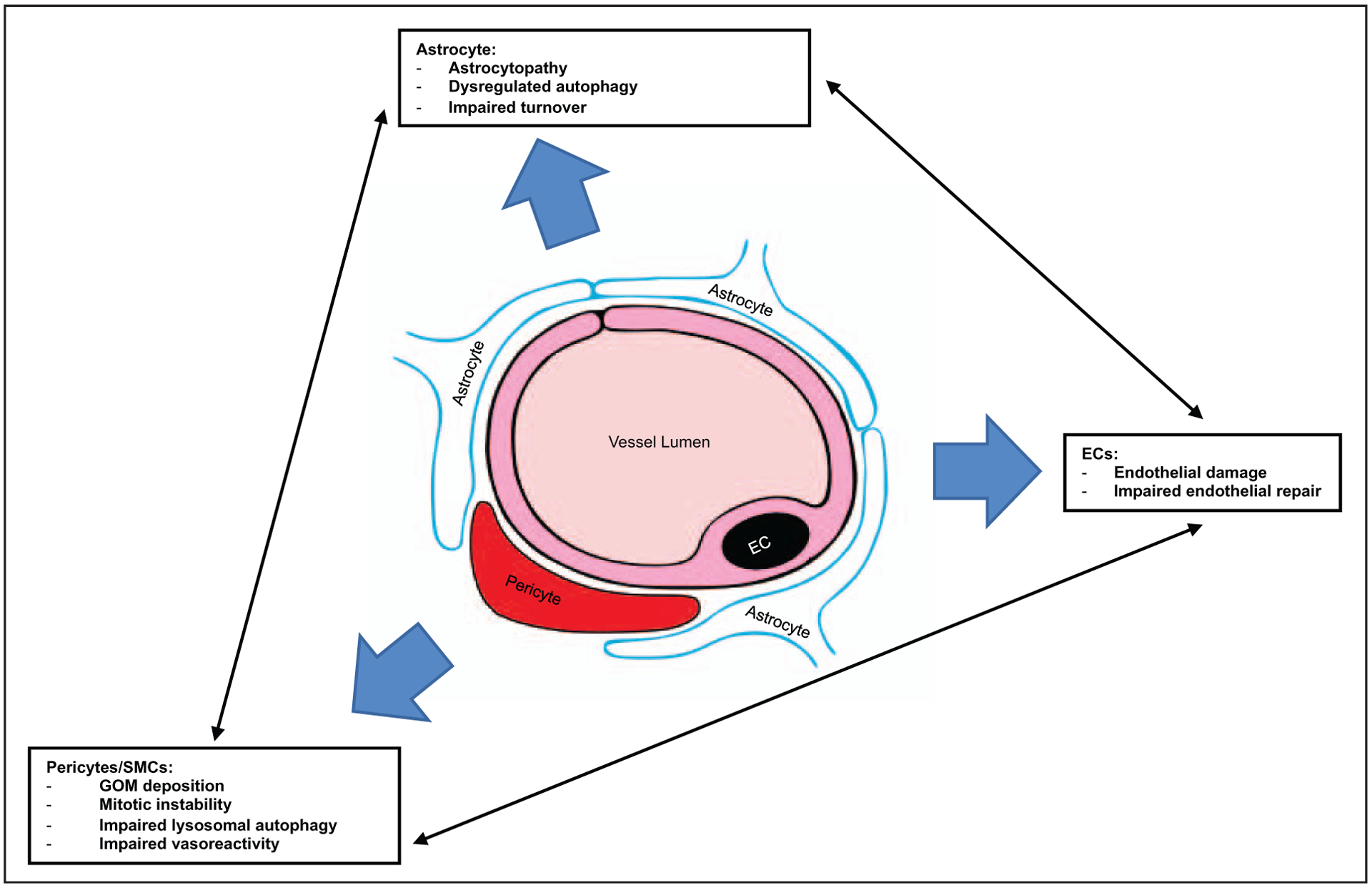
Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) vessel disorder: schematic of a cerebral capillary shows the effects on multiple molecular systems on the endothelial, pericyte/smooth muscle and neural cell levels because of CADASIL.
Although quick progress has been made and as we move forward, translational research combining basic science advances and clinical findings as well as long-term clinical studies enrolling large cohorts of CADASIL patients at different disease stages will be critical to help fully elucidate its pathophysiology and have the power to identify appropriate biomarkers and therapeutic agent options for patients.
KEY POINTS.
CADASIL is the most common genetic cause of stroke and dementia in adults but a genotypically and phenotypically diverse disease.
The disease mechanism has not been elucidated but significant progress has been made to better understand the genotype and phenotype of the disease, including mutation type/location, variant effect on protein function, protein accumulation into GOMs and vascular dysfunction extending to VSMC, endothelium and the gliovascular unit.
Research on biomarkers is ongoing and promising with newly identified NfL.
MRI advances allow us to discern the effect of the location within the brain of white matter changes and correlate to disease severity.
Translational research is essential for the identification of treatment options still not identified and much needed for patients with CADASIL.
Acknowledgements
Thank you to Dr Camilo Toro for providing the MRI images and his expertise. Thank you to Dr Alessandra Brofferio for her expertise and detailed feedback. Thank you to Christopher J. Roach for his help in rendering Fig. 2.
Financial support and sponsorship
National Institutes of Health Intramural program and U01HL131019.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1.Dichgans M, Mayer M, Uttner I, et al. The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol 1998; 44:731–739. [DOI] [PubMed] [Google Scholar]
- 2.Markus HS, Martin RJ, Simpson MA, et al. Diagnostic strategies in CADASIL. Neurology 2002; 59:1134–1138. [DOI] [PubMed] [Google Scholar]
- 3.Viswanathan A, Gschwendtner A, Guichard JP, et al. Lacunar lesions are independently associated with disability and cognitive impairment in CADASIL. Neurology 2007; 69:172–179. [DOI] [PubMed] [Google Scholar]
- 4.Tikka S, Baumann M, Siitonen M, et al. CADASIL and CARASIL. Brain Pathol 2014; 24:525–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Donato I, Bianchi S, De Stefano N, et al. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: update on clinical, diagnostic, and management aspects. BMC Med 2017; 15:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.■.Wang MM. CADASIL. Handbook Clin Neurol 2018; 148:733–743. [DOI] [PubMed] [Google Scholar]; This is a book chapter by Dr Wang that is an excellent comprehensive review on many aspects of CADASIL.
- 7.Rutten JW, Dauwerse HG, Gravesteijn G, et al. Archetypal NOTCH3 mutations frequent in public exome: implications for CADASIL. Ann Clin Transl Neurol 2016; 3:844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joutel A, Vahedi K, Corpechot C, et al. Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet 1997; 350:1511–1515. [DOI] [PubMed] [Google Scholar]
- 9.■■.Rutten JW, Van Eijsden BJ, Duering M, et al. The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1–6 pathogenic variants are associated with a more severe phenotype and lower survival compared with EGFr 7–34 pathogenic variant. Genet Med 2018; 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article focuses on the effects of NOTCH3 mutation location and how it correlates with disease severity.
- 10.Bray SJ. Notch signaling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 2006; 7:678–689. [DOI] [PubMed] [Google Scholar]
- 11.Wang T, Baron M, Trump D. An overview of Notch3 function in vascular smooth muscle cells. Prog Biophys Mol Biol 2008; 96:499–509. [DOI] [PubMed] [Google Scholar]
- 12.Monet-Leprêtre M, Haddad I, Baron-Menguy C, et al. Abnormal recruitment of extracellular matrix proteins by excess Notch3ECD: a new pathomechanism in CADASIL. Brain 2013; 136:1830–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.■.Lorenzi T, Ragno M, Paolinelli F, et al. CADASIL: ultrastructural insights into the morphology of granular osmiophilic material. Brain Behav 2017; 7:e00624. [DOI] [PMC free article] [PubMed] [Google Scholar]; This article focuses on structural features within GOM deposits in an effort to better characterize them.
- 14.Yamamoto Y, Craggs LJ, Watanabe A, et al. Brain microvascular accumulation and distribution of the NOTCH3 ectodomain and GOM in CADASIL. J Neuropathol Exp Neurol 2013; 72:416–431. [DOI] [PubMed] [Google Scholar]
- 15.Kast J, Hanecker P, Beaufort N, et al. Sequestration of latent TGF-β binding protein 1 into CADASIL-related Notch3-ECD deposits. Acta Neuropathol Commun 2014; 2:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.■■.Nagatoshi A, Ueda M, Ueda A, et al. Serum amyloid P component: a novel potential player in vessel degeneration in CADASIL. J Neurol Sci 2017; 379:69–76. [DOI] [PubMed] [Google Scholar]; This group found that SAP colocalizes with NOTCH3ECD and GOMs and may play a role in stabilizing these deposits in a way that sequesters other molecules from performing their normal physiologic roles.
- 17.Verwey NA, Schuitemaker A, Van Der Flier WM, et al. Serum amyloid P component as a biomarker in mild cognitive impairment and Alzheimer’s disease. Dement Geriatr Cogn Disord 2008; 26:522–527. [DOI] [PubMed] [Google Scholar]
- 18.Dziewulska D, Nycz E, Rajczewska-Oleszkiewicz C, et al. Nuclear abnormalities in vascular myocytes in cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Neuropathology 2018; 38:601–608. [DOI] [PubMed] [Google Scholar]
- 19.■■.Hanemaaijer ES, Panahi M, Swaddiwudhipong N, et al. Autophagy-lysosomal defect in human CADASIL vascular smooth muscle cells. Eur J Cell Biol 2018; 97:557–567. [DOI] [PubMed] [Google Scholar]; Work by this group strongly implicates a defect in clearance of deposited ECM materials as having a role in CADASIL.
- 20.■.Pescini F, Donnini I, Cesari F, et al. Circulating biomarkers in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy patients. J Stroke Cerebrovasc Dis 2017; 26:823–833. [DOI] [PubMed] [Google Scholar]; Important study of potential circulating biomarkers (von Willebrand factor, thrombomodulin and endothelial progenitor cells) in CADASIL patients along with clinical/functional and neuroimaging associations.
- 21.■■.Hase Y, Chen A, Bates LL, et al. Severe white matter astrocytopathy in CADASIL. Brain Pathol 2018; 28:832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]; This group determined CADASIL astrocytes apoptose by means of an authophagy-like cell death and have lower cell turnover.
- 22.■.Huneau C, Houot M, Joutel A, et al. Altered dynamics of neurovascular coupling in CADASIL. Ann Clin Transl Neurol 2018; 5:788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]; Altered neurovascular physiology in CADASIL has a profound effect on vascular function rather than cognitive function at an early stage of the disease.
- 23.Gavazzi G, Orsolini S, Salvadori E, et al. Functional magnetic resonance imaging of inhibitory control reveals decreased blood oxygen level dependent effect in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 2019; 50:69–75. [DOI] [PubMed] [Google Scholar]
- 24.Disanto G, Barro C, Benkert P, et al. Serum neurofilament light: a biomarker of neuronal damage in multiple sclerosis. Ann Neurol 2017; 81:857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattsson N, Andreasson U, Zetterberg H, et al. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol 2017; 74:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiedt S, Duering M, Barro C, et al. Serum neurofilament light: a biomarker of neuroaxonal injury after ischemic stroke. Neurology 2018; 91:e1338–e1347. [DOI] [PubMed] [Google Scholar]
- 27.■■.Duering M, Konieczny MJ, Tiedt S, et al. Serum neurofilament light chain levels are related to small vessel disease burden. J Stroke 2018; 20:228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]; Very promising work showing circulating NfL as a potential marker of SVDs.
- 28.■■.Gravesteijn G, Rutten JW, Verberk IM, et al. Serum neurofilament light correlates with CADASIL disease severity and survival. Ann Clin Transl Neurol 2019; 6:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]; This group explores the potential of NfL as a circulating biomarker specifically for CADASIL.
- 29.Kim H, Lim YM, Oh YJ, et al. Comparison of brain magnetic resonance imaging between myotonic dystrophy type 1 and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. PloS One 2018; 13:e0208620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vinciguerra C, Giorgio A, Zhang J, et al. Peak width of skeletonized mean diffusivity (PSMD) as marker of widespread white matter tissue damage in multiple sclerosis. Mult Scler Relat Disord 2019; 27:294–297. [DOI] [PubMed] [Google Scholar]
- 31.Ling Y, De Guio F, Jouvent E, et al. Clinical correlates of longitudinal MRI changes in CADASIL. J Cereb Blood Flow Metab 2018; Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.■.Duchesnay E, Selem FH, De Guio F, et al. Different types of white matter hyperintensities in CADASIL. Front Neurol 2018; 9:526. [DOI] [PMC free article] [PubMed] [Google Scholar]; As MRI can often lead to confounding diagnosis, this group focuses on better characterizing regional differences in MRI observed in CADASIL patients.
- 33.Mack JJ, Iruela-Arispe ML. NOTCH regulation of the endothelial cell phenotype. Curr Opin Hematol 2018; 25:212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]