

Abstract
Congenital generalized lipodystrophy (CGL) is an autosomal recessive disorder with two major subtypes. Variants in AGPAT2 result in CGL type 1 with milder manifestations, whereas BSCL2 variants cause CGL type 2 with more severe features. Muscle hypertrophy caused by lack of adipose tissue is present early in life in CGL patients. Our aim was to investigate 10 CGL patients from 7 different countries and report genotype–phenotype relationships. Genetic analysis identified disease-causing variants in AGPAT2 (five patients) and in BSCL2 (five patients), including three novel variants; c.134C>A (p.Ser45*), c.216C>G (p.Tyr72*) in AGPAT2 and c.458C>A (p.Ser153*) in BSCL2. We also report possible novel clinical features such as anemia, breast enlargement, steatorrhea, intraventricular hemorrhage and nephrolithiasis in CGL patients. Generalized lipodystrophy and muscular hypertrophy were the only features in all of our patients. Hepatomegaly was the second common feature. Some manifestations were exclusively noticed in our CGL2 patients; hypertrichosis, high-pitched voice and umbilical hernia. Bone cysts and history of seizures were noticed only in CGL1 patients. The findings of this study expand our knowledge of genotype–phenotype correlations in CGL patients. These results have important clinical applications in diagnosis and management of the CGL patients as well as in genetic counseling in families at-risk.
Keywords: AGPAT2, BSCL2, CGL1, CGL2, congenital generalized lipodystrophy, genotype–phenotype correlations, novel variants
Congenital generalized lipodystrophy (CGL, Berardinelli–Seip syndrome) is an autosomal recessive disease, characterized by almost complete absence of adipose tissue at birth or during early infancy (1–3). The worldwide prevalence of CGL is estimated to be 1 in 10 million (4); however, the prevalence varies among different ethnicities (from 1 in 0.2 million in Lebanon to 1 in 12 million in United States ) (5, 6).
The main clinical features of this rare disorder are generalized muscular hypertrophy and consequently a markedly muscular appearance, severe insulin resistance and hypertriglyceridemia (3). Affected patients might have an accelerated growth along with an avid appetite in early childhood, and develop acromegaloid features such as enlarged mandible, hands and feet in late childhood. Other reported clinical manifestations of CGL patients include acanthosis nigricans, hepatomegaly due to hepatic steatosis, splenomegaly, intellectual disability and hypertrophic cardiomyopathy, and multiple focal lytic lesions in the appendicular bones (7).
CGL has been linked with variants in four genes, causing four different phenotypes with some overlapping clinical manifestations. CGL type 1 is caused by variants in the AGPAT2 gene (8), characterized by near total absence of the ‘metabolically active’ adipose tissue located in most subcutaneous regions of the body, intraabdominal and intrathoracic areas but preservation of ‘mechanically active’ adipose tissue in orbital region, in the palms, soles and periarticular areas. The patients also have lytic bone lesions, which are sparse in other types of CGL (9, 10). AGPAT2 encodes 1-acyl-glycerol-3-phosphate-acyltransferase 2, an enzyme involved in triglyceride synthesis, and predominantly expressed in adipose tissue. Therefore, variants in AGPAT2 lead to decreased biosynthesis of triglyceride and phospholipids (11). The most severe form of CGL, CGL2, is the result of variants in BSCL2 gene (12). Affected individuals with type 2 disease lack both ‘mechanical’ and ‘metabolically active’ adipose tissue (10). In addition, CGL2 patients show a higher prevalence of intellectual disability and cardiomyopathy in comparison to the other types of CGL (9, 13). BSCL2 gene encodes a protein called ‘Seipin’, an integral endoplasmic reticulum (ER) membrane protein mainly expressed in the adipose tissue, brain, and testis (12, 14). It has been reported that Seipin is an essential regulator of adipogenesis (14), and plays an important role in lipid droplet formation and adipocyte differentiation (15). CGL types 3 and 4 are caused by homozygous variants in CAV1 (16) and PTRF genes, respectively (17–19).
We previously identified a novel nonsense variant and a missense substitution in AGPAT2, in two Persian families with CGL (20). In this study, we describe clinical and molecular features of 10 patients, of both type 1 and type 2 CGL, from 7 different countries.
Methods and patients
Patients and families
Ten patients with CGL from eight families were investigated. All members of families underwent comprehensive physical examinations, including endocrinological, neurological, cardiac, urogenital and psychiatric assessments. Written informed consent for clinical and molecular investigation (by consent forms of Institute of Pathology and Genetics, Gosselies, Belgium) was obtained from all members of the families or their legal guardians. The study was conducted in accordance with the Helsinki Declaration.
Molecular analysis
Genomic DNA was extracted from peripheral blood collected from all participating family members. Entire coding region and splice junction sites of AGPAT2 and BSCL2 genes were amplified by polymerase chain reaction (PCR) and Sanger sequenced. Sequences were compared with the published reference sequence (BSCL2: NM_032667.6 and AGPAT2: NM_006412.3). The details of methods are described in Supporting Information (Appendix S1).
Results
Clinical findings
We evaluated 10 patients with CGL from 7 different countries. Five patients had CGL type 1 (AGPAT2 variants) and five had CGL type 2 (BSCL2 variants). Six cases (with CGL1 and CGL2) in this study were male. The youngest patient was a 3-month-old girl from Oman (Case 9, Table 1), whereas the oldest case was a 33-year-old man from Sri Lanka (Case 10, Table 1). One of our patients (Case 4, Table 1) was diagnosed in adulthood (25 years of age) by genetic testing after clinical confirmation of CGL diagnosis in her niece. The only clinical features of this patient (Case 4, Table 1) were triangular face and muscular appearance.
Table 1.
Clinical features of the patients
| CGL Type | CGL1 (AGPAT2) |
CGL2 (BSCL2) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Clinical features | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | Case 8 | Case 9 | Case 10 |
| Mutation | Homozygous, (c.134C>A, p.Ser45*), exon 1 (N) | Homozygous, (c.589-2A>G, p.Gln19 6fs*228), intron 4 | Homozygous, (c.202C>T, p.Arg68*), exon 2 | Homozygous, (c.202C>T, p.Arg68*), exon 2 | Homozygous, (c.216C>G, p.Tyr72*), exon 2 (N) | Compound heterozygous, (c.412C>T, p.Arg138*), exon 4 (c.458C>A, p.Ser153*), exon 5 (N) | Homozygous, (c.317_321 del, p.Tyr106 Cysfs*6), exon 4 | Homozygous, (c.672-2A>C), intron 6 | Homozygous, (c.672-2A>C), intron 6 | Homozygous, (c.636del, p.Tyr213 Thrfs*20), exon 6 |
| Mutation effect | Stop-gained | Splice site | Stop-gained | Stop-gained | Stop-gained | Stop-gained | Frame shift | Splice site | Splice site | Frame shift |
| Gender | Male | Female | Female | Male | Male | Male | Male | Female | Female | Male |
| Age | 4.5 months | 3 years | 5 months | 25 years | 3 years | 9 years | 5 years | 7 years | 3 months | 33 years |
| Ethnicity | Iranian | Afrocaribbean (Dominican Rep) | Iranian | Iranian | Indian (from Sri Lanka) | Lithuanian | Lebanese (from Australia) | Arab (from Oman) | Arab (from Oman) | Sinhalese (from Sri Lanka) |
| Muscle hypertrophy | + | + | + | + | + | + | + | + | + | + |
| Hepatomegaly | + | + | + | − | N/A | + | + | + | + | + |
| Steatohepatitis | + | − | + | − | N/A | + | + | + | mild | N/A |
| Splenomegaly | + | + | + | − | − | + | + | + | + | − |
| Hernia | Inguinal | Inguinal | Inguinal | − | − | Umbilical | Inguinal | Umbilical | Umbilical | − |
| Acromegaloid features | + | − | + | − | + | + | + | + | N/A | Mild |
| Large ears | + | − | + | − | − | + | + | + | + | + |
| Triangular facies | + | − | + | Mild | − | + | + | Mild | Mild | + |
| Acanthosis nigricans | − | + | − | − | + | + | − | + | + | + |
| Bone cysts | − | N/A | − | + | N/A | − | no- | − | N/A | N/A |
| Breast enlargement | − | N/A | − | − | − | − | − | + | N/A | N/A |
| Changes in genitalia | − | − | + | − | Penis enlargement | + | Retractile testes | Clitoromegaly | N/A | − |
| Hypertrichosis | − | − | − | − | N/A | − | + | Mild | Mild | N/A |
| Hypercholesterolemia | − | − | − | − | − | − | − | − | + | N/A |
| High-pitched voice | − | − | − | − | N/A | + | − | + | N/A | − |
| Cardiomyopathy | − | + | − | − | N/A | − | + | + | − | N/A |
| Hypertension | − | − | − | − | N/A | + | − | + | N/A | − |
| Nephrolithiasis | + | − | + | − | N/A | − | − | + | N/A | − |
| Nephropathy | − | − | − | − | N/A | + | − | − | N/A | Microalbuminuria |
| Growth retardation | − | − | Mild | Mild | − | − | IUGR | − | + | N/A |
| BMI (kg/m2) | 15.1 | 16 | 20 | N/A | 19.1 | 14 | 18.3 | 10.2 | N/A | |
| Intellectual disability | − | − | − | − | N/A | Mild | + | + | N/A | N/A |
| Seizures | − | − | + | − | − | − | −(abnormal EEG) | − | N/A | − |
| Anemia | + | − | − | − | − | − | − | − | − | − |
| Hypertriglyceridemia | + | + | + | − | + | + | + | Borderline high | + | N/A |
| Elevated insulin levels | + | − | + | − | N/A | + | + | + | N/A | + |
| Elevated liver enzymes | + | − | + | − | N/A | + | + | + | mild | − |
| Diabetes | − | − | − | − | − | − | − | + | N/A | + |
+, presence of the feature; −, absence of the feature; BMI, body mass index; EEG, electroencephalogram; IUGR, intrauterine growth restriction; N (mutation), novel; N/A, not available.
The only constant clinical features in CGL type 1 patients were generalized lipodystrophy and their muscular appearance (all patients), followed by hypertriglyceridemia (four patients), splenomegaly (three patients) and hepatomegaly (three patients), triangular facies (three patients), acromegaloid changes (three patients) and inguinal hernia (three patients), large ears (two patients), acanthosis nigricans (two patients), nephrolithiasis (two patients), increased liver enzymes (two patients) and growth retardation (two patients). Changes in external genitalia were observed in two patients, mild clitoromegaly in one girl and penis enlargement in a boy. Increased plasma insulin levels, anemia, cardiomyopathy, bone cyst and seizure disorders, each were seen in only one patient. None of CGL1 patients in this study had diabetes, hypertriglyceridemia, nephropathy, intellectual disability, hypertrichosis or high-pitched voice.
All CGL2 patients had generalized lipodystrophy and muscular appearance. The next most frequent features in CGL2 patients were triangular faces with large ears and enlarged liver. Intellectual disability was also reported in all three of those who had available data. The other most prevalent clinical features in CGL2 patients included muscle hypertrophy, hernia, splenomegaly, steatohepatitis, acanthosis nigricans and large hands and feet, elevated liver enzymes and increased plasma insulin levels (four patients), hypertrichosis (three patients), high-pitched voice (two patients), diabetes (two patients), hypertension (two patients), cardiomyopathy (two patients) and nephropathy (two patients). External genitalia changes were observed in three patients. One boy had retractile testes, another boy had advanced pubertal status. One girl had clitoromegaly. Hypertriglyceridemia, breast enlargement and anemia, each was detected in only one patient. The clinical and biochemical features of the patients have been summarized in Table 1.
CGL1 patients
Case 1.
A 4.5-month-old Iranian male presented with abdominal distension and severe perspiration, hepatomegaly and inguinal hernia. He was the third child of first cousin healthy parents with negative family history for this condition. He presented with hypertrophy of skeletal muscles, with large hands and feet, enlarged liver and spleen (Fig. 1a). He had hypertriglyceridemia, bilateral microlithiasis and microcytic anemia (hemoglobin: 9 g/dl). His anemia was due to iron deficiency and improved by iron supplement.
Fig. 1.
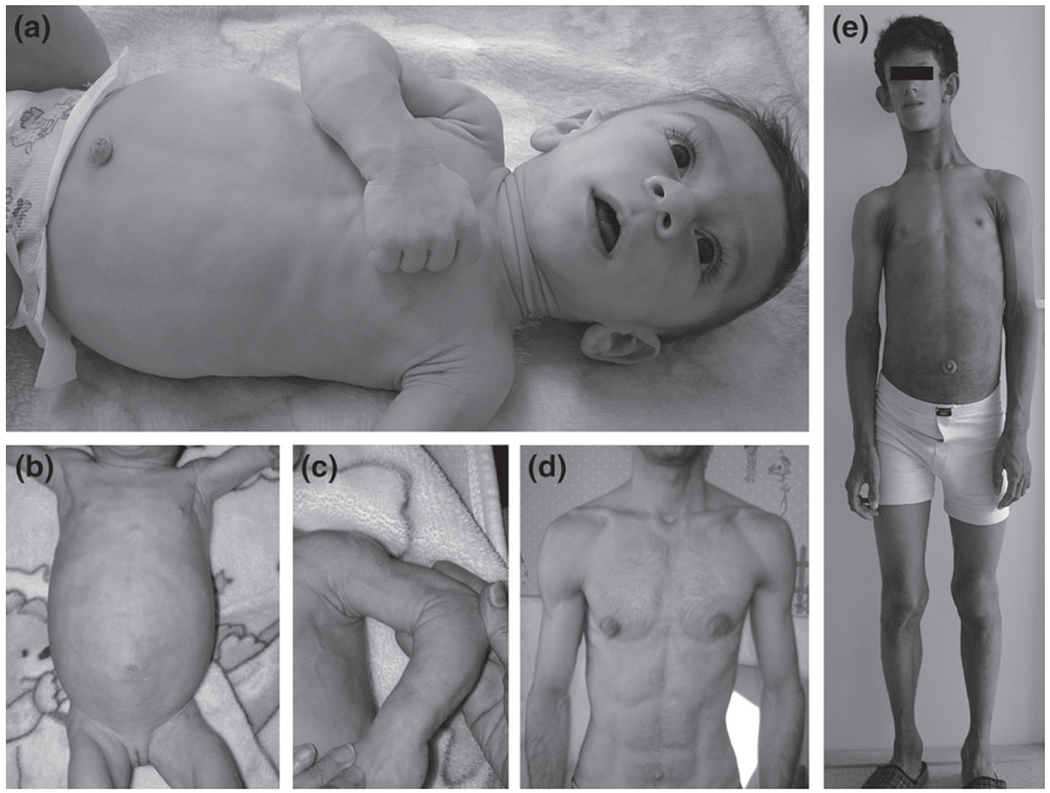
Clinical features of the patients. (a) Case 1. Absence of subcutaneous fat, muscular hypertrophy, marked umbilical prominence, large hands and feet, enlarged liver and spleen. (b, c) Case 3. Hypertrophy of skeletal muscles, enlarged liver and spleen. (d) Case 4. Mild generalized loss of subcutaneous fat and muscular stature, marked umbilical prominence. (e) Case 6. Acromegalic appearance, protruding abdomen, marked umbilical prominence, velvety thickening and hyperpigmentation of the skin around neck, in axillae and antecubital fossae (acanthosis nigricans).
Case 2.
A 3-year-old female from Dominican Republic presented with lipodystrophic features, hepatosplenomegaly, acanthosis nigricans and inguinal hernia. At 3 months of age, she presented with hepatomegaly, being initially investigated for a metabolic storage disorder, which could not be confirmed. At the age of 7 months, she was referred to us, and lipodystrophy was noted with normal facial phenotype, generalized lipodystrophy affecting the trunk, arms and legs, sparing the face, hands and feet, dry skin with marked acanthosis nigricans and big hands and feet without clear acromegaloid features. She had a mild left ventricular hypertrophy.
Case 3.
A 5-month-old Iranian girl of nonconsanguineous healthy parents whose parents were not known to be related but came from the same region of Iran. The patient presented first with severe steatorrhea, many fat droplets in stool and negative sweat test. Then Wolman disease was suspected because of hepatosplenomegaly (Fig. 1b) and steatorrhea, but no adrenal calcinosis was detected. She looked muscular (Fig. 1c) with broad hands and feet, distended abdomen along with phlebomegaly. She had triangular faces with curly hairs. Further examination revealed mild clitromegaly, mildly abnormal liver enzymes and severe hypertriglyceridemia. No fat-soluble vitamin deficiency was detected. A needle liver biopsy showed macrovesicular steatosis. Endocrine workup was normal. Medium chain triglyceride based formula was introduced along with reduction of breast feeding. She was given fat-soluble vitamins supplementation. Steatorrhea stopped shortly after nutritional manipulation and serum triglyceride level returned to normal after one month. The parents mentioned that one of the paternal adolescent cousins died with diabetes mellitus and fatty liver. Her uncle (Case 4) had similar facial features, therefore he was also investigated.
Case 4.
A 25-year-old Iranian male (uncle of Case 3) with muscular appearance (Fig. 1d) and mildly lipodystrophic changes. He had a history of chronic diarrhea since childhood as well as repeated fractures diagnosed as having bone cysts (Fig. 2). He was otherwise normal upon physical examination. He also showed normal liver function tests, normal lipid profile, normal fasting and 2 h postprandial glucose and hemoglobin A1C level. His stool examination showed no fat droplet.
Fig. 2.
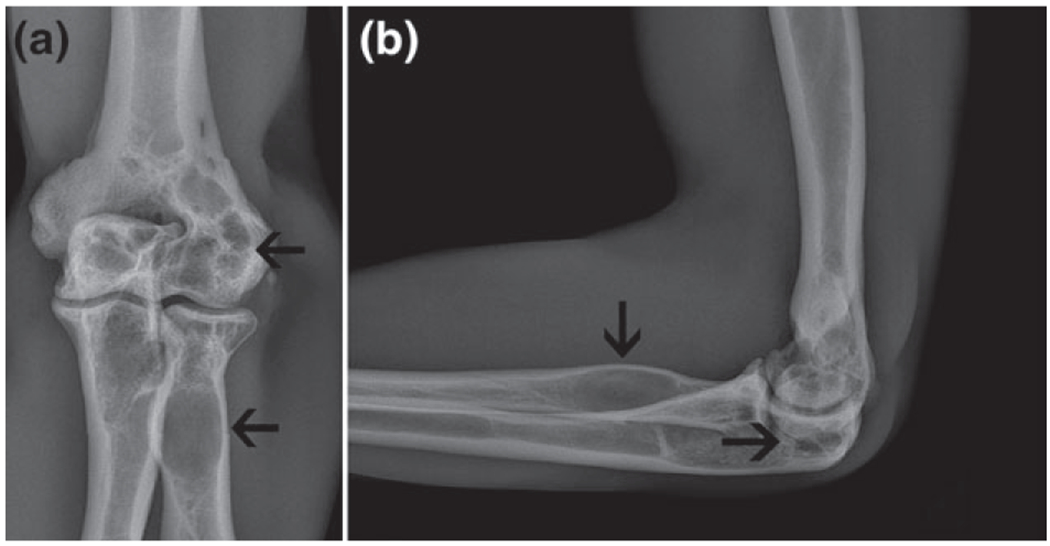
Antero-posterior (a) and lateral views of the roentgenograms of the elbow region of Case 4. Multiple radiolucent lesions (cysts) are seen in the distal end of humerus, and in the proximal region of radius and ulna bones.
Case 5.
A 3-year-old Indian male (residing in Sri Lanka) with mild lipodystrophic changes since 2 month of age presented with large hands and feet, acanthosis nigricans and penis enlargement. He had hypertriglyceridemia.
CGL2 patients
Case 6.
A 9-year-old Lithuanian male offspring of nonconsanguineous healthy parents. He had triangular facies, muscular appearance, acromegaloid features with a long neck, low set ears and curly hair (Fig. 1e). Marked acanthosis nigricans, phlebomegaly, umbilical hernia, severe hepatosplenemagaly, strabismus, hyperhidrosis, hypertension, dyslipidemia and proteinuria were detected in further investigations. He showed stage two Tanner pubertal status and also advanced bone age of 14 years. This patient suffered from intellectual disability (Case 6, Table 1).
Case 7.
A 5-year-old Lebanese boy (residing in Australia) with a history of intrauterine growth retardation, intraventricular hemorrhage and secondary hydrocephalus. He was the first child of consanguineous healthy parents. He suffered from a quadriplegic cerebral palsy. Clinical examination revealed triangular facies, large ears, epiphoria, acromegaloid features and enlarged superficial veins along with hepatosplenomegaly. In addition, he had a history of incarcerated inguinal hernia that was surgically repaired.
Case 8.
A 7-year-old Arab girl from Oman who presented with hypertension, cardiomyopathy, severe intellectual disability, diabetes and precocious puberty with breast enlargement and pubic hair. She also had clitoromegaly. She had mild triangular facies, low set ears, marked acanthosis nigricans, broad hands and feet, large tongue, adenoids hypertrophy, hypo pigmented patches on trunk, deep seated nails, hypertrophic sacral tail, hepatomegaly, steatohepatitis and nephrolithiasis.
Case 9.
A 3-month-old girl from Oman (sister of Case 8) with hepatosplenomegaly, cardiomyopathy, steatohepatitis and large tongue. She had very high level of serum triglycerides. A liver biopsy revealed steatotic changes consistent with non-alcoholic fatty liver disease (Fig. 3).
Fig. 3.
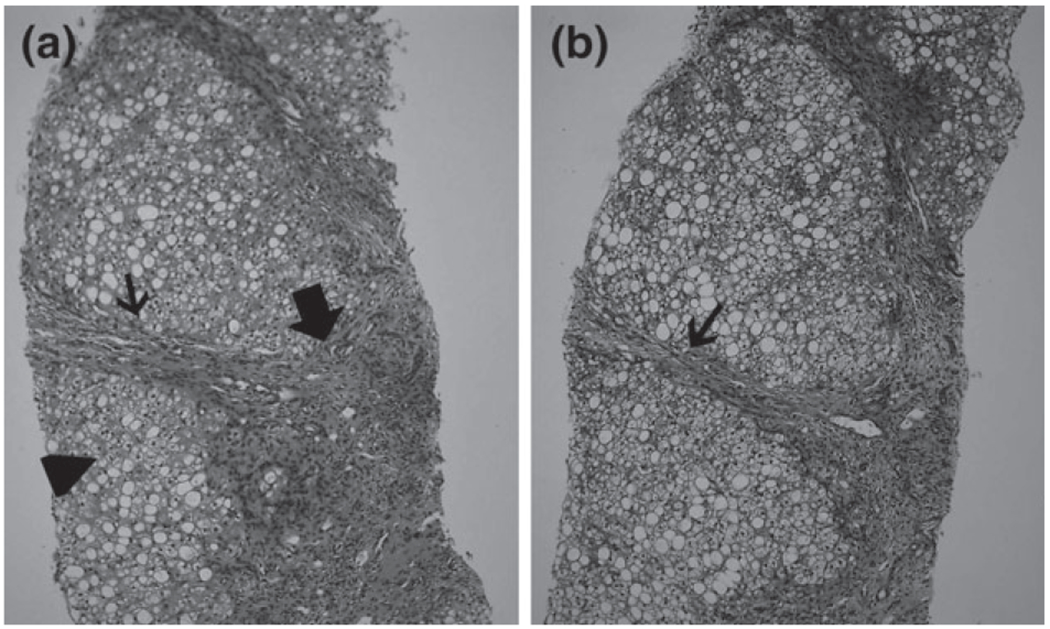
Histopathology of liver biopsy of Case 9. (a) Portal space fibrosis and mononuclear infiltrate (large arrow), fibrous expansion of portal space with portal to portal bridging (small arrow) and macro vesicular steatosis of hepatic parenchyma (arrow head) (hematoxylin and eosin). (b) Portal fibrosis and bridging (arrow) as shown here in the trichrome staining system.
Case 10.
A 32-year-old Sinhalese male, from Sri Lanka, presented with triangular facies, mild acromegaloid changes and diabetes, acanthosis nigricans and hepatomegaly was also noted to have generalized loss of subcutaneous fat upon his physical examination.
Genetic analysis
Sequencing identified disease-causing variants in AGPAT2 (in five patients) and in BSCL2 (in five patients), including some novel variants. The results of molecular analysis are presented in Table 1.
The sequencing analysis identified three novel variants; c.134C>A (p.Ser45*), c.216C>G (p.Tyr72*) in AGPAT2 and c.458C>A (p.Ser153*) in BSCL2. In addition, six previously reported mutations were identified; c.589-2A>G (p.Gln196fs*228) (8, 21–23), c.202C>T (p.Arg68*) (8, 21, 24) in AGPAT2 and c.412C>T (p.Arg138*) (12, 25–27), c.317_321del (p.Tyr106Cysfs*6) (12, 21, 28), c.672-2A>C (29, 30) and c.636del (p.Tyr213Thrfs*20) (12, 26, 31) in BSCL2. The details of these results are described in Appendix S1.
Variant analysis of AGPAT2 and BSCL2 genes was performed in other members of the families. All parents were heterozygous for the relevant identified variants and unaffected siblings were either wild type or carried variant on one allele.
Discussion
We investigated genotype and phenotype relationship in CGL in 10 patients with variants in AGPAT2 (CGL1, 5 patients) and BSCL2 (CGL2, 5 patients) genes (Table 1). Muscular hypertrophy was detected in all patients of this study (both CGL1 and CGL2). Other findings included acanthosis nigricas, curly and frizzy hair, dry and thick skin, hypertrichosis, hyperhidrosis, and large prominent superficial veins. In our study, acanthosis nigricans was more prevalent in CGL2 (five patients, 100%) than CGL1 (in two of five patients). Interestingly, one of the CGL1 patients (Case 2, Table 1) who presented acanthosis nigricans had normal levels of insulin. Hypertrichosis was only seen only in three of five CGL2 patients in our study, while none of the CGL1 patients showed hypertrichosis. All CGL2 patients had large ears, whereas only two CGL1 patients had this feature. High-pitched voice is a progeroid feature that was seen in two CGL2 patients, while none of the CGL1 patients presented this feature.
Hernia was observed in both CGL1 and CGL2 patients in our study. Sixty percent of CGL1 patients had hernia, which was inguinal, while the occurrence of hernia in CGL2 group was slightly higher (four of five patients). Four patients had umbilical and only one patient had inguinal hernia in CGL2 patients (Case 2, Table 1), whereas none of the CGL1 patients had umbilical hernia.
Studies have suggested a link between insulin resistance and uric acid nephrolithiasis, through a defect in ammonia genesis in the proximal tubular cell and reduced ammonium excretion in the tubular lumen (32). This study, for the first time, reports nephrolithiasis in CGL patients (Cases 1 and 3, Table 1). Two CGL1 patients had nephrolithiasis in presence of elevated insulin levels. The only CGL2 patient who presented nephrolithiasis had normal level of insulin (Case 8, Table 1).
Genital enlargement has been previously reported in CGL patients (3, 4). In our study, changes in external genitalia were prevalent in both CGL2 (three patients) than CGL1 (two patients). Three CGL2 patients had penis enlargement, retractile testes and clitoromegaly. These features were found in two CGL1 patients; phallic hypertrophy and clitromegaly. One CGL patient (Case 8, Table 1), homozygous for a BSCL2 variant, had premature puberty with breast enlargement and pubic hair. To the best of our knowledge, breast enlargement was not previously reported in CGL.
Little is known about the cardiovascular manifestations of generalized lipodystrophy. Hypertrophic cardiomyopathy is a known manifestation of the CGL, which is present in 20–25% of patients (33). The previously reported mean age for diagnosis of cardiomyopathy in CGL1 and CGL2 were 23 and 17 years, respectively (34). Early cardiomyopathy has been rarely reported in infancy. At least three studies have reported CGL2 patients with early onset cardiomyopathy (27, 35, 36). There are a few reports of early infantile onset of cardiomyopathy in CGL1 patients, including hypertrophic cardiomyopathy in a 1-year-old patient (37) and obstructive cardiomyopathy in a 2 months old Pakistani boy (38). The mechanism of cardiomyopathy in CGL patients is not fully understood, but trophic effect of insulin, in presence of insulin resistance and hyperinsulinemia, may play a role. Therefore, CGL must be kept in mind in infantile cardiomyopathy with liver disease.
Our study showed that cardiomyopathy was more frequent in CGL2 (2 patients), in comparison with CGL1 patients (1 patient) which was in accordance with previous studies. Hypertension was only noted in CGL2 patients (2 patients), while none of the CGL1 patients (with AGPAT2 variants) showed this feature. Faria et al. studied blood pressure of 18 CGL patients and 19 control individuals, and concluded that CGL patients have higher pressure compared with normal controls (39). Roth et al. reported hypertension in a 14-year-old Yemeni girl with CGL2 (40). She also suffered from diabetes mellitus, hypertrichosis, dysmorphic features, developmental delay and hepatosplenomegaly.
Studies reported that approximately 80% of CGL2 patients have mild to moderate intellectual impairment, whereas only 10% of CGL1 cases have intellectual disability (9). In this study, all CGL1 patients had normal intelligence quotient (IQ) but three CGL2 patients (60%) had some degree of intellectual disability.
Only one of our CGL1 patients (Case 3, Table 1) suffered from seizures. However, seizures have been previously reported in a CGL patient, but no genetic study was performed to confirm the CGL type (41). To the best of our knowledge, this is the first report of seizures in CGL1 patients (Case3, Table 1). Seizure disorders have been reported in the CGL2 patients (42). In this study, although one CGL2 patient had abnormal EEG pattern, no patient had a history of seizures.
On radiologic examination, bone cysts were detected in one of our CGL1 patients (Case 4 Table 1, Fig. 2), but none of the CGL2 patients had bone cyst. Radiological evidence of bone cysts has been previously reported in patients with AGPAT2 variants (23). Fleckenstein (43) reported skeletal abnormalities in three female African-American CGL patients. One of the cases was radiologically normal in first 5 years of life, but showed pathologic fractures after a fall at the age of 13 years. Her sister with CGL showed no bone lesion on the examination at the age of 8 years, but at 18 years of age, multiple focal bony lytic lesions were detected. The other female patient had no previously detected radiologic sign, but her radiologic studies, at the age of 21 years, showed bone lesions. All these patients were subsequently reported to harbor AGPAT2 mutations (9).
Accumulation of triglyceride and glycogen in hepatocytes may cause hepatosplenomegaly, accompanied by abnormal levels of liver enzymes that may lead to cirrhosis (4, 5). All of our CGL2 patients had hepatomegaly, and splenomegaly was detected in four patients. In CGL1 patients, hepatosplenomegaly was present in three cases.
Impaired carbohydrate metabolism is characterized by primary peripheral insulin resistance associated with hyperinsulinemia, which, after puberty, results in secondary diabetes mellitus. In patients with CGL, overt clinical diabetes mellitus usually develops during the second decade of life (4, 9,10). Four (80%) of our CGL2 patients showed hyperinsulinemia, and one patient was not evaluated for serum insulin level. The incidence of hyperinsulinemia in CGL1 group was lower (one of four evaluated patients). In regard of overt clinical diabetes mellitus, only two of the hyperinsulinemic CGL2 patients showed diabetes, while none of the CGL1 patients had this condition.
In summary, CGL is a rare syndrome that illustrates the importance of adipose tissue for the majority of metabolic processes. The phenotypic characteristics of this disease are well identified and in most cases lead to the clinical diagnosis. BSCL2 variants usually lead to more severe symptoms in comparison to AGPAT2 variants.
Understanding the molecular defects leading to various phenotypes of the disease is useful in the genetic counseling and prenatal diagnosis of affected families and help in improving specific therapeutic interventions. We suggest that CGL should be considered as a differential diagnosis in children with bony lytic lesions or pathological fracture, cardiomyopathy, hypertension, seizure, hepatic steatosis, nephropathy, external genitalia changes such as penis enlargement and clitoromegaly, especially in those who show lipodystrophic changes.
Supplementary Material
Acknowledgements
This study was undertaken as part of GENE-ME (a research initiative for investigation of inherited diseases from the Middle-East).
Footnotes
Conflict of interest
The authors declare that they have no competing interests.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- 1.Berardinelli W. An undiagnosed endocrinometabolic syndrome: report of 2 cases. J Clin Endocrinol Metab 1954: 14: 193–204. [DOI] [PubMed] [Google Scholar]
- 2.Seip M. Lipodystrophy and gigantism with associated endocrine manifestations. A new diencephalic syndrome? Acta Paediatr 1959: 48: 555–574. [PubMed] [Google Scholar]
- 3.Seip M, Trygstad O. Generalized lipodystrophy, congenital and acquired (lipoatrophy). Acta Paediatr Suppl 1996: 413: 2–28. [DOI] [PubMed] [Google Scholar]
- 4.Garg A. Acquired and inherited lipodystrophies. N Engl J Med 2004: 350: 1220–1234. [DOI] [PubMed] [Google Scholar]
- 5.Garg A. Lipodystrophies. Am J Med 2000: 108: 143–152. [DOI] [PubMed] [Google Scholar]
- 6.van Maldergem L. Berardinelli-Seip congenital lipodystrophy. Orphanet encyclopedia, Paris, France. 2001, https://www.orpha.net/data/patho/GB/uk-berard.pdf. [Google Scholar]
- 7.Agarwal AK, Garg A. Genetic disorders of adipose tissue development, differentiation, and death. Annu Rev Genomics Hum Genet 2006: 7: 175–199. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal AK, Arioglu E, De Almeida S et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat Genet 2002: 31: 21–23. [DOI] [PubMed] [Google Scholar]
- 9.Van Maldergem L, Magre J, Khallouf TE et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet 2002: 39: 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simha V, Garg A. Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy caused by mutations in the AGPAT2 or seipin genes. J Clin Endocrinol Metab 2003: 88: 5433–5437. [DOI] [PubMed] [Google Scholar]
- 11.Agarwal AK, Garg A. Congenital generalized lipodystrophy: significance of triglyceride biosynthetic pathways. Trends Endocrinol Metab 2003: 14: 214–221. [DOI] [PubMed] [Google Scholar]
- 12.Magre J, Delepine M, Khallouf E et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet 2001: 28: 365–370. [DOI] [PubMed] [Google Scholar]
- 13.Agarwal AK, Simha V, Oral EA et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab 2003: 88: 4840–4847. [DOI] [PubMed] [Google Scholar]
- 14.Payne VA, Grimsey N, Tuthill A et al. The human lipodystrophy gene BSCL2/seipin may be essential for normal adipocyte differentiation. Diabetes 2008: 57: 2055–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szymanski KM, Binns D, Bartz R et al. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci USA 2007: 104: 20890–20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim CA, Delepine M, Boutet E et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab 2008: 93: 1129–1134. [DOI] [PubMed] [Google Scholar]
- 17.Shastry S, Delgado MR, Dirik E et al. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet A 2010: 152A: 2245–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hayashi YK, Matsuda C, Ogawa M et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest 2009: 119: 2623–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajab A, Straub V, McCann LJ et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet 2010: 6: e1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haghighi A, Razzaghy-Azar M, Talea A et al. Identification of a novel nonsense mutation and a missense substitution in the AGPAT2 gene causing congenital generalized lipodystrophy type 1. Eur J Med Genet 2012: 55: 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boutet E, El Mourabit H, Prot M et al. Seipin deficiency alters fatty acid Delta9 desaturation and lipid droplet formation in Berardinelli-Seip congenital lipodystrophy. Biochimie 2009: 91: 796–803. [DOI] [PubMed] [Google Scholar]
- 22.MacArthur DG, Balasubramanian S, Frankish A et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012: 335: 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu M, Kazlauskaite R, Baracho Mde F et al. Mutations in Gng3lg and AGPAT2 in Berardinelli-Seip congenital lipodystrophy and Brunzell syndrome: phenotype variability suggests important modifier effects. J Clin Endocrinol Metab 2004: 89: 2916–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Magre J, Delepine M, Van Maldergem L et al. Prevalence of mutations in AGPAT2 among human lipodystrophies. Diabetes 2003: 52: 1573–1578. [DOI] [PubMed] [Google Scholar]
- 25.Sim MF, Talukder MM, Dennis RJ et al. Analysis of naturally occurring mutations in the human lipodystrophy protein seipin reveals multiple potential pathogenic mechanisms. Diabetologia 2013: 56: 2498–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rahman OU, Khawar N, Khan MA et al. Deletion mutation in BSCL2 gene underlies congenital generalized lipodystrophy in a Pakistani family. Diagn Pathol 2013: 8: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miranda DM, Wajchenberg BL, Calsolari MR et al. Novel mutations of the BSCL2 and AGPAT2 genes in 10 families with Berardinelli-Seip congenital generalized lipodystrophy syndrome. Clin Endocrinol 2009: 71: 512–517. [DOI] [PubMed] [Google Scholar]
- 28.Guillen-Navarro E, Sanchez-Iglesias S, Domingo-Jimenez R et al. A new seipin-associated neurodegenerative syndrome. J Med Genet 2013: 50: 401–409. [DOI] [PubMed] [Google Scholar]
- 29.van Maldergem L Berardinelli–Seip congenital lipodystrophy In: Pagon RA, Bird TD, Dolan CR, et al. eds. GeneReviews, Vol. 2015 Seattle, WA: University of Washington, 2003. [PubMed] [Google Scholar]
- 30.Heathcote K, Rajab A, Magre J et al. Molecular analysis of Berardinelli-Seip congenital lipodystrophy in Oman: evidence for multiple loci. Diabetes 2002: 51: 1291–1293. [DOI] [PubMed] [Google Scholar]
- 31.Mandal K, Aneja S, Seth A et al. Berardinelli-Seip congenital lipodystrophy. Indian Pediatr 2006: 43: 440–445. [PubMed] [Google Scholar]
- 32.Abate N, Chandalia M, Cabo-Chan AV Jr et al. The metabolic syndrome and uric acid nephrolithiasis: novel features of renal manifestation of insulin resistance. Kidney Int 2004: 65: 386–392. [DOI] [PubMed] [Google Scholar]
- 33.Van Maldergem L Berardinelli-Seip congenital lipodystrophy In: Pagon RA, Adam MP, Ardinger HH, et al. , eds. GeneReviews®, Vol. 1993–2015 Seattle, WA: University of Washington, 2003. [PubMed] [Google Scholar]
- 34.Lupsa BC, Sachdev V, Lungu AO et al. Cardiomyopathy in congenital and acquired generalized lipodystrophy: a clinical assessment. Medicine 2010: 89: 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhayana S, Siu VM, Joubert GI et al. Cardiomyopathy in congenital complete lipodystrophy. Clin Genet 2002: 61: 283–287. [DOI] [PubMed] [Google Scholar]
- 36.Friguls B, Coroleu W, del Alcazar R et al. Severe cardiac phenotype of Berardinelli-Seip congenital lipodystrophy in an infant with homozygous E189X BSCL2 mutation. Eur J Med Genet 2009: 52: 14–16. [DOI] [PubMed] [Google Scholar]
- 37.Ben Turkia H, Tebib N, Azzouz H et al. Congenital generalized lipodystrophy: a case report with neurological involvement. Acta Paediatr 2009: 16: 27–31. [DOI] [PubMed] [Google Scholar]
- 38.Debray FG, Baguette C, Colinet S et al. Early infantile cardiomyopathy and liver disease: a multisystemic disorder caused by congenital lipodystrophy. Mol Genet Metab 2013: 109: 227–229. [DOI] [PubMed] [Google Scholar]
- 39.Faria CA, Moraes RS, Sobral-Filho DC et al. Autonomic modulation in patients with congenital generalized lipodystrophy (Berardinelli-Seip syndrome). Europace 2009: 11: 763–769. [DOI] [PubMed] [Google Scholar]
- 40.Roth T, Nair S, Kumar A. Monogenic diabetes secondary to congenital lipodystrophy in a 14-year-old Yemeni girl. J Clin Res Pediatr Endocrinol 2010: 2: 176–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Babu P, Sharma R, Jayaseelan E et al. Berardinelli-Seip syndrome in a 6-year-old boy. Indian J Dermatol Venereol Leprol 2008: 74: 644–646. [DOI] [PubMed] [Google Scholar]
- 42.Rajab A, Heathcote K, Joshi S et al. Heterogeneity for congenital generalized lipodystrophy in seventeen patients from Oman. Am J Med Genet 2002: 110: 219–225. [DOI] [PubMed] [Google Scholar]
- 43.Fleckenstein JL, Garg A, Bonte FJ et al. The skeleton in congenital, generalized lipodystrophy: evaluation using whole-body radiographic surveys, magnetic resonance imaging and technetium-99m bone scintigraphy. Skelet Radiol 1992: 21: 381–386. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.