

Summary
The generation of functional genomics datasets is surging, as they provide insight into gene regulation and organismal phenotypes (e.g., genes upregulated in cancer). The intent behind functional genomics experiments is not necessarily to study genetic variants, yet they pose privacy concerns due to their use of next-generation sequencing. Moreover, there is a great incentive to broadly share raw reads for better statistical power and general research reproducibility. Thus, we need new modes of sharing beyond traditional controlled-access models. Here, we develop a data-sanitization procedure allowing raw functional genomics reads to be shared while minimizing privacy leakage, enabling principled privacy-utility trade-offs. Our protocol works with traditional Illumina-based assays and newer technologies such as 10x single-cell RNA-sequencing. It involves quantifying the privacy leakage in reads by statistically linking study participants to known individuals. We carried out these linkages using data from highly accurate reference genomes and more realistic environmental samples.
Keywords: genome privacy, functional genomics, linkage attacks, surreptitious DNA sequencing, data sanitization
Graphical Abstract

In Brief
Growing functional genomics data puts individual privacy at risk via linkage attacks, the risk of which is quantified and can be sanitized using a privacy-preserving data format.
1. Introduction
Advances in sequencing technologies and laboratory techniques have enabled researchers to probe epigenetic and transcriptomic states of the cell comprehensively, such as gene expression levels or DNA-binding protein levels, the majority of which are clinically actionable [e.g., The Cancer Genome Atlas (TCGA)]. With the availability of more advanced techniques, such as single-cell RNA sequencing (scRNA-Seq) and single-cell assay for transposase-accessible chromatin (scATAC-Seq) using sequencing, we can now functionally annotate tissues even at the single-cell level. This increased resolution will soon bring a surge of new functional genomics datasets from large cohorts of individuals, surpassing the available sequenced genomes. For example, let us consider a human biosample being assayed for all aspects of its biology. We can sequence the DNA of the sample once, but numerous functional genomics assays can be performed on the same sample. Moreover, as more data are collected from a larger number of study individuals, we can boost statistical power for future discoveries–that is, if the collected data are made available to the wider research community.
Privacy is a barrier to the open sharing of functional genomics datasets, as individuals’ genetic variants can be inferred from the data. Studies on genomic privacy have traditionally focused on DNA, partly due to the dominance of DNA sequencing in genomics research (Erlich and Narayanan, 2014, Homer et al., 2008, Erlich et al., 2018, Kim et al., 2018, Im et al., 2012, Gymrek et al., 2013). However, as the balance in data acquisition shifts toward large-scale functional genomics data, privacy studies must shift as well (Joly et al., 2016). From a privacy standpoint, functional genomics data have a two-sided nature, in contrast to DNA sequencing data. Although these experiments are usually performed to understand the biology of cells, rather than to reveal identifying genetic variants, raw reads are nonetheless tagged by the genetic variants of the individuals due to the experimental design. Moreover, in addition to including genetic variant information that can be inferred from raw reads, functional genomics data provide the conditions under which the assay was performed (e.g., linking a specific phenotype to the sample). Furthermore, lower barriers to sequencing data enable a wider range of people, including “citizen scientists”, to access genomic data that can be overlapped with functional genomics data to infer sensitive information about study participants.
These privacy concerns should be addressed while considering the unique aspects of functional genomics data in order to maximize open sharing. To answer critical biological questions, researchers must generate key properties from functional genomics data, such as gene expression quantifications or transcription factor (TF) binding enrichments. These properties must be generated from the raw reads; however, such calculations do not require genetic variants in the reads. Sharing the raw reads, as opposed to derived quantities, is essential because this sharing provides researchers with the necessary level of control over their analyses and helps advance biomedical science by permitting a rapid assessment of tools and methods, thus enhancing reproducibility. Currently, large consortia such as TCGA (The Cancer Genome Atlas Research Network et al., 2013), the Genotype-Tissue Expression (GTEx) project (The GTEx Consortium, 2013), and PsychENCODE (PsychENCODE Consortium, 2018) store raw functional genomics reads behind controlled access following traditional DNA-sharing protocols, while providing open access to summary-level data such as gene expression levels. These large consortia and other researchers are aware that a number of genotypes can be inferred from summary-level data (Schadt et al., 2012, Schadt, 2012, Harmanci and Gerstein, 2016). Yet, this approach makes sense as the scale of the genotype leakage from functional genomics reads is much larger, far beyond an acceptable level. Meanwhile, controlled-access sharing protocols delay data access for average researchers by creating bureaucratic bottlenecks and technical challenges. Furthermore, these protocols were developed with a focus on DNA-sequencing data, which are obtained for the purpose of identifying the genetic variants of an individual. Thus, a new model for sharing raw functional genomics reads is needed, one that better fits the privacy and utility needs of functional genomics data.
In this study, we developed read-sanitization techniques that enable public sharing of minimally manipulated functional genomics reads, while protecting sensitive information and minimizing the amount of private data requiring special access and storage. Our methods are based on an optimized balance between privacy requirements and the inherent utility of functional genomics reads. Our aim is to provide reads to the community that can be easily used as inputs to any functional genomics data processing pipeline.
To develop data-sanitization techniques to reduce and even eliminate privacy risk, a comprehensive assessment of private information leakage from different types of functional genomics reads under varying noise conditions and sequencing coverage levels is necessary. Accordingly, we performed common privacy breaches using publicly available functional genomics and genome sequencing datasets. To quantify the privacy loss in a more realistic setting, we conducted DNA-sequencing and RNA-sequencing (RNA-Seq) assays on blood tissue and environmental samples, such as used coffee cups (to mimic surreptitious DNA testing) collected from consented individuals. We demonstrated that our statistical measures can link individuals to functional genomics datasets under different noise levels and sequencing coverage levels, hence revealing sensitive phenotype information about the individuals. We then showed that our data-sanitization protocol minimizes this privacy risk, while providing accurate estimations of key summary quantities, such as gene expression levels. Finally, we demonstrated that easy access to a large amount of data that are otherwise locked behind controlled access can be achieved with a principled balance between privacy and utility.
2. Results
2.1. Privacy Breach Scenarios for Quantification of Privacy Loss
A “linkage attack” is a common breach of privacy in which one can quantify the private information leakage in an anonymized database by using publicly available information about the individuals in the database (Narayanan and Shmatikov, 2008, Erlich and Narayanan, 2014). A famous example of a linkage attack is the de-anonymization of Netflix Prize challenge data using publicly available IMDB user comments (Narayanan and Shmatikov, 2008). Within genomics, researchers have shown that genetic information from publicly available genealogy websites can be overlapped with short tandem repeats on the Y chromosomes of anonymized genomes to infer the surnames of participants in genomics databases (Gymrek et al., 2013); likewise, expression quantitative trait locus (eQTL) data can be applied to de-anonymize gene expression databases (Schadt et al., 2012, Schadt, 2012, Harmanci and Gerstein, 2016). Linkage techniques have been used outside of the privacy context as well, such as to resolve sample swap problems during omics data production (Yoo et al., 2014, Lee et al., 2017, Westphal et al., 2019, Lee et al., 2019).
Here, we define three types of linkage attacks (Figure S1), which differ by the nature of the data in and , i.e., whether the data is perfect (P) or noisy (N):
(I) Case P-P involves obtaining publicly available perfect information about a known individual (e.g., date of birth, zip code, gender) and overlapping it with perfect information in anonymized database (e.g., zip code, gender) to reveal sensitive information (e.g., political affiliation). This can be done by cross-referencing two datasets, and often does not require any statistical heuristics. A famous example in medicine is the use of cross-referencing birthdate and zip code information present in medical records and voter list databases to reveal the addresses of patients (Sweeney, 2000).
(II) Case P-N involves obtaining publicly available perfect information about a known individual (e.g., whole-genome sequencing reads) and overlapping it with noisy information from anonymized database (e.g., ChIP-Seq reads) to reveal sensitive information (e.g., psychiatric disease status). Because it involves noisy data, simple overlaps often do not work and statistical analysis is required.
(III) Case N-N involves obtaining publicly available noisy information about a known individual (e.g., DNA gathered from a coffee cup) and overlapping it with noisy information from anonymized database (e.g., ChIP-Seq reads) to reveal sensitive information (e.g., psychiatric disease status). This can require more involved statistical techniques to sort through the signal in two noisy datasets.
In order to quantify the private information leakage in functional genomics datasets, we adopted Narayanan and Shmatikov’s statistical techniques of privacy breaches (Narayanan and Shmatikov, 2008). The privacy threat models are as follows. One can imagine a scenario in which an adversary gains illicit access to a known individual’s WGS data and performs linkage attacks on functional genomics data in order to infer private phenotypes associated with them. Note that, in this scenario, the private information that will be leaked are the sensitive phenotypes (e.g., HIV status, bipolar disorder status, etc.), not the genotypes. The genotypes in the functional genomics reads are merely used as a means to infer the phenotype. This is somewhat different from traditional privacy attacks, in which the leaked information is the genotype data. The genotypes obtained from functional genomics data are noisy; thus functional genomics reads constitute a noisy database , and genomes can be thought of as perfect information about the individual. Therefore, we mimicked this scenario with a Case P-N attack. One can also imagine a more realistic, “real-world” scenario: an adversary takes advantage of the DNA trail individuals leave behind in everyday life, such as saliva on a stamp (Flynn, 2018), a used facial tissue, or a cup of coffee. The DNA information extracted from environmental samples (e.g., coffee cups) is noisy due to possible contamination by multiple individuals other than the owner. Therefore, we mimicked this scenario with a Case N-N attack.
2.2. Sensitive phenotypes can reliably be inferred by linking perfect genotypes to noisy genotypes called from functional genomics data
Let us assume a study that aims to understand the changes in gene expression of individuals with bipolar disorder (BPD). This study assays the gene expression of a cohort of individuals with BPD-positive (BPD+) and -negative (BPD−) phenotypes and publicly releases the RNA-Seq reads (database ). By lawful or unlawful means, an adversary obtains access to the WGS or genotyping array data (information ) of a known individual (henceforth referred to as the query individual) and aims to predict whether this individual’s phenotype is BPD+ or BPD−.
The adversary uses traditional variant callers (e.g., GATK (DePristo et al., 2011, der Auwera et al., 2013)) to obtain genotypes from the RNA-Seq reads. They create a database of genotypes from all the individuals in the study. The genotypes obtained from information of the query individual are denoted as . contains partial (i.e., missing some alleles) and noisy (i.e., containing some misidentified alleles) genotypes, as genotyping from RNA-Seq reads may contain errors and cannot cover the entire genome. By the design of the functional genomics study, the genotypes of each individual in are linked to the phenotype of these individuals (i.e., BPD+ and BPD−). The adversary then statistically matches the genotypes of the query individual, , to the genotypes of the individuals, , as follows (see Figure 1a and STAR Methods): The adversary calculates a “linking score” as the sum of the log of the inverse of the genotyping frequency of each genotype at the intersection of and for each entry in . The adversary then ranks the linking scores and calculates a gap value for the top-ranked entry in by determining the ratio between the highest and second-highest linking scores, and then determines a p-value for the significance of it (see STAR Methods for calculation of the statistical significance of gap). Finally, the adversary denotes the best match as the list of genotypes of the query individual and this reveals the BPD status of the known query individual to the attacker.
Figure 1:
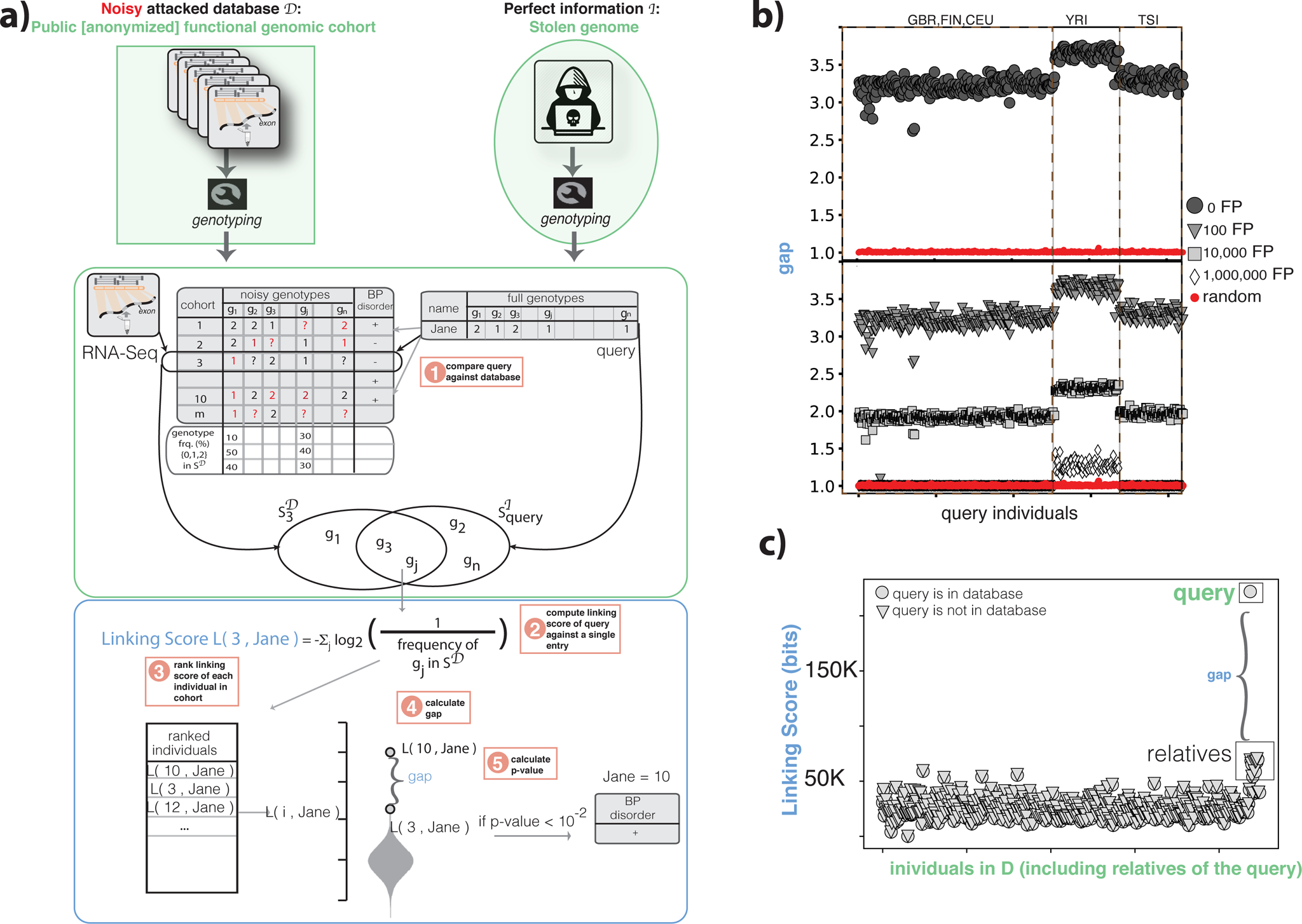
Functional genomics data de-anonymization scheme with perfect genomes. (a) Anonymized functional genomics data from a cohort of individuals can be seen as a database to be attacked, which contains functional genomics reads and phenotypes for every individual in the cohort. The perfect information about an individual can be the genome of an individual. After obtaining genotypes from the functional genomics reads, the attacker scores each individual in the cohort based on the overlapping genotypes between the known individual’s genome and the noisy genotypes called from functional genomics. These scores are then ranked and the top-ranked individual in the cohort is selected as the known individual. See also Figure S1. (b) gap values for the 1,000 Genomes individuals in the gEUVADIS RNA-Seq cohort. Red circles are the gap values obtained by linking a random set of genotypes to the RNA-seq panel. gap values are also shown after adding false-positive genotypes to the genotype set of each individual in the database. (c) The linking scores for each individual in the functional genomics cohort after the addition of genetically related individuals to the query, with and without the query individual present in the database.
In this study, we used RNA-Seq data of 421 individuals from the gEUVADIS project (Lappalainen et al., 2013) as the functional genomics database , and high-coverage WGS of the same individuals from the 1,000 Genomes Project (The 1000 Genomes Project Consortium, 2010) as the information (Figure 1a). After creating the genotype panel and query genotypes , we successfully linked all 421 query individuals to database and revealed sensitive phenotypes (p-value < 10−2).
To further increase the noise levels, we added an increasing number of randomly picked false-positive genotypes to each genotype call set and kept the number of true positive variants constant. We linked 418 out of 421 individuals to the cohort even after adding 100,000 false-positive variants to each entry (Figure 1b). The gap values become non-significant only after adding one million false-positive variants (Figure 1b). The queries from individuals with African ancestry (denoted as YRI in Figure 1b), despite having the same coverage as the rest of the RNA-Seq data (from European ancestries, i.e., GBR, FIN, CEU, and TSI), were more vulnerable to this linkage attack (Figure 1b). This result is likely due to a higher number of alternative alleles in the African genomes compared to the reference genome.
Effect of genetically related individuals:
We next assessed the effect of including related individuals in the attacked database. We added RNA-Seq data from 14 individuals related to NA12878 (Li et al., 2014) to the attacked database, and found that querying NA12878 did not return any of the relatives, whether NA12878 was present in the database or not (see STAR Methods and Figure 1c).
Effect of sequencing depth:
The number of reads in a typical functional genomics dataset can vary depending on various factors such as the study design, number of assayed samples, assay type, or tissue type. These variations can greatly affect the number and quality of the genotypes that can be called. To understand the effect of the sequencing coverage, we subsampled reads from all individuals in the gEUVADIS cohort and created new attacked databases for each level of subsampling (Figure 2a, see STAR Methods). As shown in Figure 2b, we found that the quality of genotypes decreased with a decreasing number of reads. However, linking accuracy was not affected by the sequencing depth even after the addition of 10,000 false-positive genotypes (Figure 2c).
Figure 2:
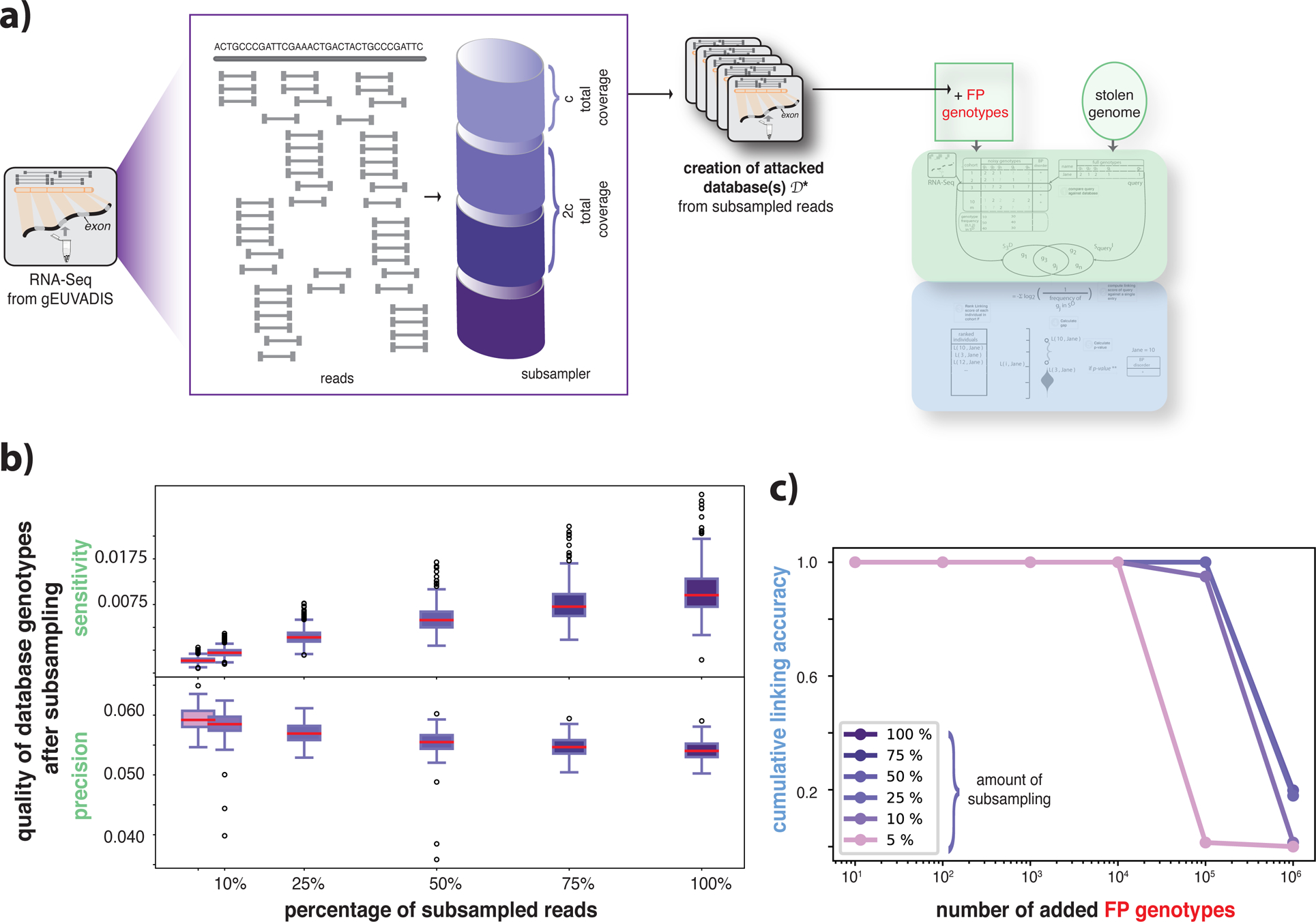
Impact of sequencing depth on genotyping and linking accuracy. (a) The process of pooling reads from functional genomics data to generate different attacked databases. c amount of reads were pooled and a new cohort was generated. A linkage attack was performed for each new database using genomes as perfect information. (b) Variant calling was performed for pooled reads for each individual in the database. The panel shows the distribution of precision and sensitivity of the called genotypes over all individuals at different subsampling percentages using their genomes as the ground truth. (c) Cumulative linking accuracy before and after adding the false-positive genotypes. Linking accuracy was calculated as the ratio between the total number of correctly linked individuals.
2.3. Sensitive phenotypes can be reliably inferred by linking surreptitiously gathered, noisy genotypes to noisy genotypes called from functional genomics data
Let us assume that the adversary is not able to access existing WGS or genotyping array datasets, and instead obtains environmental DNA samples (e.g., used coffee cups). The adversary aims to identify the disease status of the coffee cups’ owners by linking their DNA to a functional genomics cohort. To mimic this scenario, we performed RNA-Seq experiments on blood tissues from two consented individuals A and B. We then combined these raw RNA-Seq reads with the gEUVADIS RNA-Seq cohort to create the noisy attacked database. We then collected six used coffee cups from the same individuals. After extracting and amplifying the DNA from the surface of coffee-cup lids using known forensic techniques with commercially available kits, we performed WGS at 10x sequencing coverage (see STAR Methods, Tables S1&S2). Each of these WGS data was used as the noisy information . We successfully linked all 12 coffee-cup samples to the correct individuals in the database with an average gap of 1.82 and 2.70 for individuals A and B, respectively, with p-values < 10−2 (Figure 3b).
Figure 3:
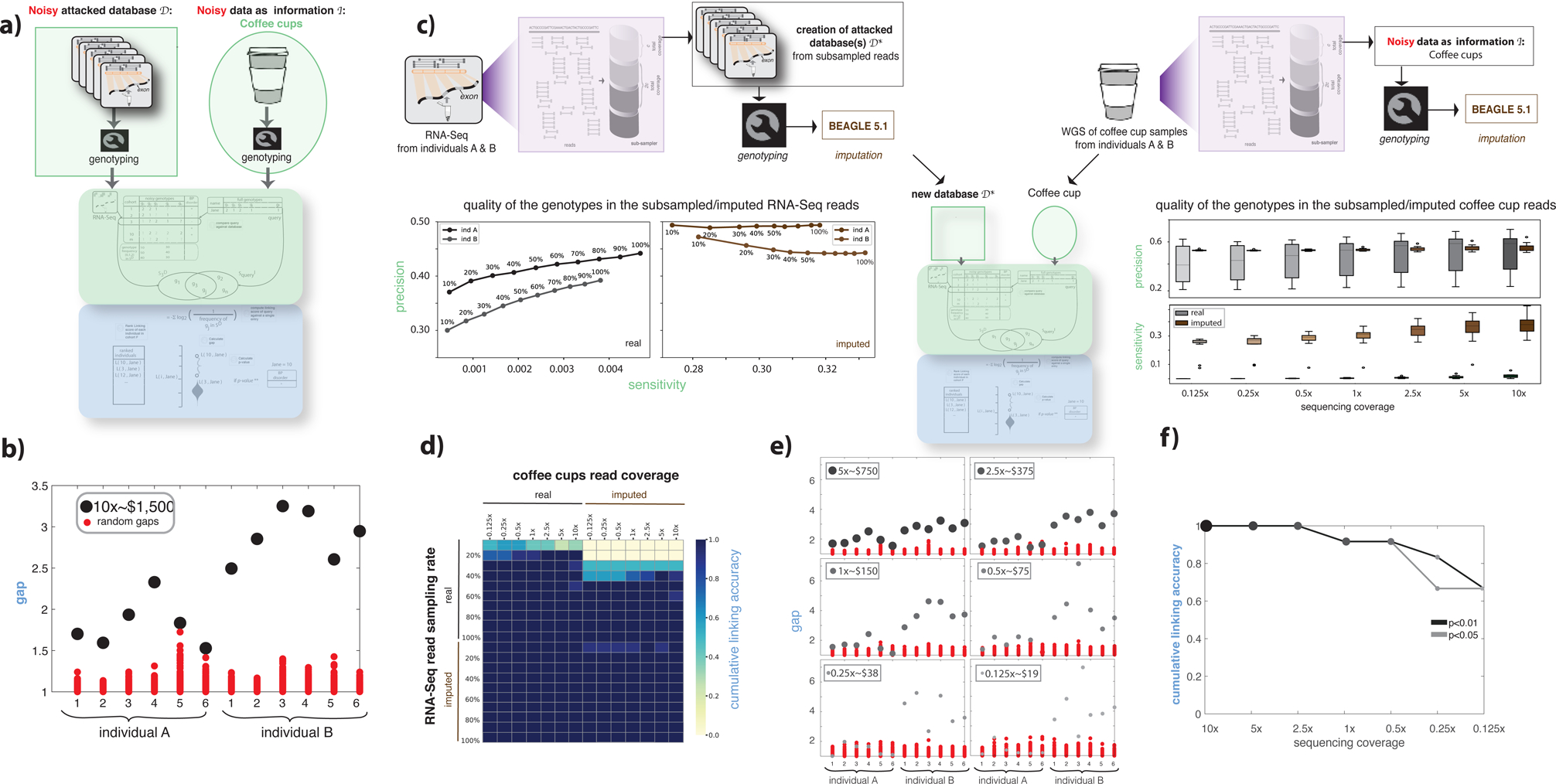
Functional genomics data de-anonymization scheme with noisy genomes. (a) Anonymized functional genomics data from a cohort of individuals can be seen as a database to be attacked. The noisy information can be assumed to be DNA surreptitiously gathered from a known individual’s used coffee cup. The procedure described in Figure 1a was repeated. See also Figure S1. (b) gap values for two individuals and six coffee cups, each at 10x sequencing coverage. (c) The process of generating different databases and noisy information by (i) subsampling the reads from functional genomics data and the coffee cup sequences and (ii) imputing the genotypes obtained from functional genomics data and the coffee-cup sequences at different subsampling rates. The precision and sensitivity of the called genotypes are shown for each subsampling/imputation level. A total of 2,880 linkage attacks were performed using each subsampled database with and without imputation and using each subsampled coffee-cup reads with and without imputation. (d) Linking accuracy was calculated for each of the 2,880 linking attacks and depicted as a heatmap. (e) gap values for 2 individuals and 12 coffee cups, each at different sequencing coverage, compared with gap values obtained using random sets of genotypes. The RNA-Seq data used in this plot corresponds to a 20% sampling rate (second row in panel d). (f) Cumulative linking accuracy after subsampling the sequencing coverage calculated using the information in panel e.
Effect of imputation and sequencing depth:
Genotypes missing from RNA-Seq due to low coverage can be imputed using population genetics-based genotype imputation tools. We first subsampled reads from the RNA-Seq data of two study individuals and created cohort genotype databases for each level of subsampling in each RNA-Seq experiment similar to above. We then imputed genotypes for each subsampling using BEAGLE (see STAR Methods) (Browning et al., 2018). We found that imputation on the genotypes from RNA-Seq reads improved the quality of the genotypes and hence, the linking accuracy. Conversely, imputation decreased the accuracy of genotypes from coffee-cup reads and linking (see STAR Methods, Figure 3c,d).
Depth-dependent sequencing cost:
The cost of sequencing is directly related to the coverage. We investigated the total monetary cost for an adversary to sequence DNA from the coffee cups in order to perform a successful linking attack on a moderately sequenced RNA-Seq cohort. We found that the coffee cups could be linked with statistical significance to the correct RNA-Seq samples when we spent as little as $19 on sequencing (Figure 3e), with a linking accuracy of 60% (see STAR Methods, Figure 3f).
Different genotyping techniques:
An ideal and cheap alternative genotyping method for the adversary could be exome-based genotyping arrays, as RNA-Seq captures reads overwhelmingly from exons. Another genotyping method popular for its low cost and portability is the Oxford Nanopore. We were not able to obtain accurate genotypes from the coffee-cup reads using either of these technologies, and therefore were not able to link the owners of the coffee cups to their RNA-Seq data (see STAR Methods, Table S2).
2.4. Different functional genomics techniques with varying sequencing depth can be reliably linked to genome databases
We assessed whether linkage attacks yield similar results when data from different functional genomics techniques (e.g., ChIP-Seq, Hi-C, or ChIA-PET) are used as the noisy information and linked to a perfect genome database. We used cell line functional genomics data as information about individuals represented in the 1,000 Genomes genotype database (the attacked database). The ENCODE data portal contains data from a range of functional genomics assays (Table S4) on the GM12878 cell line (individual NA12878 in the 1,000 Genomes database). We performed linkage attacks at varying sequencing coverages using data from a number of these assays. We were able to link NA12878 to the database successfully, even at low coverages with many different types of functional genomics data (Figure 4a). We then calculated the linking score per bp and found that some of the TF ChIP-Seq experiments may leak more information per bp than even WGS does (Figure 4b). We also found differences in linking score and gap values for different RNA-Seq assays, which may be due to the differences in genotyping frequency distribution of the called genotypes (Figure 4c). We repeated this analysis for other individuals from different ancestries and found similar trends in gap and linking score (Figure 4d–f, S2).
Figure 4:
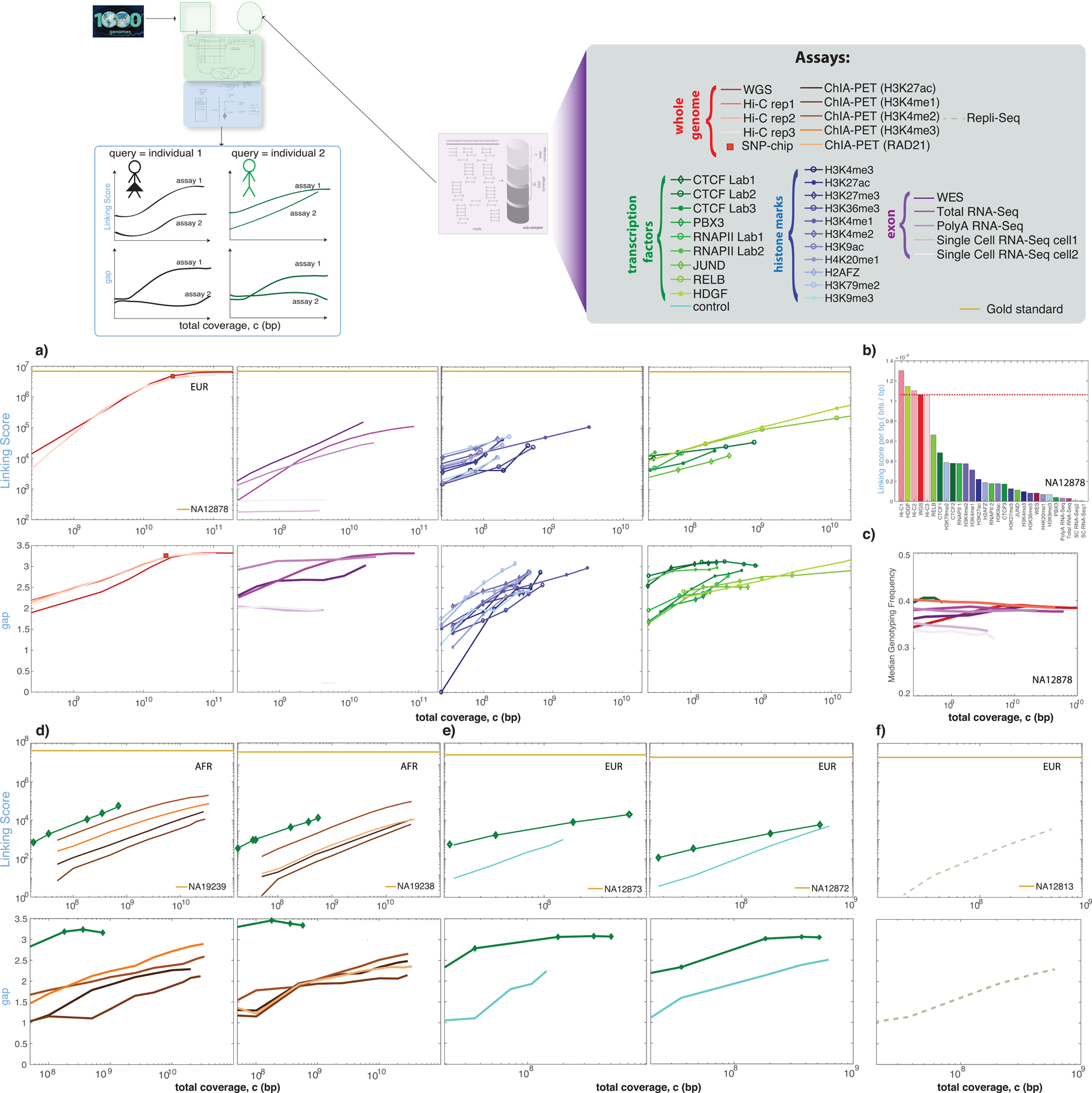
Effect of functional genomics assay type on de-anonymization. See also Figure S2 and S3. (a) The linking scores and gap as a function of sequencing coverage for individual NA12878 for different functional genomics assays. (b) Linking score per base pair for each assay was calculated by normalizing the linking score values per subsampling by the total number of nucleotides in the sample. (c) Mean genotyping frequency per sequencing coverage for different assays. (d) The linking score and gap values from two individuals for ChIA-PET and ChIP-Seq experiments, (e) ChIP-Seq experiments, and (f) repli-Seq experiments. See also Figure S2.
We also assessed whether the ancestral composition of the database affected linking. We created a database with the genotypes from an African (AFR) population (108 individuals) and added two European (EUR) individuals including NA12878. We were able to link NA12878 to this new database (p-value < 0.01). However, when we removed the query individual from the attacked database, we misidentified the remaining EUR individual as NA12878 (Figure S3).
2.5. Practical data sanitization techniques can reduce the amount of private information leakage while preserving the utility of functional genomics data
Overview:
Sharing read alignment files (SAM/BAM/CRAM) from functional genomics experiments is extremely important for developing analysis methods and discovering novel mechanisms operating on the human genome. Ideally, one would share the maximal amount of information with minimal utility loss while largely maintaining an individual’s privacy. To do so, one must balance the efficiency and effectiveness of the data anonymization process with the utility of the anonymized dataset. Thus, we propose a versatile data sanitization approach such that privacy and utility can be tuned (Figure 5a).
Figure 5:

Functional genomics data sanitization. (a) The schematic of the privacy-preserving transformation of an alignment file and the difference between the signal calculated from the original BAM and transformed pBAM files. (b) A schematic of how different reads and corresponding CIGAR strings are treated in pBAM files. See also Figure S4 and S7. (c) The change in error with an increasing number of manipulated reads for different operations Q. (d) The numerical bounds for the privacy-utility relationship for different ancestries using the average number of variants in 1,000 Genomes data.
A raw alignment file (BAM) can be thought of as a dataset that stores information for each read. Let us assume a BAM file is a dataset B, where each entry is a read. The desire is to release dataset B in the form of a privacy-preserving BAM (pBAM, say dataset B*) such that it does not leak variants from the reads, but for which any calculation f based on B and B* returns almost the same result. For example, f could be a function that calculates gene expression levels from an input BAM file. The aim is to ensure that the expression levels will be similar, whether BAM (B) or pBAM (B*) is used. Now let us perform the privacy-preserving transformation through a sanitizer function PQ,r such that PQ,r(B) = B*. Q is an operation such as “removal of variants” and r is a parameter representing the number of variants to be removed.
Read sanitization protocol:
We can construct a sanitized “pBAM” file from a BAM/SAM/CRAM file using generalization (a technique commonly used in data sanitization) for the BAM features. Some of the features in BAM files contain large amounts of variant information that directly reveal the location of a variant (CIGAR and SEQ strings). There are also features that cause subtle leakages (alignment scores, string for mismatching positions, and string for distance to the reference), which allow us to infer the presence of a variant in a read. In general, we address the BAM tags that leak the presence of a variant by generalizing them. For example, we replace the sequence with the corresponding sequence from the reference genome, convert the CIGAR into a format that does not contain variant information, and generalize the other tags. This generalization adds quantifiable noise to the reads depending on the type (mismatches, indels) of the sanitized variants (see Figure 5b, Figure S4, and STAR Methods). Our theoretical (see STAR Methods) and empirical (Figure 5c) quantifications demonstrated that pBAM adds noise to the number of reads at certain loci when there are indels, and does not alter this number with mismatches.
We store removed information in a compressed file format called “.diff” (STAR Methods). These .diff files are small files to be kept behind controlled access. We report only the differences between BAM and pBAM in the .diff file and avoid printing any sequence information that can be found in the reference human genome. This allows us to share the majority of the data with minimal utility loss. If the users find the data useful for their research, they can then apply to access the information in the “.diff” files. We provide an easy-to-use software suite that can convert pBAM files to their original BAM format.
pBAM provides privacy while maintaining utility.
Let us assume that the number of variants to remove is r′. Our sanitizer PQ,r will in fact remove r number of variants (r ≥ r′), because the variants in linkage disequilibrium (LD) with the r′ variants need to be removed as well (as one could impute variants within LD blocks). However, note that if the goal is to sanitize all variants from the BAM file, then the LD is irrelevant since all the observable variants will be removed from the BAM file.
We define key quantities to measure privacy and utility. Here, we define privacy of a pBAM with a parameter δ and consider the resulting pBAM to be δ-private. δ is the proportion of sanitized variants to the total number of observable variants (δ = 1 means 100% privacy, i.e., all the observable variants are sanitized; see STAR Methods). We define the utility of a pBAM with another parameter ε and consider the resulting pBAM to have ε-utility. ε is the proportion of unchanged genomic units to the total number of units based on an error per unit (ei) threshold γ. Error per unit (ei) is the log-fold difference between the value of the unit in the pBAM vs. BAM formats (see STAR Methods). For example, if we calculate the signal depth profile from a BAM and a corresponding pBAM, some of the bases in the genome (i.e., units) will have different values. This difference is quantified as added error (ei, Figure 6a, see STAR Methods). If this error is above a threshold value (γ), then a given base will be considered as changed. The total proportion of unchanged bases to the length of the genome will then be the ε value for this pBAM. Error and utility can also be defined at the level of genes or functional elements. The difference between a BAM and pBAM file at nucleotide resolution gives us the upper bound for the utility. ε = 1 means 100% utility, i.e., the results obtained from a BAM and a PBAM are identical.
Figure 6:

pBAM protects privacy while preserving data utility. (a) The empirical values for the privacy-utility balance using NA12878 total RNA-Seq BAM files. The linking attacks were repeated using the partially masked pBAMs and gap values were calculated. The genotypes from these pBAMs were also overlapped with GWAS SNPs. The error between the pBAM and BAM files at the base resolution was calculated. (b) The difference between the BAM and pBAM files is shown at the read level, when the reads were mapped to a reference genome. The added noise to the depth signals due to the BAM-to-pBAM transformation is shown for an example read with a deletion, insertion, and mismatch. The difference in the depth signal when calculated from BAM and pBAM is shown in the signal tracks for Chr 6. Log-fold differences of the signal of each nucleotide between the BAM vs. pBAM and BAM of replica 1 vs. replica 2 were calculated using deeptools. (Ramírez et al., 2016). (c) The difference between the BAM and pBAM files is shown at the read level, when the reads were mapped to a reference transcriptome. The BAM files from two replicas and pBAM were then used to quantify the expression of all 60,699 genes. Transcripts per million (TPM) values were compared between the replicas and between BAM and pBAM. See also Figure S5. (d) The difference between the BAM and pBAM files is shown when the aligned reads were converted back to fastq files for 10x v2 single-cell RNA-Seq data. Fastq and pFastq files were processed using the HCA’s optimus pipeline. The comparison between Fastq and pFastq processing is shown for the number of genes, total number of counts, and cell clustering levels.
We derived a mathematical relationship between the privacy parameter δ and the utility parameter ε to clarify the trade-off between the privacy and utility of a pBAM (see STAR Methods). This relationship depends on the type of sanitized variant, the number of sanitized variants, and the read length. Hence, the privacy-utility relationship will be different for different BAM files as well as for individuals with different ancestries. Therefore, to give one sense of the trade-off, we empirically calculated this relationship for the upper bound as follows: We first assumed that all of the genotypes of an individual can be observed by any functional genomics data. This is a generous assumption as, for instance, one can call around 0.5% of all the genotypes from a typical RNA-Seq BAM file. We then calculated the mean number of SNPs and indels of all individuals from the same ancestry using the 1,000 Genomes database. Finally, we calculated the change in utility as a function of the change in privacy (unit = nucleotide, γ=0) (Figure 5d). We found that, at 100% privacy, the utility loss is at most less than 10% for all the ancestries (Figure 5d). We found a small difference in utility loss (~2%) when we used the number of variants in the African population. This can be explained by the large difference in the number of variants in the African population compared to other populations (Figure 5d).
One way to understand how to interpret the values of the key utility quantities–e, γ, and ε–is to compare them against the discrepancies between replicates. It is well known that high-throughput experiments such as functional genomics assays are subject to great variability. This is remedied by using biological replicates in experiments and further performing irreproducibility discovery rate analysis (Li et al., 2011). One can calculate the ei values for the discrepancy between the replicates and use them as the γ threshold, i.e., the tolerable error between BAM and pBAM. In particular, if the difference between replicates per unit is then a pBAM can be considered to have no utility loss up to this quantity (see STAR Methods).
Empirical calculations validate privacy provided by pBAM:
To test the assumption that variants are masked in pBAM files, we performed variant calling of the pBAMs at different privacy levels on an RNA-Seq data of individual NA12878. We first systematically ramped up the total number of masked variants by thresholding using the genotyping frequencies in the 1,000 Genomes database. We then performed variant calling and subsequent linkage attacks on the database and calculated the gap values. We also calculated the number of overlapping GWAS SNPs with the called variants from pBAM set as another privacy metric. Figure 6a shows that variant calling yields a lower number of genotypes, hence a lower number of overlapping GWAS SNPs and fewer gap values with pBAM at different privacy levels. The observed gap value decreases as we remove more genotypes and crosses the random gap, meaning that we can no longer link the query to the RNA-Seq database.
Empirical calculations validate utility provided by pBAM:
We further calculated the total amount of error introduced in the signal depth profiles when we convert the BAM file to a pBAM file at each genotyping frequency thresholding level. Figure 6a shows how risk of privacy loss decreases with increasing loss of utility. To further understand utility loss, we calculated the difference (log2 ratio) between the signal depth profiles (BigWig files) obtained from pBAM and BAM (ei) files when we masked all of the variants. In addition, we performed the same comparison between the signal depth profiles obtained from BAM files belonging to two different biological replicates. As shown in Figure 6b, the difference between BAM and pBAM files is smaller than the difference between the replicates at base resolution, suggesting that the noise added to the signal is within the biological noise levels. We zoomed in to the highly polymorphic HLA locus to show the similarity between the signal in biological replicates and their corresponding pBAMs. We further quantified the gene expression levels and found minimal difference between pBAM and BAM files (Figure 6c). We performed similar calculations for ChIP-Seq data (Figure S5).
Implementation:
We implemented our pipeline for converting between BAM and pBAM+.diff files (Figure 7a) in bash, awk, and Python. The .diff files are encoded in a compressed format to save disk space. For convenience, pBAM files are saved as BAM files with manipulated content and with a p.bam extension. That is, any pipeline that uses BAM as an input can take p.bam as an input as well. CPU times (calculated using a single 2.3 GHz AMD Opteron processor) and associated file sizes for alignments from RNA-Seq and ChIP-Seq experiments are shown in Table S4. Our data sanitization pipeline has been adopted by the ENCODE Consortium Data Coordination Center and deployed in the ENCODE Uniform Pipeline Framework using workflow description language scripts and docker images, accompanied by appropriate documentation for computational reproducibility on multiple platforms (Google Cloud, Slurm Scheduler, LINUX servers, etc.) under ENCODE Data Processing pipelines. Codes for calculating information leakage, scripts for file manipulations, examples, and file specifications of BAM, pBAM, pCRAM, and .diff files can be found at privaseq3.gersteinlab.org and github.com/ENCODE-DCC/ptools.
Figure 7:

Software and fitting pBAM into the existing system of functional genomics data storage. (a) Schematic of how p-tools work with different file formats. (b) Different layers of data produced from functional genomics experiments with the associated number of leaking variants and bits of information. Table shows the leakage associated with RNA-Seq reads and gene expression quantification. See also Figure S6.
pFastqs as an alternative for alignment-free expression quantification and 10x scRNA-Seq reads:
There are many different ways to quantify expression levels using RNA-Seq reads. Some software such as RSEM (Li and Dewey, 2011) use aligned reads (BAM files), while others such as kallisto (Bray et al., 2016) use reads directly from fastq files. While converting pBAM files to pFastqs for bulk functional genomics data is trivial with available tools (e.g., samtools), 10x scRNA-Seq reads have different fastq structures. To address this, we added a module to our software suite that first maps the 10x scRNA-Seq reads to the reference genome and transcriptome, and generates a BAM file that includes the unaligned reads (following a typical 10x scRNA-Seq pipeline), as well as the tags that contain information about barcodes and unique molecular identifiers (UMI). Our software then converts this BAM file into pFastq files based on UMI, barcode, and sequence identifiers. By using a 10x (v2) scRNA-Seq dataset from the Human Cell Atlas project, we calculated the key quantities using fastq and pFastq files as inputs (see STAR Methods). We found that both fastq and pFastq returned similar results in terms of total gene count, total counts per cell, and cell clustering. Note that clustering algorithms are often stochastic and may return slightly different results even when the same input is used (Figure 6d).
3. Discussion
Functional genomics experiments are increasingly leveraging tissues or cells from donors due to their clinical significance. For example, large-scale studies such as TCGA and PsychENCODE provide functional genomics data on thousands of human subjects with known phenotypes such as cancer or psychiatric diseases. We expect to have a surge of functional genomics data on human subjects in the near future, which will require data access solutions beyond the traditional approaches. Moreover, sharing “raw” functional genomics data is important because it allows researchers to uncover biological insights by leveraging their own tools and analyses. The availability of raw data also helps address the reproducibility crisis in scientific research. However, the majority of functional genomics experiments use next-generation sequencing-based assays. Hence, their raw form includes sequence fragments from donors’ genomes. Here, we aimed to provide privacy- and utility-preserving solutions to the functional genomics data access problem.
Deriving important quantities such as gene expression values does not require the knowledge of the genetic variants of a sample. We took advantage of this, and designed a data sanitization protocol that masks the private variants in the reads while maintaining the utility. Our protocol is flexible and based on principled trade-offs between privacy and utility. While the most privacy-conservative option is to mask all variants, the tool allows study participants to opt for a more or less conservative approach. For example, some participants may wish to mask only the variants that leak information about their susceptibility to stigmatizing phenotypes that can be used against them by insurers or employers. In such a case, our protocol can be used to mask only the desired variants and those in LD. To help guide study participants and researchers, we developed a formalism to show utility loss under different resolutions along the genome. For example, the genomic coordinates of a non-expressed gene will not be sequenced in RNA-Seq, so removing any indel overlapping with these locations will not result in any utility loss. Our formalism can aid researchers in finding an optimal combination of variants to be masked in order to maintain high utility.
To design an effective data-sanitization protocol, one first needs to quantify the private information leakage in the data, which can be done via linkage attacks. Using this approach, we systematically quantified variant leakage in the data and analyzed the robustness of the leakage under different circumstances. We demonstrated how potentially private phenotypes can be inferred, which is particularly important as surreptitious sequencing is becoming more accessible; in particular, for just $19, we were able to link genotypes obtained from a used coffee cup to their RNA-Seq reads.
We addressed the most obvious leakage from functional genomics data, and provided solutions for quick quantification and safe data sharing. Other sources of information leakage from functional genomics experiments are possible at different stages of the overall data summarization process (Figure 7b). Subtle leakages can come from the quantification of expression values; given a population of individuals, these gene expression values can be related to variants through eQTLs, and hence can create leakage (Schadt, 2012, Schadt et al., 2012, Harmanci and Gerstein, 2016). We calculated the potential number of variants one can obtain from a typical RNA-Seq experiment in order to get a sense of the contribution each type of leakage makes to the total leakage (Figure 7 and Figure S6). While inferring the leakage from gene expression values through eQTLs is interesting and non-trivial, eQTLs are not a main source of genotype information in functional genomics data. The amount of genotype leakage from reads is almost 1,000 times that from gene expression levels, and can be avoided with pBAMs. Another source of subtle private information leakage could be the presence of viral (e.g., HPV/EBV) or bacterial reads. Inference of potentially stigmatizing phenotypes from these can be avoided by simply removing these reads from the BAM files during the data sanitization process.
9. STAR Methods
9.1. RESOURCE AVAILABILITY
9.1.1. Lead Contact:
Further information and requests for resources should be directed to and will be fulfilled by Lead Contact, Mark Gerstein (mark@gersteinlab.org).
9.1.2. Materials Availability:
This study did not generate new unique reagents.
9.1.3. Data and Code Availability:
The information quantification and linkage attack code; pTools; intermediate data processing scripts; test BAM, PBAM and .diff files; file format specifications; a Jupyter notebook for single-cell analysis on BAM and pBAM and links to publicly available data used in this paper can be found at privaseq3.gersteinlab.org. The pTools code can also be found at https://github.com/ENCODE-DCC/ptools. There are restrictions to the availability of the sequencing data generated from coffee cups due to their containing information that could compromise the privacy of research participants and other individuals who may have contaminated the samples.
9.2. METHOD DETAILS
9.2.1. Linkage Attack Details
Linking score:
Let us assume that there are n variants that can be observed from our query individual with known identity, either by using deeply sequenced WGS data (perfect information ) or by using DNA left on a coffee cup (noisy information ). is then the set of genotypes for each variant (gi = {0,1,2} for the homozygous reference allele, heterozygous alternative allele, and homozygous alternative allele, respectively). is the set of genotypes for an individual i in database for the same variants in . Note the genotype set is observed from functional genomics data of an anonymized individual i and some of the genotypes may be missing due to the coverage and experimental bias in the functional genomics reads. For each individual i in , we find the intersection and calculate a linking score
where f(gt) is the ratio of the number of individuals whose tth variant has the genotype gt to the total number of individuals in the cohort.
gap:
To find the individual in database that matches the query individual, we then rank all the L(i,query) scores for a given query individual in decreasing order as
where m is the total number of individuals in the attacked database. We denote the individual with the highest score as the queried individual. To assess the statistical robustness of this prediction, we defined a measure called gap, which is the ratio between the L(i,query) score of the first-ranked individual (max = L(i,query)1) and that of second-ranked individual (max2 = L(i,query)2 and gap = max/max2). The goal is to determine how separated the matching individual is from the rest of the individuals in the cohort. The gap value is similar to the eccentricity parameter in the IMDB-Netflix attack (Narayanan and Shmatikov, 2008) used to determine the reliability of the linkage attack. The difference between gap and eccentricity is that gap is the fold change, which can be compared across samples.
Sensitivity and precision of input datasets:
We calculated the sensitivity as the ratio between the called true-positive genotypes and the total (ground-truth) genotypes belonging to the individual. We calculated precision as the ratio between the called true-positive genotypes and all called genotypes.
Addition of false-positive genotypes to the attacked database:
Using the 1,000 Genomes dataset, we randomly selected genotypes and appended them to the genotype set of each individual i . These random genotypes may or may not be found in the genotype set of , hence increasing the chance of matching the query individual to any individual i in .
Addition of genetically related individuals:
We genotyped the RNA-Seq reads obtained from 14 individuals genetically related to the 1,000 Genomes individual NA12878, including the parents and children (Li et al., 2014) using GATK (DePristo et al., 2011, der Auwera et al., 2013), and added them to the database . We repeated the linking attack and calculated associated linking scores and gap values with and without NA12878 present in . The gap values for both cases were statistically tested. The goal was to asses whether the linking approach used in this study is robust to the addition of first-degree genetically related individuals to the attacked database.
Subsampling gEUVADIS RNA-Seq reads and creation of new attacked databases:
We randomly subsampled reads from all the individuals in the gEUVADIS cohort and created new attacked databases for each level of subsampling by using 5, 10, 20, …, up to 100% of the reads in each RNA-Seq data (Figure 2a). We first calculated the sensitivity and precision of the called genotypes by using the genotypes from high-coverage WGS data as the ground truth. We then performed the linking attack by a) using the genotypes obtained from reads under different coverages and b) adding random false-positive variants to the genotypes of each entry in the attacked database.
Subsampling RNA-Seq and coffee cup reads and imputation of missing genotypes:
We first randomly subsampled reads from the RNA-Seq data of two study individuals and created cohort genotype databases for each level of subsampling by using 10, 20, …, 100% of the reads in each RNA-Seq experiment. We then imputed genotypes for each subsampling using BEAGLE (Browning et al., 2018). This process resulted in ten databases from the genotypes of subsampled RNA-Seq reads and ten databases from the imputed genotypes of subsampled RNA-Seq reads. We also randomly subsampled the WGS coverage of coffee-cup samples from 10x to 0.125x and performed genotype imputation for each subsampling. In total, we had six query sets from subsampling and six query sets from imputation for each coffee cup. Next, we linked 144 queries (2 individuals × 6 coffee cups × 12 subsample-imputation combinations) to 20 different databases, totalling 2,880 different linkage attacks (Figure 3c). For each coffee-cup sample (6 cups × 2 individuals), we calculated the linking accuracy for all subsampling and imputation combinations (240 total) as the ratio between the number of correctly linked coffee cups and the total number of coffee cups (Figure 3e).
Linkage attacks with genotypes from genotyping arrays:
We used Illumina exome-based genotyping arrays on the coffee cup samples. We found that arrays had a relatively low call rate for our samples, likely due to fragmented, degraded, or damaged DNA (Table S2). Although there were many correctly genotyped SNPs in the coffee-cup samples, their overlap with RNA-Seq genotypes was low, resulting in only 2 out of 12 samples successfully linked to the RNA-Seq database (gap=1.98 and 1.73, p-value< 10−2).
Linkage attacks with genotypes from Oxford Nanopore:
SNP and indel genotype quality from a typical Oxford Nanopore run is quite low with single-pass sequencing using standard protocols. We used a standard sequencing kit recommended by the Oxford Nanopore with multiplexing. The intent of this approach was to minimize the cost and to mimic the act of a curious scientist surreptitiously gathering DNA. We were only able to identify a few genotypes and failed to link the coffee cups to RNA-Seq data (Table S2).
Linkage attacks with different functional genomics data:
We performed linkage attacks using reads from different functional genomics techniques (e.g., ChIP-Seq, Hi-C, or ChIA-PET) as the noisy information and 1,000 Genomes dataset as the perfect attacked database . The goal was to empirically quantify and compare the amount of sensitive information in various functional genomics datasets. This is particularly important as different assays target different regions of the genome with different coverage profiles. For example, RNA-Seq targets expressed exons, whereas H3K27ac ChIP-Seq targets the non-coding genome of the promoter and enhancer regions. Different assays also have different coverage profiles (i.e., some assays have spread-out peaks while others are more punctate). We used cell line functional genomics data from the ENCODE consortium as information about individuals represented in the 1,000 Genomes genotype database (the attacked database ). We treated the genotypes in the attacked database as the gold standard when comparing different metrics such as linking score and gap values.
Comparison between assays:
The ENCODE data portal contains data from a range of functional genomics assays (Table S4) on the GM12878 cell line (individual NA12878 in the 1,000 Genomes database). We performed linkage attacks at varying sequencing coverage using data from a number of these assays. We compared our results from functional genomics data to those from WGS, whole exome sequencing (WES), and genotyping arrays. Overall, ChIP-Seq for histone modification data showed lower gap values at low coverage compared to other assays (Figure 4a). We also calculated the linking score per nucleotide by normalizing the linking scores with the total number of base pairs in an assay. Although the total linking score obtained using scRNA-Seq data was lower compared to other assays, the gap values were surprisingly high even at the lowest coverage (Figure 4a). In general, experiments targeting exons (WES, RNA-Seq) demonstrated comparable gap values to whole-genome approaches even though the linking scores were lower. To investigate the reasons behind this, we calculated the median frequency of genotypes called from different functional genomics assays at different coverage (Figure 4c). We found that genotypes from assays targeting exons (especially scRNA-Seq) were slightly more rare in the cohort than genotypes from other assays (Hi-C, WGS, ChIP-Seq TF binding and histone modification), and hence the comparable gap values despite lower linking scores (Figure 4c, see STAR Methods subsection for contributions of rare and common genotypes to linking scores). The gap value of the polyA RNA-Seq sample to the correct individual at low coverage is higher than the gap value obtained from WES and total RNA-Seq at the same coverage. We think the reason is as follows: polyA RNA-Seq sequences contain highly expressed exons, WES sequences contain all exons, and total RNA-Seq data contain sequences from non-exonic parts of the genes. Compared to all the exons on a gene or other intragenic regions, highly expressed exons contain more rare variants due to selection pressure. Therefore, polyA RNA-Seq data contain more individual specific variants. Thus, although polyA RNA-Seq BAM files typically contain fewer reads than total RNA-Seq or WES BAM files, they can be highly prone to re-identification attacks, which is not clear at first glance. We repeated this analysis for individuals for whom we had less functional genomics data. We found that non-obvious data types such as ChIP-Seq control experiments and Repli-Seq data can be used for linking purposes above coverage of around 10 million bp; if we assume a typical experiment has on average a 100 bp read length, then this would correspond to roughly 100,000 reads. For some of the ChIP-Seq TF binding experiments (information ) it was not possible to link the individuals to the 1,000 Genomes database (attacked database ) despite their relatively high depth (Figure S2).
9.2.2. Sample Selection
We present short variant calls on 478 samples. Of these, 16 were newly sequenced for this study (two DNA samples from two individuals, two RNA samples from the same individuals, six coffee cup DNA samples from two individuals), and the remaining 462 RNA-Seq data were obtained from the gEUVADIS study (Lappalainen et al., 2013). Genomic materials for newly sequenced samples were obtained by collecting blood samples and used coffee cups. DNA samples from the blood were sequenced using high-coverage Illumina sequencing (30x) and used as the gold standard. RNA samples from the blood were sequenced using the Illumina total RNA-Seq protocol. Extracted DNA from coffee cups were sequenced using low-coverage Illumina sequencing (10x), Oxford Nanopore Technologies (ONT), and genotyped with the Illumina Infinium OmniExpressExome-8 v1.6.
9.2.3. Contribution of very rare and unique genotypes to the L(i,query) score
We calculated the number of unique, very rare, and common genotypes for every individual in the 1,000 Genomes panel. We observed around 15,000 unique genotypes per individual. This contributes around 11×15,000 = 165,000 bits of information. We estimated 11 from −log2(1/2503), as 1 in ~2,503 individuals in 1,000 Genomes have these unique genotypes. We observed around 670,000 very rare genotypes, which contribute 7×670,000 = 4,690,000 bits of information on average. We estimated 7 from −log2(20/2503), as 20 in ~2,503 individuals in 1,000 Genomes have these unique genotypes. In total, unique and very rare genotypes contribute 4,855,000 bits of information. We then calculated the information in the genomes of all the individuals in the 1,000 Genomes Phase III panel. The mean information per individual is around 2×107 bits. The contribution of unique and very rare variants then becomes around 24% of the total information in an individual’s genome, despite the fact that the number of unique and very rare variants is only 3% of the total number of variants in an individual’s genome. Note that this calculation is based on our scoring system adopted from Narayanan and Shmatikov (Narayanan and Shmatikov, 2008), which assumes independence between variants.
9.2.4. Experimental protocols
DNA extraction protocol from coffee cup lids:
We used the QIAamp DNA Investigator Kit from QIAGEN. This kit is designed to purify DNA from forensic and human identity samples. We first swabbed the surface of the coffee cups using a cotton swab dipped in 1 μL purified water. We followed the QIAamp DNA Investigator kit protocol for isolating DNA from surface-swab samples without modification. The final amount of DNA isolated from coffee cups was around 0.9 to 1 ng.
Whole-genome amplification:
Due to the very low starting amount of purified DNA, we used a single-cell whole-genome amplification kit (REPLI-g Single Cell Kit), which allows uniform PCR amplification from single cells or limited sample materials for use in next-generation sequencing applications. We then used the Monarch PCR and DNA Cleanup kit to purify the DNA from PCR reactions.
Illumina sequencing:
Amplified DNA samples from coffee cups as well as purified PCR-free DNA from blood (as the gold standard) were sent to the Yale Center for Genome Analysis for Illumina WGS. Coffee cup samples were sequenced at a 10x coverage and blood samples were sequenced at a 30x coverage.
Illumina genotyping arrays
We used an Infinium OmniExpressExome-8 BeadChip for the amplified DNA samples from coffee cups. Infinium OmniExpressExome-8 array surveys tag SNPs located on exons from all three HapMap phases, which includes 273,000 exonic markers. Each SNP is represented on these chips by 30 beads, on average. The Yale Center for Genome Analysis performed the BeadChip protocol and calculated the call rates using an Illumina BeadStudio.
Nanopore Sequencing:
We used the rapid sequencing kit with multiplexing (due to the low amounts of input DNA) without the additional steps of DNA repair or suggested quality control. Due to the low quality of the DNA obtained from coffee cups, we did not perform size selection of the fragments in the PCR-based libraries we obtained using the ONT rapid sequencing kit. A total of 12 libraries from six coffee cups per individual were barcoded using the ONT rapid barcoding kit. Libraries were sequenced across an individual R9.4 flow cell on a single MinION instrument. A total of 844,599 reads were successfully base-called and demultiplexed using Guppy. The recommended MinION run-time was 48 h; therefore, the run was terminated after 48 h. SNP calling was performed using Nanopolish software.
RNA extraction protocol and RNA-Seq:
Blood samples from individuals were sent to the Yale Center for Genome Analysis for RNA purification and Illumina high coverage total RNA-Seq analysis following the suggested protocols by Illumina. Note that the RNA-Seq data we generated is at a higher sequencing coverage than those in the gEUVADIS cohort. Therefore, our RNA-Seq data yielded more genotypes than the gEUVADIS data. We found that 20% of the total reads in our experiment was equivalent to the total number of reads in a sample of the gEUVADIS cohort. To do a fair comparison for the linkage attacks, we downsampled the total number of captured variants to the average number of variants observed in the gEUVADIS dataset.
9.2.5. Genotyping
Blood tissue and coffee cups
Illumina sequencing:
The DNA extracted from two blood tissues and 12 coffee cup samples were sequenced using Illumina. Raw fastq files were processed by mapping them to the hg19 reference genome (b37 assembly) using bwa (Li and Durbin, 2009). The resulting BAM files were processed using Picard tools to remove PCR duplicates. De-duplicated files were then genotyped using GATK best practices (DePristo et al., 2011, der Auwera et al., 2013).
Genotyping Arrays:
The 12 coffee cup samples were genotyped using Illumina Infinium OmniExpressExome-8 v1.6 and UV-coated chips were scanned using IScan. Scanned output files were analyzed and call rates were calculated using Illumina BeadStudio.
ONT:
The 12 coffee cup samples were prepared and sequenced using the rapid barcoding kit and minION following the manufacturer’s suggestions. After converting fast5 files to the fastq format using Guppy software for base-calling and de-multiplexing, each sample was aligned to the hg19 reference genome (b37 assembly) using bwa “mem -x ont2d” options (Li and Durbin, 2009). Aligned reads were used for variant calling using Nanopolish software.
Functional genomics data:
Raw RNA-Seq fastq files (from this study, gEUVADIS, and ENCODE) were processed by mapping them to the hg19 reference genome (b37 assembly) and gen-code v19 transcriptome assembly using STAR (Dobin et al., 2013). Other raw functional genomics (e.g., Hi-C, ChIP-Seq) fastq files were mapped to the hg19 reference genome (b37 assembly) using bwa (Li and Durbin, 2009). The resulting BAM files were processed using Picard tools to remove PCR duplicates. De-duplicated files were then genotyped using GATK best practices (DePristo et al., 2011, der Auwera et al., 2013) for RNA and DNA for RNA-Seq and other functional genomics data, respectively.
9.2.6. pBAM details
The privacy-preserving BAM (pBAM) format is a variant of the BAM format. The genomic variants in BAM files are masked using data-sanitization techniques as outlined below. pBAMs are generated after aligning the functional genomics reads to the reference genome. The alignment step can be performed in a privacy-preserving manner as well (Chen et al., 2012), however the resulting alignment files still need to be sanitized to maintain patient privacy. The goal is to create files that can be openly shared and utilized in any downstream analysis pipeline. Our data sanitization is agnostic to the type of variant. Any difference between the sample and the reference genome will be covered regardless of whether they are somatic or germline SNPs or large structural variants. We simply compare the read in a BAM file to the reference genome and remove the difference. This will prevent genotyping and variant calling from the reads (Poplin et al., 2018).
Procedure:
Below is a practical guide on how to convert BAM files to pBAM files.
Let us assume the variants that need to be sanitized from the BAM file areVs ={s1, .., si, ..sn} and the variants that are in LD with the variants in Vs are . The total number of variants that need to be sanitized are then r = n+k.
We first find all the reads that contain the variants in the such that R = {R1, ..,RT}.
- We apply sanitization techniques (i.e., generalization) to the BAM fields so that an adversary cannot infer the existence of these variants:
- CIGAR: Convert the CIGAR of each Ri to a perfectly mapped read CIGAR (e.g., 100M, where 100 is the read length and M denotes that each base on the read perfectly mapped to reference genome).
- SEQ: Replace the sequence of each Ri with the sequence in the reference genome.
- QUAL: Convert all the base qualities of Ri to perfectly called base phred scores.
- There are also optional tags in the BAM files such as AS (alignment scores), MD (string for mismatching positions), and NM (edit distance to reference) that should be sanitized. They can be generalized to the values for perfectly mapped reads if they are present in the BAM files.
- We demonstrated that there might be extremely subtle leakages through MAPQ scores (mapping quality, see Figure S7). In particular, if the goal is to prevent large leakages such as structural variants, then a data-sanitization procedure such as suppression or generalization might be suitable for this field as well.
In addition, we treat intronic reads differently to be able to capture the splicing accurately.
Privacy:
If one can observe t number of variants from a functional genomics BAM file, then the resulting B* = PQ,r(B) can be viewed as δ-private with respect to operation Q, if δ = r/t, which is the ratio of non-observable variants in a pBAM file to all observed variants in a BAM file. As mentioned earlier, PQ,r is the data sanitizer. We can reach 100% privacy when r = t. Here, variants are defined as homozygous and heterozygous alternative alleles. This definition assumes that all r number of variants are guaranteed to be deleted in the resulting pBAM file.
Utility:
We defined the the utility of pBAMs as such that any calculation f based on a BAM file should result in similar results when the pBAM is used instead. A calculation f can be a signal depth profile calculation, TF binding peak detection, or gene expression quantification (Figure 5a–5b). Then, we can reconstruct an equation for each unit i as
where a unit i can be a single base pair, an exon, or a gene depending on the function f. Accordingly, ei can be calculated as the absolute value of the log-fold change between the results derived from the BAM and pBAM file. Note that ei is a measure of the error in the new dataset B*. B* = PQ,r(B) can be viewed as having ε-utility with respect to operation Q if ε = (G−m)/G, where m is the total number of units with ei > γ and G is the total number of the units. We can obtain 100% utility if the error is smaller than the threshold γ for every unit in the genome.
Concordance between replicates vs. BAM and pBAM:
In an effort to avoid assigning the γ threshold ad hoc, we can use the concordance between biological replicates of a functional genomic experiment as guidance. Let us denote R1 and R2 as the BAM files from two biological replicates of the same experiment. Then, we can repurpose the equation above as
is the absolute value of the log-fold change between the results derived from R1 and R2. Then, the γ for every unit can be chosen as , such that the error tolerated between BAM and pBAM never exceeds the biological noise levels.
Privacy-Utility Relationship:
The relationship between privacy and utility can be derived through the mathematical relationship between δ and ε. Let us assume the units for our utility calculation are single bases in the genome, as this will give us the upper bound (i.e., more utility loss at higher resolution). Let us also assume that the function f for which we want to measure the utility loss is the signal depth calculation. Sanitization is done over three kinds of variants: SNPs, insertions, and deletions. (1) In the case of SNPs, when we change a letter from the alternative allele to the reference allele, the resulting signal profile at that location does not change. (2) In the case of an insertion at position x, when we delete the insertion from a read (since an insertion is not represented in the reference), we have to append lins number of bases to the end of the read (lins is the length of the insertion). This adds error to all of the bases between position x and x+LR, where LR is the length of the reads. That is, for each insertion, the total number of bases with ei > γ will be at most LR, when γ is equal to 0 for the upper bound. (3) In the case of a deletion at position x, when we fill the deletion with the reference, we have to delete ldel number of bases from the end of the read (ldel is the length of the deletion). This adds error to all of the bases between position x and x+LR +ldel −1, where LR is the length of the reads. The maximum detected indel length varies by the aligner settings. In the most extreme cases, ldel can be as large as LR−1. That is, for each deletion, the total number of bases with ei > γ will be at most 2·LR−2, when γ is equal to 0 and ldel is equal to LR−1 for the upper bound.
If r is the total number of variants to be sanitized, then r = rsnp+rins+rdel. The number of bases with m such that ei > 0 are at most
Since ε = (G−m)/G, then m = −ε ·G+G. We can then say
If we replace rins with r−rsnp−rdel and r with δ ·t, then our relationship becomes
Utility bounds
The goal is to consider the impact of sanitizing BAM files through modifications applied to identified variants. The sanitization procedure used here is inherently asymmetric, as bases are added or removed at only one end of the read. We make a distinction between a personal genome and the reference genome: the personal genome includes all of the SNPs and indels; the reference genome is the standard external metric. While the personal genome may not actually be constructed, it serves as a useful conceptual tool to understand the impact of the transformation involved in the sanitization procedure. We discuss three types of variants and the chosen sanitization method applied in the pBAM format:
SNP:
A single-nucleotide variant/polymorphism is changed by mutating the variant to the reference allele in every read in which the mutation is observed.
Insertion:
An insertion is sanitized by removing any fraction of the new inserted segment and adding the equivalent number of reference nucleotides to one end of the corresponding read.
Deletion:
A deletion is sanitized by filling the reference nucleotides into the part of the deleted segment occurring on any read, and then removing the equivalent number of nucleotides from that read.
Definitions
The genome is indexed by discrete positions i. The coverage prior to and after sanitization are functions of the genomic position, and are labeled as cpre(i) and cpost(i), respectively. The read length is fixed at LR. The size of the insertion is labeled , while the size of the deletion is labeled as , where in both cases the position i marks the start position of the indel. In addition, N(i) = the number of reads that start at position i in the mapping to the personal genome.
SNPs:
Every read containing a SNP will be modified, with the alternate allele replaced by the corresponding reference allele. Under the assumption that the presence of this SNP does not alter the mapping efficiency (say, if the other variants within a particular read sum to mmis−1, then this SNP will lead to that read being dropped) and thus read dropout, we see that cpre(i) = cpost(i). Therefore, no impact will be observed, unless one looks through the mapping quality control (QC) and finds all the reads overlapping a given locus have slightly lower quality. This might be possible, unless the QC metadata is being modified explicitly in the sanitization procedure (see Quantifications and Statistical Analysis).
Short Indels:
For indels, we consider the mapping changes due to the sanitization procedure in the following.
- Insertions: The variant is indexed by position i, where the insertion occupies the base pairs from . We consider the following cases:
-
: No individual read will dropout in this case due to the presence of the insertion. Consider a case in which the added nucleotides are on the end of a higher genomic position for the sake of clarity. The process of sanitization leads to an additional build-up of reads downstream of the insertion point. This happens due to the replacement process discussed above. Certain reads that would have been mapped to the insertion in the personal genome of the individual are now added downstream of the insertion in the mapping to the reference genome. This allows us to quantify the read build-up in terms of the start positions of the reads. Thus, for all reads that overlap with the insertion the following transformation occurs:
- , all reads starting at position x in the personal genome are newly mapped to the reference in the interval [i+1,x+LR+1].
- , all reads starting at position x in the personal genome are newly mapped to the reference in the interval .
- , all reads starting at position x in the personal genome are newly mapped to the reference in the interval .
The genomic footprint of the sanitization procedure is the interval [i+1,i+LR]. The resultant read build-up of the sanitized BAM relative to the original BAM is thus given by the integral/discrete sum over all the accumulated contributions described above (again, x is the position along the personal genome, with the insertion in place; α in the equation below is the position along the reference genome in the downstream interval [i+1,i+LR−1] that is impacted by read build-up due to post-sanitization remapping):For example, imagine an ideal case where all the reads are uniformly distributed in the given region. This means that c(x) = constant = c and across the original mapping. Note that in the ideal case of uniformly distributed reads, the number of reads that begin at a locus is the total number of reads that overlap with a locus, divided by the length of each read. Thus,Thus, the fold-change in the coverage is given by in the interval i+1 ≤ α ≤ i+L If we define, FC(α) = exp(eα), we end up with in the interval i+1 ≤ α ≤ i+L. The general formula is - : This case will be slightly different from the above case, as some of the reads will completely overlap with the insertion region and never contribute to the remapping build-up downstream. The calculation would then ignore the reads that overlap significantly with the insertion. We do not discuss this situation here, as these reads will have soft or hard clipping in their CIGARs (split reads) and will be treated differently as discussed above.
-
- Deletions: The variant is indexed by position i, where the deletion removes the reference base pairs from . We exclusively consider in this case, with the understanding that longer deletions would require slight modifications of the following calculations. The mapping to the reference genome after sanitization results in loss of coverage from regions downstream of the deletion, and an equal gain in regions of the deletion. The mapping changes are as follows:
- , all reads starting at position x in the personal genome are removed from and newly mapped to the reference in the interval [i+1,x+LR−1].
- , all reads starting at position x in the personal genome are removed from and newly mapped to the reference in the interval .
The genomic footprint of the sanitization procedure is the interval . The change in coverage can be calculated in a manner similar to the case of insertions, with the additional notion that reads that are remapped to the deleted segment are drawn from downstream portions of the genome:
If the gain in the raw read count in the deleted segment of the reference genome is Gain, then
If the loss in the raw read count downstream of the deleted segment is Loss, then
It is not possible to compute the fold change in the coverage in the deleted segment, as the coverage is 0 pre-sanitization. However, it can be calculated by adding a pseudo count to the coverage pre-sanitization. In the downstream segment, the fold change in the coverage is given by
for , and with FC(α) = eeα, we have
Again, considering the example of constant coverage discussed for the insertions, we have
The upper bound on the fold change is given by . Note that the formula and the genomic footprint are different from those in the insertion case.
Suggestions for a higher utility pBAM while preserving privacy:
After quantifying gene expression or other functional genomics quantities such as transcription factor binding enrichment from a BAM and corresponding pBAM file, one can pinpoint particular indels that overlap with the functional regions of the genome. We can then calculate the joint genotyping probability of these functional indels in a cohort of individuals (e.g., 1,000 Genomes individuals) in order to determine how rare or common they are. We can then systematically mask rare indels one by one until the difference between BAM and pBAM files are within noise levels (e.g., less than the difference between two replicates).
Transcriptome alignments:
pTools searches the reference transcriptome for the position of the transcripts and reports the reference transcriptome sequences in the pBAM. We used the reference transcriptome files that are generated by STAR software (Dobin et al., 2013).
.diff files:
.diff files contain the difference between the original BAM files and the pBAM files in a compact form. If the information is already available in the reference human genome such as sequence of the fragment, then the .diff file does not report it. This approach keeps the .diff files as small as possible. These files require special permission for access and contain the private information about the individual. To be able to go back and forth between BAM and pBAM files using the .diff files, the BAM and pBAM files must be coordinate sorted.
9.2.7. Functional genomics data processing
RNA-Seq data were processed using STAR (Dobin et al., 2013) for alignment and RSEM (Li and Dewey, 2011) for quantification. ChIP-Seq data were processed using bwa (Li and Durbin, 2009) for alignment and MACS2 (Zhang et al., 2008) for peak calling. ScRNA-Seq data were processed using HCA’s optimus pipeline for quantifications and further analyzed using the Scanpy (Wolf et al., 2018) (A jupyter notebook. The figures and the necessary input files can be found at privaseq3.gersteinlab.org).
9.3. QUANTIFICATION AND STATISTICAL ANALYSIS
9.3.1. Statistical significance of gap
We estimated the empirical p-values for a particular gap value by linking a set of random genotypes that do not belong to a particular individual to the database, as follows.
We selected n random genotypes from a database of nearly 50 million genotypes (taken from 2,504 individuals in the 1,000 Genomes database). n is the total number of genotypes that were linked to the database for the gap calculation.
We calculated the L(i,query) between these random n genotypes and every individual i in the database.
We calculated the gap as the ratio between the first-ranked and second-ranked L(i,query).
We repeated the above steps 1,000 times and obtained a distribution of random gap values.
The total number of random gap values that are equal to or greater than the real gap divided by 1,000 is the probability of observing the real gap value by chance.
We empirically calculated the probability of observing the real gap value or a larger gap value by chance by randomly subsetting n variants (the same number of variants as in ) observed from the information , performing the linkage attack, and calculating the associated gap value. If this p-value is statistically significant, then the attacker can rely on the prediction.
9.3.2. Quantifications related to the data sanitization
Calculation of average and maximum leakage per variant:
We first overlapped the 1,000 Genomes variants with the exon annotations. We classified the variants into the following categories: exonic variants, exonic SNVs (excluding indels), exonic indels, and exonic small deletions. For each category, we calculated the self-information of the variant for all three possible genotypes (0, 1 and 2) as h(s0),h(s1) and h(s2). The average of self-information for each variant in each category is the average information leakage and the max(h(s0),h(s1),h(s2) is the maximum information leakage for that particular variant. We then calculated the mean and standard deviation for all the variants in each category. Total information leakage is calculated as the product of the total number of accessible variants and the average information leakage per variant. The distributions of the leakage can be seen in Figure S6. These distributions do not follow the normal distribution and are skewed, thus calculating the average is not the best approach. Therefore, mean leakage can be thought of an approximation that might be overestimated and since raw reads leak a lot more information than the signal profiles and eQTLs, the conclusion does not change with this approximation.
Leakage from MAPQ:
We found that reads with MAPQ values below the mean MAPQ contain insertions, deletions, and soft and hard clipping at a higher rate than expected (Figure S7), hence they might leak the location of large structural variants. In Figure S7, we analyzed a subsampled BAM file from a WGS dataset. The BAM files from functional genomics data are noisier than WGS, however the MAPQ values could still potentially be a source of variant leakage.
Supplementary Material
Figure S3: Linking NA12878 to a panel with 108 AFR individuals and one EUR individual, with and without NA12878 in the database, related to Figure 4.
Figure S2: gap values for the remaining seven individuals with different functional genomics assays, related to Figure 4
Figure S4: Visual representation of mapped fragments before and after converting the CIGARs to a pBAM file format, related to Figure 5. The insertions, deletions, soft and hard-clipping, and intronic reads are depicted. The noise that is added to the pBAM file to enhance privacy is also depicted in the fragments.
Figure S5: The difference between ChIP-Seq peak calling using BAM and pBAM as input for the fold change compared to control, the number of reads that pile up on the location of peak, and the location of the peak summit, related to Figure 6. Peak calling was performed using MACS2 (Zhang et al., 2008).
Figure S6: The distributions of the information leakage per variant in different levels of the data stack, related to Figure 7. Individual characterizing information (ICI) is calculated based on ref (Harmanci and Gerstein, 2016).
Figure S7: Potential variant leakage from MAPQ scores, related to Figure 5. As shown, the reads with potential large structural variants have smaller than expected MAPQ scores. An adversary can sort the MAPQ scores in a BAM file and guess the location of these structural variants that are mapped with low MAPQs.
KEY RESOURCES TABLE
Highlights.
Surging functional genomics data necessitates improved data-sharing modes.
Quantification of private information in these data is done via linkage attacks.
A data sanitization protocol grounded in privacy and utility is developed.
The sanitized format is compatible with existing file formats and pipelines.
4. ACKNOWLEDGMENTS
We thank the Yale Center for Genome Analysis for assistance. We thank Jennifer Chien and Jason Hilton for the assistance with the single-cell RNA-Seq data. This study was supported by grants from the NIH (R01 HG010749 to M.G.; K99 HG010909 to G.G.). This work is also supported by AL Williams Professorship fund and the Chan Zuckerberg Initiative Donor-Advised Fund.
Footnotes
DECLARATION OF INTERESTS
All authors declare no competing interests.
References
- Bray NL, Pimentel H, Melsted P and Pachter L (2016), ‘Near-optimal probabilistic RNA-seq quantification.’, Nature Biotechnology 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Browning BL, Zhou Y and Browning SR (2018), ‘A one-penny imputed genome from next generation reference panels.’, Am J Hum Genet. 103, 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Peng B, Wang X and Tang H (2012), ‘Large-Scale Privacy-Preserving Mapping of Human Genomic Sequences on Hybrid Clouds.’, Proceeding of the 19th Network and Distributed System Security Symposium. [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D and Daly MJ (2011), ‘A framework for variation discovery and genotyping using next-generation DNA sequencing data.’, Nature Genetics 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- der Auwera GAV, Carneiro MO, Hartl C, Poplin R, Angel GD, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S and DePristo MA (2013), ‘From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline.’, Current Protocols in Bioinformatics 43, 1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR (2013), ‘STAR: ultrafast universal RNA-seq aligner.’, Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich Y and Narayanan A (2014), ‘Routes for breaching and protecting genetic privacy.’, Nat. Rev. Genet 15, 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich Y, Shor T, Peer I and Carmi S (2018), ‘Identity inference of genomic data using long-range amilial searches.’, Science 362, 690–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn M (2018), ‘The culprit’s name remains unknown. But he licked a stamp, and now his DNA stands indicted.’, Washington Post. URL: https://www.washingtonpost.com/news/morning-mix/wp/2018/10/17/the-culprits-name-remains-unknown-but-he-licked-a-stamp-and-now-his-dna-stands-indicated?utm_term=.25eba675732b
- Gymrek M, McGuire AL, Golan D, Halperin E and Erlich Y (2013), ‘Identifying personal genomes by surname inference.’, Science 339, 321–324. [DOI] [PubMed] [Google Scholar]
- Harmanci A and Gerstein M (2016), ‘Quantification of private information leakage from phenotype-genotype data: linking attacks.’, Nature Methods 13, 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homer N, Szelinger S, Redman M, Duggan D, Tembe W, Muehling J, Pearson JV, Stephan DA, Nelson SF and Craig DW (2008), ‘Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays.’, PLoS Genet. 8, e1000167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im HK, Gamazon ER, Nicolae DL and Cox NJ (2012), ‘On sharing quantitative trait GWAS results in an era of multiple-omics data and the limits of genomic privacy.’, Am. J. Hum. Genet 90, 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly Y, Dyke SO, Knoppers BM and Pastinen T (2016), ‘Are Data Sharing and Privacy Protection Mutually Exclusive?’, Cell 167, 1150–1154. [DOI] [PubMed] [Google Scholar]
- Kim J, Edge MD, Algee-Hewitt BF, Li Z, and Rosenberg J,NA (2018), ‘Statistical Detection of Relatives Typed with Disjoint Forensic and Biomedical Loci.’, Cell 175, 848–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, Hoen PAC, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, van Iterson M, Almlof J, Ribeca P, Pulyakhina I, Esser D, Giger T, Tikhonov A, Sultan M, Bertier G, MacArthur DG, Lek M, Lizano E, Buermans HPJ, Padioleau I, Schwarzmayr T, Karlberg O, Ongen H, Kilpinen H, Beltran S, Gut M, Kahlem K, Amstislavskiy V, Stegle O, Pirinen M, Montgomery SB, Donnelly P, McCarthy MI, Flicek P, Strom TM, Consortium TG, Lehrach H, Schreiber S, Sudbrak R, Ángel Carracedo, Antonarakis SE, Häsler R, Syvanen A-C, van Ommen G-J, Brazma A, Meitinger T, Rosenstiel P, Guigo R, Gut IG, Estivill X and Dermitzakis ET (2013), ‘Transcriptome and genome sequencing uncovers functional variation in humans.’, Nature 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Yoo S, Wang W, Tu Z and Zhu J (2019), ‘A probabilistic multi-omics data matching method for detecting sample errors in integrative analysis.’, Gigascience 8, giz080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee S, Ouellette S, Park W-Y, Lee EA and Park PJ (2017), ‘NGSCheckMate: software for validating sample identity in next-generation sequencing studies within and across data types.’, Nucleic Acids Res. 45, e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B and Dewey CN (2011), ‘RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome.’, BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H and Durbin R (2009), ‘Fast and accurate short read alignment with Burrows-Wheeler Transform.’, Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennel T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009), ‘The Sequence Alignment/Map format and SAMtools.’, Bioinformatics 25, 2078–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H and Bickel PJ (2011), ‘Measuring reproducibility of high-throughput experiments.’, Annals of Applied Statistics 5, 1752–1779. [Google Scholar]
- Li X, Battle A, Karczewski KJ, Zappala Z, Knowles DA, Smith KS, Kukurba KR, Wu E, Simon N and Montgomery SB (2014), ‘Transcriptome sequencing of a large human family identifies the impact of rare noncoding variants.’, Am J Hum Genet. 95, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Quick J, Simpson JT (2015), ‘A complete bacterial genome assembled de novo using only nanopore sequencing data’, Nat Methods 12, 733–735 [DOI] [PubMed] [Google Scholar]
- Narayanan A and Shmatikov V (2008), ‘Robust De-anonymization of Large Sparse Datasets.’, Proceedings of 2008 IEEE Symposium on Security and Privacy pp. 111–125. [Google Scholar]
- Poplin R, Ruano-Rubio V, Mark A DePristo TJF, Carneiro MO, der Auwera GAV, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, Shakir K, Thibault J, Chandran S, Whelan C, Lek M, Gabriel S, Daly MJ, Neale B, MacArthur DG and Banks E (2018), ‘Scaling accurate genetic variant discovery to tens of thousands of samples.’, Biorxiv. URL: 10.1101/201178 [DOI] [Google Scholar]
- PsychENCODE Consortium (2018), ‘Revealing the brain’s molecular architecture.’, Science 362, 1262. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F and Manke T (2016), ‘deepTools2: A next Generation Web Server for Deep-Sequencing Data Analysis.’, Nucleic Acids Research 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE (2012), ‘The changing privacy landscape in the era of big data.’, Molecular systems biology 8, 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Woo S and Hao K (2012), ‘Bayesian method to predict individual SNP genotypes from gene expression data.’, Science 44, 603–608. [DOI] [PubMed] [Google Scholar]
- Sweeney L (2000), ‘Simple Demographics Often Identify People Uniquely.’, Data Privacy Working Paper 3 URL: https://dataprivacylab.org/projects/identifiability/paper1.pdf [Google Scholar]
- The 1000 Genomes Project Consortium (2010), ‘A map of human genome variation from population-scale sequencing.’, Nature 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C and Stuart JM (2013), ‘The Cancer Genome Atlas Pan-Cancer analysis project.’, Nature Genetics 10, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium (2013), ‘The Genotype-Tissue Expression (GTEx) project.’, Nature Genetics 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal M, Frankhouser D, Sonzone C, Shields PG, Yan P and Bundschuh R (2019), ‘SMaSH: Sample matching using SNPs in humans.’, BMC Genomics 20, 1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf A, Angerer P and Theis FJ (2018), ‘SCANPY: large-scale single-cell gene expression data analysis’, Genome Biology 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, Huang T, Campbell JD, Lee E, Tu Z, Geraci MW, Powell CA, Schadt EE, Spira A and Zhu J (2014), ‘MODMatcher: multi-omics data matcher for integrative genomic analysis.’, PLoS Comput Biol. 10, e1003790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W and Liu XS (2008), ‘Model-based analysis of ChIP-Seq (MACS).’, Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S3: Linking NA12878 to a panel with 108 AFR individuals and one EUR individual, with and without NA12878 in the database, related to Figure 4.
Figure S2: gap values for the remaining seven individuals with different functional genomics assays, related to Figure 4
Figure S4: Visual representation of mapped fragments before and after converting the CIGARs to a pBAM file format, related to Figure 5. The insertions, deletions, soft and hard-clipping, and intronic reads are depicted. The noise that is added to the pBAM file to enhance privacy is also depicted in the fragments.
Figure S5: The difference between ChIP-Seq peak calling using BAM and pBAM as input for the fold change compared to control, the number of reads that pile up on the location of peak, and the location of the peak summit, related to Figure 6. Peak calling was performed using MACS2 (Zhang et al., 2008).
Figure S6: The distributions of the information leakage per variant in different levels of the data stack, related to Figure 7. Individual characterizing information (ICI) is calculated based on ref (Harmanci and Gerstein, 2016).
Figure S7: Potential variant leakage from MAPQ scores, related to Figure 5. As shown, the reads with potential large structural variants have smaller than expected MAPQ scores. An adversary can sort the MAPQ scores in a BAM file and guess the location of these structural variants that are mapped with low MAPQs.
Data Availability Statement
The information quantification and linkage attack code; pTools; intermediate data processing scripts; test BAM, PBAM and .diff files; file format specifications; a Jupyter notebook for single-cell analysis on BAM and pBAM and links to publicly available data used in this paper can be found at privaseq3.gersteinlab.org. The pTools code can also be found at https://github.com/ENCODE-DCC/ptools. There are restrictions to the availability of the sequencing data generated from coffee cups due to their containing information that could compromise the privacy of research participants and other individuals who may have contaminated the samples.