

Abstract
Pleiotrophin (PTN) is a potent mitogenic cytokine with a high affinity for the polysaccharide glycosaminoglycan (GAG). Although it is most strongly associated with neural development during embryogenesis and the neonatal period, its expression has also been linked to a plethora of other physiological events including cancer metastasis, angiogenesis, bone development, and inflammation. A considerable amount of research has been carried out to understand the mechanisms by which PTN regulates these events. In particular, PTN has now been shown to bind a diverse collection of receptors including many GAG-containing proteoglycans. These interactions lead to the activation of many intracellular kinases and, ultimately, activation and transformation of cells. Structural studies of PTN in complex with both GAG as well as domains from its non-proteoglycan receptors reveal a binding mechanism that relies on electrostatic interactions and points to PTN-induced receptor oligomerization as one of the possible ways PTN uses to control cellular functions.
Keywords: pleiotrophin, PTPRZ, neural regeneration, angiogenesis, glycosaminoglycan
1. Introduction
Pleiotrophin (PTN) is a small cationic protein with potent mitogenic and angiogenic activity. It has been associated with a wide range of important biological events, including neural regeneration, bone development, inflammation, cancer metastasis, and tissue repair. Its discovery was reported almost simultaneously by several groups around 1990 [1–4]. As a result, it was initially known by different names (HARP; HBBM; HBNF; HBGF8; NEGF1; OSF-1; HB-GAM; HBGF-8; HBNF-1, etc.). However, the name pleiotrophin is now the most widely adopted nomenclature in the literature. The word pleiotrophin is derived from pleiotropy, a reference to the phenomenon that a single gene can be related to several distinct phenotypes. This is indeed an apt name for PTN, which has, so far, been connected with physiological events ranging from ocular development to adipocyte differentiation and Alzheimer’s Disease. In the thirty years since its discovery, close to 800 articles have been published on the protein, providing detailed descriptions of its connections to the development of normal and pathological physiologies. Many excellent review articles on PTN [5–9] as well as an encyclopedic web page for PTN (http://atlasgeneticsoncology.org//Genes/PTNID41904ch7q33.html) are also in existence. The scope of PTN’s involvement in biology is too vast to cover comprehensively in one article. As a result, this chapter will focus mostly on recent developments as well as placing an emphasize on understanding the mechanism of the protein, which, although still not completely elucidated, is sufficiently clear to shed some insights into the principles behind PTN’s activities and reveals a complex picture of interactions among PTN and its receptors.
2. The ptn gene and its expression
2.1. Structure of the ptn gene
PTN is encoded by the ptn gene, which is approximately 116 kb in size and is located on chromosome seven in the human genome (band 7q33). Analysis of the latest tissue RNA sequencing results shows that there are as many as nine splicing variants of the ptn gene and up to seven exons in the gene [6], resulting in mRNAs approximately 1.6 kb long. However, the ORF is usually only 507 bases long. Figure 1 lists the six complete splicing variants identified by AceView [10]. Three of the variants encode the same 168-residue protein but differ in the sequence of the untranslated regions. One of the transcripts contains an extra coding exon at the 5’ end, thus contains an additional segment at its N-terminus. Another variant contains an alternative 3’ exon, resulting in a different C-terminus [11]. Because the additional residues are in the unstructured N- and C-terminal tails, it is unclear whether these proteins behave differently than the wild type protein. The protein sequence of PTN is highly conserved among mammals with sequence identity well over 90 % even in the unstructured regions of the protein. Analogs of PTN are also commonly found in other vertebrates and even in insects. It has been postulated that ancestral PTN existed as an antimicrobial peptide because of the cationic nature of the protein and acquired other functions as it gained the ability to interact with other receptors [12].
Figure 1.
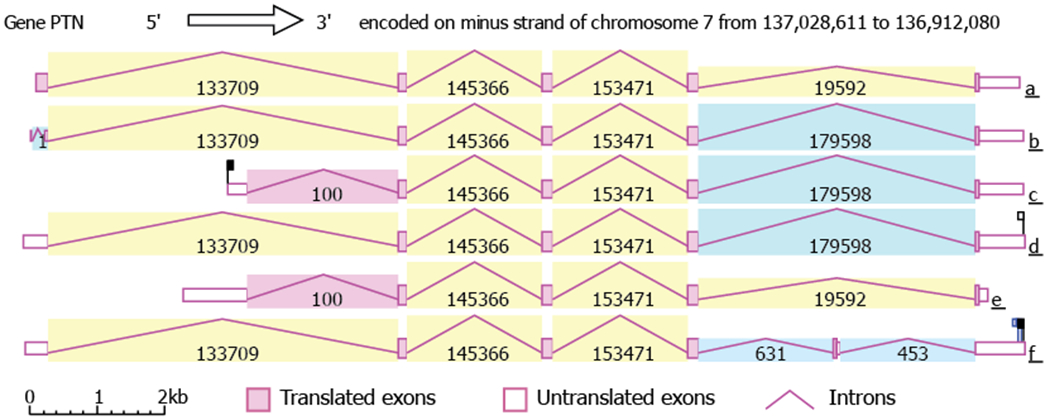
The ptn gene structure and six complete splicing variants identified by AceView. Introns are colored according to sequence identity such that identical introns have the same color. The number displayed with each intron is the number of reads for the intron. Variant a codes for a 222-residue protein; variants b, c, and d code for a 168-residue protein; variant e codes for a 167-residue protein; variant f codes for a 166-residue protein.
2.2. Expression of the ptn gene and its regulation
Although PTN is produced at detectable levels in adult human by many organs including brain, prostate, and testis, the highest expression level is found in the central nervous system (CNS) during embryonic and neonatal periods [1, 13, 14]. This is consistent with the role of PTN in promoting neural development. A more recent study increased our understanding of the temporal resolution of PTN expression by showing that both PTN mRNA and protein are present at high concentrations in the first 12 days after birth but fall dramatically by day 21 [15]. PTN expression level also increases in response to hypoxia, ischemia, and inflammatory triggers such as hydrogen peroxide [16–18]. A number of growth factors and signaling proteins are known to increase PTN expression level, including fibroblast growth factor 2 [19], platelet-derived growth factor [20], epidermal growth factor [21], and various hormones [22, 23]. Effect of mechanical force on the expression of PTN by the skeletal system has also been studied extensively. In particular, these studies showed that mechanical force loading significantly increases the expression of PTN in the skeletal system [24–28]. Finally, drugs such as doxorubicin are also known to increase PTN levels [29].
PTN is also perceived as having an oncogenic potential. PTN’s expression has been shown to be elevated in many different cancer cells, including cancers of breast [30, 31], prostate [32–34], pancreas [35], stomach [36], lung [37–39], colon [40, 41], and ovary [42, 43]. However, the biggest increase in PTN level is found in glioblastomas [44–46], making PTN a promising target of therapy for this type of cancer. In agreement with these observations, a more systematic expression profiling of a large collection of tumors and normal tissues carried out by GEPIA2 showed that PTN levels are most consistently elevated in glioblastoma multiforme and more benign low grade glioma (Figure 2).
Figure 2.
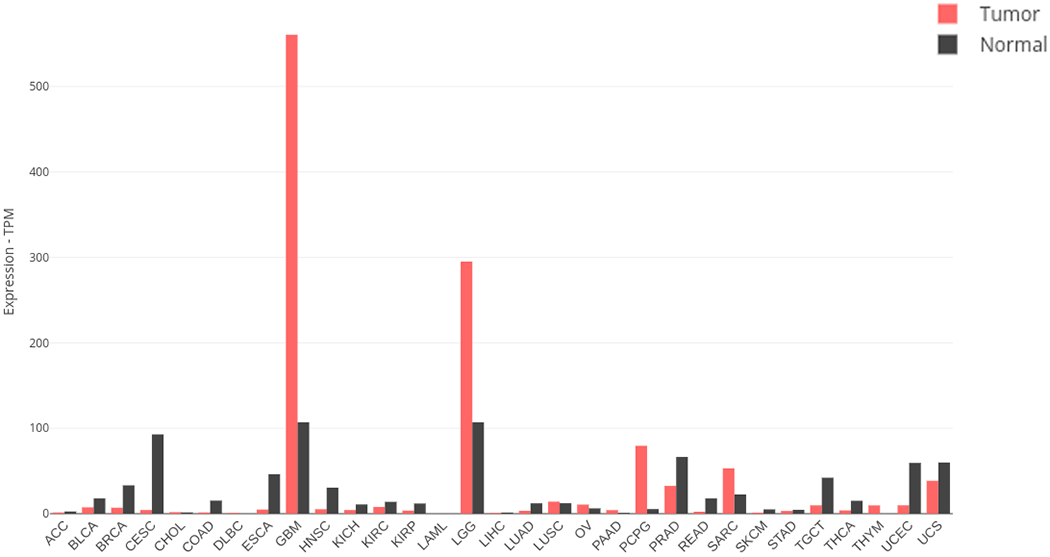
Expression levels of PTN in tumor and normal tissue. Data are tabulated by the GEPIA database (gepia2.cancer-pku.cn). Tumor types are ACC: adrenocortical carcinoma, BLCA: bladder urothelial carcinoma, BRCA: breast invasive carcinoma, CESC: cervical squamous cell carcinoma and endocervical adenocarcinoma, CHOL: cholangio carcinoma, COAD: colon adenocarcinoma, DLBC: lymphoid neoplasm diffuse large b-cell lymphoma, ESCA: esophageal carcinoma, GBM: glioblastoma multiforme, HNSC: head and neck squamous cell carcinoma, KICH: kidney chromophobe, KIRC: kidney renal clear cell carcinoma, KIRP: kidney renal papillary cell carcinoma, LAML: acute myeloid leukemia, LGG: brain lower grade glioma, LIHC: liver hepatocellular carcinoma, LUAD: lung adenocarcinoma, LUSC: lung squamous cell carcinoma, MESO: mesothelioma, OV: ovarian serous cystadenocarcinoma, PAAD: pancreatic adenocarcinoma, PCPG: pheochromocytoma and paraganglioma, PRAD: prostate adenocarcinoma, READ: rectum adenocarcinoma, SARC: sarcoma, SKCM: skin cutaneous melanoma, STAD: stomach adenocarcinoma, TGCT: testicular germ cell tumors, THCA: thyroid carcinoma, THYM: thymoma, UCEC: uterine corpus endometrial carcinoma, UCS: uterine carcinosarcoma, UVM: uveal melanoma.
Recently, several studies have also shown that PTN’s expression level can be regulated by micro interfering RNA (miR). In particular, miR-143, miR-182, miR-384, and miR-137 have all been shown to target PTN and reduce its expression [47–50]. These miRs have led to changes in adipocyte differentiation, endometrium development, cancer cell metastasis, and growth of hypertrophic scars. Clearly, miRs are important yet still under appreciated regulators of PTN expression.
3. Biological activities of PTN
PTN has been associated with numerous important physiological events including, hematopoietic stem cell maintenance, adipocyte differentiation, chondrocyte development, endothelial cell migration, and leukocyte activation. Despite these connections, the first study of PTN knockout mice indicated the animals developed no obvious defects [51]. The one exception is that the weight-bearing bone in young pups showed growth retardation and osteopenia [52]. However, subsequent analysis of these mice’s cognitive behavior showed PTN knockout mice also demonstrated defects in cognition characterized by cognitive rigidity, heightened anxiety, and behavioral reticence in novel spatial or social contexts. The mice also showed neuroanatomical abnormalities in the lateral entorhinal cortex [53]. In addition, PTN knockout animals show decreased resistance to addictive drugs such as amphetamine and ethanol [54, 55]. More recently, PTN knock out mice have also been shown to have lower body fat and better lipid metabolism [56]. These observations point to PTN’s essential roles in regulating CNS, bone formation, and lipid metabolism. The following sections outline PTN’s known activities in neural development, angiogenesis, inflammation, and other areas.
3.1. Neural development
PTN cDNA and protein were originally isolated from neural tissues because it is specifically expressed in the CNS during development and has potent neurite growth stimulating activity. Although it has been associated with many crucial biological phenomena since then, by far the most prominent role played by PTN is in the area of neural development. Its effects in the CNS have been demonstrated in the following areas:
Neurite growth. PTN is a potent stimulator of neurite outgrowth in CNS neurons [14, 57], which may explain the cognition abnormalities developed by PTN knockout mice and their inability to resist addictive drugs. Surprisingly, a recent study also showed that PTN can convert the neurite growth inhibitory effect of chondroitin sulfate proteoglycans (CSPG) into an activating effect and, most impressively, application of PTN to severed spinal cords of mice significantly enhanced the neural regeneration in the wound, making PTN a promising therapy for treating spinal cord injuries [58]. The authors of the study also showed that the effect is most likely mediated by glypican, a heparan sulfate proteoglycan (HSPG). In addition, PTN also seems to enhance the neurite stimulating activity of the peptide Y-P30, which is a peptide expressed during brain development and known to stabilize axon growth and dendritic spine maturation [59–62].
Neuron development. Several different types of cells in the CNS are now known to express PTN. One particular type is the neural stem cell (NSC), which is found in the subgranular zone of the hippocampus and the subventricular zone of the lateral ventricle in adults. These stem cells are responsible for producing newborn neurons throughout a person’s life. Recently, a study showed that NSCs secrete PTN into the niche to facilitate the maturation of the newborn neurons [63]. Without NSC-secreted PTN, the newborn neurons show defective neurite development and arborization. These defects were only avoided if PTN was supplied exogenously. Moreover, PTN secreted by amniotic epithelial cells is known to induce the differentiation of human umbilical cord mesenchymal stem cells into dopaminergic neuron-like cells [64]. Combined together, these studies demonstrate the importance of PTN in inducing maturation and differentiation of neurons.
Oligodendrocyte differentiation. Besides facilitating the development of neurons, PTN has also been shown to be important in the differentiation of oligodendrocytes in the CNS [65–67]. This effect is primarily attributed to the silencing of the phosphatase activity of receptor-type tyrosine phosphatase ζ (PTPRZ) in oligodendrocyte precursor cells and is consistent with the fact that PTN knockout mice develop delays in myelination [66]. PTPRZ is a CSPG and one of the most studied receptors of PTN. Interestingly, removal of chondroitin sulfate through applications of the enzyme chondrotinase ABC, or addition of other GAG-binding proteins, including midkine and IL-34, can have the same inhibitory effect on PTPRZ .
Sources of PTN in CNS. PTN is now known to be produced by many types of cells in the CNS. In particular, pericyte is known to produce PTN and ablation of pericyte resulted in significant neuronal deaths that can only be rescued by exogenous sources of PTN [68]. Neural stem cells and neural precursor cells in the subventricular zone also produce PTN, which plays a role in the maturation of neurons but can also serve to promote chemotaxis of gliomas cells into the subventricular zone [63, 69]. Finally, tumor-associated microglial cells in the CNS are also known to secrete PTN, leading to the promotion of glioma metastasis [45].
3.2. Angiogenesis
PTN has long been known to activate proliferation and migration of endothelial cells [70–73]. In particular, PTN has been shown to induce the formation of tube-like structures by endothelial cells in several different types of matrices in vitro [72, 73]. It is also known to induce angiogenesis in model systems such as embryoid bodies [74] and in chick embryo chorioallantoic membrane in vivo [73]. The main receptor for PTN’s angiogenic activity is believed to be PTPRZ [75]. However, PTN also binds αVβ3, the primary integrin expressed by endothelial cells. In fact, this interaction has been shown to be important to the PTN-PTPRZ signaling axis [76]. In addition, VEGF is also a ligand for αVβ3. This complex network of interactions can lead to moderation of both PTN’s and VEGF’s effects on angiogenesis [77]. Greater details on the effects of PTN in angiogenesis can be found in the reviews by Papadimitriou and co-workers [6, 9].
3.3. Inflammation and injury
PTN expression is elevated after tissue injury in numerous organs. These include brain [18, 78, 79], heart [80], liver [81], and bone marrow [82]. PTN’s effect on neural damage induced by drug use is especially well studied [83]. In these situations, PTN is responsible for promoting regeneration of damaged neurons and angiogenesis in the injured area to facilitate recovery. However, PTN is also known to activate macrophages. In particular, transgenic animal overexpressing PTN produced a more robust microglia response after lipopolysaccharide treatment [78]. Mechanistically, this activation effect maybe the result of PTN’s interactions with the integrin αMβ2 (Mac-1), a receptor vital to the activation and migration of both macrophages and microglial cells [84].
3.4. Bone development
The fact that PTN knockout mice showed lower bone density and growth in weight-bearing bones demonstrates the importance of PTN to bone development. In fact, PTN’s expression in osteoclasts played a crucial role in the protein’s discovery [4]. It is now known that PTN is found in cell matrices that are precursors to bone formation [85]. Interestingly, PTN is believed to promote the attachment of osteoclasts to these matrices through its C-terminal domain [86]. This indicates PTN may play a prominent role in recruiting osteoclasts during bone formation. PTN is also found to provide protection against bone mass loss during microgravity [28], and mechanical force loading increases PTN expression in the endplate of vertebrae under certain situations [87].
3.5. Adipocyte differentiation
PTN is expressed in significant quantities in adipocytes [88, 89]. It has also been shown to have effects on adipocyte differentiation. In particular, PTN inhibits preadipocyte maturation, but the receptor is not believed to be PTPRZ [89]. Interestingly, despite its inhibitory effects on preadipocyte differentiation, PTN does stimulate the proliferation of preadipocytes [90]. More recently, PTN knockout mice were shown to have significantly lower total body fat, enhanced lipolytic response, and improved thermogenesis. These changes were attributed to higher expression of deiodinase and uncoupling protein 1 in brown fat in the absence of PTN [56].
3.6. Mammary epithelial cell differentiation
PTN’s association with breast cancer also prompted interest in its role in mammary development and growth. Initial studies demonstrated that PTN is expressed in epithelial cells in both human and mouse breast [91, 92]. A more detailed analysis showed that, in mouse, expression of PTN is exclusive to the epithelial cells of the mammary glands [93]. Interestingly, the same study found expression of PTN decreased dramatically during lobular-alveolar differentiation, which is an essential step in lactation. In agreement with this observation, treatment of mice with anti-PTN antibody produced enhanced ductal development in the mammary glands by activating the ERK1/2 pathway, implying PTN suppresses epithelial cell differentiation in vivo. However, anti-PTN antibody treatment elicited faster proliferation and cell migration of epithelial cells in vitro [93]. These seemingly contradictory results can only be explained by differences in the environment in vitro and in vivo. It has also been observed that PTN expression levels are permanently depressed in mammary glands of mice after multiple pregnancies. The authors speculated that this maybe advantageous toward suppressing breast cancer, which often arises in immature precursor cell populations.
4. PTN in cancer
PTN’s expression is altered in a number of pathological situations, including ischemia, diabetes, and Alzheimer’s Disease. However, the most well studied PTN-associated pathological condition is cancer. Levels of PTN have been shown to be increased in a number of cancer cell lines and PTN overexpression is usually an indicator of poor prognosis. Despite these observations, PTN does not play a prominent role in all types of cancers and a deeper understanding of the role of PTN is needed before PTN-based therapy for cancer can be effective. Here we summarize PTN’s role in four types of cancers with whom it is strongly linked. Readers interested in additional details of PTN in these and other cancers should consult reviews by Papadimitriou et al. [6]
4.1. Glioblastoma
Although PTN is known to be overexpressed in a number of cancer cell lines, systematic comparison of normal and cancerous tissues’ gene expression patterns using the XENA database and GEPIA2 showed that glioblastoma is more likely to overexpress PTN than any other types of cancer (Figure 2). PTN overexpression has also been shown in a number of glioblastoma cell lines [46] and suppression of PTN level either through anti-PTN agents or RNAi has led to reductions in cancer cell proliferation, colony formation in glioblastoma cell lines in vitro and tumor growth in vivo [94, 95]. PTN may help with the growth of glioblastoma in several ways. In particular, PTN’s ability to promote angiogenesis has been shown to help vascularization in glioblastoma tumors [96]. Moreover, tumor-associated microglia are known to secret PTN, which promotes the growth and migration of glioblastoma. Finally, PTN secreted by neural precursor cells in the subventricular zone can facilitate the migration of glioblastoma cells into the subventricular zone, resulting in additional metastasis [69]. In all three instances, PTPRZ is believed to be a major receptor for PTN. This makes the PTN-PTPRZ signaling axis a promising target of glioblastoma therapy.
4.2. Breast cancer
PTN has also been shown to be overexpressed in several breast cancer cell lines [31, 97, 98]. However, a systematic examination of breast cancer samples showed PTN levels in many breast cancer samples are not higher than normal (Figure 2). Nevertheless, overexpression of PTN in some breast cancer models resulted in rapid growth and increased angiogenesis in the tumor [99]. Interestingly, one breast cancer cell line, MCF-7, showed no growth advantage when overexpressing PTN in vitro, but did show faster growth in vivo [100]. This demonstrates PTN’s role in breast cancer cells maybe context-dependent and heterogeneous. More recently, a study showed that the PTN-PTPRZ axis promoted chemoresistance in triple negative breast cancers. In particular, the study found that doxorubicin treatment activated PTN-PTPRZ signaling through the NF-κB pathway [101]. The authors also noted that PTN and PTPRZ expression form a positive feedback loop that can exaggerate the chemoresistant effect of PTN. This may explain why PTPRZ and PTN have very similar expression profiles in cancerous tissues.
4.3. Lung cancer
PTN is overexpressed in some lung cancer cell lines [102]. Once again, examination of a larger sample size using TCGA and GTEx data showed such a trend is not universal (Figure 2). Nevertheless, several studies have indicated that serum PTN level is significantly elevated in a majority of lung cancer samples [103, 104], making it a useful biomarker for the disease. In addition, menin, a lung tumor suppressor, is believed to exert most of its effect by suppressing the expression of PTN, resulting in reduced tumor cell proliferation and migration [37, 105]. Because lung cancer cells are also known to highly express PTPRZ, PTPRZ is believed to be the main receptor through which PTN acts in lung cancer.
4.4. Ovarian cancer
PTN is known to be expressed in ovary and female PTN/MDK double knockout mice are mostly sterile. PTN level is also elevated in a majority of both human and chicken ovarian cancer [42, 43]. In addition, PTN has been identified as a high-impact target for the treatment of ovarian cancer by an RNA-interference lethality screen study [43]. In particular, knockdown of PTN expression through RNAi resulted in significant apoptosis of an epithelial ovarian cancer cell line. Like in lung cancers, serum PTN has been shown to be a potential biomarker for the diagnosis of the disease [106].
4.5. Pancreatic cancer
According to the GEPIA2 database, PTN is overexpressed in pancreatic cancer samples. However, the overall level remains well below normal values found in the CNS. High PTN levels, together with high levels of syndecan-3, also correlates with perineural invasion, which is commonly found in pancreatic cancer, and poor prognosis [35]. Knocking down PTN levels in pancreatic cancer cell lines using ribozymes reduced cells’ proliferation rate, colony formation capability, and the size of tumors they form in animals [107]. A similar study using lentivirus to reduce PTN levels in pancreatic cancer cells also resulted in a significant reduction in dorsal root ganglion neurite outgrowth when the dorsal root ganglion cells are co-cultured with pancreatice cancer cells [108]. These studies point to PTN as a possible target for pancreatic cancer treatment.
5. Receptors for PTN
The highly positively charged nature of PTN means it has the potential to interact with numerous cell surface receptors that have an affinity for positively charged molecules. Because electrostatic forces are effective over a long range and require minimal geometric constraint, such a binding mechanism most likely underlies the promiscuity of PTN in receptor binding. So far, PTN has been shown to interact with many cell surface receptors, including PTPRZ , syndecans, nucleolin, neuropilin-1, integrin αVβ3 and αMβ2 (Figure 3). ALK has also been identified as one of the functional receptors early on [109]. However, some studies indicate PTN only influences ALK signaling in an indirect way [110]. A significant fraction of these PTN receptors are proteoglycans, which are extracellular glycoproteins containing the polysaccharide glycosaminoglycan (GAG). GAG is a linear polysaccharide composed of repeating disaccharide units. One of the sugar in the disaccharide unit is either a glucosamine (GlcN) or a galactosamine (GalN) while the other is usually a uronic acid. GAGs are classified based on the monosaccharides found in the repeating disaccharide units. Figure 4 shows the structures of some disaccharides found in different types of GAGs. GAGs containing disaccharide units of iduronate (IdoA) and N-sulfated glucosamine (GlcNS) are known as heparan sulfate (HS). Highly sulfated HS that are secreted without core proteins by mast cells are known as heparin (HP). GAGs containing disaccharide units of glucuronate (GlcA) and N-acetylgalactosamine (GalNAc) are known as chondroitin sulfate (CS). CS containing IdoA rather than GlcA are known as dermatan sulfate (DS). Finally, keratan sulfate (KS) contains galactose (Gal) and N-acetylglucosamine (GlcNAc) in its repeating disaccharide units. In addition to the normal carbohydrate backbones, most of the GAGs found in proteoglycans also contain extensive sulfation on both the amino sugar and the uronic acid sugar. HS and HP contain N-sulfated as well as O-sulfated GlcN. IdoA in HS and HP can also be O-sulfated. GalNAc and GlcNAc in CS, DS, and KS can be O-sulfated. These modifications give GAG molecules high negative charge densities, making them ideal receptors for cationic proteins such as PTN. It should be noted that there are non-sulfated GAG such as hyaluronan. However, PTN’s interactions with non-sulfated GAGs are not known to be biologically relevant.
Figure 3.
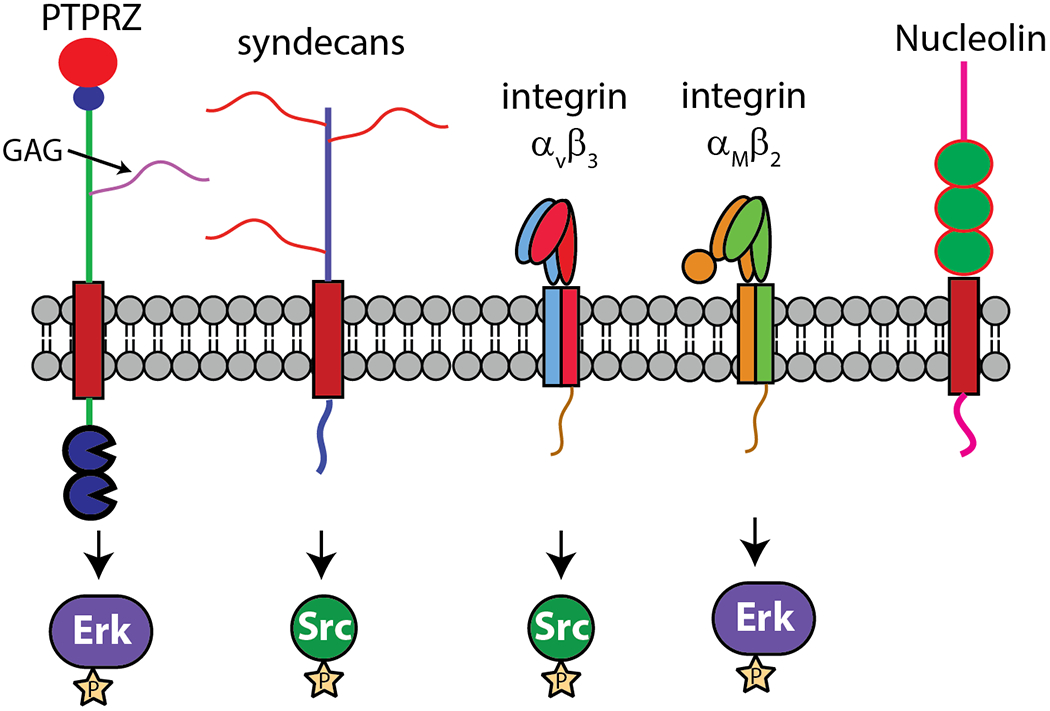
Major receptors of PTN and the downstream kinases they activate. PTPRZ and syndecans are proteoglycans, but αVβ3, αMβ2, and nucleolin do not contain GAG.
Figure 4.
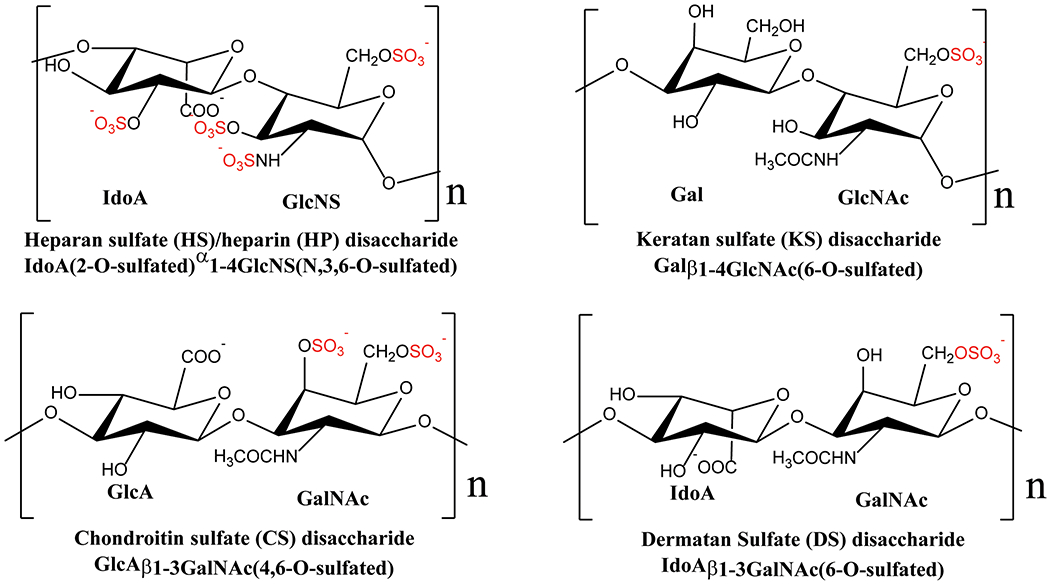
Chemical structures of fully sulfated disaccharides found in HS/HP, CS, DS, and KS. All common sulfation positions are shown even though most monosaccharides are not fully sulfated.
Although PTN is an avid binder of GAG, GAG-free receptors of PTN have also been identified. These include integrins αVβ3 and αMβ2, nucleolin, and neuropilin-1. In most cases, the mechanisms used by these non-proteoglycan receptors to recognize PTN are not absolutely clear.
5.1. PTPRZ
PTPRZ, or receptor-type tyrosine phosphatase ζ, is one of the first PTN receptors to be identified. It is a CSPG with two phosphatase domains in its cytoplasmic portion. However, only one of its phosphatase domains is believed to be an active phosphatase. The terminus of its ectodomain is composed of a carbonic anhydrase domain followed by a fibronectin type III domain. The rest of the ectodomain is likely to be unstructured and contains at least five GAG chains. A vast majority of the GAG chains are believed to be CS, but KS has also been found on PTPRZ recently [111]. Three isoforms of PTPRZ exist: PTPRZ-A is the full length form with a full sized ectodomain; PTPRZ-B is a short form with a truncated ectodomain containing no GAG or only a small amount of GAG; PTPRZ-S is a soluble form consisting of only the full-sized ectodomain and is released into the extracellular matrix. Interestingly, only PTPRZ-B stimulates neurite growth in neurons whereas both PTPRZ-A and PTPRZ-S inhibit neurite growth [112]. This maybe a consequence of the presence of CS on PTPRZ-A and PTPRZ-S since CSPG is known to inhibit neurite growth. Similar to PTN, PTPRZ is highly expressed in the brain, but mostly by glia, astrocytes, and oligodendrocytes. PTPRZ expression is also highly elevated in glioblastoma. Interestingly, GAG is not the only type of glycan modification PTPRZ is known to have. PTPRZ has also shown to contain O-mannosylation, which plays a role in the regulation of PTPRZ activity by carbohydrate-binding proteins such as galectins [113].
PTPRZ’s function and mechanism have been carefully dissected by the group of Masaharu Noda in a series of papers over the last two decades. PTPRZ is believed to exist as a monomer with an active phosphatase domain normally. The default monomeric state of PTPRZ is consistent with the fact that the large, highly charged GAG chains should prevent receptor oligomerization through electrostatic repulsion. In agreement with this observation, PTPRZ oligomerization in oligodendrocyte can be induced by either removing the GAG chains on the protein using chondroitinase ABC, or by adding proteins with a high affinity for GAGs, such as PTN [66]. The oligomerization, which is evidently driven by oligomerization of the ectodomain [114], triggers auto-inhibition of the phosphatase domain and increases phosphorylation of a number of proteins including β-catenin. Surprisingly, the soluble ectodomain of PTPRZ has a biological function independent of the phosphatase domain. In particular, a recent study conducted using PTPRZ knockout mice expressing a phosphatase-inactive variant of PTPRZ showed the ectodomain alone can control mice’s ability to respond to novelty and formation of aversive memories [115]. This is most likely the result of the presence of CS on the protein, which is known to be a potent regulator of neural development.
PTPRZ is also the receptor most often associated with some of the vital biological functions of PTN. Inhibition of PTPRZ’s phosphatase activity increases phosphorylation of several proteins, including β-catenin, ALK, c-Src, PKC, integrin αVβ3, p190RhoGAP, PI3K, and ERK1/2. Many of these proteins are connected with the activation of cell migration and proliferation. The ectodomain of PTPRZ binds many ligands. Its GAG chains are believed to be the target of PTN, midkine and many other GAG-binding proteins. However, GAG-binding proteins are by no means the only protein ligand for PTPRZ. The carbonic anhydrase domain at the N-terminus of PTPRZ is known to bind a number of cell adhesion molecules expressed in neurons, including the contactin family of proteins [116, 117]. These interactions with contactins are believed to be crucial to PTPRZ’s activity in facilitating myelination of neurons. Interestingly, PTN is also known to bind PTPRZ after its CS chains have been removed [118]. This indicates PTN may bind to the protein component of PTPRZ. However, the exact site of binding has not been identified. Consistent with that observation is the fact that PTPRZ expressed in cancer cells is often not glycosylated, but PTN is still capable of signaling through the unglycosylated form of the receptor [119].
5.2. Syndecans
Syndecans are a family of proteoglycans containing a single transmembrane helix, a large ectodomain, and a small but highly conserved cytoplasmic domain. There are four members in the family: syndecan-1 and syndecan-3 contain GAG chains on both membrane-distal and membrane-proximal ends of the ectodomain while syndecan-2 and syndecan-4 contain GAG chains only on their membrane-distal ends. GAG chains on syndecans can either be HS or CS, depending on the cell and tissue type. Syndecan-1 is mostly expressed in epithelial cells, syndecan-2 is expressed in cells derived from mesenchymal stem cells, syndecan-3 is expressed mainly in brain and cartilage, and syndecan-4 is expressed by many different types of tissues [120]. Syndecan-3 was the first PTN receptor identified and is associated with PTN’s neurite promoting activity [121]. However, all four syndecans have now been reported to interact with PTN either directly or as a complex with other binding partners [59, 122]. The cytoplasmic domain of a syndecan is rather small and is made up of two highly conserved regions (C1 and C2) separated by a variable region. Segment C1 is known to bind ezrin while C2 is known to bind proteins with PDZ domains [123]. Syndecan’s interactions with PTN is mostly through its GAG component, and PTN binding is suspected to induce oligomerization of the receptor and change its interactions with other receptors on the surface, thereby changing intracellular signaling. PTN’s interaction with syndecans has been shown to be enhanced by the peptide Y-P30, resulting in enhanced axonal growth [61]. In addition, PTN’s interactions with syndecan play an important role in cancer. In particular, PTN’s binding of syndecan-3 can activate focal adhesion kinase and ERK1/2 in prostate cancer, leading to greater cancer cell metastasis [124]. Finally, the PTN-syndecan signaling axis also contributes to perineural invasion in pancreatic cancer, which correlates strongly with poor prognosis [35, 125, 126].
5.3. Nucleolin
Nucleolin is a protein mainly found in the nucleolus and is involved in the maturation of ribosomes. However, nucleolin has also been found on the cell surface of normal as well as cancer cells. Although the transport mechanism that allows nucleolin to be displayed on cell surfaces is not well understood, it is believed to depend on pathways that are independent of the traditional endoplasmic reticulum/Golgi apparatus export system. In particular, the export of nucleolin is dependent on intact actin cytoskeletons and other cytoplasmic factors [127] and involves intracellular vesicles [128]. Once on the cell surface, it interacts with both PTN and midkine with low affinity [129]. One hypothesis is that shuttling of nucleolin between the cell surface and the nucleus helps to deliver PTN into the nucleus where it antagonizes bromodomain proteins and produces changes in gene expression patterns [130]. Nucleolin also forms a complex with the integrin αVβ3 and PTPRZ [131], both of which are also known to bind PTN. PTN’s interactions with these three receptors may strengthen the interactions among them, leading to the activation of intracellular signaling networks.
5.4. Integrin αVβ3
Integrins are heterodimeric cell surface receptors that play important roles in cell migration, transformation, and adhesion. They connect extracellular environments with intracellular signaling pathways and are vital to tissue development, cancer metastasis, inflammation, and various other physiological phenomena. Integrins are made up of an α subunit and a β subunit. There are 18 different α subunits and 8 different β subunits in mammals. The pairing of different subunits creates 24 unique integrins expressed selectively in various cell types. PTN’s close analog midkine is known to bind a number of β1 integrins. However, αVβ3 was the first integrin known to bind PTN [76]. αVβ3 is highly expressed in endothelial cells and plays a crucial role in angiogenesis. Not surprisingly, PTN-induced cell migration is highly dependent on αVβ3 and PTN can bind to αVβ3 through the β3 I-domain. In particular, a peptide corresponding to residues 177 to 184 of β3, a segment in β3 I-domain known to modulate ligand binding, can completely eliminate the PTN-induced cell migration, indirectly validating the importance of the segment in PTN interactions [76]. In addition, treatment of human umbilical vein endothelial cells with PTN resulted in phosphorylation of the cytoplasmic domains of β3, a sign of integrin activation. Interestingly, αVβ3 forms a multi-receptor complex with both PTPRZ and nucleolin. Moreover, PTPRZ is necessary for the phosphorylation of the β3 cytoplasmic domain [76]. This demonstrates the potential importance of PTN in activating signaling through this complex network of receptors.
5.5. Integrin αMβ2 (Mac-1)
Integrin αMβ2 (Mac-1, CR3, CD11b/CD8) is expressed mostly in myeloid cells, especially macrophages and neutrophils. It is an essential receptor needed for migration and activation of these cells. It is also one of the most promiscuous integrin receptors with a broad specificity for ligands [132–134]. A recent study showed that the ligand-binding domain of Mac-1, the αM I-domain, has specificity for basic peptides depleted in acidic amino acids [133]. The result from the study enabled the discovery of several other Mac-1 ligands, including PTN [84]. Immobilized PTN was shown to trigger adhesion, migration and spreading of macrophages in a Mac-1 dependent fashion. These effects were completely eliminated by the anti-Mac-1 antibody. Treatment of Mac-1-expressing cells with soluble PTN also activated the kinase ERK1/2. In addition, the structured domains of PTN were shown to be the binding site for αMI-domain, with the CTD conferring a higher Mac-1 affinity than the NTD. αMI-domain’s binding site for PTN and other cationic ligands has not yet been identified. However, PTN is a highly basic peptide whose interactions with receptors are most likely dominated by electrostatic interactions. This implies that highly electronegative regions of αM I-domain should be ideal binding sites for PTN. Interestingly, the most electronegative site in αMI-domain is around its metal ion dependent adhesion site (MIDAS), a metal-binding site that is also important to ligand binding. Although the negative charges in MIDAS are largely neutralized when the metal is bound, αMI-domain’s metal affinity is low [135]. As a result, a large fraction of αMI-domain should remain metal-free under physiological conditions.
6. Structure of PTN
PTN is expressed as a 168-residue protein. However, the first 32 residues at the N-terminus is the secretory signal and are removed in the mature form. The 136-residue mature PTN is normally secreted into the extracellular space via the normal secretory pathways in cells. Interestingly, histology studies of PTN expression have also identified the presence of PTN in the nucleus and cytoplasm of chondrocyte [9], glioma [129], and cardiomyocyte [136]. The exact physiological significance of these intracellular PTN is unclear. However, nuclear localizations maybe the result of interactions between PTN and nucleolin, which is known to transport ligands from the cell surface to the nucleus. Proteoglycans are also found in the nucleus. Given PTN’s high affinity for proteoglycans and the fact that the same organelles are used to process both PTN and proteoglycans, it is also possible that PTN can be transported into the nucleus together with proteoglycans.
PTN contains no N-glycosylation sequon and there is no report of glycosylation or other post-translational modifications in the protein. The protein is highly basic with 36 positively charged amino acids and only 17 negatively charged amino acids, resulting in a predicted pI of 9.66. The protein contains two small structured domains surrounded by long unstructured termini. Both structured domains are about forty residues long and adopt the same thrombospondin type-1 repeat (TSR) fold. TSR domains are small domains consisted of a simple three-stranded β-sheet stabilized by disulfide bonds. The hallmark of TSR domains is the existence of extensive cation-pi interactions created by aromatic residues on one strand intercalated by basic amino acids on a neighboring strand. Cation-pi interactions are attractive forces that allow aromatic rings to have favorable interactions with positively charged functional groups. Such an arrangement is essential to the folding of the protein because of the lack of a strong hydrophobic core in such a small domain. In the N-terminal TSR domain of PTN (NTD), these cation-pi interactions are created between W18 and W20 on one strand and R35, R39 as well as K49 on other strands (Figure 5). In the C-terminal TSR domain of PTN (CTD), these cation-pi interactions are created between W74 and F71 on one strand and R86 and K84 on another strand. Besides cation-pi interactions, the structure of PTN is further stabilized by the five disulfide bonds that covalently link different strands. It is important to point out that both cation-pi interactions and disulfide bonds are necessary but not sufficient on their own to produce the correctly folded structure. This is most likely because the cation-pi interactions are required for the initial folding, but disulfide bonds are needed to stabilize the fold. Although there is no significant hydrophobic core in PTN, a small hydrophobic cluster is found in CTD that contains residues F71, V103, and I105. Interestingly, residue F63 from the linker between NTD and CTD is also part of the hydrophobic cluster. This allows the linker to have significant interactions with the CTD and may explain the lack of flexibility in the linker. One of the most notable features of the TSR domains in PTN is that opposite faces of each domain have drastically different electrostatic surface potentials. This is because the folding of the protein has allowed basic amino acids to cluster on a single face, creating strong basic patches on one side of the domain. These patches are crucial to the interactions of PTN with many of its receptors. However, these patches are not the only basic segments in PTN. Both the N-terminus and the C-terminus of PTN are rich in basic amino acids. The C-terminus, in particular, is known to have potent biological activity [137, 138]. In addition, although the termini are not structured, their sequences are highly conserved among different species. This provides additional support to the theory that the termini play important roles in PTN’s activity.
Figure 5.
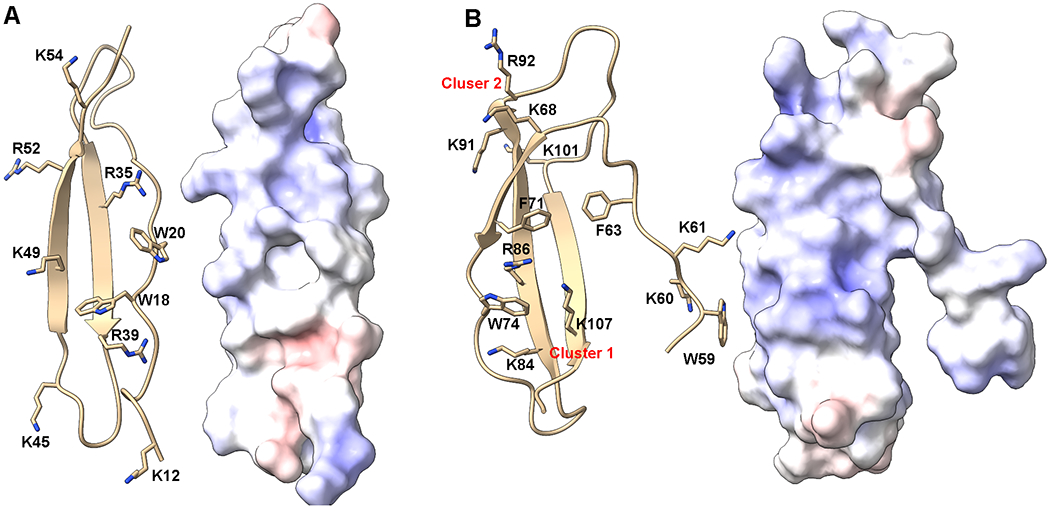
Structure of PTN. Ribbon representations and electrostatic surface potentials of NTD (A) and CTD (B) of PTN. Sidechains of positively charged residues that may be involved in electrostatic interactions with receptors as well as aromatic residues that may be involved in cation-pi interactions are shown and labeled. Blue surfaces represent positively charged patches while red surfaces represent negatively charged patches.
In the CTD, two clusters of basic amino acids can be identified (Figure 5). Residues K68, K91 and R92 form one cluster near the C-terminal tail. This cluster is commonly referred to as cluster 1. Residues K84, R86 and K107 form another cluster on the opposite side of the domain. This cluster is commonly referred to as cluster 2. In the NTD, several basic amino acids can be found on the same side of the NTD, giving the face considerable electropositivity. It should be noted that the aromatic rings of W74 and F71 intercalate cluster one basic amino acids while W18 and W20 in the NTD intercalate the basic amino acid cluster on the NTD. These interactions between the basic amino acids and the aromatic amino acids most likely play an important role in the folding of the protein. However, it is also highly likely that these interactions compete with the binding of GAG to these amino acids. The effect of the aromatic amino acids on GAG binding of PTN is yet to be investigated.
As a highly basic peptide, auto-oligomerization of PTN is unlikely because of the strong electrostatic repulsion between the molecules. However, both PTN and the homologous cytokine midkine have been shown to exist as dimers in vivo [139, 140]. The mechanism of midkine oligomerization is well studied and is believed to be triggered by GAG-binding followed by covalent linkage through transglutaminase activity [139]. There is yet no study on the oligomerization mechanisms of PTN. However, given the similarities between PTN and midkine, a comparable mechanism may also apply to PTN. The physiological significance of such oligomerization has remained unclear. It is possible that this is no more than a consequence of GAG-induced aggregation followed by promiscuous transglutaminase activity. However, it is also likely that such oligomers may have much higher affinities for its receptors than the monomeric forms as a result of multivalency. In such cases, the oligomerization maybe an important part of PTN activity.
7. Interactions of PTN with its receptors
7.1. PTN’s Interactions with GAG
One of the most important receptors for PTN is the polysaccharide GAG, which is crucial to PTN’s interactions with proteoglycans such as PTPRZ and syndecans. Two different types of GAGs are found in PTPRZ: CS, which are consisted of GalNAc-GlcA disaccharide units, and KS, which are consisted of GlcNAc-Gal disaccharide units. Syndecans contain both CS and HS. The sizes of these GAG chains are highly heterogeneous. These GAGs are also sulfated in a template-independent manner, making their structures complex and polyanionic. PTN’s interactions with these GAGs have been studied intensely [141–143]. It is now known that PTN binds the native GAG chains with extremely high affinity (Kd ~ 1 nM) [141]. However, the affinity decreases greatly once the size of the chain has been reduced to less than ten monosaccharides [142]. Such a trend is common among GAG binding proteins and is a consequence of the multivalent nature of these interactions. Solution NMR studies of PTN-GAG interactions show that residues in cluster 2 of PTN CTD are highly perturbed by the presence of these oligosaccharide ligands, as are residues in the linker between CTD and NTD. However, residues in cluster 1 of CTD, NTD, and the unstructured tails are not perturbed as strongly [143]. In addition, different types of GAGs produce the same perturbation pattern on PTN. Specifically, HP, CS, and DS hexasaccharides all induce NMR signal chemical shift changes in the same directions in the 15N-edited heteronuclear single quantum coherence (HSQC) NMR spectrum of PTN (Figure 6). 15N-edited HSQC spectrum is commonly used to identify chemical shifts of backbone amide nitrogen and hydrogen on a residue-specific basis. Because chemical shifts of backbone amide nitrogens and hydrogens of a protein are very sensitive to changes in the magnetic environment as a result of ligand binding, ligand-induced changes in these chemical shifts are usually seen as indications that the atoms are involved in ligand binding. The fact that all GAGs induced the same changes in the NMR spectrum of PTN implies PTN’s interactions with GAG are similar for all GAGs and not specific to GAG types. However, there are significant differences in PTN’s affinity for different types of GAG. In particular, PTN has the highest affinity for HP oligosaccharide (Kd ~ 20 µM for hexasaccharides) while its affinity for CS and DS are weaker (Kd ~ 300 µM for CS and DS hexasaccharides). This correlates nicely with the sulfation density on these GAGs and shows affinity is determined mostly by the sulfation density of the GAG. In addition, these NMR data also allowed us to measure the affinity of each domain for GAG. Specifically, the NTD’s affinity for GAG is weaker than that of CTD (Kd of ~ 170 µM in NTD vs ~ 20 µM in CTD for HP hexasaccharides). This is also consistent with differences in electrostatic surface potential observed on the domains.
Figure 6.
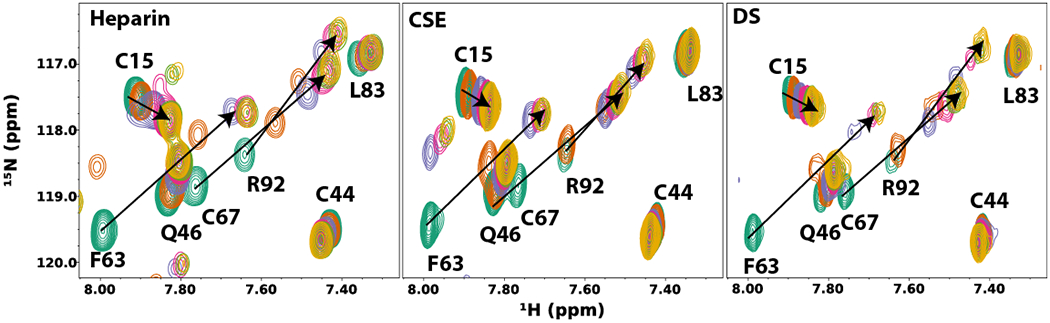
HP, CS, and DS hexasaccharide-induced changes in the 15N-edited HSQC spectrum of PTN. HSQC spectra of PTN in the presence of different concentrations of GAG hexasaccharides were acquired and superimposed. The concentration of the PTN was 0.1 mM. The concentrations of GAG hexasaccharides were 0 (dark green), 0.1 (brown), 0.2 (blue), 0.4 (pink), 0.6 (light green), and 0.8 (yellow) mM, respectively. All GAGs induced similar changes in the chemical shifts of PTN’s backbone amide nitrogens and hydrogens.
Because CTD and NTD are linked by an unstructured linker, one central question is whether the two domains co-operatively bind the same GAG chain or bind GAGs independently. To study this, we have purified truncated PTN containing either NTD or CTD. The domains remain folded and their 15N-edited HSQC spectra are superimposable with the intact PTN. This result implies the domains fold and behave independently. Titrations of the domains with HP hexasaccharides showed the chemical shift changes induced by the GAG oligosaccharides on the domains are virtually identical to those observed in intact PTN (Figure 7). This is consistent with the fact that interactions of PTN with GAG are preserved even when the domains are separated. In addition, these chemical shift changes can also be used to calculate the Kd of interaction if the plot of ligand-induced chemical shift changes at different ligand concentrations are fitted to the equation , where Δδ(L) is the measured chemical shift change at oligosaccharide concentration L, Δδmax is the maximum change, P is the PTN concentration, Kd is the dissociation constant of interaction. Performing such fittings on chemical shift changes of residue Q52 in NTD and residue C67 in CTD showed the HP hexasaccharide affinities of NTD and CTD are not changed by the removal of the other domain. In other words, the domains can bind hexasaccharides independently. However, whether this is true for longer GAGs remains to be examined. In particular, one concern is that a hexasaccharide is too short to connect both domains simultaneously since each hexasaccharide is only about the size of the domain and incapable of binding two domains simultaneously. The report that GAG oligosaccharides containing ten or more monosaccharides have a dramatically higher affinity for PTN than shorter GAG oligosaccharides [144] is consistent with the hypothesis that longer GAG oligosaccharides maybe sufficiently large to bind two domains simultaneously.
Figure 7.
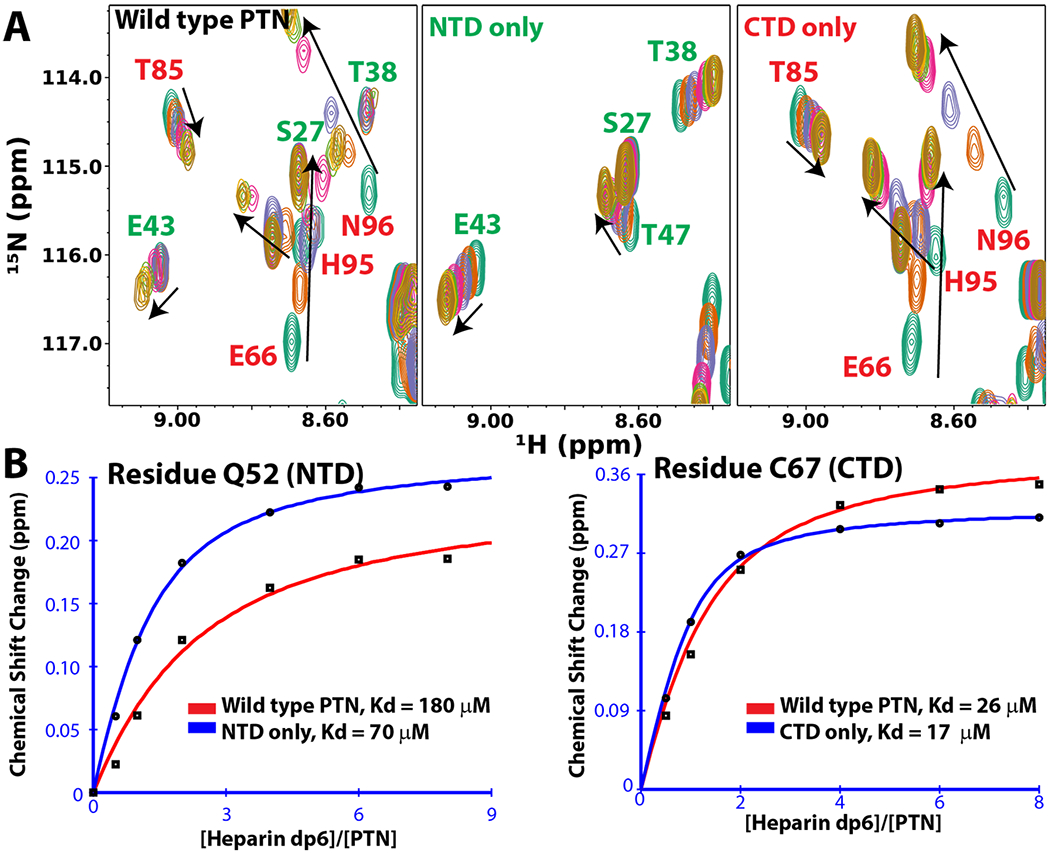
PTN domains bind HP hexasaccharides (dp6) independently. A) HP hexasaccharides induced chemical shift changes in the 15N-edited HSQC spectrum of wild type PTN, NTD, & CTD. HSQC spectra of PTN variants in the presence of different concentrations of HP hexasaccharides were acquired and superimposed. The concentration of the PTN was 0.1 mM. The concentrations of HP hexasaccharides are 0 (dark green), 0.1 (brown), 0.2 (blue), 0.4 (pink), 0.6 (light green), and 0.8 (yellow) mM, respectively. Separating the domains did not change the chemical shifts of the amide nitrogens and hydrogens in each domain. This indicates the domains can bind hexasaccharides independently. B) Plots of HP hexasaccharides-induced chemical shift changes in amide nitrogens and hydrogens at different oligosaccharide concentrations for residues Q52 in NTD and C67 in CTD. The curves can be fitted to the following equation to obtain the value of Kd: , where Δδ(L) is the measured chemical shift change at oligosaccharide concentration L, Δδmax is the maximum change, P is the PTN concentration, Kd is the dissociation constant of interaction.
PTN’s interactions with GAG have also been studied with structurally homogeneous HP hexasaccharides conjugated to paramagnetic nitroxides. These ligands can be easily synthesized by reductively aminating the reducing end of the oligosaccharides with 4-amino TEMPO [143]. The paramagnetic tag induces additional NMR signal relaxations in nearby atoms in a distance-dependent manner. This effect is commonly referred to as paramagnetic relaxation enhancement (PRE). PRE decreases intensities of NMR signals and the magnitude of the decrease can be used to estimate the distance between the paramagnetic tag and the atoms on the protein, thereby yielding information on the binding site and binding orientation of the ligand. Perturbation of PTN with paramagnetically labeled CS hexasaccharides produced NMR signal changes mostly around cluster 2, the linker and the end of CTD [143]. Because the paramagnetic tag on the oligosaccharide is only conjugated to its reducing end, this result shows that oligosaccharides can bind to PTN in at least two orientations or at two binding sites, which implies that PTN’s interactions with GAGs involve multiple binding conformations. Similar experiments have also been carried out using a suite of structurally defined HP hexasaccharides which differ in the total number as well as the location of their sulfation modifications (Figure 8). Not surprisingly, these hexasaccharides induced similar PRE perturbation patterns and had only minor differences in their affinity for PTN. These results provide additional proof that PTN’s interactions with GAGs are very dynamic, flexible, adaptable, and insensitive to small differences in sulfation density and sulfate positions.
Figure 8.
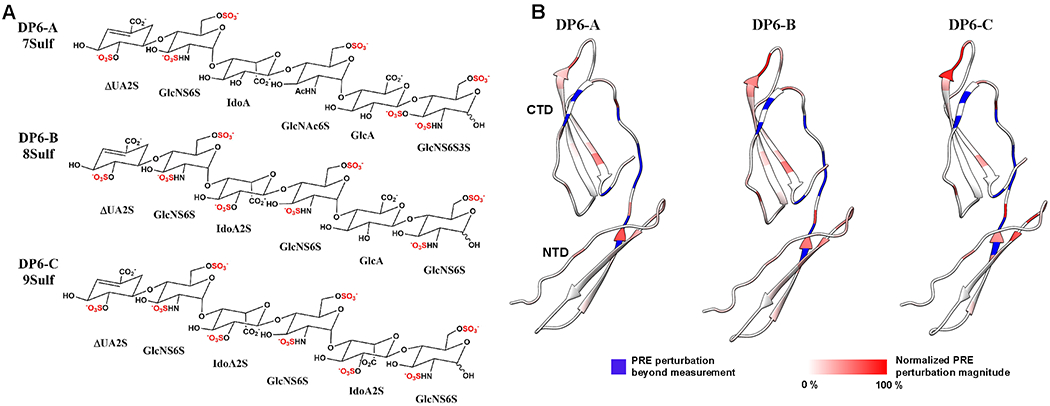
HP hexasaccharides-induced PRE perturbations in PTN. A) structures of the three structurally defined HP hexasaccharides used to create the paramagnetically tagged oligosaccharides used to study PTN-GAG interactions. They contain 7, 8, and 9 sulfates, respectively. B) Ribbon representations of PTN colored according to the magnitudes of PRE perturbations produced by each of the paramagnetically labeled hexasaccharides. Residues colored in blue experience large PRE that are beyond measurement. Residues that experience quantifiable PRE perturbations are colored with a red gradient.
Compared to the structured NTD and CTD, the C-terminal tail showed little perturbation either in chemical shifts or in PRE. However, several studies have indicated that the C-terminal tail plays a role in PTN’s biological activity. In particular, the C-terminal tail was shown to be necessary for PTN-dependent signaling involving PTPRZ [46, 138]. The NMR data seemed to contradict these functional studies. This maybe because most of the NMR studies were carried out with HP, which is a highly sulfated variant of GAG with significant physical differences compared to CS, the most common GAG found on PTPRZ. The effect of GAG type on C-terminal tail-mediated GAG binding was demonstrated in an experiment conducted by Ryan et al.[143] In this study, solid-phase binding assays were carried out with CS-A, whose disaccharide units are consisted mostly of GlcA and 4-O-sulfated GalNAc, and CS-E, whose disaccharide units are consisted of GlcA and 4,6-O-disulfated GalNAc. This assay showed that although the C-terminal tail is inconsequential to the binding of PTN to CS-E, the GAG with high sulfation density, it is necessary to maintain stable interactions between PTN and CS-A, the GAG with low sulfation density. Specifically, truncated PTN missing the C-terminal tail showed no binding to CS-A at all while the same mutant had an nanomolar affinity for CS-E. Given the fact that greater than 90 % of disaccharide units in PTPRZ GAG chains are believed to be CS-A, the essential nature of PTN’s tail to its interactions with CS-A explains the observation that PTN without the C-terminal tail is unable to initiate signaling through PTPRZ.
7.2. The mechanism of PTN-induced inhibition of the PTPRZ phosphatase activity
Binding of PTN to PTPRZ has been shown to increase phosphorylated forms of several PTPRZ ligands [75, 145]. This is consistent with PTN-induced inhibition of PTPRZ’s phosphatase activity. The mechanism of the inhibition was elucidated by a series of studies by the group of Masaharu Noda. Specifically, similar to other tyrosine phosphatase receptors, the homodimerization of PTPRZ leads to auto-inhibition of its phosphatase activity [75]. Microscopy studies indicate that the treatment of PTPRZ-expressing cells with PTN leads to large scale clustering of PTPRZ [65, 75]. This triggers the dimerization and auto-inhibition of PTPRZ and increases the phosphorylation level of cellular proteins [114]. How PTN induce clustering of PTPRZ has also been studied by Noda and colleagues. One possibility was that, because PTPRZ contains several GAG chains and PTN contains two domains that may bind to different chains independently, PTN may crosslink different PTPRZ and induce clustering. However, not all GAG-binding proteins aggregate proteoglycans and there is no evidence that PTN can crosslink PTPRZ receptors [146]. In addition, an insightful study conducted by Kuboyama et al. showed that the removal of CS chains alone was sufficient to trigger PTPRZ clustering [66]. This indicates PTPRZ’s core protein has an innate affinity to homo-oligomerization that is inhibited by GAG chains attached to it, which presumably prevents oligomerization through strong electrostatic repulsion. However, strong GAG-binding proteins, such as PTN, effectively neutralizes the repulsion, leading to clustering. Despite these important results, many questions regarding PTN’s interaction with PTPRZ remain unanswered. In particular, PTPRZ-B, the short form of PTPRZ that is believed to be GAG free, does not cluster on its own and still required PTN to trigger signaling [101]. The reason behind PTPRZ’s intrinsic ability to oligomerize also remains unexplained. In addition, the soluble form of PTPRZ, PTPRZ-S, has its own biological function in controlling neural development [115]. Because it is not attached to any cell, its activity is independent of the activation of intracellular signaling pathways. How PTN regulates neural development through interactions with PTPRZ-S is still not well understood.
7.3. PTN’s interactions with αMβ2 (Mac-1)
Besides proteoglycans receptors, PTN is also known to bind non-proteoglycan receptors such as nucleolins and integrins αMβ2 (Mac-1) as well as αVβ3. The mechanisms by which PTN recognize these receptors are largely unclear. A segment in the β I-domain of αVβ3 has been shown to inhibit the binding of PTN to αVβ3, therefore it may be part of the binding site on αVβ3 [76]. Interactions of PTN with Mac-1 is better understood compared to the others. NMR studies have shown that PTN’s structured domains play an important role in binding Mac-1. In particular, both structured domains can bind Mac-1, but CTD has a higher affinity for Mac-1 than NTD [84]. The unstructured tails may also have weak interactions with Mac-1 because they also contain sequence motifs that are recognized by αMI-domain. Mac-1’s binding site for PTN is also better characterized than other PTN-binding integrins. Mac-1 binds more than 90 % of its ligands through the αM I-domain, which is a Rossmann-fold domain positioned at the end of the integrin, allowing it to contact extracellular ligands easily. Like I-domains from other integrins, the domain has a metal ion-dependent adhesion site (MIDAS), which is an electronegative pocket capable of binding divalent cations such as Ca2+ or Mg2+. In a conventional ligand-binding scheme, an acidic amino acid in the ligand will coordinate the divalent cation in MIDAS, allowing it to bind to the domain. However, αM I-domain’s metal affinity is significantly weaker than other α I-domains ( Kd ~ 1 mM) [135]. Such an affinity leaves approximately half of the I-domain in the metal-free state at physiological concentrations of these metals. This weaker metal affinity most likely lies at the root of Mac-1’s unique ligand specificity, which has a high propensity for binding ligands rich in basic amino acids while eschewing acidic amino acids [133]. Such a unique ligand preference completely contradicts the canonical ligand-binding mechanism of α I-domains, but is most likely the result of a metal-free MIDAS, whose acidic amino acids are available to bind basic ligands in the absence of the metal. It should also be noted that, besides a weaker metal affinity, αM also possesses two extra acidic amino acid clusters compared to αL I-domain, which does not bind cationic ligands despite having high sequence homology with αM I-domain. PTN, being a highly basic protein, most likely binds to Mac-1 in a similar manner as the other ligands. In particular, several sequences in PTN have been identified as having a high potential of binding αMI-domain and inhibitory antibody targeting αM I-domain was able to eliminate adhesion of Mac-1-expressing cells to immobilized PTN [84]. What is less clear is how the activation of Mac-1 changes its affinity for cationic ligands. In particular, activation of Mac-1 changes the conformation of αMI-domain’s MIDAS, dramatically increasing the domain’s affinity for ligands that rely on metal-mediated interactions. However, because PTN and other cationic ligands do not rely on metal in their interactions with Mac-1. It is unclear how activation of the domain would affect their interactions. More interestingly, αM I-domain’s affinity for Mg2+ is dependent on its conformation. Specifically, αM I-domain in the active conformation binds Mg2+ ten times stronger than αM I-domain in the inactive conformation [135]. Because MIDAS is postulated to be an important PTN-binding site, whether this change in metal affinity leads to changes in PTN affinity still needs to be examined.
8. Differences between PTN and midkine
Midkine and PTN are homologous proteins sharing ~ 50 % sequence identity. The two proteins are the only members of this cytokine family and have almost identical three-dimensional structures [143, 147, 148]. Moreover, mice lacking either midkine or PTN do not display observable defects [51]. This implies midkine and PTN maybe mutually compensating. Interestingly, PTN expression in midkine deficient mice is elevated significantly, but not vice versa [149]. The two proteins have been shown to have similar biological activities. For instance, both are known to play important roles in development, tissue repair, and cancer. However, expression profiles of the two proteins in GEPIA2 are different. Unlike PTN, which is mostly expressed in the brain, midkine is mostly expressed by myeloid cells, cervix, gut, and ovary. Interestingly, GEPIA2 analysis of the gene expression pattern of PTN and midkine showed that midkine is overexpressed in far more cancers than PTN [150]. This shows midkine maybe more important in pathogenesis of cancer, but PTN maybe more important in regulating neural development. Developmentally, midkine expression is known to be increased by retinoic acid and its expression is also closely related to ocular development, which PTN also plays a role in.
Given the structural similarities between PTN and midkine, it should not be a surprise that the two proteins also share common receptors. Both have a strong affinity for GAGs and both bind proteoglycan receptors such as syndecans and PTPRZ. Both also bind integrins. However, midkine has been identified as a ligand for β1 integrins such as α4β1 and α6β1 [151]. These interactions are closely associated with midkine’s activity in activating monocytes during inflammation and angiogenesis [152, 153]. Interestingly, midkine has not been shown to bind αVβ3 or αMβ2, the two integrins known to bind PTN. The combination of midkine’s interactions with these monocyte integrins and its expression by myeloid cells point to the important role midkine plays in regulating inflammation. Midkine also binds to the protein LDL receptor related protein 1 (LRP1), which is often expressed by leucocytes. Midkine’s interactions with LRP1 is believed to lead to activation of β2 integrins and activation of leukocytes during inflammation [154]. Given the fact that many midkine receptors are involved in modulating inflammation, it should not be a surprise that midkine overexpression is associated with many chronic inflammatory diseases, including atherosclerosis and diabetic nephropathy [155, 156]. In comparison, PTN’s role in inflammation is less prominent.
Consistent with the higher expression levels of midkine found in many tumors, midkine is associated with poor prognosis in many cancers. In particular, midkine promotes tumor growth and invasion by enhancing tumor angiogenesis [157], and midkine is a mediator of chemotherapy resistance in gastric cancer models [158]. These activities are similar to those reported for PTN. The fact that midkine is overexpressed in more cancers than PTN is most likely the result of differences in the promoters of the two genes. In particular, the midkine gene promoter is likely to be more responsive to the presence of other oncogenic genes than the ptn gene. Interestingly, unlike PTN, the midkine expression profile does not have any correlation with the expression profile of PTPRZ, the receptor shared by both cytokines.
Overall, despite subtle differences in the structures of midkine and PTN, the two protein should share similar affinities for various receptors. The differences in their activity are most likely due to differences in the temporal and spatial expression levels of the two proteins, which are regulated differently.
9. Detecting PTN in vitro
PTN has not yet been included in any FDA approved clinical test for diseases. However, reliable antibodies against PTN have been available commercially for more than twenty years. Many antibodies are polyclonal antibodies that are not well characterized, thus may have large quality variations from batch-to-batch. Several monoclonal antibodies from different hosts are also available. One of the most well-characterized of these is the mouse monoclonal antibody 3B10 developed by Wellstein and co-workers [93]. Not only is the antibody available commercially, the hybridoma cells are also available through ATCC, and the variable region sequences of heavy and light chains in the antibody are also known. This allows researchers to acquire and modify the sequences of the antibody easily. These antibodies have been used routinely to detect PTN in histology studies as well as in westerns. The epitope of the antibody can be readily determined using solution NMR. Using similar techniques as those described in section 7.1 to study PTN-GAG interactions, amino acids at the interface of protein-protein interactions can be identified by comparing antibody-induced changes in chemical shift or intensity of the signals. In the case of 3B10, the presence of the antibody-induced severe decreases in the NMR signal intensities of CTD residues, but not the NTD residues (Figure 9). This is consistent with the binding of a large protein to CTD, but not NTD, thereby implicating CTD as the epitope for the 3B10 antibody.
Figure 9.
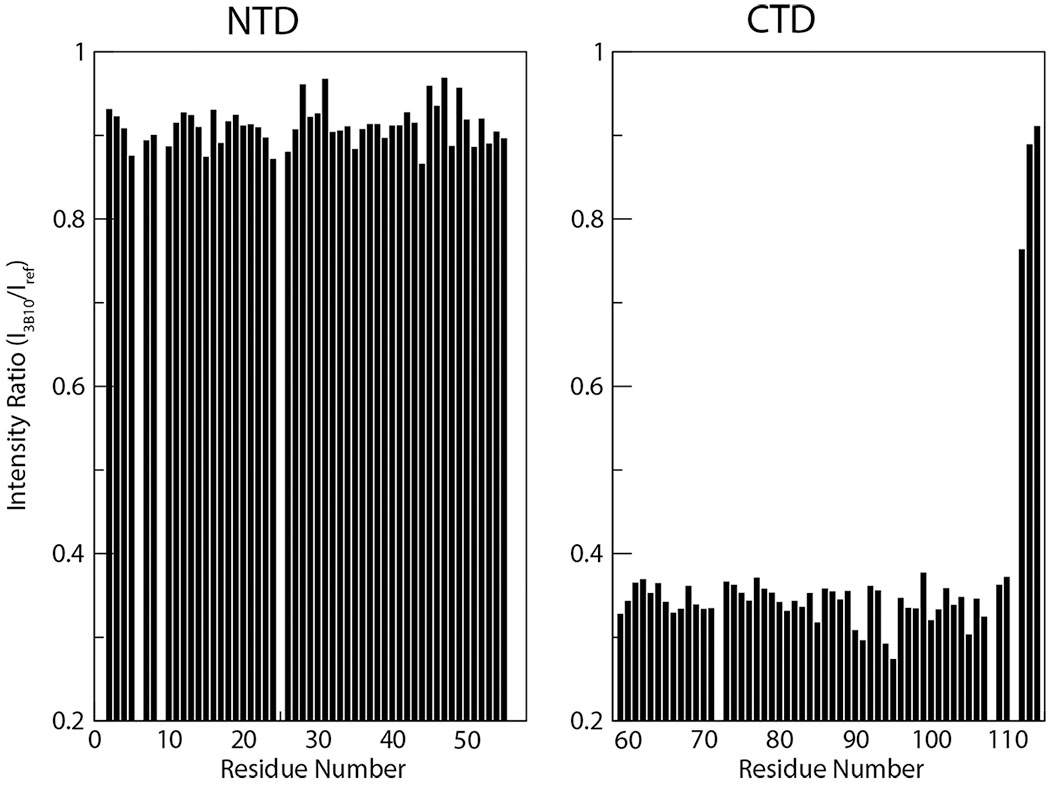
3B10-induced 15N-edited HSQC signal intensity decreases in PTN domains. Ratios of 15N-edited HSQC signal intensities of PTN in the presence (I3B10) and absence (Iref) of the antibody 3B10. The molar ratio of PTN to 3B10 is 4:1.
Several ELISA kits for detecting PTN are also available. In one published protocol for a sandwich style ELISA, BSA-conjugated HP was first coated on the wells to capture PTN. PTN antibody was then added to detect these immobilized PTN [159]. The sensitivity of such assays is usually in the ~ 50 ng /mL range. One particular kit increases the detection sensitivity even further by conjugating DNA to the PTN antibody so a PCR can be carried out to amplify the surface DNA. Such an implementation reduces the detection threshold down to pictogram level without losing specificity.
Besides characterizing the protein concentration in samples, PTN mRNA level can also be characterized using conventional techniques such as Northern hybridization or quantitative RT-PCR. The spatial distribution of these RNAs can also be obtained using fluorescence in situ hybridization [15].
10. Conclusion
Thirty years of research carried out on PTN has produced a rich yet complex picture of PTN’s activity and mechanisms. PTN’s ability to regulate many seemingly disparate biological events is the result of its promiscuity in interacting with a diverse set of receptors. In most cases, this promiscuity is likely a consequence of its numerous charged amino acid side chains. However, its promiscuity also makes understanding and predicting PTN’s effects difficult. Nevertheless, it is now clear that PTN’s potent mitogenic activity can be a double-edged sword. When utilized correctly, it is capable of stimulating neural regeneration at times of severe neural damage. However, it is also capable of stimulating the growth and spread of glioblastoma. The next phase in PTN research should be in devising methods to harvest its activity at times of need and inhibiting PTN specifically so as not to aid the proliferation of tumors. Developing these methods presents a significant challenge to the research community, but it will open new arenas for the application of PTN in medicine.
Reference
- 1.Kovesdi I, et al. , Heparin-binding neurotrophic factor (HBNF) and MK, members of a new family of homologous, developmentally regulated proteins. Biochem Biophys Res Commun, 1990. 172(2): p. 850–4. [DOI] [PubMed] [Google Scholar]
- 2.Li YS, et al. , Cloning and expression of a developmentally regulated protein that induces mitogenic and neurite outgrowth activity. Science, 1990. 250(4988): p. 1690–4. [DOI] [PubMed] [Google Scholar]
- 3.Merenmies J and Rauvala H, Molecular cloning of the 18-kDa growth-associated protein of developing brain. J Biol Chem, 1990. 265(28): p. 16721–4. [PubMed] [Google Scholar]
- 4.Tezuka K, et al. , Isolation of mouse and human cDNA clones encoding a protein expressed specifically in osteoblasts and brain tissues. Biochem Biophys Res Commun, 1990. 173(1): p. 246–51. [DOI] [PubMed] [Google Scholar]
- 5.Herradon G, Ramos-Alvarez MP, and Gramage E, Connecting Metainflammation and Neuroinflammation Through the PTN-MK-RPTPbeta/zeta Axis: Relevance in Therapeutic Development. Front Pharmacol, 2019. 10: p. 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papadimitriou E, et al. , Pleiotrophin and its receptor protein tyrosine phosphatase beta/zeta as regulators of angiogenesis and cancer. Biochim Biophys Acta, 2016. 1866(2): p. 252–265. [DOI] [PubMed] [Google Scholar]
- 7.Sorrelle N, Dominguez ATA, and Brekken RA, From top to bottom: midkine and pleiotrophin as emerging players in immune regulation. J Leukoc Biol, 2017. 102(2): p. 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez-Pinera P, Berenson JR, and Deuel TF, Pleiotrophin, a multifunctional angiogenic factor: mechanisms and pathways in normal and pathological angiogenesis. Curr Opin Hematol, 2008. 15(3): p. 210–4. [DOI] [PubMed] [Google Scholar]
- 9.Lamprou M, et al. , The role of pleiotrophin in bone repair. Injury, 2014. 45(12): p. 1816–23. [DOI] [PubMed] [Google Scholar]
- 10.Thierry-Mieg D and Thierry-Mieg J, AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol, 2006. 7 Suppl 1: p. S121–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kretschmer PJ, et al. , Genomic organization of the human HBNF gene and characterization of an HBNF variant protein as a splice mutant. Biochem Biophys Res Commun, 1993. 192(2): p. 420–9. [DOI] [PubMed] [Google Scholar]
- 12.Svensson SL, et al. , Midkine and pleiotrophin have bactericidal properties: preserved antibacterial activity in a family of heparin-binding growth factors during evolution. J Biol Chem, 2010. 285(21): p. 16105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohlen P and Kovesdi I, HBNF and MK, members of a novel gene family of heparin-binding proteins with potential roles in embryogenesis and brain function. Prog Growth Factor Res, 1991. 3(2): p. 143–57. [DOI] [PubMed] [Google Scholar]
- 14.Rauvala H, et al. , Expression of HB-GAM (heparin-binding growth-associated molecules) in the pathways of developing axonal processes in vivo and neurite outgrowth in vitro induced by HB-GAM. Brain Res Dev Brain Res, 1994. 79(2): p. 157–76. [DOI] [PubMed] [Google Scholar]
- 15.Basille-Dugay M, et al. , Spatio-temporal characterization of the pleiotrophinergic system in mouse cerebellum: evidence for its key role during ontogenesis. Exp Neurol, 2013. 247: p. 537–51. [DOI] [PubMed] [Google Scholar]
- 16.Antoine M, et al. , Upregulation of pleiotrophin expression in rat hepatic stellate cells by PDGF and hypoxia: implications for its role in experimental biliary liver fibrogenesis. Biochem Biophys Res Commun, 2005. 337(4): p. 1153–64. [DOI] [PubMed] [Google Scholar]
- 17.Polytarchou C, Hatziapostolou M, and Papadimitriou E, Hydrogen peroxide stimulates proliferation and migration of human prostate cancer cells through activation of activator protein-1 and up-regulation of the heparin affin regulatory peptide gene. J Biol Chem, 2005. 280(49): p. 40428–35. [DOI] [PubMed] [Google Scholar]
- 18.Yeh HJ, et al. , Upregulation of pleiotrophin gene expression in developing microvasculature, macrophages, and astrocytes after acute ischemic brain injury. J Neurosci, 1998. 18(10): p. 3699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatziapostolou M, et al. , Heparin affin regulatory peptide/pleiotrophin mediates fibroblast growth factor 2 stimulatory effects on human prostate cancer cells. J Biol Chem, 2006. 281(43): p. 32217–26. [DOI] [PubMed] [Google Scholar]
- 20.Li YS, Gurrieri M, and Deuel TF, Pleiotrophin gene expression is highly restricted and is regulated by platelet-derived growth factor. Biochem Biophys Res Commun, 1992. 184(1): p. 427–32. [DOI] [PubMed] [Google Scholar]
- 21.Pufe T, et al. , Expression of pleiotrophin, an embryonic growth and differentiation factor, in rheumatoid arthritis. Arthritis Rheum, 2003. 48(3): p. 660–7. [DOI] [PubMed] [Google Scholar]
- 22.Tamura M, et al. , 1alpha,25-Dihydroxyvitamin D(3) down-regulates pleiotrophin messenger RNA expression in osteoblast-like cells. Endocrine, 1995. 3(1): p. 21–4. [DOI] [PubMed] [Google Scholar]
- 23.Vacherot F, et al. , Upregulation of heparin-affin regulatory peptide by androgen. In Vitro Cell Dev Biol Anim, 1995. 31(9): p. 647–8. [DOI] [PubMed] [Google Scholar]
- 24.Liedert A, et al. , Mechanical regulation of HB-GAM expression in bone cells. Biochem Biophys Res Commun, 2004. 319(3): p. 951–8. [DOI] [PubMed] [Google Scholar]
- 25.Camerino GM, et al. , Effects of pleiotrophin overexpression on mouse skeletal muscles in normal loading and in actual and simulated microgravity. PLoS One, 2013. 8(8): p. e72028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarville JL, et al. , Spaceflight influences both mucosal and peripheral cytokine production in PTN-Tg and wild type mice. PLoS One, 2013. 8(7): p. e68961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albi E, et al. , Loss of parafollicular cells during gravitational changes (microgravity, hypergravity) and the secret effect of pleiotrophin. PLoS One, 2012. 7(12): p. e48518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tavella S, et al. , Bone turnover in wild type and pleiotrophin-transgenic mice housed for three months in the International Space Station (ISS). PLoS One, 2012. 7(3): p. e33179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu D, et al. , Pleiotrophin promotes chemoresistance to doxorubicin in osteosarcoma by upregulating P-glycoprotein. Oncotarget, 2017. 8(38): p. 63857–63870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma J, et al. , Pleiotrophin as a potential biomarker in breast cancer patients. Clin Chim Acta, 2017. 466: p. 6–12. [DOI] [PubMed] [Google Scholar]
- 31.Riegel AT and Wellstein A, The potential role of the heparin-binding growth factor pleiotrophin in breast cancer. Breast Cancer Res Treat, 1994. 31(2–3): p. 309–14. [DOI] [PubMed] [Google Scholar]
- 32.Tsirmoula S, et al. , Implications of pleiotrophin in human PC3 prostate cancer cell growth in vivo. Cancer Sci, 2012. 103(10): p. 1826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang XY, et al. , Meta-analysis of gene expression data identifies causal genes for prostate cancer. Asian Pac J Cancer Prev, 2013. 14(1): p. 457–61. [DOI] [PubMed] [Google Scholar]
- 34.Hatziapostolou M, et al. , Heparin affin regulatory peptide is a key player in prostate cancer cell growth and angiogenicity. Prostate, 2005. 65(2): p. 151–8. [DOI] [PubMed] [Google Scholar]
- 35.Yao J, et al. , Pleiotrophin expression in human pancreatic cancer and its correlation with clinicopathological features, perineural invasion, and prognosis. Dig Dis Sci, 2009. 54(4): p. 895–901. [DOI] [PubMed] [Google Scholar]
- 36.Hu H, et al. , Increased expression of pleiotrophin is a prognostic marker for patients with gastric cancer. Hepatogastroenterology, 2014. 61(133): p. 1478–82. [PubMed] [Google Scholar]
- 37.Feng ZJ, et al. , Lung cancer cell migration is regulated via repressing growth factor PTN/RPTP beta/zeta signaling by menin. Oncogene, 2010. 29(39): p. 5416–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du ZY, et al. , Serum pleiotrophin could be an early indicator for diagnosis and prognosis of non-small cell lung cancer. Asian Pac J Cancer Prev, 2015. 16(4): p. 1421–5. [DOI] [PubMed] [Google Scholar]
- 39.Wang HQ and Wang J, Expression of pleiotrophin in small cell lung cancer. Mol Neurobiol, 2015. 29(1): p. 175–9. [PubMed] [Google Scholar]
- 40.Kong Y, et al. , Pleiotrophin is a potential colorectal cancer prognostic factor that promotes VEGF expression and induces angiogenesis in colorectal cancer. Int J Colorectal Dis, 2012. 27(3): p. 287–98. [DOI] [PubMed] [Google Scholar]
- 41.Yao Y, et al. , Luteolin suppresses colorectal cancer cell metastasis via regulation of the miR384/pleiotrophin axis. Oncol Rep, 2019. 42(1): p. 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JY, et al. , Chicken pleiotrophin: regulation of tissue specific expression by estrogen in the oviduct and distinct expression pattern in the ovarian carcinomas. PLoS One, 2012. 7(4): p. e34215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sethi G, et al. , An RNA interference lethality screen of the human druggable genome to identify molecular vulnerabilities in epithelial ovarian cancer. PLoS One, 2012. 7(10): p. e47086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu LQ, et al. , CREB3L1 and PTN expressions correlate with prognosis of brain glioma patients. Biosci Rep, 2018. 38(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi Y, et al. , Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. J Clin Pathol, 2017. 8: p. 15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu KV, et al. , Differential induction of glioblastoma migration and growth by two forms of pleiotrophin. J Biol Chem, 2005. 280(29): p. 26953–64. [DOI] [PubMed] [Google Scholar]
- 47.Yi C, et al. , MiR-143 enhances adipogenic differentiation of 3T3-L1 cells through targeting the coding region of mouse pleiotrophin. FEBS Lett, 2011. 585(20): p. 3303–9. [DOI] [PubMed] [Google Scholar]
- 48.Bai PS, et al. , Pleiotrophin, a target of miR-384, promotes proliferation, metastasis and lipogenesis in HBV-related hepatocellular carcinoma. 2017. 21(11): p. 3023–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang L, et al. , miR-182 aids in receptive endometrium development in dairy goats by down-regulating PTN expression. Reprod Domest Anim, 2017. 12(7): p. e0179783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Q, et al. , miR-137 Inhibits Proliferation and Metastasis of Hypertrophic Scar Fibroblasts via Targeting Pleiotrophin. Cell Physiol Biochem, 2018. 49(3): p. 985–995. [DOI] [PubMed] [Google Scholar]
- 51.Muramatsu H, et al. , Female infertility in mice deficient in midkine and pleiotrophin, which form a distinct family of growth factors. Genes Cells, 2006. 11(12): p. 1405–17. [DOI] [PubMed] [Google Scholar]
- 52.Imai S, et al. , Osteocyte-derived HB-GAM (pleiotrophin) is associated with bone formation and mechanical loading. Bone, 2009. 44(5): p. 785–94. [DOI] [PubMed] [Google Scholar]
- 53.Krellman JW, et al. , Behavioral and neuroanatomical abnormalities in pleiotrophin knockout mice. PLoS One, 2014. 9(7): p. e100597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gramage E, et al. , The neurotrophic factor pleiotrophin modulates amphetamine-seeking behaviour and amphetamine-induced neurotoxic effects: evidence from pleiotrophin knockout mice. Addict Biol, 2010. 15(4): p. 403–12. [DOI] [PubMed] [Google Scholar]
- 55.Vicente-Rodriguez M, et al. , Pleiotrophin differentially regulates the rewarding and sedative effects of ethanol. J Neurochem, 2014. 131(5): p. 688–95. [DOI] [PubMed] [Google Scholar]
- 56.Sevillano J, et al. , Pleiotrophin deletion alters glucose homeostasis, energy metabolism and brown fat thermogenic function in mice. Diabetologia, 2019. 62(1): p. 123–135. [DOI] [PubMed] [Google Scholar]
- 57.Yanagisawa H, et al. , Pleiotrophin induces neurite outgrowth and up-regulates growth-associated protein (GAP)-43 mRNA through the ALK/GSK3beta/beta-catenin signaling in developing mouse neurons. Neurosci Res, 2010. 66(1): p. 111–6. [DOI] [PubMed] [Google Scholar]
- 58.Paveliev M, et al. , HB-GAM (pleiotrophin) reverses inhibition of neural regeneration by the CNS extracellular matrix. Sci Rep, 2016. 6: p. 33916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Landgraf P, et al. , The survival-promoting peptide Y-P30 enhances binding of pleiotrophin to syndecan-2 and −3 and supports its neuritogenic activity. J Biol Chem, 2008. 283(36): p. 25036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Landgraf P, et al. , Binding of Y-P30 to syndecan 2/3 regulates the nuclear localization of CASK. PLoS One, 2014. 9(2): p. e85924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Neumann JR, et al. , Y-P30 promotes axonal growth by stabilizing growth cones. Brain Struct Funct, 2015. 220(4): p. 1935–50. [DOI] [PubMed] [Google Scholar]
- 62.Neumann JR, et al. , The primate-specific peptide Y-P30 regulates morphological maturation of neocortical dendritic spines. 2019. 14(2): p. e0211151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang C, et al. , Neural Stem Cells Behave as a Functional Niche for the Maturation of Newborn Neurons through the Secretion of PTN. Neuron, 2019. 101(1): p. 32–44.e6. [DOI] [PubMed] [Google Scholar]
- 64.Yang S, et al. , Pleiotrophin is involved in the amniotic epithelial cell-induced differentiation of human umbilical cord blood-derived mesenchymal stem cells into dopaminergic neuron-like cells. Neurosci Lett, 2013. 539: p. 86–91. [DOI] [PubMed] [Google Scholar]
- 65.Kuboyama K, et al. , Inactivation of Protein Tyrosine Phosphatase Receptor Type Z by Pleiotrophin Promotes Remyelination through Activation of Differentiation of Oligodendrocyte Precursor Cells. Journal of Neuroscience, 2015. 35(35): p. 12162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kuboyama K, et al. , Role of Chondroitin Sulfate (CS) Modification in the Regulation of Protein-tyrosine Phosphatase Receptor Type Z (PTPRZ) Activity: Pleiotrophin-PTPRZ-A Signaling is Involved in Oligodendrocyte Differentiation. J Biol Chem, 2016. 291(35): p. 18117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tanga N, et al. , The PTN-PTPRZ signal activates the AFAP1L2-dependent PI3K-AKT pathway for oligodendrocyte differentiation: Targeted inactivation of PTPRZ activity in mice. PLoS One, 2019. 67(5): p. 967–984. [DOI] [PubMed] [Google Scholar]
- 68.Nikolakopoulou AM, et al. , Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. 2019. 22(7): p. 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qin EY, et al. , Neural Precursor-Derived Pleiotrophin Mediates Subventricular Zone Invasion by Glioma. Cell, 2017. 170(5): p. 845–859.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Courty J, et al. , Mitogenic properties of a new endothelial cell growth factor related to pleiotrophin. Biochem Biophys Res Commun, 1991. 180(1): p. 145–51. [DOI] [PubMed] [Google Scholar]
- 71.Fang W, et al. , Pleiotrophin stimulates fibroblasts and endothelial and epithelial cells and is expressed in human cancer. J Biol Chem, 1992. 267(36): p. 25889–97. [PubMed] [Google Scholar]
- 72.Souttou B, Raulais D, and Vigny M, Pleiotrophin induces angiogenesis: involvement of the phosphoinositide-3 kinase but not the nitric oxide synthase pathways. J Cell Physiol, 2001. 187(1): p. 59–64. [DOI] [PubMed] [Google Scholar]
- 73.Papadimitriou E, et al. , HARP induces angiogenesis in vivo and in vitro: implication of N or C terminal peptides. Biochem Biophys Res Commun, 2001. 282(1): p. 306–13. [DOI] [PubMed] [Google Scholar]
- 74.Magnusson PU, et al. , FGFR-1 regulates angiogenesis through cytokines interleukin-4 and pleiotrophin. Blood, 2007. 110(13): p. 4214–22. [DOI] [PubMed] [Google Scholar]
- 75.Fukada M, et al. , Protein tyrosine phosphatase receptor type Z is inactivated by ligand-induced oligomerization. FEBS Lett, 2006. 580(17): p. 4051–6. [DOI] [PubMed] [Google Scholar]
- 76.Mikelis C, et al. , Integrin alpha(v)beta(3) is a pleiotrophin receptor required for pleiotrophin-induced endothelial cell migration through receptor protein tyrosine phosphatase beta/zeta. Faseb j, 2009. 23(5): p. 1459–69. [DOI] [PubMed] [Google Scholar]
- 77.Poimenidi E, et al. , Vascular endothelial growth factor A (VEGF-A) decreases expression and secretion of pleiotrophin in a VEGF receptor-independent manner. Vascul Pharmacol, 2016. 80: p. 11–9. [DOI] [PubMed] [Google Scholar]
- 78.Fernandez-Calle R, et al. , Pleiotrophin regulates microglia-mediated neuroinflammation. J Neuroinflammation, 2017. 14(1): p. 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fernandez-Calle R, et al. , Endogenous pleiotrophin and midkine regulate LPS-induced glial responses. Neurosci Lett, 2018. 662: p. 213–218. [DOI] [PubMed] [Google Scholar]
- 80.Christman KL, et al. , Enhanced neovasculature formation in ischemic myocardium following delivery of pleiotrophin plasmid in a biopolymer. Biomaterials, 2005. 26(10): p. 1139–44. [DOI] [PubMed] [Google Scholar]
- 81.Ochiai K, et al. , The role of midkine and pleiotrophin in liver regeneration. Liver Int, 2004. 24(5): p. 484–91. [DOI] [PubMed] [Google Scholar]
- 82.Himburg HA, et al. , Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med, 2010. 16(4): p. 475–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Herradon G and Perez-Garcia C, Targeting midkine and pleiotrophin signalling pathways in addiction and neurodegenerative disorders: recent progress and perspectives. Br J Pharmacol, 2014. 171(4): p. 837–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shen D, et al. , Pleiotrophin, a multifunctional cytokine and growth factor, induces leukocyte responses through the integrin Mac-1. 2017. 292(46): p. 18848–18861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Imai S, et al. , Osteoblast recruitment and bone formation enhanced by cell matrix-associated heparin-binding growth-associated molecule (HB-GAM). J Cell Biol, 1998. 143(4): p. 1113–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gieffers C, et al. , Receptor binding of osteoblast-specific factor 1 (OSF-1/HB-GAM) to human osteosarcoma cells promotes cell attachment. Eur J Cell Biol, 1993. 62(2): p. 352–61. [PubMed] [Google Scholar]
- 87.Kaspiris A, et al. , Effects of mechanical loading on the expression of pleiotrophin and its receptor protein tyrosine phosphatase beta/zeta in a rat spinal deformity model. Cytokine, 2016. 78: p. 7–15. [DOI] [PubMed] [Google Scholar]
- 88.Marzan CV, et al. , Adipocyte derived paracrine mediators of mammary ductal morphogenesis controlled by retinoic acid receptors. Dev Biol, 2011. 349(2): p. 125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gu D, et al. , The effect of pleiotrophin signaling on adipogenesis. FEBS Lett, 2007. 581(3): p. 382–8. [DOI] [PubMed] [Google Scholar]
- 90.Wong JC, et al. , A glucocorticoid- and diet-responsive pathway toggles adipocyte precursor cell activity in vivo. Sci Signal, 2016. 9(451): p. ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ledoux D, et al. , Cellular distribution of the angiogenic factor heparin affin regulatory peptide (HARP) mRNA and protein in the human mammary gland. J Histochem Cytochem, 1997. 45(9): p. 1239–45. [DOI] [PubMed] [Google Scholar]
- 92.Kouros-Mehr H and Werb Z, Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev Dyn, 2006. 235(12): p. 3404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rosenfield SM, et al. , Pleiotrophin (PTN) expression and function and in the mouse mammary gland and mammary epithelial cells. PLoS One, 2012. 7(10): p. e47876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grzelinski M, et al. , Ribozyme-targeting reveals the rate-limiting role of pleiotrophin in glioblastoma. Int J Cancer, 2005. 117(6): p. 942–51. [DOI] [PubMed] [Google Scholar]
- 95.Grzelinski M, et al. , RNA interference-mediated gene silencing of pleiotrophin through polyethylenimine-complexed small interfering RNAs in vivo exerts antitumoral effects in glioblastoma xenografts. Hum Gene Ther, 2006. 17(7): p. 751–66. [DOI] [PubMed] [Google Scholar]
- 96.Zhang L, et al. , Pleiotrophin promotes vascular abnormalization in gliomas and correlates with poor survival in patients with astrocytomas. Sci Signal, 2015. 8(406): p. ra125. [DOI] [PubMed] [Google Scholar]
- 97.Wellstein A, et al. , A heparin-binding growth factor secreted from breast cancer cells homologous to a developmentally regulated cytokine. J Biol Chem, 1992. 267(4): p. 2582–7. [PubMed] [Google Scholar]
- 98.Zhang N, et al. , Human breast cancer growth inhibited in vivo by a dominant negative pleiotrophin mutant. J Biol Chem, 1997. 272(27): p. 16733–6. [DOI] [PubMed] [Google Scholar]
- 99.Chang Y, et al. , Secretion of pleiotrophin stimulates breast cancer progression through remodeling of the tumor microenvironment. Proc Natl Acad Sci U S A, 2007. 104(26): p. 10888–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Choudhuri R, et al. , An angiogenic role for the neurokines midkine and pleiotrophin in tumorigenesis. Cancer Res, 1997. 57(9): p. 1814–9. [PubMed] [Google Scholar]
- 101.Huang P, et al. , Chemotherapy-driven increases in the CDKN1A/PTN/PTPRZ1 axis promote chemoresistance by activating the NF-kappaB pathway in breast cancer cells. Cell Commun Signal, 2018. 16(1): p. 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jager R, et al. , Differential expression and biological activity of the heparin-binding growth-associated molecule (HB-GAM) in lung cancer cell lines. Int J Cancer, 1997. 73(4): p. 537–43. [DOI] [PubMed] [Google Scholar]
- 103.Jager R, et al. , Serum levels of the angiogenic factor pleiotrophin in relation to disease stage in lung cancer patients. Br J Cancer, 2002. 86(6): p. 858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu C, et al. , Serum pleiotrophin as a diagnostic and prognostic marker for small cell lung cancer. J Cell Mol Med, 2019. 23(3): p. 2077–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gao SB, et al. , Suppression of lung adenocarcinoma through menin and polycomb gene-mediated repression of growth factor pleiotrophin. Oncogene, 2009. 28(46): p. 4095–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sethi G, et al. , PTN signaling: Components and mechanistic insights in human ovarian cancer. Mol Carcinog, 2015. 54(12): p. 1772–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weber D, et al. , Pleiotrophin can be rate-limiting for pancreatic cancer cell growth. Cancer Res, 2000. 60(18): p. 5284–8. [PubMed] [Google Scholar]
- 108.Yao J, et al. , Recombinant lentivirus targeting the pleotrophin gene reduces pleotrophin protein expression in pancreatic cancer cells and inhibits neurite outgrowth of dorsal root ganglion neurons. Mol Med Rep, 2014. 9(3): p. 999–1004. [DOI] [PubMed] [Google Scholar]
- 109.Stoica GE, et al. , Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J Biol Chem, 2001. 276(20): p. 16772–9. [DOI] [PubMed] [Google Scholar]
- 110.Deuel TF, Anaplastic lymphoma kinase: “Ligand Independent Activation” mediated by the PTN/RPTPbeta/zeta signaling pathway. Biochim Biophys Acta, 2013. 1834(10): p. 2219–23. [DOI] [PubMed] [Google Scholar]
- 111.Narentuya, et al. , GlcNAc6ST3 is a keratan sulfate sulfotransferase for the protein-tyrosine phosphatase PTPRZ in the adult brain. Scientific Reports, 2019. 9(1): p. 4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peles E, Schlessinger J, and Grumet M, Multi-ligand interactions with receptor-like protein tyrosine phosphatase β: implications for intercellular signaling. Trends in Biochemical Sciences, 1998. 23(4): p. 121–124. [DOI] [PubMed] [Google Scholar]
- 113.Abbott KL, Matthews RT, and Pierce M, Receptor tyrosine phosphatase beta (RPTPbeta) activity and signaling are attenuated by glycosylation and subsequent cell surface galectin-1 binding. J Biol Chem, 2008. 283(48): p. 33026–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fujikawa A, et al. , A head-to-toe dimerization has physiological relevance for ligand-induced inactivation of protein tyrosine receptor type Z. J Biol Chem, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tanga N, et al. , Behavioral and neurological analyses of adult mice carrying null and distinct loss-of-receptor function mutations in protein tyrosine phosphatase receptor type Z (PTPRZ). PLoS One, 2019. 14(6): p. e0217880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bouyain S and Watkins DJ, The protein tyrosine phosphatases PTPRZ and PTPRG bind to distinct members of the contactin family of neural recognition molecules. Proceedings of the National Academy of Sciences, 2010. 107(6): p. 2443–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Peles E, et al. , The carbonic anhydrase domain of receptor tyrosine phosphatase β is a functional ligand for the axonal cell recognition molecule contactin. Cell, 1995. 82(2): p. 251–260. [DOI] [PubMed] [Google Scholar]
- 118.Maeda N, et al. , 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase zeta/RPTPbeta, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM). J Biol Chem, 1996. 271(35): p. 21446–52. [DOI] [PubMed] [Google Scholar]
- 119.Fu F, et al. , Expression of receptor protein tyrosine phosphatase zeta is a risk factor for triple negative breast cancer relapse. Biomed Rep, 2016. 4(2): p. 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Szatmári T and Dobra K, The Role of Syndecan-1 in Cellular Signaling and its Effects on Heparan Sulfate Biosynthesis in Mesenchymal Tumors. Frontiers in Oncology, 2013. 3(310). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Raulo E, et al. , Isolation of a neuronal cell surface receptor of heparin binding growth-associated molecule (HB-GAM). Identification as N-syndecan (syndecan-3). J Biol Chem, 1994. 269(17): p. 12999–3004. [PubMed] [Google Scholar]
- 122.Deepa SS, et al. , Chondroitin sulfate chains on syndecan-1 and syndecan-4 from normal murine mammary gland epithelial cells are structurally and functionally distinct and cooperate with heparan sulfate chains to bind growth factors. A novel function to control binding of midkine, pleiotrophin, and basic fibroblast growth factor. J Biol Chem, 2004. 279(36): p. 37368–76. [DOI] [PubMed] [Google Scholar]
- 123.Essner JJ, Chen E, and Ekker SC, Syndecan-2. The International Journal of Biochemistry & Cell Biology, 2006. 38(2): p. 152–156. [DOI] [PubMed] [Google Scholar]
- 124.Diamantopoulou Z, et al. , Loss of receptor protein tyrosine phosphatase beta/zeta (RPTPbeta/zeta) promotes prostate cancer metastasis. J Biol Chem, 2012. 287(48): p. 40339–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yao J, et al. , Pleiotrophin promotes perineural invasion in pancreatic cancer. World J Gastroenterol, 2013. 19(39): p. 6555–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yao J, et al. , Pleiotrophin and N-syndecan promote perineural invasion and tumor progression in an orthotopic mouse model of pancreatic cancer. J Cell Physiol, 2017. 23(21): p. 3907–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Koutsioumpa M and Papadimitriou E, Cell surface nucleolin as a target for anti-cancer therapies. Recent Pat Anticancer Drug Discov, 2014. 9(2): p. 137–52. [DOI] [PubMed] [Google Scholar]
- 128.Fujiki H, Watanabe T, and Suganuma M, Cell-surface nucleolin acts as a central mediator for carcinogenic, anti-carcinogenic, and disease-related ligands. Journal of cancer research and clinical oncology, 2014. 140(5): p. 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Koutsioumpa M, et al. , Pleiotrophin expression and role in physiological angiogenesis in vivo: potential involvement of nucleolin. Vasc Cell, 2012. 4: p. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Garcia-Gutierrez P, et al. , Pleiotrophin antagonizes Brd2 during neuronal differentiation. J Cell Sci, 2014. 127(Pt 11): p. 2554–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Koutsioumpa M, et al. , Interplay between alphavbeta3 integrin and nucleolin regulates human endothelial and glioma cell migration. J Biol Chem, 2013. 288(1): p. 343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Davis GE, The Mac-1 and p150,95 beta 2 integrins bind denatured proteins to mediate leukocyte cell-substrate adhesion. Experimental Cell Research, 1992. 200: p. 242–252. [DOI] [PubMed] [Google Scholar]
- 133.Podolnikova NP, et al. , Ligand recognition specificity of leukocyte integrin alphaMbeta2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry, 2015. 54(6): p. 1408–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yakubenko VP, et al. , A molecular basis for integrin alphaMbeta 2 ligand binding promiscuity. J Biol Chem, 2002. 277(50): p. 48635–42. [DOI] [PubMed] [Google Scholar]
- 135.Ajroud K, et al. , Binding Affinity of Metal Ions to the CD11b A-domain Is Regulated by Integrin Activation and Ligands. J Biol Chem, 2004. 279(24): p. 25483–8. [DOI] [PubMed] [Google Scholar]
- 136.Li J, et al. , The pro-angiogenic cytokine pleiotrophin potentiates cardiomyocyte apoptosis through inhibition of endogenous AKT/PKB activity. J Biol Chem, 2007. 282(48): p. 34984–93. [DOI] [PubMed] [Google Scholar]
- 137.Hamma-Kourbali Y, et al. , The synthetic peptide P111-136 derived from the C-terminal domain of heparin affin regulatory peptide inhibits tumour growth of prostate cancer PC-3 cells. BMC Cancer, 2011. 11: p. 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mikelis C, et al. , A peptide corresponding to the C-terminal region of pleiotrophin inhibits angiogenesis in vivo and in vitro. J Cell Biochem, 2011. 112(6): p. 1532–43. [DOI] [PubMed] [Google Scholar]
- 139.Kojima S, et al. , Dimerization of midkine by tissue transglutaminase and its functional implication. J Biol Chem, 1997. 272(14): p. 9410–6. [DOI] [PubMed] [Google Scholar]
- 140.Bernard-Pierrot I, et al. , Glycosaminoglycans promote HARP/PTN dimerization. Biochem Biophys Res Commun, 1999. 266(2): p. 437–42. [DOI] [PubMed] [Google Scholar]
- 141.Mizumoto S, Fongmoon D, and Sugahara K, Interaction of chondroitin sulfate and dermatan sulfate from various biological sources with heparin-binding growth factors and cytokines. Glycoconj J, 2013. 30(6): p. 619–32. [DOI] [PubMed] [Google Scholar]
- 142.Li F, et al. , Structure of pleiotrophin- and hepatocyte growth factor-binding sulfated hexasaccharide determined by biochemical and computational approaches. J Biol Chem, 2010. 285(36): p. 27673–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ryan E, Shen D, and Wang X, Structural studies reveal an important role for the pleiotrophin C-terminus in mediating interactions with chondroitin sulfate. Febs j, 2016. 283(8): p. 1488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Maeda N, Fukazawa N, and Hata T, The binding of chondroitin sulfate to pleiotrophin/heparin-binding growth-associated molecule is regulated by chain length and oversulfated structures. J Biol Chem, 2006. 281(8): p. 4894–902. [DOI] [PubMed] [Google Scholar]
- 145.Meng K, et al. , Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A, 2000. 97(6): p. 2603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Migliorini E, et al. , Cytokines and growth factors cross-link heparan sulfate. Open Biol, 2015. 5(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Iwasaki W, et al. , Solution structure of midkine, a new heparin-binding growth factor. EMBO J, 1997. 16(23): p. 6936–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lim J, et al. , Structure-function analysis of full-length midkine reveals novel residues important for heparin binding and zebrafish embryogenesis. Biochem J, 2013. 451(3): p. 407–15. [DOI] [PubMed] [Google Scholar]
- 149.Herradon G, et al. , Midkine regulates pleiotrophin organ-specific gene expression: evidence for transcriptional regulation and functional redundancy within the pleiotrophin/midkine developmental gene family. Biochem Biophys Res Commun, 2005. 333(3): p. 714–21. [DOI] [PubMed] [Google Scholar]
- 150.Tang Z, et al. , GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res, 2017. 45(W1): p. W98–w102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Muramatsu H, et al. , alpha4beta1- and alpha6beta1-integrins are functional receptors for midkine, a heparin-binding growth factor. J Cell Sci, 2004. 117(Pt 22): p. 5405–15. [DOI] [PubMed] [Google Scholar]
- 152.Jin H, et al. , Integrin alpha4beta1 promotes monocyte trafficking and angiogenesis in tumors. Cancer Res, 2006. 66(4): p. 2146–52. [DOI] [PubMed] [Google Scholar]
- 153.Randolph GJ, Ochando J, and Partida-Sanchez S, Migration of dendritic cell subsets and their precursors. Annu Rev Immunol, 2008. 26: p. 293–316. [DOI] [PubMed] [Google Scholar]
- 154.Weckbach LT, et al. , The cytokine midkine supports neutrophil trafficking during acute inflammation by promoting adhesion via beta2 integrins (CD11/CD18). Blood, 2014. 123(12): p. 1887–96. [DOI] [PubMed] [Google Scholar]
- 155.Kosugi T, et al. , Midkine is involved in tubulointerstitial inflammation associated with diabetic nephropathy. Lab Invest, 2007. 87(9): p. 903–13. [DOI] [PubMed] [Google Scholar]
- 156.Narita H, et al. , Midkine is expressed by infiltrating macrophages in in-stent restenosis in hypercholesterolemic rabbits. J Vasc Surg, 2008. 47(6): p. 1322–9. [DOI] [PubMed] [Google Scholar]
- 157.Kadomatsu K, et al. , Midkine induces the transformation of NIH3T3 cells. Br J Cancer, 1997. 75(3): p. 354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mirkin BL, et al. , Identification of midkine as a mediator for intercellular transfer of drug resistance. Oncogene, 2005. 24(31): p. 4965–74. [DOI] [PubMed] [Google Scholar]
- 159.Soulie P, et al. , Immunoassay for measuring the heparin-binding growth factors HARP and MK in biological fluids. J Immunoassay Immunochem, 2002. 23(1): p. 33–48. [DOI] [PubMed] [Google Scholar]