

Abstract
Background
The utility of O6-methylguanine-DNA methyltransferase (MGMT) gene promoter methylation status as a prognostic marker in patients with glioblastoma (GBM) has been established. However, the number of CpG sites that must be methylated to cause transcriptional silencing remains unclear, and no significant consensus exists on the optimal method of assessing MGMT methylation. We developed a new high-performance liquid chromatography (HPLC) method that enables accurate analysis of DNA methylation levels using long PCR products. In the present study, we analyzed the MGMT methylation status of 28 isocitrate dehydrogenase–wild-type GBMs treated with temozolomide using ion-exchange HPLC and set the optimal cutoff values.
Results
We designed three primers for separate regions (regions 1–3) that had 21 to 38 CpGs for PCR and validated the MGMT promoter methylation status using frozen samples. There was a strong correlation between HPLC and bisulfite sequencing results (R = 0.794). The optimal cutoff values for MGMT methylation in HPLC were determined to allow differentiation of patient prognosis by receiver operating characteristic curve analysis. The cutoff values were 34.15% for region 1, 8.84% for region 2, and 36.72% for region 3. Kaplan–Meyer curve analysis estimated that the most differentiated prognosis was enabled in the setting of 8.84% methylation of MGMT in region 2. Progression-free survival and overall survival were significantly longer for patients in this setting of region 2 methylation (p = 0.00365 and p = 0.00258, respectively).
Conclusions
The combination of our HPLC method and the original primer setting provides a new standard method for determination of MGMT methylation status in patients with GBM and is useful for refining MGMT-based drug selection.
Keywords: MGMT, DNA methylation, Glioblastoma, HPLC, Prognostic factor
Background
Glioblastoma (GBM) is the most common brain tumor in adults; it has an aggressive lethal nature, with a median survival of only 12–15 months, despite the standard established therapy of maximum resection followed by radiation and chemotherapy [1–3]. GBM chemotherapy is based on drugs that alkylate the DNA in the O6-position of guanine, such as temozolomide (TMZ). O6-methylguanine-DNA methyltransferase (MGMT) is a protein that repairs the naturally occurring mutagenic DNA lesion O6-methylguanine back to guanine and prevents mismatch and errors during DNA replication and transcription. Since epigenetic silencing of the MGMT gene via promoter methylation is associated with loss of MGMT expression [4, 5], analysis of MGMT methylation status has become a key prognostic marker of TMZ.
In GBM patients, the relevance of MGMT promoter methylation as a predictive marker for TMZ effectiveness has been strengthened by a randomized trial [6]. Furthermore, it has been reported that MGMT methylation is significantly correlated with prognosis of glioma patients treated with TMZ, with or without adjustment of isocitrate dehydrogenase (IDH) 1/2 status [7, 8]. Based on these results, MGMT methylation analysis has been intensely investigated in several clinical trials [6, 8, 9].
Despite considerable research, consensus regarding the optimal method for MGMT gene promoter methylation assessment and the optimal promoter region for MGMT methylation analysis is lacking. The CpG island of MGMT exhibits heterogeneous methylation patterns, and it is therefore possible to fail to detect methylation when only analyzing a specific area [10]. Genome sequencing analysis can overcome this problem; however, it is costly. Previous studies have identified two regions (the promoter and gene body) that are significantly correlated with MGMT expression [11]. However, due to the difficulty of designing a primer for the GC-rich promoter area and the lack of a method that enables analysis of long PCR products at low cost, the region of the gene body is generally selected as the target for MGMT methylation analysis [12]. To demonstrate the effectiveness of methylation analysis in the promoter region, a new method for analyzing longer PCR products is required.
We developed an anion-exchange, high-performance liquid chromatography (HPLC) column for the detection of methylated DNA, thus enabling the analysis of long PCR products. HPLC provides rapid and accurate quantification of DNA methylation levels within 10 min without the need for time-consuming pretreatment of the PCR products [13]. In the present study, we identified three original primers and determined the MGMT promoter methylation status in newly diagnosed GBM patients using this HPLC method and set the optimal cutoff values.
Results
We identified three separate primer regions: region 1 (198 bp, 21 CpGs), region 2 (294 bp, 38 CpGs), and region 3 (259 bp, 27 CpGs) (Fig. 1, Table 1). Previous studies identified two distinct regions of MGMT CpG islands, known as differentially methylated region (DMR) 1 and 2, which were demonstrated to correlate with transcriptional silencing [10, 14, 15]. Our region 2 encompassed DMR 1, whereas region 3 encompassed DMR 2 (Fig. 1).
Fig. 1.

Schematic representation of the MGMT promoter analyzed in the present study
Table 1.
List of bisulfite PCR primers
| Primer sequence (5′–3′) | Amplicon size (bp) | |
|---|---|---|
| Region 1 | F: GGTAAATTAAGGTATAGAGTTTTAGG | 198 |
| R: AAAACCTAAAAAAAACAAAAAAAC | ||
| Region 2 | F: GGTTTGGGGGTTTTTGATTAG | 294 |
| R: CCTTTTCCTATCACAAAAATAATCC | ||
| Region 3 | F: GGATATGTTGGGATAGTT | 259 |
| R: ACAACACCTAAAAAACACTTAAAAC |
The chromatograms of three regions are shown in Fig. 2. The peaks derived from synthetic DNA fragments corresponding to 0% and 100% methylation were completely separated and detected in region 1 between 6.5 and 7.5 min (Fig. 2a), in region 2 between 3.5 and 4.5 min (Fig. 2b), and in region 3 between 7.5 and 8.5 min (Fig. 2c).
Fig. 2.
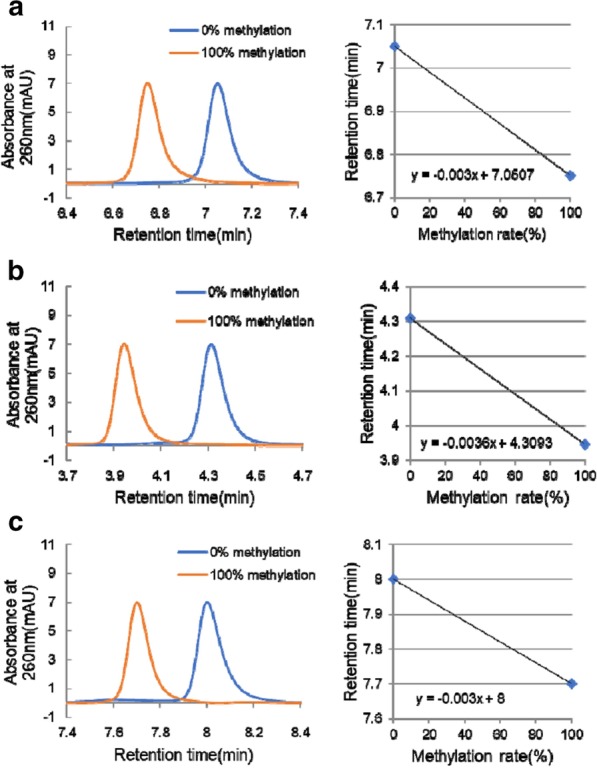
DNA methylation analysis by HPLC. Chromatograms of synthetic DNA fragments corresponding to 0% (TPG) and 100% (CpG) methylation (a–c)
To verify the validity of the HPLC method for DNA methylation analysis, we compared the methylation status by HPLC and bisulfite sequencing using a next-generation sequencing (NGS) approach. A strong correlation in average MGMT methylation was observed for the frozen samples analyzed by HPLC and NGS for each primer pair (Fig. 3).
Fig. 3.
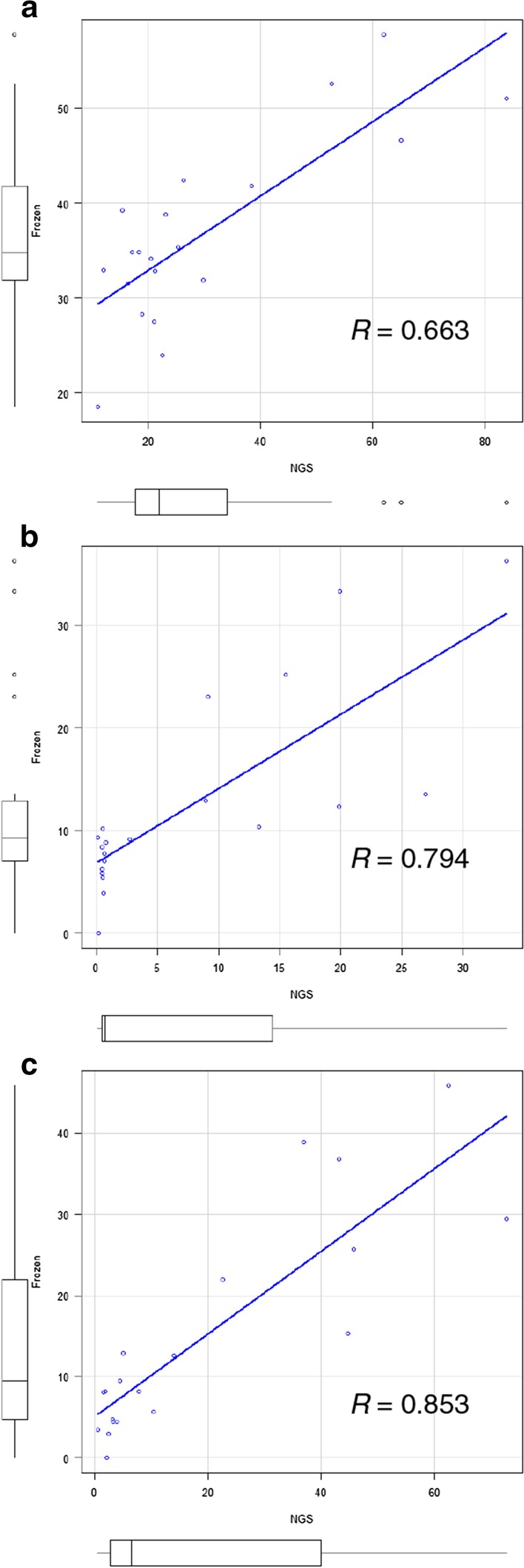
Correlation of results obtained on HPLC and NGS. Agreement between HPLC and NGS in MGMT methylation using Spearman’s rank correlation for region 1 (a), region 2 (b), and region 3 (c)
HPLC analysis was performed using frozen tumor samples from 21 patients, and MGMT methylation status could be determined for all patients. To determine the prognostic capability of the HPLC method, receiver operating characteristic (ROC) curve analysis was used to estimate the optimal cutoff values. The cutoff values for discriminating methylated and unmethylated MGMT were 34.15% for region 1, 8.84% for region 2, and 36.72% for region 3. Using these cutoff values, MGMT promoter methylation was observed in 61.9% of region 1; 61.9% of region 2, and 14.3% of region 3. Unadjusted Kaplan–Meier plots of overall survival (OS) and progression-free survival (PFS) are shown in Fig. 4. For region 1, patients whose tumors were methylated exhibited significantly longer PFS than patients with unmethylated tumors (p = 0.00108) (Fig. 4a). For region 2, patients whose tumors were methylated exhibited significantly longer OS (p = 0.00258) and PFS (p = 0.00365) than patients with unmethylated tumors (Fig. 4b).
Fig. 4.

Kaplan–Meier curve based on MGMT promoter methylation status determined using frozen samples. For region 1, patients whose tumors were methylated exhibited significantly longer PFS than patients with unmethylated tumors (a). For region 2, patients whose tumors were methylated exhibited significantly longer OS and PFS than patients with unmethylated tumors (b). For region 3, no statistically significant differences in survival were observed between methylated and unmethylated tumors (c)
Next, HPLC analysis was performed using formalin-fixed paraffin-embedded (FFPE) tumor samples from 28 GBM patients. MGMT methylation status could be determined for 26 of the 28 tumors (92.9%) in region 1, 8 of the 28 tumors (28.6%) in region 2, and 21 of the 28 tumors (75%) in region 3. Unadjusted Kaplan–Meier plots of OS and PFS are shown in Fig. 4. Log-rank test results revealed no significant differences between the groups with methylated and unmethylated MGMT for regions 1 and 3 (Fig. 5a, b). MGMT promoter methylation was observed in 46.2% of region 1 and 19.0% of region 3.
Fig. 5.

Kaplan–Meier curve based on MGMT promoter methylation status determined using FFPE sections. Log-rank test results showed no significant difference between groups with methylated and unmethylated MGMT for regions 1 and 3 (a, b)
We compared the results obtained using frozen and FFPE samples to determine the usefulness of the HPLC method for analysis of both sample types. Spearman’s rank correlation analysis indicated a moderate correlation between frozen and FFPE samples in region 1 (Fig. 6a). There was no correlation in region 3 (Fig. 6b). We could not determine the methylation status of region 2 for the majority of FFPE samples.
Fig. 6.
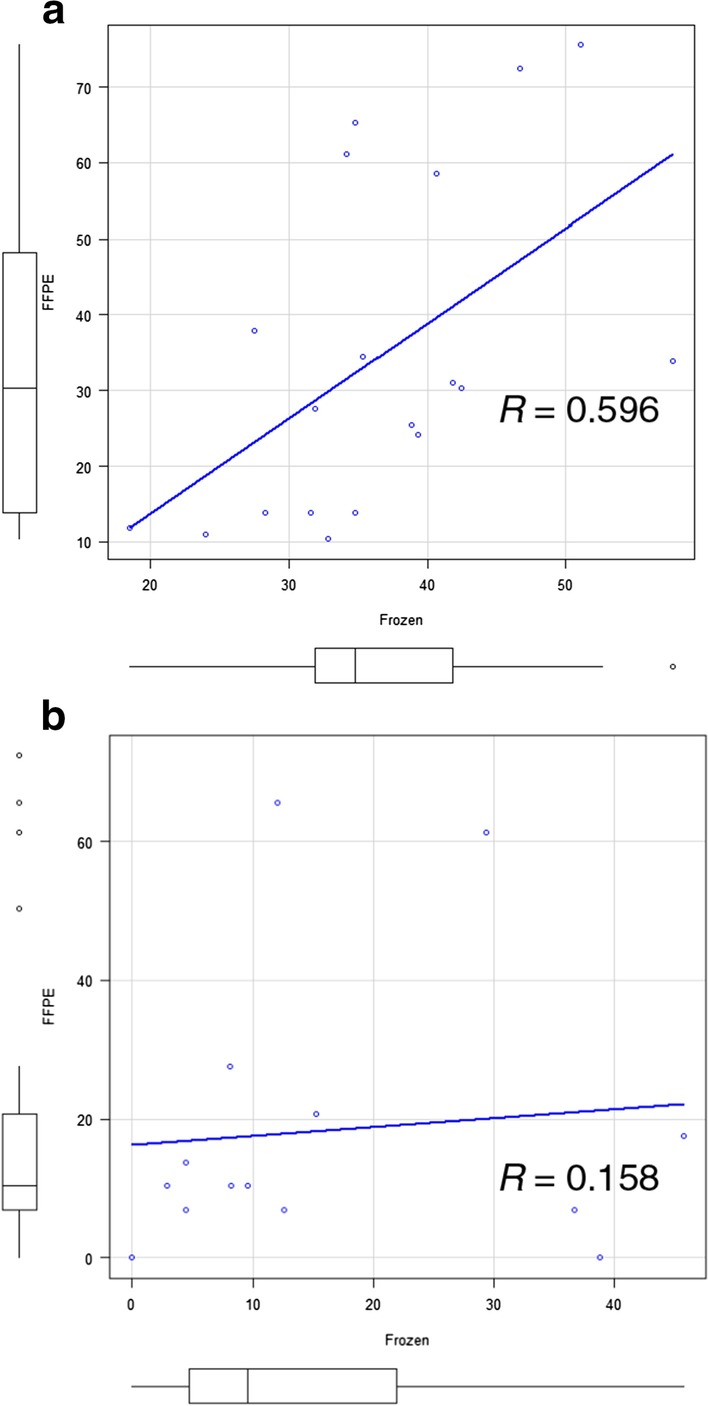
Correlation of results obtained using frozen samples versus FFPE. Agreement between frozen and FFPE samples in terms of MGMT methylation in region 1 based on Spearman’s rank correlation (a). There was no agreement for region 3 (b)
We also analyzed clinical factors affecting OS (Table 2). Univariable analysis revealed that age < 70 years (p = 0.0003) and MGMT methylation in region 2 (p = 0.0026) were associated with better OS. A preoperative Karnofsky Performance Status score of < 70 affected OS positively (p = 0.06).
Table 2.
Association of clinical and genetic factors with overall survival
| Factor | Group | n | Median survival (d) | p value |
|---|---|---|---|---|
| Age (y) | > 70 | 20 | 660 (413–876) | 0.00030 |
| ≤ 70 | 8 | 333 (105–432) | ||
| Sex | Male | 17 | 475 (385–735) | 0.39 |
| Female | 11 | 412 (175–841) | ||
| Pre-KPS | > 70 | 16 | 409 (314–611) | 0.060 |
| ≤ 70 | 12 | 689 (432–881) | ||
| EOR | Gross total resection | 25 | 475 (406–689) | 0.50 |
| Partial resection | 3 | 432 (105-NA) | ||
| BCNU wafer | No | 19 | 660 (381–876) | 0.11 |
| Yes | 9 | 441 (149–611) | ||
| Bevacizumab | No | 20 | 453.5 (335–674) | 0.75 |
| Yes | 8 | 565 (314–881) | ||
| Region 1 | Methylated | 13 | 674 (406–876) | 0.27 |
| Unmethylated | 8 | 422 (105–881) | ||
| Region 2 | Methylated | 13 | 841 (413–961) | 0.0026 |
| Unmethylated | 8 | 363 (105–660) | ||
| Region 3 | Methylated | 3 | 876 (406-NA) | 0.65 |
| Unmethylated | 18 | 521.5 (331–735) |
Pre-KPS Preoperative Karnofsky Performance Status
Discussion
The present study examined the diagnostic power of MGMT methylation analysis using ion-exchange HPLC for GBM. The patients included in the study were all newly diagnosed with IDH–wild-type GBM and treated with a standard regimen at a single center. The results of the present study provide evidence of a new MGMT promoter region (region 2) that encompasses DMR 1 and can be prognostic in IDH–wild-type GBM.
One of the most commonly used methods for MGMT methylation analysis is methylation-specific PCR (MSP) [16], which has been adopted in many clinical trials [6, 8, 9]. A major limitation of MSP, however, is that it detects only fully methylated promoters and cannot detect heterogeneous patterns of methylation [17]. Pyrosequencing (PSQ) can overcome this disadvantage, and several studies reported that PSQ is the optimal technique [12, 18]. Disadvantages of PSQ include a requirement for expensive equipment and capacity to analyze only a limited number of sequences up to 100 bp in length [19]. Therefore, analysis of MGMT methylation using PSQ could cover only a relatively short section of heterogeneous CpG island methylation. Even using PSQ, a relationship between methylation and expression of MGMT could be confirmed for only 85% of patients [14].
Other epigenetic mechanisms could explain the factors affecting MGMT transcription. Recently, single CpG methylation of heat shock protein B2 (HSPB2) was reported to have predictive value as a novel epigenetic signature for IDH–wild-type GBM [20]. Therefore, inconsistencies in MGMT methylation testing results can be explained not only by differences in methodology, but also by other epigenetic factors. As such, we cannot exclude borderline or gray-zone results in MGMT methylation tests. However, in clinical settings, more accurate and cost-effective methods are needed.
In this study, we used three novel bisulfite PCR primers to determine average MGMT methylation. We tested methylation in 21 to 38 CpGs of longer length compared to those analyzed in previous studies [10, 12, 21, 22]. In most previous studies, MGMT methylation was analyzed in DMR 2, which is located in the gene body, because the GC-rich MGMT promoter region limits bisulfite primer design. Our HPLC method can analyze relatively long PCR products compared to PSQ, and thus, we were able to verify three regions including the promoter. In tumors exhibiting heterogeneous methylation, it could be better to determine the average methylation rate of long CpG islands than the methylation of a specific CpG site. Our HPLC method enables accurate calculation of the average methylation rate.
Furthermore, for region 2, which encompasses DMR 1, the cutoff value that best correlated with prognostic outcome was 8.84% methylation of MGMT. The cutoff value of 8.84% and the ratio of methylated to unmethylated MGMT (62:38) were consistent with the results of a previous study [23]. However, methylation analysis in region 3, which encompasses DMR 2 and was thought to be the most reliable region, was not correlated with prognosis. The increase in the number of CpGs with increasing length of the PCR product was suggested as the cause of this result. Although previous studies assessed methylation levels of less than 10 CpGs in DMR 2, our HPLC method allowed analysis of the average methylation rate for 38 CpGs in region 3. The higher number of CpGs could be related to differential methylation levels. We demonstrated that methylation in regions 1 and 2 is associated with prognosis, and ours is thus the first report showing the usefulness of analyzing such a relatively long CpG island in the MGMT promoter area.
The methylation status could not be determined using our HPLC method for several samples from FFPE sections, particularly for region 2. Although we analyzed the correlation of methylation status between frozen and FFPE samples, FFPE samples were found to be unsuitable, as they contain degraded DNA, which results in even shorter fragment lengths after digestion. Furthermore, the extraction efficiency and DNA quality are poor for older FFPE samples, which precludes high-quality molecular analyses and could affect the results of MGMT methylation analyses [24]. The present results thus suggest that frozen samples are most appropriate for HPLC analysis of MGMT methylation.
Table 3 shows a comparison of established protocols with the present study protocols in MGMT methylation analysis. Simple, robust, and cost-effective methods are required for commercial MGMT methylation testing. Our HPLC method costs only a few dollars per test, and the procedure only involves transferring the PCR product to a vial before HPLC analysis, which takes 10 min. The equipment is small, and the HPLC column can be used thousands of times. Our new method is thus useful and provides excellent quality and cost-effectiveness in comparison with conventional methods.
Table 3.
Comparison of established protocols with the present study protocols in MGMT methylation analysis
| Assay | Target CpGs | Strengths | Weaknesses | References |
|---|---|---|---|---|
|
MSP (qMSP) |
DMR2 (CpG 71–86, 76–87) |
Simple Low cost |
Unable to analyze heterogeneous methylation | [12, 17] |
| PSQ | DMR2 (CpG 74–78) | High accuracy |
High cost Limitation of Amplicon length |
[12, 23] |
| MS-HRM | DMR2 (CpG 72–83, 72–89, 84–89) | Capable of analyzing heterogeneous methylation |
Low accuracy Limitation of Amplicon length |
[10, 12, 22] |
| HPLC |
500 bp upstream of TSS (CpG 1–20) DMR1 (CpG 22–59) DMR2 (CpG 72–98) |
Simple Low cost Capable of analyzing long amplicons |
Unstable measurement of paraffin-embedded specimen | Present study |
CpG numbers on MGMT CpG islands were defined by Harris et al. [16]
DMR differentially methylated region, MSP methylation-specific polymerase chain reaction, PSQ pyrosequencing,
qMSP quantitative MSP, MS-HRM methylation-sensitive high-resolution melting analysis, TSS transcription start site
One limitation of this study was the small number of patients. As such, we could not perform a multivariate analysis of MGMT methylation. However, the patients included in this study had undergone standardized therapy at a single center, and the significant differences observed among the small number of patients underscore the accuracy of our HPLC method and the strong prognostic impact of MGMT methylation testing in GBM patients.
Conclusions
In neuro-oncology, molecular biomarker–based diagnosis and decision making are routine in clinical settings. MGMT promoter methylation is one of the most important markers in patients with GBM. The combination of our HPLC method and original primers enabled the stratification of GBM patients into two groups based on MGMT promoter methylation status. Further analyses involving a larger number of patients are needed, however, to confirm our HPLC method as the gold standard for MGMT methylation testing.
Methods
Patients and tissue specimens
The study included patients newly diagnosed with GBM and treated at the Department of Neurosurgery, University of Yamanashi, between 2007 and 2018. Patients had histologically proven GBM based upon WHO 2016 criteria (International Agency for Research on Cancer [IARC], 4th edition) [25]. IDH1 mutation status was verified by immunohistochemical staining with monoclonal anti-R132H-IDH1 antibody (1:4000; Dianova, Hamburg, Germany). The expression of IDH1 mutant protein was determined semiquantitatively by assessing the proportion of positively stained tumor cells. Cases in which > 10% of cells were positive were defined as involving IDH1 mutation.
HPLC analysis was performed on 28 newly diagnosed GBM patients who underwent maximum resection, followed by radiotherapy plus concomitant and adjuvant TMZ using the Stupp regimen [26]. IDH1 R132H mutation status was verified by immunohistochemical staining and was found to be negative in these patients.
Tissue specimens were acquired after surgical removal, immediately frozen, and stored at − 80 °C until analysis. For FFPE tissue specimens, removed tumors were fixed in 10% buffered formalin and embedded in paraffin. A total of 28 FFPE GBM tumor samples were obtained, and frozen sections were prepared for 23 tumors. All tumor samples were obtained at the time of surgery after patients provided informed consent. This study was approved by the University of Yamanashi (authorization no. 1800) and performed in accordance with all relevant guidelines and regulations.
DNA extraction
DNA was extracted from fresh frozen tissue samples using a DNeasy Blood & Tissue kit (QIAGEN) or from FFPE tissues using a QIAamp DNA FFPE Tissue kit (QIAGEN) according to the manufacturer’s instructions.
Bisulfite modification and PCR
A total of 200 ng of genomic DNA was modified with sodium bisulfite using an EZ DNA Methylation-Lightning kit (Zymo Research). The DNA was amplified by PCR using TaKaRa EpiTaq™ HS (Takara Bio). PCR primers for bisulfite PCR were designed using MethPrimer (https://www.urogene.org/methprimer/) (Table 1).
HPLC
DNA methylation analysis using HPLC was performed as described previously [13]. Briefly, HPLC was performed on an LC-20A system (Shimadzu Corp., Kyoto, Japan) equipped with a stainless steel column (150 × 4.6 mm I.D.) filled with anion-exchange packing material, an oven, an auto-injector, a photodiode array detector, a degassing module, and a data analysis system. Eluent A was 25 mmol/L MES-NaOH buffer (pH 6.0), and eluent B was the same buffer containing 2 mol/L guanidine sulfate. Bisulfited PCR products were separated on a gradient of 30–50% eluent B for 10 min at a flow rate of 1.0 mL/min. The separated PCR products were detected at 260 nm. HPLC analysis was performed at a column temperature of 70 °C. Methylation rate was calculated based on a calibration curve using synthetic DNA fragments of three regions corresponding to 0% (TPG) and 100% (CpG) methylation (Fig. 2a–c).
Bisulfite NGS
Amplicon libraries were generated using an Ion Plus Fragment Library kit (ThermoFisher Scientific, MA, USA) as described previously [27]. Sequencing was performed using an Ion PGM Hi-Q View Sequencing kit (ThermoFisher Scientific) with 850 flows on an Ion 318 Chip kit v2 (ThermoFisher Scientific), according to the manufacturer’s protocol. After sequencing, single processing and base calling were performed using Torrent Suite 5.12.1 (ThermoFisher Scientific). Methylation analysis was performed using Methylation Analysis_Amplicon plug-in v2.1 (ThermoFisher Scientific).
Statistical analysis
All statistical analyses were performed using EZR, version 2.3-1 (Saitama Medical Centre, Jichi Medical University, Saitama, Japan), a graphical user interface for R (R Foundation for Statistical Computing, Vienna, Austria) [28]. Average methylation level was calculated for both HPLC and NGS analyses. MGMT methylation levels in the same tumor samples were correlated using Spearman rank correlation. The threshold enabling detection of a favorable outcome, defined as longer OS, was identified by ROC curve analysis. PFS and OS were calculated from the time of surgery until recurrence, death, or last follow-up. PFS and OS were estimated using the Kaplan–Meier method, and data were compared using the two-sided log-rank test. Univariate Cox regression analysis was applied to assess the prognostic and predictive values of methylation status of the MGMT promoter. A p value < 0.05 was considered statistically significant.
Acknowledgements
We are grateful to the patients and staff who participated in this study. We thank Wakaba Iha for technical support and Dr. Zentaro Yamagata for providing helpful comments.
Abbreviations
- DMR
Differentially methylated region
- DNA
Deoxyribonucleic acid
- GBM
Glioblastoma
- HPLC
High-performance liquid chromatography
- HSPB2
Heat shock protein B2
- IDH
Isocitrate dehydrogenase
- MGMT
O6-methylguanine-DNA methyltransferase
- MSP
Methylation-specific polymerase chain reaction
- NGS
Next-generation sequencing
- OS
Overall survival
- PFS
Progression-free survival
- PSQ
Pyrosequencing
- TMZ
Temozolomide
Authors’ contributions
KM, TI, and HK conceived and designed the study. MH and TK1 acquired the data. MH and AW analyzed and interpreted the data. MH and KM wrote and revised the manuscript. YY, NO, and TK5 provided administrative, technical, or material support. All authors read and approved the final manuscript.
Funding
This work was funded by the Sekisui Medical Co., Ltd. (Ibaraki, Japan).
Availability of data and materials
The datasets used and analyzed during the present study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Written informed consent about research use of samples was obtained from all participants from Yamanashi University Hospital. All procedures performed in the study involving human subjects were in accordance with the ethical standards of the institutional research committee and with the 1964 declaration of Helsinki and its later amendments or comparable ethical standards.
Consent for publication
Not applicable.
Competing interests
Sekisui Medical Co., Ltd. (Ibaraki, Japan) provided support for the work in the current study and could be influenced by a secondary interest such as future financial gain.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kohler BA, Ward E, McCarthy BJ, et al. Annual report to the nation on the status of cancer, 1975–2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst. 2011;103:714–736. doi: 10.1093/jnci/djr077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol. 2005;64:479–489. doi: 10.1093/jnen/64.6.479. [DOI] [PubMed] [Google Scholar]
- 3.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 4.Qian XC, Brent TP. Methylation hot spots in the 5' flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1997;57:3672–3677. [PubMed] [Google Scholar]
- 5.Watts GS, Pieper RO, Costello JF, et al. Methylation of discrete regions of the O6-methylguanine DNA methyltransferase (MGMT) CpG island is associated with heterochromatinization of the MGMT transcription start site and silencing of the gene. Mol Cell Biol. 1997;17:5612–5619. doi: 10.1128/MCB.17.9.5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 7.Bell EH, Zhang P, Fisher BJ, et al. Association of MGMT promoter methylation status with survival outcomes in patients with high-risk glioma treated with radiotherapy and temozolomide: an analysis from the NRG oncology/RTOG 0424 trial. JAMA Oncol. 2018;4:1405–1409. doi: 10.1001/jamaoncol.2018.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weller M, Tabatabai G, Kästner B, et al. MGMT promoter methylation is a strong prognostic biomarker for benefit from dose-intensified temozolomide rechallenge in progressive glioblastoma: the DIRECTOR trial. Clin Cancer Res. 2015;21:2057–2064. doi: 10.1158/1078-0432.CCR-14-2737. [DOI] [PubMed] [Google Scholar]
- 9.Wick W, Hartmann C, Engel C, et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol. 2009;27:5874. doi: 10.1200/JCO.2009.23.6497. [DOI] [PubMed] [Google Scholar]
- 10.Malley DS, Hamoudi RA, Kocialkowski S, et al. A distinct region of the MGMT CpG island critical for transcriptional regulation is preferentially methylated in glioblastoma cells and xenografts. Acta Neuropathol. 2011;121:651–661. doi: 10.1007/s00401-011-0803-5. [DOI] [PubMed] [Google Scholar]
- 11.Shah N, Lin B, Sibenaller Z, et al. Comprehensive analysis of MGMT promoter methylation: correlation with MGMT expression and clinical response in GBM. PLoS ONE. 2011;6:e16146. doi: 10.1371/journal.pone.0016146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johannessen LE, Brandal P, Myklebust TA, et al. MGMT gene promoter methylation status—assessment of two pyrosequencing kits and three methylation-specific PCR methods for their predictive capacity in glioblastomas. Cancer Genom Proteomics. 2018;15:437–446. doi: 10.21873/cgp.20102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yotani T, Yamada Y, Arai E, et al. Novel method for DNA methylation analysis using high-performance liquid chromatography and its clinical application. Cancer Sci. 2018;109:1690–1700. doi: 10.1111/cas.13566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Everhard S, Tost J, El Abdalaoui H et al. Identification of regions correlating MGMT promoter methylation and gene expression in glioblastomas. Neuro-oncology 2009; 11: 348–356. [DOI] [PMC free article] [PubMed]
- 15.Harris LC, Potter PM, Tano K, et al. Characterization of the promoter region of the human O6-methylguanine-DNA methyltransferase gene. Nucleic Acids Res. 1991;19:6163–6167. doi: 10.1093/nar/19.22.6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esteller M, Hamilton SR, Burger PC, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59:793–797. [PubMed] [Google Scholar]
- 17.Tetzner R. Prevention of PCR cross-contamination by UNG treatment of bisulfite-treated DNA. Methods Mol Biol. 2009;507:357–370. doi: 10.1007/978-1-59745-522-0_26. [DOI] [PubMed] [Google Scholar]
- 18.Quillien V, Lavenu A, Karayan-Tapon L, et al. Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, MethyLight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer. 2012;118:4201–4211. doi: 10.1002/cncr.27392. [DOI] [PubMed] [Google Scholar]
- 19.Mikeska T, Felsberg J, Hewitt CA, Dobrovic A. Analysing DNA methylation using bisulphite pyrosequencing. In: Epigenetics protocols. Springer 2011; 33–53. [DOI] [PubMed]
- 20.Yin A-A, He Y-L, Etcheverry A, et al. Novel predictive epigenetic signature for temozolomide in non-G-CIMP glioblastomas. Clin Epigenet. 2019;11:76. doi: 10.1186/s13148-019-0670-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamashita S, Yokogami K, Matsumoto F, et al. MGMT promoter methylation in patients with glioblastoma: is methylation-sensitive high-resolution melting superior to methylation-sensitive polymerase chain reaction assay? J Neurosurg. 2018;130:780–788. doi: 10.3171/2017.11.JNS171710. [DOI] [PubMed] [Google Scholar]
- 22.Quillien V, Lavenu A, Sanson M, et al. Outcome-based determination of optimal pyrosequencing assay for MGMT methylation detection in glioblastoma patients. J Neurooncol. 2014;116:487–496. doi: 10.1007/s11060-013-1332-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wick W, Weller M, van den Bent M, et al. MGMT testing–the challenges for biomarker-based glioma treatment. Nat Rev Neurol. 2014;10:372–385. doi: 10.1038/nrneurol.2014.100. [DOI] [PubMed] [Google Scholar]
- 24.Quillien V, Lavenu A, Ducray F, et al. Validation of the high-performance of pyrosequencing for clinical MGMT testing on a cohort of glioblastoma patients from a prospective dedicated multicentric trial. Oncotarget. 2016;7:61916–61929. doi: 10.18632/oncotarget.11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 26.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe A, Miyake K, Nordlund J et al. Association of aberrant ASNS imprinting with asparaginase sensitivity and chromosomal abnormality in childhood BCP-ALL. Blood 2020. [DOI] [PMC free article] [PubMed]
- 28.Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transpl. 2013;48:452–458. doi: 10.1038/bmt.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and analyzed during the present study are available from the corresponding author on reasonable request.