

Abstract
Bacterial resistance to β-lactam antibiotics is largely mediated by β-lactamases, which catalyze the hydrolysis of these drugs and continue to emerge in response to antibiotic use. β-lactamases that hydrolyze the last resort carbapenem class of β-lactam antibiotics (carbapenemases) are a growing global health threat. Inhibitors have been developed to prevent β-lactamase-mediated hydrolysis and restore the efficacy of these antibiotics. However, there are few inhibitors available for problematic carbapenemases such as Oxacillinase-48 (OXA-48). A DNA-encoded chemical library approach was used to rapidly screen for compounds that bind and potentially inhibit OXA-48. Using this approach, a hit compound, CDD-97, was identified with sub-micromolar potency (Ki = 0.53 ± 0.08 μM) against OXA-48. X-ray crystallography showed that CDD-97 binds non-covalently in the active site of OXA-48. Synthesis and testing of derivatives of CDD-97 revealed structure-activity relationships and informed the design of a compound with a two-fold increase in potency. CDD-97, however, synergizes poorly with β-lactam antibiotics to inhibit the growth of bacteria expressing OXA-48 due to poor accumulation into E. coli. Despite the low in vivo activity, CDD-97 provides new insights into OXA-48 inhibition and demonstrates the potential of using DNA-encoded chemistry technology to rapidly identify β-lactamase binders and to study β-lactamase inhibition, leading to clinically useful inhibitors.
Keywords: β-lactam antibiotic, β-lactamase, carbapenemase, OXA-48, drug discovery, DNA-encoded library, DECL, DEC-Tec
Multidrug-resistant bacteria are associated with 2 million infections and approximately 23,000 deaths per year in the U.S. alone and thus are a serious threat to public health1. Growing resistance to the invaluable β-lactam antibiotic family is a concern that is elevated by the slowed development of novel therapeutic agents. β-lactam antibiotics make up approximately 65% of the prescribed antibiotics worldwide and reduced efficacy of these drugs due to resistance would severely impact the treatment of bacterial infections2–4. β-lactams act by covalently inhibiting essential cell wall biosynthesis transpeptidase enzymes called penicillin-binding proteins (PBPs), which leads to cell death 5,6. Bacteria can circumvent the bactericidal effect of β-lactams by expressing β-lactamase enzymes that hydrolyze the pharmacophoric β-lactam ring of the antibiotics to confer resistance7–9. β-lactamase inhibitors have been developed and used in conjunction with β-lactams to suppress β-lactamase activity and prolong antibiotic efficacy2,10. However, nearly 5000 different β-lactamases have been identified11 and the potency of inhibitors varies among enzymes. Inhibitor-resistant β-lactamases that hydrolyze the last resort carbapenem class of β-lactam antibiotics are a particular concern.
Carbapenem-resistant Enterobacteriaceae (CRE) infections are associated with increased patient morbidity and mortality and new treatment options are needed1,12,13. The Gram-negative Enterobacteriaceae family frequently causes multidrug-resistant nosocomial infections and the presence of carbapenem-hydrolyzing β-lactamases, or carbapenemases, in these strains severely limits treatment options. Most β-lactamases are unable to hydrolyze carbapenems due to the unique stereochemistry of these antibiotics14. The emergence and dissemination of carbapenemases has resulted in increased β-lactamase-mediated resistance to the drugs13,15–17.
β-lactamases are grouped into classes A – D based on primary amino acid sequence homology and enzyme mechanism. Classes A, C, and D inactivate β-lactam antibiotics through a serine hydrolase mechanism while members of class B are metalloenzymes that utilize zinc ions for hydrolysis18,19. The most prevalent carbapenemases in Enterobacteriaceae are the class A KPC-2 β-lactamase, the NDM, IMP, and VIM enzymes of class B, and the class D enzyme OXA-48 13. OXA-48 was first reported from a Klebsiella pneumoniae isolate in Turkey in 2008. However, it has now been identified worldwide in a range of Enterobacteriaceae. OXA-48 is resistant to all clinically available β-lactamase inhibitors except avibactam20–22.
β-lactam-based molecules such as clavulanic acid were the first clinically available inhibitors and their ability to synergize with antibiotics proved an effective strategy to manage resistance10. Unfortunately, these inhibitors are largely ineffective on OXAs13 and are susceptible to hydrolysis and resistance due to the β-lactam ring23–25. Non-β-lactam-based inhibitors have the prospect of slowing β-lactamase-mediated resistance as new mechanisms of inhibitor inactivation will likely be required26. Inhibitors that do not contain a β-lactam ring such as avibactam, relebactam (both diazabicyclooctanes) and vaborbactam (boronic acid-based) have unique scaffolds yet covalently inhibit a range of β-lactamases. Despite having variable activity against the Class D enzymes, avibactam is the only clinically available inhibitor with significant efficacy against OXA-4821. Relebactam and vaborbactam are ineffective against OXA-4827–29, highlighting the need for novel OXA-48 inhibitors. Most β-lactamase inhibitors are directed towards class A β-lactamases but OXA enzymes share low sequence homology with class A30. No OXA β-lactamase specific inhibitors have been clinically approved, but the β-lactam-based penicillin sulfones31,32 and non-β-lactams (phosphates/phosphonates, diazabicyclooctanes, and boronic acid-based compounds) have shown promise as inhibitors of OXA enzymes33,34. Although the scaffolds of non-β-lactam-based inhibitors are unique, they are still ‘mechanism-based’ and similar to β-lactams in how they covalently inactivate β-lactamases. The ability of the KPC-2 β-lactamase to hydrolyze avibactam suggests variants can occur to inactivate non-β-lactam-based inhibitors21.
Although clinically available β-lactamase inhibitors utilize a covalent inactivation mechanism, non-covalent inhibitors may also be clinically viable. Promising non-mechanism-based OXA-48 inhibitors have been identified using fragment-based approaches35,36. An orthogonal fragment screen using both SPR and inhibition assays was used to find new scaffolds and aided the development of an OXA-48 inhibitor with micromolar potency35. This inhibitor was developed by merging the alternate conformations of a weaker pyridylbenzoic acid inhibitor seen in a co-crystal structure with OXA-48. This fragment study was continued with a focused library of monosubstituted benzoic acid fragments to further examine the OXA-48 binding pocket36. Nevertheless, more potent inhibitors of OXA-48 inhibition are needed
DNA-encoded chemical libraries (DECLs) are combinatorial chemical libraries in which each compound is attached to a unique DNA molecule37–39. The DNA molecule serves as a barcode, in which the different chemical building blocks are encoded by short, unique DNA sequences. This allows all the compounds to be pooled and screened against a target and subsequently, binding hits can be identified by sequencing the DNA tags using next-generation sequencing. DELs are typically synthesized with a split and pool approach to create combinatorial libraries with up to millions of compounds to efficiently scan chemical space39–42. DNA-encoded chemistry technology has been used to identify inhibitors for various targets including the essential InhA enzyme in Mycobacterium tuberculosis43, suggesting it could be expanded to other bacterial enzymes.
In this study, we describe the identification of two non-β-lactam OXA-48 inhibitors with sub-micromolar potency by using a DECL screening approach. Additionally, the X-ray crystal structure of one inhibitor in complex with OXA-48 was determined, and though in vivo potential is currently limited due to low accumulation in bacteria, this property can potentially be improved. DECLs have been used to identify inhibitors for a variety of proteins such as kinases, phosphatases, among others39,44,45. To date, however, no β-lactamase inhibitors have been reported using the DECL approach. This work suggests DECLs can provide new chemotypes for β-lactamase inhibition and may be a useful means to rapidly study the inhibition of β-lactamases and aid in the design of new inhibitors.
Results
DNA-encoded library synthesis and screening against OXA-48
A split and pool approach was used to create a combinatorial triazine-based library (Figure 1). Library members contained a double-stranded DNA portion, which functioned as the encoding DNA tag39.
Figure 1.
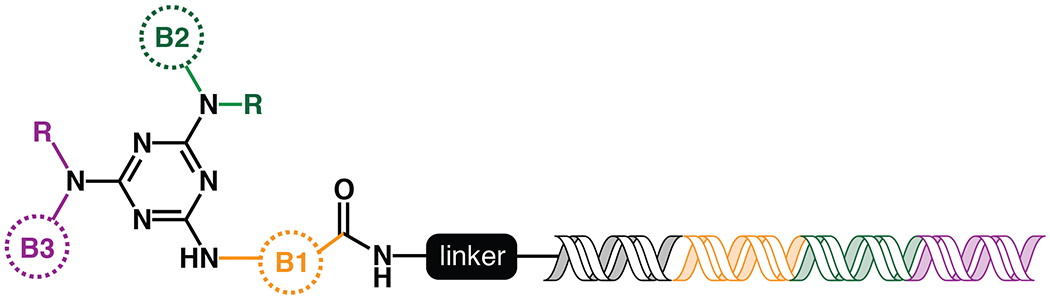
Topology of the DNA-encoded library synthesized and screened against OXA-48. For each library, the colored circles indicate building block 1 (orange), 2 (green) and 3 (purple). The “R” on positions B2 and B3 indicates both primary and secondary amines were added at these positions. The sequences within the DNA tag are color-coded to correspond to which building block it is encoding.
To build the triazine library, an initial array of building blocks (B1s) were linked to a short unique oligonucleotide sequence, coded to represent the specific building block added. All the unique DNA-linked compounds were pooled and attached to a triazine scaffold. This pool, containing the B1 building blocks attached to the triazine scaffold, was split and the set of B2 building blocks was added. A short oligonucleotide encoding the specific B2 building block was also ligated to the primary oligonucleotide. The compounds and associated DNA were then pooled again, split, and B3 building blocks were added and encoding oligonucleotides were ligated to the primary oligonucleotide to create a unique tag that encodes all attached building blocks for a given compound. After ligation, the compounds were pooled to form the final library, which contained approximately 162 million unique compounds. Details of library synthesis can be found in Supporting Information (Figures S1–S3).
For the selection of compounds that bind OXA-48, His-tagged OXA-48 was immobilized onto Ni-NTA agarose beads and incubated with the triazine library. A non-target control (NTC) with no enzyme that contained only the Ni magnetic beads was also screened against the library. After incubation, the beads were isolated and washed and the bound ligands were eluted. Two subsequent rounds of selection were performed with fresh immobilized OXA-48 to enrich for specific binders. The DNA tags of the recovered DECL compounds were amplified using PCR and NGS sequencing was performed to delineate the tag sequences. The identities of enriched compounds were determined by decoding DNA barcodes from the sequencing data set and translating them into their associated molecular components. Subsequently, the enrichment of each observed chemical species was evaluated. In our enrichment analysis strategy, a specific combination of building blocks is called an n-synthon, where n denotes the number of building blocks in the combination46. Since the triazine library has three cycles of combinatorial synthesis, it contains mono-, di-, and tri-synthons. The enrichment of each observed n-synthon was analyzed by comparing its observed population to its expected population from a reference distribution44. N-synthon classification is advantageous for both statistical and chemical analysis. Features with fewer synthons are expected to appear in the sampled data more frequently and thus are typically well-sampled. Such classification also allows rapid insight into structure-activity relationships and can suggest minimum pharmacophores necessary to yield enrichment in the affinity selection. OXA-48 hits were assessed by comparing the enrichment of n-synthons from the OXA-48 selection versus their corresponding enrichment in the non-target control selection. Enrichment was quantified using normalized z-scores47 and depicted by plotting the NTC z-score against the OXA-48 z-scores (Figure 2). Such screening provided a rapid means of identifying compounds enriched for binding to OXA-48. Enriched n-synthons from the library were used to guide the design and synthesis of compounds without DNA tags for biochemical evaluation.
Figure 2.
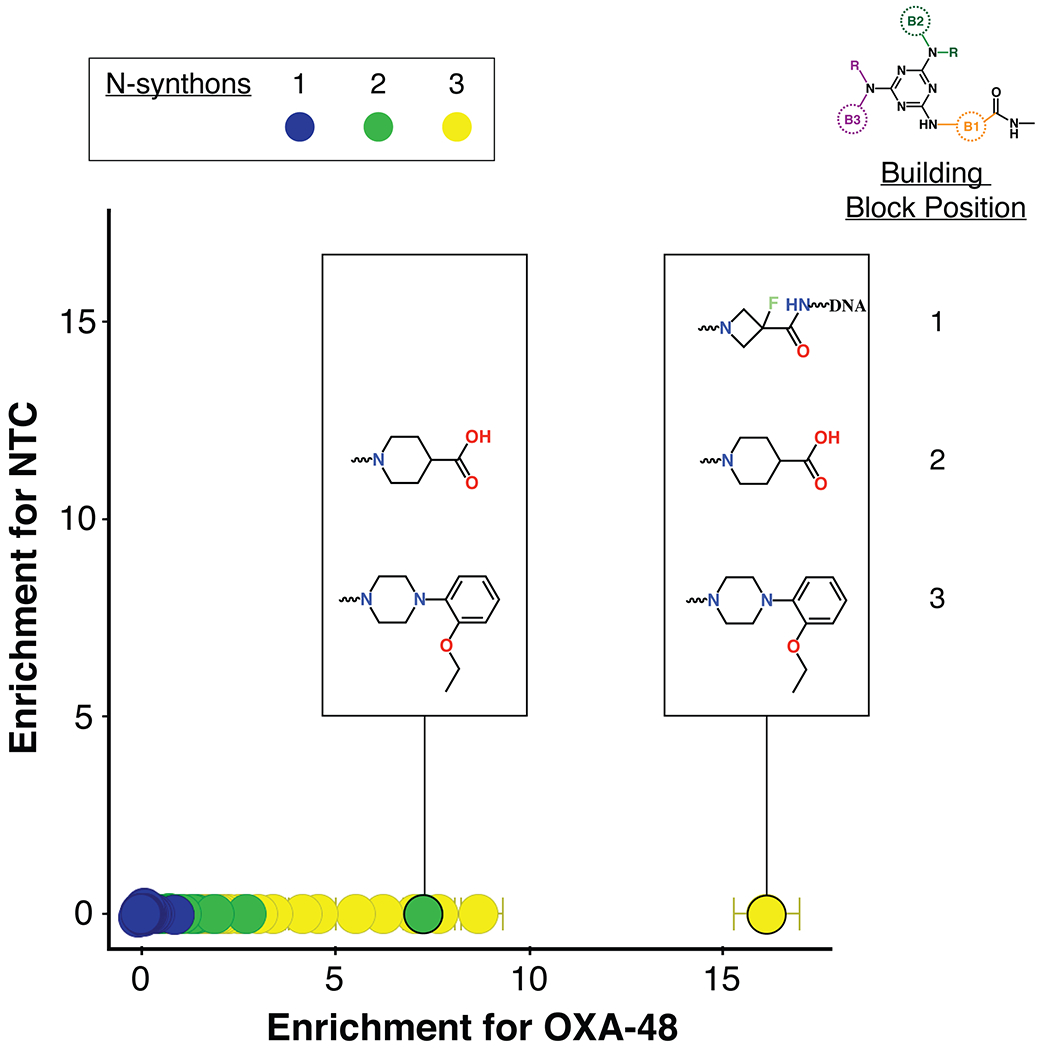
Enrichment plot of triazine DECL compounds for binding to OXA-48 versus the control. Points represent observed n-synthons and are color-coded by the number of component building blocks: mono-synthons are blue, di-synthons are green and tri-synthons are yellow. The y-axis is the normalized z-score of enrichment for the non-target control selection versus the x-axis which represents this score for the OXA-48 selection.
Biochemical evaluation of synthon-derived compounds and lead identification
Compounds that were identified from DECL screening were re-synthesized without DNA tags. The activity of these compounds was assessed by in vitro inhibition assays with purified OXA-48. Compounds were tested for their ability to inhibit OXA-48 mediated hydrolysis of nitrocefin, a chromogenic β-lactam substrate routinely used in β-lactamase inhibition assays. Nitrocefin contains a conjugated ring system and produces a visible color shift from yellow to red upon hydrolysis48. The rate of nitrocefin hydrolysis in the presence of a constant amount of OXA-48 and increasing concentrations of compound was plotted against compound concentration to determine the inhibition constant (Ki) towards OXA-48.
The most enriched n-synthon in the screen was a single tri-synthon compound with an enrichment score of 16 (Figure 2). These building blocks were assembled on to a triazine scaffold to create Compound 1 (Cmpd 1) (Figure 3). Cmpd 1 was enriched ~ 2-fold more than other highly enriched compounds that shared identical or similar building blocks at positions 2 and 3 (Figure 2). This suggested that either the unique building block at position 1 led to the increased enrichment of Cmpd 1 compared to similar tri-synthons or, alternatively, its enrichment was due to other reasons such as higher synthetic yield during library synthesis. Cmpd 1 showed near micromolar activity against purified OXA-48 (Ki = 0.9 ± 0.1μM) (Figure 3). Another interesting finding was that the di-synthon incorporating the building blocks from cycles 2 and 3 from Cmpd 1 was the fourth most enriched feature with a normalized enrichment of 7.2. Based on this finding, a series of compounds were synthesized using only building blocks in these positions on the triazine scaffold. These all contained the ethoxyphenyl-piperazine group, which was found in several highly enriched di- and tri-synthons. To rule out the possibility that the synthon at position 1 of Cmpd 1 (the fluoro-N-methylazetidine-3-carboxamide group) contributes to binding, Cmpds 2 and 3 were synthesized. Cmpd 3 contained the original B1 building block of Cmpd 1 with the ethoxyphenyl-piperazine while Cmpd 2 was similar but with a carboxylated B1 building block. (Figure 3). The OXA-48 active site, like that of all β-lactamases, contains a carboxylate binding pocket. For OXA-48, this pocket consists of residues Ser118, Thr209 and Arg250. Assuming the compounds bind the active site, it was predicted that the carboxamide group would be near this pocket. Therefore, Cmpd 2, a carboxylate derivative of Cmpd 3, was also synthesized. Both compounds showed minimal activity with Ki values > 50 μM (Figure 3). These results suggest that the fluoro-N-methylazetidine-3-carboxamide group of Cmpd 1 does not contribute to inhibition. Next, the original highly enriched di-synthon was used to design Cmpd 4 (Figure 3). Cmpd 4 was the most potent of these derivatives with a Ki of 0.53 ± 0.08 μM against OXA-48. This compound contained a carboxylate group that could potentially bind the carboxylate binding pocket of OXA-48. To test this hypothesis, Cmpd 5, which is an alkylated form of Cmpd 4, was also evaluated. Cmpd 5 exhibited a loss of activity (Ki > 50 μM), showing that the carboxylate in showing that the carboxylate in CDD-97 is indeed important for potency, consistent with an interaction with the carboxylate binding pocket. Cmpd 4 showed the most promise against OXA-48 and was further investigated as the lead compound (CDD-97). Details of chemical synthesis can be found in Supporting Information.
Figure 3.
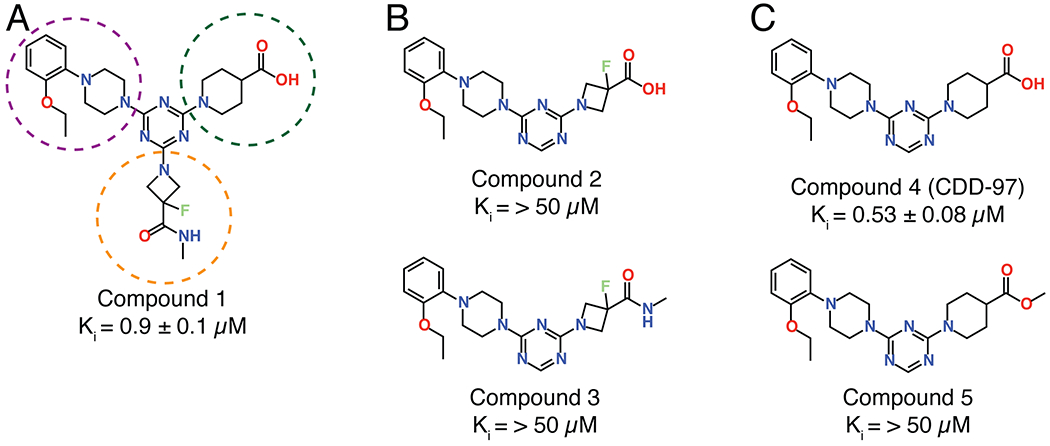
Compounds designed and synthesized based on the synthons enriched for binding OXA-48 in the DECL screening. Compound 1 represents the most enriched compound in the screen and showed a submicromolar Ki against OXA-48. The building blocks of compound 1 are circled based on the color-coding of building block position shown in Figure 2; orange is building block 1, green is building block 2 and purple is building block 3. To assess the importance of the B1 building block, Compound 2 was created with the acid form of the most enriched cycle 1 building block along with a methylated version (Compound 3), however potency was low for both compounds. CDD-97 (Compound 4) was designed based on the most enriched di-synthon. It showed the highest activity with a sub-micromolar Ki. The methylated version (Compound 5) showed low activity revealing the importance of the acid for potency.
Crystallization of CDD-97 with OXA-48
The structure of CDD-97 in complex with OXA-48 was determined to assess the binding site and facilitate medicinal chemistry modifications to enhance potency. The structure was determined by soaking OXA-48 apo crystals with CDD-97. The structure was solved in the P65 space group with 4 molecules in the asymmetric unit, each bound to a molecule of CDD-97, with occupancies of 85, 87, 88, and 89% (Table S1). The structure shows that CDD-97 binds in the active site of OXA-48 and makes several interactions, consistent with its relatively high potency (Figure 4). A polder mFo-Fc OMIT map confirmed the presence of the CDD-97 in the OXA-48 structure (Figure S5). The orientation of CDD-97 in the active site of all 4 monomers was identical, with small variations seen in the position of the rotatable carboxylate group, which was oriented to hydrogen bond with either 2 or 3 active site residues. The carboxylate of CDD-97 was confirmed to lie in the carboxylate binding pocket of OXA-48. This carboxylate group hydrogen bonds with Ser118, Thr209, and a salt bridge with Arg250, although its rotation leads to loss of the Ser118 interaction in some chains in the asymmetric unit. OXA-48, like all β-lactamases of its class, possesses a largely hydrophobic active site49. The structure of the complex reveals that CDD-97 makes several hydrophobic interactions with OXA-48 including the triazine ring interacting with Ile102 and Tyr211, the piperazine ring interacting with Trp105, Leu158, and Thr213, and the terminal 2-ethoxyphenyl group interacting with Trp105, Val120, Leu158 (Figure 4). A comparison of the CDD-97/OXA-48 structure with the previously solved apo OXA-48 structure (PDB: 3HBR), the overall RMSD value of 0.39Å, which suggests there is not a significant change in overall residue positions between the two structures. In the apo OXA-48 structure, Arg214 forms hydrogen bonds with Asp159 and several water molecules, one of which additionally binds Gln124. In the CDD-97/OXA-48 complex, the inhibitor fills the active site and ultimately displaces Arg214, which is found at the base of the active site. Due to this movement, the electron density for the side chain of Arg214 is relatively weak, however it is clear that the stabilizing interactions that typically hold Arg214 in place in the apo structure are lost in the structure of the complex due to CDD-97 binding (Figure S6).
Figure 4.
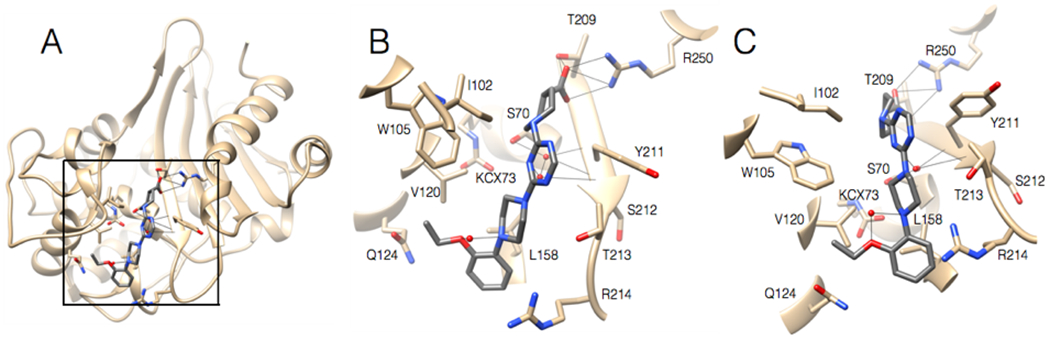
A. Ribbon diagram of 2.2 Å structure of OXA-48 (tan) bound with CDD-97 (gray). The box indicates the region viewed in 4B and 4C. B. Active site region of OXA-48 with CDD-97 shown in gray with nitrogen and oxygen atoms colored in blue and red, respectively. OXA-48 residues are shown as stick models and labeled. C. View of OXA-48 with CDD-97 tilted 30 degrees from the position shown in 4B.
Structure-activity relationship studies
Several small-molecule derivatives of CDD-97 were created based on the inhibition and X-ray data to better understand the necessity of the different chemical moieties and gain insight for increasing potency (synthesis details are provided in Supporting Information). Derivatives were categorized by which ring was modified on CDD-97 (Figure 5) and are shown alongside their Ki values in Table 1. Structures of selected compounds in complex with OXA-48 were predicted by docking to gain insight into the molecular basis of changes in potency (Figure 6). The removal of ring 1 (ethoxyphenyl group), to create Cmpd 6, resulted in significant loss of potency (Ki > 50 μM). Docking results show the carboxylate group from the 4-carboxypiperidine ring retains strong interactions with Ser118, Lys208, Thr209, and Arg250 residues of the carboxylate binding pocket, suggesting the lack of the hydrophobic interactions resulting from the removal of the terminal 2-ethoxyphenyl ring adversely affects binding (Figure 6). Other derivatives with modifications of this ring, Cmpds 7, 8 and 9, showed little change in potency (Ki values of 0.76, 1.7 and 2.2 μM respectively) compared to CDD-97 despite the addition of electronegative atoms. These results suggest ring 1 can tolerate additional substituents in its ortho-position and still maintain sufficient hydrophobic interactions to retain potency.
Figure 5.
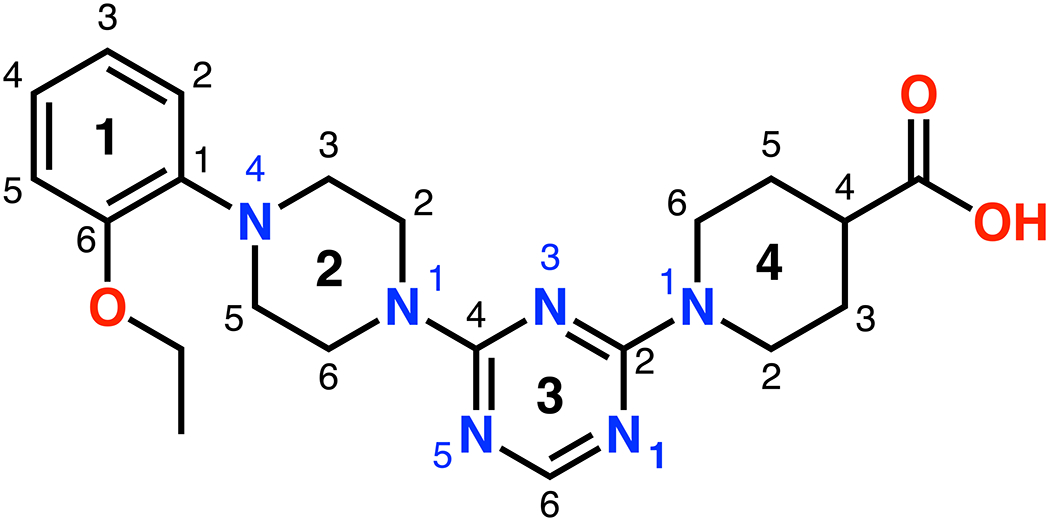
Schematic showing the ring and atom numbering of the lead compound CDD-97.
Table 1.
Inhibition of CDD-97 and CDD-97 derivatives for structure-activity relationship studies (modified portion of derivatives is in bold).
| Compound I.D. | Structure | Ki (μM) |
|---|---|---|
| 6 | 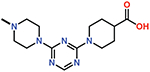 |
> 50 |
| 7 | 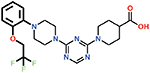 |
0.76 ± 0.03 |
| 8 | 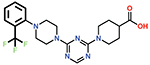 |
1.7 ± 0.3 |
| 9 | 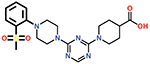 |
2.2 ± 0.2 |
| 10 | 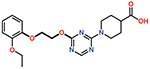 |
18.8 ± 2.5 |
| 11 | 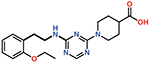 |
7.8 ± 0.1 |
| 12 | 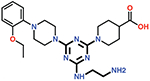 |
0.84 ± 0.03 |
| 13 | 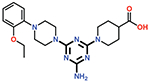 |
0.61 ± 0.02 |
| 14 | 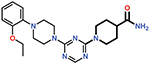 |
> 50 |
| 15 | 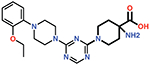 |
29 ± 4 |
| 16 | 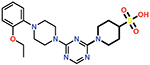 |
0.26 ± 0.04 |
Figure 6.
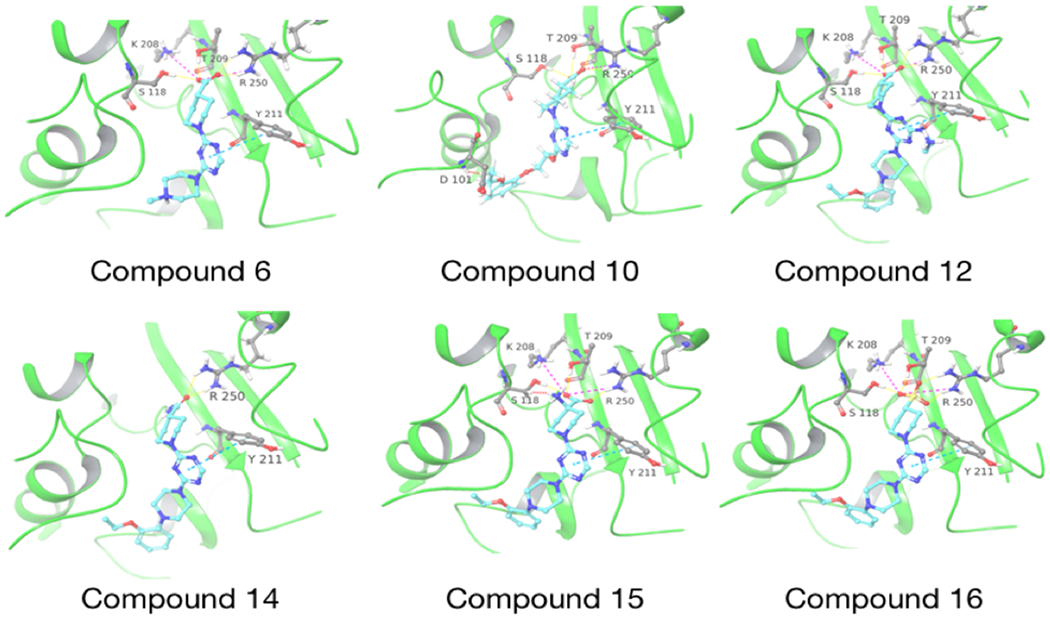
Molecular docking of CDD-97 derivatives in the active site of OXA-48. The docked compound is indicated below each structure. OXA-48 is colored in green and the compounds are colored with carbon-cyan, oxygen-red, nitrogen-blue. Hydrogen bonds are shown as dashed yellow lines, salt bridges are pink dashed lines and clashes are red dashed lines.
The piperazine ring (ring 2) also makes important hydrophobic interactions (Figures 4,5). Cmpds 10 and 11 were synthesized to test the effect of a linear chain replacing the piperazine ring. The introduction of flexibility from the linear chains at this position resulted in approximately a 16 to 40-fold reduction in potency for these derivatives, suggesting the shape and rigidity of the piperazine contributes to the inhibition potency. Docking results support this conclusion in that the carboxylate group from the 4-carboxypiperidine ring in Cmpd 10 retains strong interactions with Ser118, Lys208, Thr209, and Arg250 (Figure 6). However, the lack of the rigid piperazine ring gives rise to a more mobile terminal part of the ligand and with this atomic clash between the ligand and the binding pocket are possible. The triazine core (ring 3) makes hydrophobic interactions with Ile102 and pi-pi stacking interactions with the similarly aromatic Tyr211 residue (Figure 4). The addition of an amine group at the 6-position of the ring (Cmpd 13) does not have a large effect on potency even if followed by a longer ethylamine chain (Cmpd 12). Docking of Cmpd 12 shows the amine group is oriented towards the solvent and does not clash with residues in OXA-48 (Figure 6). This is consistent with the structures of the CDD97/OXA-48 complexes, which reveal this position is exposed to solvent (Figure 4). It is also consistent with the DECL selection data, as the triazine library had cycle 1 building blocks and the DNA linker in this position (Figures 1,2).
Finally, the 4-carboxypiperidine group (ring 4) showed the highest sensitivity to chemical modification (Table 1). This ring is situated above the Ser70 catalytic nucleophile in the OXA-48/CDD-97 structure, although it is not in direct contact with Ser70 (Figure 4). The carboxylate group of CDD-97 makes several hydrogen bonds to OXA-48 residues, which likely contributes to its potency. Substitution of the carboxylate group with an ester (Cmpd 5) (Figure 3) or a primary amide (Cmpd 14) resulted in a significant loss of inhibitory activity, as expected due to the loss of charge and hydrogen bonding potential (Table 1). It was not possible to obtain viable poses of Cmpd 5 by docking due to steric clashes while Cmpd 14 loses interactions with Ser118, Lys208, and Thr209 in the carboxylate binding pocket (Figure 6). Since the carboxypiperidine ring is close to Ser70, Cmpd 15 was designed with a primary amine added at position 4 to examine if the amine could interact with Ser70 or the carboxylated Lys73, which serves as the general base in OXA-48 catalysis. This resulted in a significant decrease in potency, suggesting the added amine may perturb the hydrogen bonding contribution of the carboxylate group. Docking results suggest that the added amino group clashes with the Ser118 side chain (Figure 6). The amino group also decreases the electron density of the carboxylate to weaken the hydrogen bonding and the salt bridge interactions with the residues in the carboxylate binding pocket.
Of all the derivatives, Cmpd 16, which substitutes the hydrogen bonding carboxylate group with sulfonic acid, showed an increase in potency (Ki = 0.27 ± 0.02 μM). This two-fold increase in potency may be due to enhanced interactions of the sulfonic acid with the carboxylate binding pocket. Consistent with this idea, docking results show salt bridges between the sulfonic acid and residues Lys208 and Arg250, as well as hydrogen bonds to Ser118, Thr209, and Arg250 (Figure 6).
Spectrum of activity with other OXA enzymes
The high sequence diversity of class D β-lactamases has made it difficult to identify a class-wide inhibitor10,50. We tested the inhibition activity of CDD-97 against other OXA enzymes of varying sequence identity to assess its spectrum of activity (Table 2). OXA-10 is regarded as the canonical OXA enzyme, however, it is not a major clinical threat as it mainly hydrolyzes penicillins30,51,52. OXA-24 and OXA-58 are also carbapenemases and, similar to OXA-48, they have poor activity against extended-spectrum cephalosporins53–55. Unlike OXA-48, the OXA-24 and OXA-58 enzymes have a hydrophobic bridge in their active sites, which contributes to a preference for carbapenems with bulkier, more hydrophobic tails56,57. OXA-48 is prevalent in Enterobacteriaceae (mainly Klebsiella pneumoniae) while OXA-24 and OXA-58 are prevalent in the Moraxellaceae family (mainly Acinetobacter baumannii)54,58. While K. pneumoniae is more widespread in the clinics and most OXA-mediated carbapenem resistance is due to OXA-48, A. baumannii nosocomial infections are a growing problem. OXA-163 is an OXA-48-like enzyme also prevalent in Enterobacteriaceae59. OXA-163 only differs from OXA-48 by a 4 amino acid deletion (Arg214 – Pro217), which gives it reduced carbapenemase activity but increased activity against extended-spectrum cephalosporins60,61. OXA-163, as expected based on the high similarity to OXA-48, was inhibited by CDD-97 with a similar Ki as observed with OXA-48. OXA-10 and OXA-58 have lower sequence identities to OXA-48 (49.4% and 36.2% respectively) and were inhibited by CDD-97 with Ki values about 100-fold less potent than observed for OXA-48. CDD-97 displayed only a 26-fold decrease in potency for OXA-24 compared to OXA-48. Similar to OXA-58, OXA-24 has low sequence identity to OXA-48 (36.1%) and a hydrophobic bridge in the active site and yet is inhibited more potently by CDD-97 compared to OXA-58. Both enzymes contain a 2-residue hydrophobic bridge found to act as an inducible substrate binding cleft55,62. However, OXA-58 additionally has a phenylalanine residue at the base of the active site which may further occlude the active site and perturb CDD-97 binding. Ultimately, CDD-97 inhibits other OXA enzymes but is much more potent towards OXA-48 and OXA-48-like enzymes.
Table 2.
Inhibition constants (Ki) for CDD-97 with various OXA β-lactamases.
| Enzyme | Sequence Identity (%) | CDD-97 Ki (μM) |
|---|---|---|
| OXA-48 | 100.0 | 0.53 ± 0.08 |
| OXA-10 | 49.4 | 61 ± 27 |
| OXA-24 | 36.1 | 14 ± 1 |
| OXA-58 | 36.2 | 45 ± 0.8 |
| OXA-163 | 97.9 | 0.44 ± 0.02 |
Bacterial susceptibility and efficacy in E. coli
Clinically effective β-lactamase inhibitors synergize with β-lactam antibiotics and ultimately lower the minimum inhibitory concentration (MIC) of antibiotic needed to kill the bacteria63. MIC studies of E. coli expressing the OXA-48 enzyme were performed with the β-lactam antibiotics ampicillin and imipenem (separately). Both of these antibiotics can be hydrolyzed by OXA-4864. The OXA-48 plasmid construct contained the endogenous signal sequence of OXA-48 for secretion to the periplasm and was expressed under the control of the trc-promoter, which exhibits leaky expression in the absence of IPTG inducer65. The construct was transformed into E. coli strain MG1655 for OXA-48 expression (MG1655OXA-48). All MICs were performed by broth microdilution with 2-fold dilutions of antibiotic and/or inhibitor. The ampicillin (AMP) and imipenem (IMP) MICs were separately determined for a range of CDD-97 and avibactam concentrations. Avibactam was used as a positive control since it has been shown to lower the MIC of β-lactams for bacteria expressing OXA-4820. With increasing avibactam concentrations (up to 4 μg/ml), there was a dose-dependent decrease in both the AMP and IMP MICs (Table 3). This confirmed that avibactam synergizes with β-lactam antibiotics to kill E. coli expressing OXA-48 and suggested that comparable inhibitors should exhibit a similar effect. The AMP MIC of E. coli MG1655OXA-48 in the absence of inhibitor was determined to be 512 μg/ml. CDD-97 was then tested for synergy by determining AMP MICs with a range of CDD-97 concentrations. Higher concentrations of CDD-97 were used (up to 256 μg/ml) due to its reduced potency against OXA-48 compared to avibactam21. At 256 μg/ml of CDD-97, which corresponds to 0.6 mM and is over 1000-fold above the Ki for OXA-48, there was no significant decrease in the AMP MIC indicating CDD-97 did not synergize with ampicillin to enhance bacterial killing. Similarly, no synergy was seen between CDD-97 and imipenem in that the IMP MIC of 0.625 μg/ml for E. coli MG1655OXA-48 did not decrease even at up to 256 μg/ml of CDD-97 (Table 3). The poor activity of CDD-97 in the MIC assays compared to avibactam could be due to lower OXA-48 potency compared to avibactam21 and/or insufficient accumulation into the bacteria.
Table 3.
Minimum inhibitory concentrations (MICs) of ampicillin (AMP) and imipenem (IMP) against E. coli MG1655OXA-48 with increasing concentrations of CDD-97 and avibactam.
| MIC (μg/mL) | ||
|---|---|---|
| AMP | IMP | |
| 0 | 512 | 0.625 |
| 4 | 1024 | 0.625 |
| 8 | 512 | 0.3125 |
| 16 | 512 | 0.3125 |
| 32 | 512 | 0.3125 |
| 64 | 512 | 0.3125 |
| 128 | 512 | 0.3125 |
| 256 | 512 | 0.625 |
| [CDD-97] (μg/mL) | ||
| MIC (μg/mL) | ||
| AMP | IMP | |
| 0 | 512 | 0.625 |
| 0.125 | 256 | 0.3125 |
| 0.25 | 128 | 0.3125 |
| 0.5 | 64 | 0.1563 |
| 1 | 32 | 0.1563 |
| 2 | 16 | 0.1563 |
| 4 | 4 | 0.0781 |
| [Avibactam] (μg/mL) | ||
Impermeability of the outer membrane to small molecules is a major challenge for the development of therapeutics for Gram-negative bacterial infections66–70. β-lactamases, including OXA-48, reside in the periplasmic space and so inhibitors must traverse the outer membrane to be effective. To determine whether low permeability caused the poor in vivo activity of CDD-97, an accumulation assay was performed to quantify the amount of CDD-97 present in E. coli cells after incubation71. Tetracycline was used as a positive control for the assay while fusidic acid and clindamycin were used as negative controls71. The antibiotic controls and CDD-97 were separately incubated with wild-type E. coli cells and the cells were then centrifuged through silicone oil to separate the free, unaccumulated compounds from the cell pellet. Cell lysates were collected from the pellets by freeze-thawing and analyzed by mass spectrometry to quantify the amount of accumulated compound. CDD-97 was present in cells at 20-fold lower levels than the positive control tetracycline (Figure 7). These results indicate CDD-97 accumulates poorly in E. coli, explaining the lack of synergy with AMP and IMP in the MIC assay. The low accumulation of CDD-97 may be due to poor porin permeability to traverse the outer membrane and/or the action of efflux pumps that drive the compound out of the cell.
Figure 7.
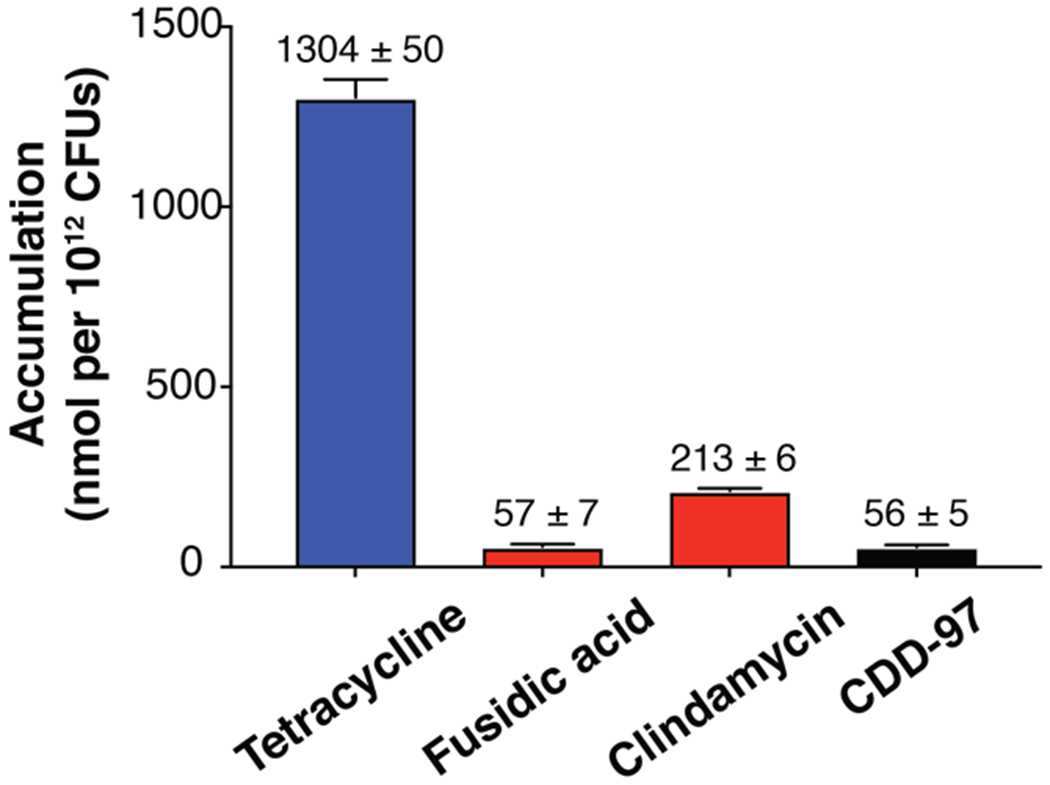
Accumulation of compounds in E. coli MG1655. The high accumulating control (tetracycline) is depicted in blue, and the low accumulating controls (clindamycin and fusidic acid) are in red. CDD-97 is shown in black. The number above each column indicates the accumulation value of the given sample.
Discussion
β-lactamase inhibitors have been pivotal in managing antibiotic resistance mediated by β-lactamases. Most β-lactamase inhibitors are focused on class A β-lactamases. Initially, class D β-lactamases were of little clinical relevance but the emergence of carbapenemases such as OXA-48 has highlighted the need for OXA specific inhibitors due to their low sequence homology with class A enzymes30. No OXA specific inhibitors have been clinically approved, but many have been developed and provide insight on OXA enzyme inhibition. The β-lactam based penicillin sulfones have efficacy against various OXA enzymes31. Specifically, the LN-1-255 inhibitor of this class is a 1000-fold more potent inhibitor of OXA-48 than is tazobactam and it increases the carbapenem susceptibility to bacteria expressing OXA-4832. Studies of phosphates/phosphonates, diazabicyclooctanes, and boronic acid-based compounds have shown promise as inhibitors of OXA enzymes33,34. However, since they maintain the typical acylation mechanism for β-lactamase inhibition, they may be susceptible to future variants that lead to rapid deacylation thereby reducing inhibitor potency.
To find more potent OXA-48 inhibitors, we utilized a DNA-encoded chemistry technology (DECL) approach. DECLs allow chemical space to be sampled more efficiently than typical high-throughput screening approaches. Additionally, tagging compounds with DNA allows rapid screening for faster identification of hits72. DECLs have helped identify hits for various targets such as kinases but there have been no reports of applications to β-lactamases45,72. We utilized a DECL approach to identify OXA-48 inhibitors and gain insight into binding and inhibition.
Harnessing the efficiency of DECLs allowed for the identification of two sub-micromolar OXA-48 inhibitors, CDD-97 and its derivative Cmpd 16. CDD-97 was identified out of a library of ~160 million compounds, showing the potential of DNA-encoded chemistry technology. CDD-97 was one of the most enriched compounds in the screen and exhibited sub-micromolar inhibition potency. The insights gained from structure-activity relationship studies and the crystal structure of CDD-97 bound to OXA-48 were used to create a more potent inhibitor (Cmpd 16). This study provides more information regarding features that may be important for inhibition of OXA-48 and OXA-48 like enzymes in general, which can be expanded upon to find clinically useful inhibitors. Like previous inhibitors, we see the importance of an acidic group in the carboxylate binding pocket of OXA-48 as observed with CDD-97 and Cmpd 16. The inhibitors also revealed how the hydrophobic active site of OXA-48 can be utilized to impact potency. The inclusion of hydrophobic moieties to interact with hydrophobic residues comprising and surrounding the OXA-48 active site enhances potency.
Despite the poor efficacy in vivo, the discovery of sub-micromolar inhibitors is an advance towards designing clinically useful OXA-48 inhibitors since it has previously been difficult to identify potent inhibitors. Permeability is an important limiting factor for the discovery of antibacterial agents for Gram-negative bacteria. The selective permeability of the outer membrane and porins limit access of compounds into the cell. Uptake of antimicrobials is further complicated by efflux pumps, which typically have a broad spectrum of activity68. Hydrophobic compounds are more susceptible to efflux pumps, which trap compounds through hydrophobic and stacking interactions73. CDD-97 is largely hydrophobic and potentially susceptible to efflux pumps but may also have low porin permeability ultimately leading to its poor accumulation. The poor activity of CDD-97 and ampicillin against E. coli expressing OXA-48 due to low access to the periplasm was corroborated by mass spectrometry studies revealing it accumulated poorly into E. coli (Figure 7). Accumulation studies have shown that compounds exhibit enhanced entry into bacteria if they are rigid, amphiphilic, flat and contain a primary amine71. Primary amines that are not sterically hindered were found to aid accumulation and even broaden the spectrum of activity of compounds to include Gram-positive bacteria71. However, the addition of amines is insufficient for increasing accumulation if the flexibility and globularity of the compound are too high. Calculations from the Entryway71 server (which provides predictions for accumulation in Gram-negative bacteria) revealed that while CDD-97 has reasonable globularity it is not sufficiently rigid. Globularity was computationally ranked on a scale from 0 – 1 with 0 representing flatness (i.e. benzene) and 1 representing sphericity (i.e. adamantane). CDD-97 yielded a globularity score of 0.061 falling within the range of low globularity (globularity ≤ 0.25) as previously described71. Accumulation studies also revealed low flexibility (having 5 or fewer rotatable bonds) was also important for accumulation and CDD-97 was found to have 6 rotatable bonds, potentially explaining its poor accumulation when the lack of primary amines is also considered. The accumulation of CDD-97 could potentially be optimized by decreasing its flexibility and by the strategic addition of 1 or more primary amines. Such modifications are predicted based on steered molecular dynamics to aid with traversing porins, particularly the major OmpF porin, which, along with OmpC, is responsible for a significant amount of drug accumulation71. A primary amine on a rigid compound is predicted to be more available and accessible and thus able to interact with the key Asp113 residue of OmpF, which can modulate the porin’s constriction site to assist passage71. Although CDD-97 may also be susceptible to efflux pumps, modifications for more efficient porin passage could potentially allow the compound to accumulate sufficiently to inhibit OXA-48 in the periplasm before being removed by efflux pumps on the cytoplasmic membrane. While many effective antibacterial agents deviate from the predicted accumulation trends, these parameters allow notable prediction of accumulation71 and show promise to aid in optimizing compounds to enter bacteria. This could be particularly powerful for potent β-lactamase inhibitors that may accumulate poorly. With the increasing number of problematic β-lactamases, expediting the identification of potent, high-accumulating inhibitors will be vital to manage resistance.
Conclusion
This work highlights DEL-based drug discovery with DNA-encoded chemistry technology as an effective approach that could be applied to various β-lactamases to study their inhibition and potentially design useful inhibitors. The power of such libraries could be amplified in the future by creating focused libraries specifically tailored to β-lactamases, particularly OXA-48 and other carbapenemases. Despite the high sequence variability between classes of serine β-lactamases, they all contain a carboxylate binding pocket that favors negatively charged moieties. This could prove to be an effective strategy to rapidly find lead compounds to aid in developing β-lactamase inhibitors. Additionally, DEL screening can be modular and include rounds of screening with different targets or allow screening of multiple targets in parallel. This could potentially be used to remove binders to a given β-lactamase from the pool of compounds to find specific binders for another β-lactamase or to uncover a broad-spectrum inhibitor with efficacy across β-lactamase classes. There is great potential for the application of DNA-encoded chemistry technology to β-lactamase inhibitor drug discovery. The speed at which leads can be found will be a useful tool to study β-lactamase inhibition to ultimately aid inhibitor design to manage growing β-lactamase-mediated resistance.
Experimental Section
Protein expression and purification
His-tagged OXA-48 was expressed from a pET28a vector under the IPTG inducible T7 promoter in E. coli BL21(DE3) cells74–76. In the pET28a vector, the thrombin site used to cleave the N-terminal His-tag was changed to a TEV cleavage site. Cultures were grown to an OD600 of 0.4 – 0.8 in media containing 25 μg/mL kanamycin, then induced for protein expression by adding IPTG to a final concentration of 0.5 mM and incubating for 18 – 20 hours at 25°C. The cell pellets were collected by centrifugation and frozen at −20°C. For cell lysis, frozen pellets were resuspended in 50 mM HEPES pH 7.5, 0.05% octyl-β-D-glucopyranoside buffer and sonicated with pauses to avoid sample heating. Cell lysates were filtered using 0.45 μm filters and then loaded onto a His Trap HP column (GE Healthcare, Pittsburg, PA). His-tagged OXA-48 was eluted using a linear gradient with 500 mM imidazole in 50 mM HEPES pH 7.5 buffer. NaCl was excluded from the buffer because a high concentration of chloride ions can inhibit OXA-48. Fractions were checked on SDS-PAGE gels and fractions with contaminants were excluded before concentrating using Amicon centrifugal filters with a 10,000 MW cut-off (Merck KGaA, Darmstadt, Germany). After concentrating, the protein was further purified by size-exclusion chromatography using a HiLoad 10/300 Superdex 75 column (GE Healthcare) in 50 mM HEPES pH 7.5, 15 mM NaHCO3. Fractions were examined by SDS-PAGE before concentrating and cleaving off the N-term His-tag using TEV protease. The His-tagged version of OXA-48 without cleaving the tag was used for compound screening and SPR assays. Inhibition assays and X-ray crystallography experiments were performed using OXA-48 with the tag removed.
Synthesis of DNA-free compounds and derivatives for inhibition assays
All compounds were procured through Sigma-Aldrich and Fisher Scientific. NMR spectra were collected using a Bruker 600 MHz NMR. LCMS data was collected using an Agilent 1290 Infinity Series LC system with 6150 MS. Column used was Agilent Eclipse Plus C18, 2.1 mm x 50 mm (8 μm). Mobile phase solvents were A: 0.05% formic acid; B: 5% water in acetonitrile. Runs consisted of 5% A for 0.5 minutes, gradient over 3 minutes of 5% A to 95% A, then 95% A over 1 minute. Synthesis of individual derivatives can be found in the Supporting Information.
Affinity selection with DNA-encoded chemistry technology
DNA-encoded chemical libraries were incubated with 1μM of His-OXA-48 in 200 μL of selection buffer (20 mM HEPES, 134 mM KOAc, 8 mM NaOAc, 4 mM NaCl, 0.8 mM MgOAc, 5 mM imidazole, 0.02% Tween-20 (pH 7.2) ) supplemented with 0.1mg/ml sheared salmon sperm DNA and 15 mM NaHCO3 for 45 min at 25 °C on a thermomixer (1000 RPM). The same library pool without protein was also incubated under the same conditions as a negative control to assess background binding of DECL molecules to the affinity resin. Before incubation, 1 μL of library molecules were set aside for quantitation using quantitative PCR (qPCR). Ni-NTA magnetic beads (25μL) were prewashed with selection buffer and the target protein–library mixture was added for affinity selection. The magnetic beads were washed with 500 μL of selection buffer to remove unbound DECL molecules. Bound compounds were eluted by incubating the beads with 100 μL of selection buffer at 80 °C for 10 min. One microliter of elution material was again set aside for quantitation by qPCR, and the entire remaining volume of the sample was subjected to an additional two rounds of affinity selection with fresh protein as mentioned above. After 3 rounds of selection, DECL molecules were quantified by qPCR which serves as a quality control measure. qPCR is done on each sample of recovered DECL material after each round of selection to monitor the total amount of DNA tags remaining. This information is used to guide both the selection process and sequencing preparation.
An appropriate number of PCR cycles was used to amplify the DNA and add DNA sequences compatible with Illumina sequencing flow cells. The PCR product was purified using Agencourt AMPure XP SPRI beads (Agencourt, Danvers, MA) according to the manufacturer’s instructions, and then quantitated on an Agilent BioAnalyzer (Santa Clara, CA) using a high-sensitivity DNA kit. The final concentration of amplicon for each sample was pooled at a concentration of between 3 and 4 nM. The final concentration of 1.8 pM library pool samples were loaded onto an Illumina Next-Seq 500 sequencer (San Diego, CA) at the Genomic and RNA Profiling Core Facility at Baylor College of Medicine.
Steady state kinetics with nitrocefin substrate
Nitrocefin, a chromogenic β-lactam substrate, was used as a reporter in all inhibition assays with the various OXA enzymes and various compounds. Nitrocefin is not stable in aqueous solution long-term. For these assays, nitrocefin stocks were prepared in DMSO, stored at −20°C and used within 2 weeks of dissolving. To avoid freeze-thawing, fresh stocks were always used. The Km of nitrocefin was determined prior to inhibition assays by performing steady state kinetics. Assays were performed using a DU800 spectrophotometer at room temperature in 50 mM HEPES pH 7.5, 15 mM NaHCO3, and 0.02% tween-20. Hydrolyzed nitrocefin was detected at 482 nm. For a single experiment, a series of at least 5 nitrocefin concentrations were tested against the given enzyme in duplicate and the initial velocities were measured and fit to the Michaelis-Menten equation. Experiments were repeated twice, and error reported is the standard error of the mean.
Inhibition assays using nitrocefin as reporter
Compounds were tested for inhibition of a given OXA enzyme based on the hydrolysis of nitrocefin using a Tecan plate reader. A nitrocefin concentration near the Km was used with a constant concentration of OXA-48 and a range of 6 concentrations of the compound of interest (10 μM with 3-fold serial dilutions) were tested in duplicate. A control with no compound was included in each run. For each compound concentration, the velocity of nitrocefin hydrolysis was determined at 482 nm and fit to the Morrison equation77. Compounds that exhibited Ki values above 50 μM were simply denoted as having a Ki value >50 μM. The average Ki of separate experiments, each of which consisted of duplicate runs, is reported and error was quantified as the standard deviation.
Crystallography and data collection
Hanging drops were set up to obtain crystals via the vapor diffusion method either manually or using an automated Mosquito robot. Crystallization conditions were screened with commercially available screens from Hampton Research (Aliso Viejo, CA) and Qiagen (Venlo, Netherlands) using a 96-well format. Initial crystallization conditions were optimized by varying pH and precipitant concentrations. The CDD-97/OXA-48 structure was determined from an apo crystal that grew from a 1:1 (protein to reservoir solution) mixture of 7.1 mg/mL OXA-48 and 0.077 M Tris pH 8, 27% PEG 550 MME, which was later soaked with a 1μL drop of 2 mM CDD-97. For soaking, a 10 mM DMSO stock of CDD-97 was diluted 1:1 in 100% 2-Methyl-2,4-pentanediol, and then diluted in the crystal buffer for a final concentration of 2 mM. The data sets were collected at the Berkeley Center for Structural Biology in the context of the Collaborative Crystallography Program on beamline 5.0.2
Crystallography data processing and refinement
Diffraction data were processed using the HKL2000 software, molecular replacement (PHASER) and initial refining (REFMAC) were performed using the CCP4 suite78,79. Molecular replacement was performed using the OXA-48 structure (PDB ID: 3HBR) as the search model. Further refinements were performed using the PHENIX software (phenix.refine)80. The structure was manually examined throughout the structure completion process after molecular replacement, using the Crystallography Object-Oriented Toolkit (COOT) program81. When appropriate, TLS groups were determined using the TLSMD server. The final refinement was done using phenix.refine and the final structure was inspected and validated with MolProbity and COOT81,82. The electron density of CDD-97 bound to OXA-48 in the crystal structure was further examined by creating a polder mFo-Fc OMIT map using the PHENIX software80.
Docking Protocol for the CDD-97 Derivatives into OXA-48
The images show the derivatives of CDD-97 docked into OXA-48 obtained from the CDD-97/OXA-48 co-crystal structure. To collect these poses, first the protein from the crystal structure was processed by the Schrodinger Suite Release 2018-383 : First, bond orders were assigned and H atoms were added by the Protein Preparation Wizard84. Then, the hydrogen bonding networks were optimized according to the protonation states determined by the program PROPKA85 at a neutral pH. Finally, the entire structure was energy-minimized where the heavy atoms were restrained to deviate from their original locations to a max. of 0.30 Å. A grid was generated at the site of the native crystal ligand for small molecule docking. The docked CDD-97 derivatives were prepared with the LigPrep program. This entails obtaining accurate 3D-structures of the ligands in their correctly assigned protonation states by the program Epik86,87. The prepared ligands were docked into the aforementioned grid using the Glide88–90 program in the extra precision (XP) mode. The shown poses were extracted from the docking results and visualized by the Maestro91 program, which was also used to render the figures.
Bacterial susceptibility in E. coli
Bacterial susceptibility was specifically tested using minimum inhibitory concentration assays. Susceptibility assays were performed with E. coli strains using broth microdilution in a microtiter plate. Two-fold dilutions of antibiotic and/or inhibitor were used. For susceptibility tests, bacterial strains were transformed with plasmids containing OXA-48 with its endogenous signal sequence for periplasmic migration (cloned into the pTP470 vector)92. E. coli MG1655 bacteria were grown in Mueller-Hinton broth overnight with 12.5 μg/mL chloramphenicol to select for the OXA-48-containing pTP470 plasmid. An overnight culture was diluted 1:104 into the final well solutions and incubated for 16-18 hours. After incubation, microplates were read using a Tecan plate reader at 600 nm absorbance. Growth was defined as yielding an absorbance ≥ 2 times the absorbance of the negative control (media and no culture).
Accumulation assays in E. coli
For the accumulation assay, the protocol from Ritcher et al.71 was followed. An overnight culture of E. coli MG1655 bacteria grown in LB Lennox broth, was inoculated into fresh media and grown to an OD600 of 0.5 - 0.6. Cells were pelleted, resuspended in 1x phosphate-buffered saline (PBS) and then incubated with the compound of interest for 10 minutes at 37°C. After incubation, the OD600 was recorded to note if compounds affected cell growth and then the cultures were centrifuged through a 9:1 mixture of silicone oil (viscosity = 20 cST) to high temperature silicone oil which was cooled at −80 °C. Cell pellets were isolated and lysed by 3 cycles of freeze-thawing using liquid nitrogen and a 65 °C water bath. The cell lysate was isolated from the cell debris (in water and methanol) and then analyzed via liquid chromatography-mass spectrometry (Agilent Technologies, 6490 QQQ Santa Clara, CA). LC-MS/MS was operated in positive mode for tetracycline, clindamycin, and CDD-97 and in negative mode for fusidic acid with electrospray ionization. Multiple reaction monitoring (MRM) was used to quantify these compounds. The precursors to production transitions of these compounds were as follows: m/z 445.16 > 153.70 (tetracycline); 425.14>126.10 (clindamycin); 515.30>455.20 (fusidic acid); 413.23>249.50 (CDD-97). The concentrations of these compounds were calculated based on their corresponding standard curves.
To determine the colony forming units (CFU) per mL based on the OD600, the OD600 of E. coli MG1655 culture in LB-Lennox was measured then the culture was diluted and plated to count colonies. Based on this, the CFU per mL for 1 O.D.600 (for MG1655 cells grown in LB-Lennox) was determined to be approximately 2.15 x 108 and this value was used for accumulation calculations.
Supplementary Material
Acknowledgements
The authors thank Dr. Ying-Chu Chen from the Center for Drug Discovery at Baylor College of Medicine (Houston, TX) for providing Figure S4.
Funding
This work was supported by NIH grants AI143832 and AI32956 (to TP) and T32 GM120011 (to DT). This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. The ALS-ENABLE beamlines are supported in part by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169-01. DNA-encoded chemical library production in the Center for Drug Discovery work is supported by a Core Facility Support Award from the Cancer Prevention Research Institute of Texas (RP160805), and the Welch Foundation (Grant H-Q-0042).
Footnotes
Supporting Information
The supporting information is available free of charge on the ACS Publications website. The following is provided in Supporting Information.
Accession Codes
The atomic coordinates and structure factors of the CDD-97/OXA-48 structure have been deposited at the Protein Data Bank under accession code 6UVK.
References
- (1).U.S. Department of Health and Human Services, C. for D. C. and P. Antibiotic Resistance Threats in the United States, 2013. Current 2013, 114 https://doi.org/CS239559-B. [Google Scholar]
- (2).Bush K; Bradford PA β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6 (8), a025247 10.1101/cshperspect.a025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Poole K Resistance to β-Lactam Antibiotics. Cell. Mol. Life Sci. 2004, 61 (17), 2200–2223. 10.1007/s00018-004-4060-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Elander RP Industrial Production of β-Lactam Antibiotics. Appl. Microbiol. Biotechnol. 2003, 61 (5–6), 385–392. 10.1007/s00253-003-1274-y. [DOI] [PubMed] [Google Scholar]
- (5).Blumer KJ; Thorner J Cutting and Stitching: The Cross-Linking of Peptidoglycan in the Assembly of the Bacterial Cell Wall. ACS Chem. Biol. 2007, 2 (12), 783–786. [DOI] [PubMed] [Google Scholar]
- (6).Kitano K; Tomasz A Triggering of Autolytic Cell Wall Degradation in Escherichia Coli by Beta-Lactam Antibiotics. Antimicrob. Agents Chemother. 1979, 16 (6), 838–848. 10.1128/AAC.16.6.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Abraham EP and Chain E An Enzyme from Bacteria Able to Destroy Penicillin. Oxford Journals 1988, 10 (4), 677–678. [PubMed] [Google Scholar]
- (8).Wright GD; Poinar H Antibiotic Resistance Is Ancient: Implications for Drug Discovery. Trends Microbiol. 2012, 20 (4), 157–159. 10.1016/j.tim.2012.01.002. [DOI] [PubMed] [Google Scholar]
- (9).Bush K Bench-to-Bedside Review: The Role of β-Lactamases in Antibiotic-Resistant Gram-Negative Infections. Crit. Care 2010, 14 (3). 10.1186/cc8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Drawz SM; Bonomo RA Three Decades of β-Lactamase Inhibitors. Clin. Microbiol. Rev. 2010, 23 (1), 160–201. 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Naas T; Oueslati S; Bonnin RA; Dabos ML; Zavala A; Dortet L; Retailleau P; Iorga BI Beta-Lactamase Database (BLDB)–Structure and Function. J. Enzyme Inhib. Med. Chem. 2017, 32 (1), 917–919. 10.1080/14756366.2017.1344235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Guh AY; Limbago BM; Kallen AJ Epidemiology and Prevention of Carbapenem-Resistant Enterobacteriaceae in the United States. Expert Rev. Anti. Infect. Ther. 2014, 12 (5), 565–580. 10.1586/14787210.2014.902306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Logan LK; Weinstein RA The Epidemiology of Carbapenem-Resistant Enterobacteriaceae: The Impact and Evolution of a Global Menace. J. Infect. Dis. 2017, 215 (Suppl 1), S28–S36. 10.1093/infdis/jiw282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Papp-Wallace KM; Endimiani A; Taracila MA; Bonomo RA Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 2011, 55 (11), 4943–4960. 10.1128/aac.00296-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Doi Yohei & Paterson, David L Carbapenemase-Producing Enterobacteriaceae. Semin. Respir. Crit. Care Med. 2016, 36 (1), 74–84. 10.1055/s-0035-1544208.Carbapenemase-Producing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nordmann P; Dortet L; Poirel L Carbapenem Resistance in Enterobacteriaceae: Here Is the Storm! Trends Mol. Med. 2012, 18 (5), 263–272. 10.1016/j.molmed.2012.03.003. [DOI] [PubMed] [Google Scholar]
- (17).Queenan AM; Bush K Carbapenemases: The Versatile β-Lactamases. Clin. Microbiol. Rev. 2007, 20 (3), 440–458. 10.1128/CMR.00001-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ambler RP The Structure of β-Lactamases. 1983, 331 (1969), 75–97. [Google Scholar]
- (19).Bush K; Jacoby GA Updated Functional Classification of β-Lactamases. Antimicrob. Agents Chemother. 2010, 54 (3), 969–976. 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Aktaş Z; Kayacan C; Oncul O In Vitro Activity of Avibactam (NXL104) in Combination with β-Lactams against Gram-Negative Bacteria, Including OXA-48 β-Lactamase-Producing Klebsiella Pneumoniae. Int. J. Antimicrob. Agents 2012, 39 (1), 86–89. 10.1016/j.ijantimicag.2011.09.012. [DOI] [PubMed] [Google Scholar]
- (21).Ehmann DE; Jahić H; Ross PL; Gu RF; Hu J; Durand-Réville TF; Lahiri S; Thresher J; Livchak S; Gao N; Palmer T; Walkup GK; Fisher SL Kinetics of Avibactam Inhibition against Class A, C, and D β-Lactamases. J. Biol. Chem. 2013, 288 (39), 27960–27971. 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gu R-F; Jahic H; Kern G; Ross PL; Ehmann DE; Hu J; Fisher SL; Walkup GK Avibactam Is a Covalent, Reversible, Non-β-Lactam β-Lactamase Inhibitor. Proc. Natl. Acad. Sci. 2012, 109 (29), 11663–11668. 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ripoll A; Galán JC; Rodríguez C; Tormo N; Gimeno C; Baquero F; Martínez-Martínez L; Cantón R Detection of Resistance to Beta-Lactamase Inhibitors in Strains with CTX-M Beta-Lactamases: A Multicenter External Proficiency Study Using a Well-Defined Collection of Escherichia Coli Strains. J. Clin. Microbiol. 2014, 52 (1), 122–129. 10.1128/JCM.02340-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Stapleton P; Wu PJ; King A; Shannon K; French G; Phillips I Incidence and Mechanisms of Resistance to the Combination of Amoxicillin and Clavulanic Acid in Escherichia Coli. Antimicrob. Agents Chemother. 1995, 39 (11), 2478–2483. 10.1128/AAC.39.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Winkler ML; Papp-Wallace KM; Taracila MA; Bonomo RA Avibactam and Inhibitor-Resistant SHV β-Lactamases. Antimicrob. Agents Chemother. 2015, 59 (7), 3700–3709. 10.1128/aac.04405-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Drawz SM; Papp-Wallace KM; Bonomo RA New β-Lactamase Inhibitors: A Therapeutic Renaissance in an MDR World. Antimicrob. Agents Chemother. 2014, 58 (4), 1835–1846. 10.1128/AAC.00826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wu J; Racine F; Wismer MK; Young K; Carr DM; Xiao JC; Si Q; Katwaru R; Harradine P; Motyl M; Bhagunde PR; Rizk ML Exploring the Pharmacokinetic/Pharmacodynamic Relationship of Relebactam (MK-7655) in Combination with Imipenem in a Hollow-Fiber Infection Model. Antimicrob. Agents Chemother. 2018, 62 (5), 1–13. 10.1128/aac.02323-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).van Duin D; Bethel CR; Kreiswirth BN; Becka SA; Bonomo RA; Taracila MA; Papp-Wallace KM; Barnes MD; Alsop J; Kaye KS Relebactam Is a Potent Inhibitor of the KPC-2 β-Lactamase and Restores Imipenem Susceptibility in KPC-Producing Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62 (6), 1–9. 10.1128/aac.00174-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lomovskaya O; Sun D; Rubio-Aparicio D; Nelson K; Tsivkovski R; Griffith DC; Dudley MN Vaborbactam: Spectrum of Beta-Lactamase Inhibition and Impact of Resistance Mechanisms on Activity in Enterobacteriaceae. Antimicrob. Agents Chemother. 2017, 61 (11), 1–15. 10.1128/AAC.01443-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Evans BA; Amyes SGB OXA β-Lactamases. Clin. Microbiol. Rev. 2014, 27 (2), 241–263. 10.1128/CMR.00117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Drawz SM; Bethel CR; Doppalapudi VR; Sheri A; Pagadala SRR; Hujer AM; Skalweit MJ; Anderson VE; Chen SG; Buynak JD; Bonomo RA Penicillin Sulfone Inhibitors of Class D β-Lactamases. Antimicrob. Agents Chemother. 2010, 54 (4), 1414–1424. 10.1128/AAC.00743-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Martínez-Guitián M; Vallejo JA; Poza M; Vázquez-Ucha JC; Bou G; González-Bello C; Bonomo RA; Beceiro A; Bethel CR; Buynak JD LN-1-255, a Penicillanic Acid Sulfone Able to Inhibit the Class D Carbapenemase OXA-48. J. Antimicrob. Chemother. 2016, 71 (8), 2171–2180. 10.1093/jac/dkw105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Majumdar S; Adediran SA; Nukaga M; Pratt RF Inhibition of Class D β-Lactamases by Acyl Phosphates and Phosphonates. Biochemistry. 2005, 44 (49), 16121–16129. 10.1128/AAC.49.10.4410. [DOI] [PubMed] [Google Scholar]
- (34).Werner JP; Mitchell JM; Taracila MA; Bonomo RA; Powers RA Exploring the Potential of Boronic Acids as Inhibitors of OXA-24/40 β-Lactamase. Protein Sci. 2017, 26 (3), 515–526. 10.1002/pro.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lund BA; Christopeit T; Guttormsen Y; Bayer A; Leiros HKS Screening and Design of Inhibitor Scaffolds for the Antibiotic Resistance Oxacillinase-48 (OXA-48) through Surface Plasmon Resonance Screening. J. Med. Chem. 2016, 59 (11), 5542–5554. 10.1021/acs.jmedchem.6b00660. [DOI] [PubMed] [Google Scholar]
- (36).Akhter S; Lund BA; Ismael A; Langer M; Isaksson J; Christopeit T; Leiros HKS; Bayer A A Focused Fragment Library Targeting the Antibiotic Resistance Enzyme - Oxacillinase-48: Synthesis, Structural Evaluation and Inhibitor Design. Eur. J. Med. Chem. 2018, 145, 634–648. 10.1016/j.ejmech.2017.12.085. [DOI] [PubMed] [Google Scholar]
- (37).Brenner S; Lerner RA Encoded Combinatorial Chemistry. Proc. Natl. Acad. Sci. 2006, 89 (12), 5381–5383. 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Melkko S; Dumelin CE; Scheuermann J; Neri D Lead Discovery by DNA-Encoded Chemical Libraries. Drug Discov. Today 2007, 12 (11–12), 465–471. 10.1016/j.drudis.2007.04.007. [DOI] [PubMed] [Google Scholar]
- (39).Clark MA; Acharya RA; Arico-Muendel CC; Belyanskaya SL; Benjamin DR; Carlson NR; Centrella PA; Chiu CH; Creaser SP; Cuozzo JW; Davie CP; Ding Y; Franklin GJ; Franzen KD; Gefter ML; Hale SP; Hansen NJV; … Morgan BA Design, Synthesis and Selection of DNA-Encoded Small-Molecule Libraries. Nat. Chem. Biol. 2009, 5 (9), 647–654. 10.1038/nchembio.211 [DOI] [PubMed] [Google Scholar]
- (40).MacConnell AB; McEnaney PJ; Cavett VJ; Paegel BM DNA-Encoded Solid-Phase Synthesis: Encoding Language Design and Complex Oligomer Library Synthesis. ACS Comb. Sci. 2015, 17 (9), 518–534. 10.1021/acscombsci.5b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Škopić MK; Bugain O; Jung K; Onstein S; Brandherm S; Kalliokoski T; Brunschweiger A Design and Synthesis of DNA-Encoded Libraries Based on a Benzodiazepine and a Pyrazolopyrimidine Scaffold. Medchemcomm 2016, 7 (10), 1957–1965. 10.1039/c6md00243a. [DOI] [Google Scholar]
- (42).Yuen LH; Franzini RM Achievements, Challenges, and Opportunities in DNA-Encoded Library Research: An Academic Point of View. ChemBioChem 2017, 18 (9), 829–836. 10.1002/cbic.201600567. [DOI] [PubMed] [Google Scholar]
- (43).Soutter HH; Centrella P; Clark MA; Cuozzo JW; Dumelin CE; Guie M-A; Habeshian S; Keefe AD; Kennedy KM; Sigel EA; Troast DM; Zhang Y; Ferguson AD; Davies G; Stead ER; Breed J; Madhavapeddi P; Read JA Discovery of Cofactor-Specific, Bactericidal Mycobacterium Tuberculosis InhA Inhibitors Using DNA-Encoded Library Technology. Proc. Natl. Acad. Sci. 2016, 113 (49), E7880–E7889. 10.1073/pnas.1610978113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Barluenga S; Zambaldo C; Ioannidou HA; Ciobanu M; Morieux P; Daguer JP; Winssinger N Novel PTP1B Inhibitors Identified by DNA Display of Fragment Pairs. Bioorganic Med. Chem. Lett. 2016, 26 (3), 1080–1085. 10.1016/j.bmcl.2015.11.102. [DOI] [PubMed] [Google Scholar]
- (45).Kleiner RE; Dumelin CE; Tiu GC; Sakurai K; Liu DR In Vitro Selection of a DNA-Templated Small-Molecule Library Reveals a Class of Macrocyclic Kinase Inhibitors. J. Am. Chem. Soc. 2010, 132 (33), 11779–11791. 10.1021/ja104903x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Faver JC; Riehle K; Lancia DR; Milbank JBJ; Christopher S Supporting Information for “ Quantitative Comparison of Enrichment from DNA-Encoded Chemical Library Selections ” Table of Contents. 1–31. [DOI] [PMC free article] [PubMed]
- (47).Faver JC; Riehle K; Yu Z; Kollmann CS; Milbank JBJ; Lancia DR; Simmons N; Matzuk MM Quantitative Comparison of Enrichment from DNA-Encoded Chemical Library Selections. ACS Comb. Sci. 2019, 1–28. 10.1021/acscombsci.8b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).O’Callaghan CH; Morris A; Kirby SM; Shingler AH Novel Method for Detection of Beta-Lactamases by Using a Chromogenic Cephalosporin Substrate. Antimicrob. Agents Chemother. 1972, 1 (4), 283–288. 10.1128/AAC.1.4.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Leonard DA; Bonomo RA; Powers RA Class D β-Lactamases: A Reappraisal after Five Decades. Acc. Chem. Res. 2013, 46 (11), 2407–2415. 10.1021/ar300327a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Poirel L; Naas T; Nordmann P Diversity, Epidemiology, and Genetics of Class D β-Lactamases. Antimicrob. Agents Chemother. 2010, 54 (1), 24–38. 10.1128/AAC.01512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Paetzel M; Danel F; De Castro L; Mosimann SC; Page MGP; Strynadka NCJ Crystal Structure of the Class D β-Lactamase OXA-10. Nat. Struct. Biol. 2000, 7 (10), 918–925. 10.1038/79688. [DOI] [PubMed] [Google Scholar]
- (52).Mobashery S; Samama J-P; Maveyraud L; Golemi D; Vakulenko S Critical Involvement of a Carbamylated Lysine in Catalytic Function of Class D β-Lactamases. Proc. Natl. Acad. Sci. 2002, 98 (25), 14280–14285. 10.1073/pnas.241442898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Santillana E; Beceiro A; Bou G; Romero A Crystal Structure of the Carbapenemase OXA-24 Reveals Insights into the Mechanism of Carbapenem Hydrolysis. Proc. Natl. Acad. Sci. 2007, 104 (13), 5354–5359. 10.1073/pnas.0607557104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Poirel L; Marque S; He C; Segonds C; Nordmann P OXA-58, a Novel Class D β-Lactamase Involved in Resistance to Carbapenems in Acinetobacter Baumannii. Society 2005, 49 (1), 202–208. 10.1128/AAC.49.1.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ishii Y; Ueno G; Miyano M; Saino H; Sugiyabu T; Yamamoto M Crystal Structure of OXA-58 with the Substrate-Binding Cleft in a Closed State: Insights into the Mobility and Stability of the OXA-58 Structure. PLoS One 2015, 10 (12), e0145869 10.1371/journal.pone.0145869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Stewart NK; Smith CA; Antunes NT; Toth M; Vakulenko SB Role of the Hydrophobic Bridge in the Carbapenemase Activity of Class D β-Lactamases. Antimicrob. Agents Chemother. 2019, 63 (2), 1–13. 10.1128/AAC.02191-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Docquier J-D; Mangani S Structure-Function Relationships of Class D Carbapenemases. Curr. Drug Targets 2015, 17 (9), 1061–1071. 10.2174/1389450116666150825115824. [DOI] [PubMed] [Google Scholar]
- (58).Gallego L; Rosales I; Fernández E; Bustamante Z; Umaran A; Zabalaga S; Sevillano E Emergence and Clonal Dissemination of Carbapenem-Hydrolysing OXA-58-Producing Acinetobacter Baumannii Isolates in Bolivia. J. Med. Microbiol. 2011, 61 (1), 80–84. 10.1099/jmm.0.032722-0. [DOI] [PubMed] [Google Scholar]
- (59).Aleo A; Mammina C; Bonura C; Fasciana T; Abdelaziz MO; El-Domany RA OXA-163-Producing Klebsiella Pneumoniae in Cairo, Egypt, in 2009 and 2010. J. Clin. Microbiol. 2012, 50 (7), 2489–2491. 10.1128/jcm.06710-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Nordmann P; Poirel L; Rodriguez CP; Carrër A; Smayevsky J; Castanheira M; Jones RN OXA-163, an OXA-48-Related Class D β-Lactamase with Extended Activity Toward Expanded-Spectrum Cephalosporins. Antimicrob. Agents Chemother. 2011, 55 (6), 2546–2551. 10.1128/aac.00022-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Stojanoski V; Chow DC; Fryszczyn B; Hu L; Nordmann P; Poirel L; Sankaran B; Prasad BVV; Palzkill T Structural Basis for Different Substrate Profiles of Two Closely Related Class D β-Lactamases and Their Inhibition by Halogens. Biochemistry 2015, 54 (21), 3370–3380. 10.1021/acs.biochem.5b00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).June CM; Vallier BC; Bonomo RA; Leonard DA; Powers A Structural Origins of Oxacillinase Specificity in Class D β-Lactamases. 2014, 58 (1), 333–341. 10.1128/AAC.01483-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Kobayashi S; Arai S; Hayashi S; Sakaguchi T In Vitro Effects of β-Lactams Combined with β-Lactamase Inhibitors against Methicillin-Resistant Staphylococcus Aureus. Antimicrob. Agents Chemother. 1989, 33 (3), 331–335. 10.1128/AAC.33.3.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Docquier JD; Calderone V; De Luca F; Benvenuti M; Giuliani F; Bellucci L; Tafi A; Nordmann P; Botta M; Rossolini GM; Mangani S Crystal Structure of the OXA-48 β-Lactamase Reveals Mechanistic Diversity among Class D Carbapenemases. Chem. Biol. 2009, 16 (5), 540–547. 10.1016/j.chembiol.2009.04.010. [DOI] [PubMed] [Google Scholar]
- (65).Brosius J; Erfle M; Storella J Spacing of the −10 and −35 Regions in the Tac Promoter. Effect on Its in Vivo Activity. J. Biol. Chem. 1985, 260 (6), 3539–3541. [PubMed] [Google Scholar]
- (66).Cox G; Wright GD Intrinsic Antibiotic Resistance: Mechanisms, Origins, Challenges and Solutions. Int. J. Med. Microbiol. 2013, 303 (6–7), 287–292. 10.1016/j.ijmm.2013.02.009. [DOI] [PubMed] [Google Scholar]
- (67).Hiroshi N Prevention of Drug Access to Bacterial Targets: Permeability Barriers and Active Efflux. Science (80-. ). 1994, 264 (April), 382–388. [DOI] [PubMed] [Google Scholar]
- (68).Delcour AH Outer Membrane Permeability and Antibiotic Resistance. Biochim. Biophys. Acta - Proteins Proteomics 2009, 1794 (5), 808–816. 10.1016/j.bbapap.2008.11.005.Outer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Masi M; Réfregiers M; Pos KM; Pagès JM Mechanisms of Envelope Permeability and Antibiotic Influx and Efflux in Gram-Negative Bacteria. Nat. Microbiol. 2017, 2(3). 10.1038/nmicrobiol.2017.1. [DOI] [PubMed] [Google Scholar]
- (70).Zgurskaya H; Lopez C; Gnanakaran S Permeability Barrier of Gram-Negative Cell Envelopes and Approaches To Bypass It. ACS Infect Dis. 2015, 1 (11), 512–522. 10.1021/acsinfecdis.5b00097.Permeability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ Predictive Compound Accumulation Rules Yield a Broad-Spectrum Antibiotic. Nature 2017, 545 (7654), 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Goodnow RA; Dumelin CE; Keefe AD DNA-Encoded Chemistry: Enabling the Deeper Sampling of Chemical Space. Nat. Rev. Drug Discov. 2017, 16 (2), 131–147. 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- (73).Sun J; Deng Z; Yan A Bacterial Multidrug Efflux Pumps: Mechanisms, Physiology and Pharmacological Exploitations. Biochem. Biophys. Res. Commun. 2014, 453 (2), 254–267. 10.1016/j.bbrc.2014.05.090. [DOI] [PubMed] [Google Scholar]
- (74).Studier FW; Moffatt BA Use of Bacteriophage T7 RNA Polymerase to Direct Selective High-Level Expression of Cloned Genes. J. Mol. Biol. 1986, 189 (1), 113–130. 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- (75).Rosenberg AH; Lade BN; Dao-shan C; Lin SW; Dunn JJ; Studier FW Vectors for Selective Expression of Cloned DNAs by T7 RNA Polymerase. Gene 1987, 56 (1), 125–135. 10.1016/0378-1119(87)90165-X. [DOI] [PubMed] [Google Scholar]
- (76).Phillip S; Chen Y Use of T7 RNA Polymerase to Direct Expression of Cloned. Methods Enzymol. 1990, 185, 60–89. 10.1109/MSP.2007.273050. [DOI] [PubMed] [Google Scholar]
- (77).Morrison JF Kinetics of the Reversible Inhibition of Enzyme-Catalysed Reactions by Tight-Binding Inhibitors. Biochim. Biophys. Acta - Enzymol. 1969, 185 (2), 269–286. 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- (78).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J. Appl. Crystallogr. 2007, 40 (4), 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Skubák P; Murshudov GN; Pannu NS Direct Incorporation of Experimental Phase Information in Model Refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 (12I), 2196–2201. 10.1107/S0907444904019079. [DOI] [PubMed] [Google Scholar]
- (80).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (2), 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Emsley P; Cowtan K Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60 (12I), 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (82).Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 (1), 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Schrödinger LLC. Schrödinger Suite 2019-1. New York, NY: 2019. [Google Scholar]
- (84).Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aid. Mol. Des 27, 221–234. [DOI] [PubMed] [Google Scholar]
- (85).Olsson MH; Sondergaard CR; Rostkowski M; Jensen JH PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical PKa Predictions J. Chem. Theory Comput 7, 525–537. [DOI] [PubMed] [Google Scholar]
- (86).Greenwood JR; Calkins D; Sullivan AP; Shelley JC Towards the Comprehensive, Rapid, and Accurate Prediction of the Favorable Tautomeric States of Drug-like Molecules in Aqueous Solution. J. Comput. Aided Mol. Des 24, 591–604. [DOI] [PubMed] [Google Scholar]
- (87).Shelley JC; Cholleti A; Frye L; Greenwood JR; Timlin MR; Uchimaya M Epik: A Software Program for PKa Prediction and Protonation State Generation for Drug-like Molecules. J. Comp. Aided Mol. Des 21, 681–691. [DOI] [PubMed] [Google Scholar]
- (88).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- (89).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes J. Med. Chem 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- (90).Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47 (7), 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- (91).Schrödinger LLC. Schrödinger Release 2019-4: Maestro. New York, NY: 2019. [Google Scholar]
- (92).Sun Z; Mehta SC; Adamski CJ; Gibbs RA; Palzkill T Deep Sequencing of Random Mutant Libraries Reveals the Active Site of the Narrow Specificity CphA Metallo-β-Lactamase Is Fragile to Mutations. Sci. Rep. 2016, 6 (May), 1–12. 10.1038/srep33195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.