

Abstract
Physicians think of mast cells and IgE primarily in the context of allergic disorders, including fatal anaphylaxis. This ‘bad side’ of mast cells and IgE is so well accepted that it can be difficult to think of them in other contexts, particularly those in which they may have beneficial functions. However, there is evidence that mast cells and IgE, as well as basophils (circulating granulocytes whose functions partially overlap with those of mast cells), can contribute to host defense as components of adaptive type 2 immune responses to helminths, ticks and certain other parasites. Accordingly, allergies often are conceptualized as “misdirected” type 2 immune responses, in which IgE antibodies are produced against any of a diverse group of apparently harmless antigens, and against components of animal venoms. Indeed, certain unfortunate patients who have become sensitized to venoms develop severe IgE-associated allergic reactions, including fatal anaphylaxis, upon subsequent venom exposure. In this review, we will describe evidence that mast cells can enhance innate resistance, and survival, to challenge with reptile or arthropod venoms during a first exposure to such venoms. We also will discuss findings indicating that, in mice surviving an initial encounter with venom, acquired type 2 immune responses, IgE antibodies, the high affinity IgE receptor (FcεRI), and mast cells can contribute to acquired resistance to the lethal effects of both honeybee venom and Russell’s viper venom. These findings support the hypothesis that mast cells and IgE can help protect the host against venoms and perhaps other noxious substances.
Keywords: Allergy, IgE, Th2 cell immunity, toxin hypothesis, venom
Mast cells, basophils and IgE in the pathology of allergic disorders.
Allergies, that currently afflict 20–30% of people worldwide, are detrimental immune responses against any of a large variety of environmental antigens [1]. Such antigens (called allergens) share the ability to elicit acquired type 2 immune responses which are orchestrated by CD4+ T helper type (Th)2 cells and include the production of allergen-specific IgE antibodies [2–4]. In such Th2 cell-associated “type 2” immune responses, IgE enables antigen-specific function of effector cells by binding to high affinity receptors for IgE (FcεRI) on the cells’ surface [5, 6]. FcεRI are expressed on mast cells, that reside in most vascularized tissues in mammals and other vertebrates, and on basophilic granulocytes (“basophils”), that ordinarily circulate in very low numbers in the blood but which can be recruited to sites of inflammation [3, 5–10].
When mast cell- or basophil-bound IgE antibodies recognize antigens that are at least bivalent, rapid aggregation of the FcεRI initiates complex intra-cellular signaling pathways. This ultimately results in the release, by such activated effector cells, of a wide variety of mediators with diverse biological effects [5, 6, 8–11]. Some of these mediators are stored in the cells’ cytoplasmic granules, ready for immediate release. These include, in mast cells, histamine, heparin and other proteoglycans, proteases such as carboxypeptidase A3 (CPA3), tryptases and chymases, and some cytokines [3, 5–7, 12, 13]. In addition, products of arachidonic acid metabolism (via the cyclo-oxidase or lipoxygenase pathways; e.g., prostaglandins and cysteinyl leukotrienes) and a diverse group of cytokines, chemokines and growth factors are secreted after upregulation of their transcription as a result of FcεRI–dependent cell activation [3, 5–7, 12, 13]. Basophils activated via FcεRI aggregation can release a panel of mediators partially overlapping with those of mast cells, but, as compared to mast cells, they contain much lower amounts of proteases and appear to produce fewer cytokines and chemokines [8–10].
Innate activation of mast cells.
In addition to IgE and specific antigen, many stimuli can activate at least some mast cell populations via innate mechansims, including products of complement activation (e.g., C3a, C5a), products of pathogens (e.g., LPS and other pathogen-associated molecular patterns [PAMPs]), certain cyokines or growth factors (including IL-33 and the Kit ligand, stem cell factor), products of other hematopoietic cells, certain endogenous peptides (including endothelin-1 [ET-1] and vasoactive intestinal polypeptice [VIP]), and components of the venoms of many different vertebrates and invertebrates [10, 14–18]. Within or among different mammalian species, individual mast cell subpopulations can very in their susceptibility to activation via these innate mechanisms, likely reflecting such factors as microenvironmentally regulated differences in levels of expression of the cognate receptors [14, 19]. Also, various stimuli can differ in their ability to elicit the release of granule-stored, lipid, or cytokine mediators. For example, certain peptides such as substance P can activate some mast cell populations to robustly release the granule-stored mediators; however, compared to the same cells activated via the FcεRI, such stimuli may less potently elicit release of lipid mediators or cytokines [14, 20, 21]. In contrast, for at least some mast cell populations, PAMPs are more effective in eliciting release of cytokines and chemokines than granule-stored mediators [16, 17]. Because mast cells or basophils particpating in innate or adative immune responses may encounter simultaneously or sequentially several different stimuli of activation, it may be difficult to predict which mast cell- or basophil-derived mediators will be released and in what amounts in these settings, and even more challenging to guess what the net effects of all such mediators might be during that particular biological response.
Possible beneficial functions of mast cells, basophils and IgE.
It is now generally accepted that mast cells and basophils can contribute importantly to the pathology associated with allergic disorders, including potentially fatal anaphylaxis [3, 22, 23]. Yet the evolutionary advantages that might by conferred by IgE, mast cells and basophils have been more difficult to define. A major hypothesis about the potential “beneficial functions” of such allergic effector mechanisms is that IgE-associated type 2 immune responses contribute to host defense against helminths and certain other parasites [4, 24–26].
It should be noted, however, that it has been challenging to prove that IgE, mast cells or basophils dramatically influence the survival of parasite-infected animals. Abnormalities in host responses to certain parasites have been observed in mice that genetically lack IgE [27, 28], mast cells [29–33], or basophils [28, 33] but such studies generally have not included an analysis of the effects of those deficiencies on the overall survival or reproductive success of the infected hosts. And some findings even suggest that, in certain settings, IgE or mast cells may have effects during host responses to parasites (e.g., effects which direclty or indirectly result in increased parasite egg production) that may favor the parasite rather than the host [34–36].
The complexity of the relationships between parasites and their hosts is not surprising, given that vertebrates have been co-evolving with such parasites for millions of years. It therefore also is not surprising that, depending on the parasites and the particular setting, immune effector mechanisms such as IgE, mast cells and basophils might be exploited by the parasites to their own advantage. For example, one can speculate that by eliciting a type 2 immune response that results in IgE-dependent mast cell activation and release of vasoactive mediators in response to parasite antigens at sites of parasite infection, the parasite could influence local blood flow and vascular permeability in ways that enhance the parasite’s nutrition.
In contrast to parasites, most allergens do not represent a direct threat to the nonsensitized host. This is why such type 2 immune responses are widely considered to be “misdirected” or “maladaptive” immune responses [37, 38]. However, Margie Profet proposed a radically different notion in 1991, based in part on the observation that the common feature of most allergens is their origin from sources such as seafood, nuts, or venoms which either might contain toxins (e.g., foods) or always do (e.g., venoms) [39]. Profet proposed that acute allergic reactions, manifested as immediately occurring symptoms in response to allergen exposure, such as sneezing, coughing, vomiting and diarrhea, evolved as defense mechanisms allowing the sensitized host to respond immediately to, and to expel, neutralize and/or avoid, noxious substances which might be indicative of potentially life-threatening situations [39]. Even before Profet’s 1991 paper, James Stebbings, Jr. hypothesized that “a major function of the immediate hypersensitivity reactions has been the protection of terrestrial vertebrates from the bites of, or invasion by, arthropods” [40]. However, until recently [41], Profet’s “toxin hypothesis” was largely ignored by the scientific community; Stebbings’ paper was even more neglected [42].
Evidence for a beneficial role of basophils in acquired immunity to the feeding of ixodid ticks.
Galli et al. generated a rabbit anti-guinea pig basophil antiserum (ABS) and showed that intravenous (i.v.) administration of ABS markedly reduced numbers of blood basophils in vivo, without reducing numbers of blood eosinophils or the total white blood cell count, or changing numbers of intradermal mast cells [43]. ABS did cause a reduction in skin mast cell numbers if injected intra-dermally, raising the possibility that the antiserum targeted an antigen shared by basophils and mast cells. Administration of this ABS i.v. to guinea pigs sensitized to express “cutaneous basophil hypersensitivity” (CBH), an antigen-induced, delayed onset, erythematous skin reaction that contains large numbers of infiltrating basophils [44], revealed that ABS could be used to reduce markedly the numbers of basophils infiltrating the skin at such sites [43].
Brown et al. then used this rabbit ABS, and a rabbit anti-eosinophil serum (AES), to investigate whether basophils or eosinophils contributed to adaptive immune response that diminish the feeding success of larval Amblyomma americanum ticks [45]. A. americanum (the Lone star tick) is an ixodid tick that is a vector for a variety of diseases including Rocky Mountain spotted fever (Rickettsia rickettsia), Q fever (Coxiella burnetii), tularemia (Francisella tularensis), granulocytic ehrlichiosis (Ehrlichia ewingii), monocytotropic ehrlichiosis (Ehrlichia chaffeensis), and others [46]. Moreover, Platts-Mills and colleagues recently demonstrated that the bites of this tick can induce the development of IgE antibodies to a tick carbohydrate, galactose-α−1,3-galactose, that is also present in non-primate meat and meat products, sensitizing such individual to develop a delayed form of urticaria or anaphylaxis after they consume such meats or meat products [47] or in response to treatment with a therapeutic antibody containing this carbohydrate [48].
It was known that when guinea pigs subjected to a single round of exposure to the feeding of larval A. americanum ticks are challenged with a second exposure to the larval ticks about a month later, tick feeding success is markedly diminished, and such acquired resistance can be transferred by administering serum from sensitized to naïve guinea pigs [49]. It also was known that tick feeding sites in sensitized guinea pigs contained large numbers of basophils and eosinophils [50]. Brown et al. found that treatment of tick-sensitized guinea pigs with rabbit ABS markedly diminished basophils in the bone marrow, blood, and skin at sites of tick feeding of the sensitized guinea pigs, and also resulted in diminished numbers of eosinophils at such feeding sites [45]. Notably, ABS treatment also essentially ablated the acquired resistance to tick feeding conferred by sensitization (Fig. 1) [45]. Treatment with rabbit AES had a lesser, but still statistically significant, effect on tick feeding success, whereas normal rabbit serum was without effect (Fig. 1) [45].
Fig. 1. Effects of treatment with anti-basophil serum (ABS), anti-eosinophil serum (AES) or normal rabbit serum (NRS) on feeding success of larval Amblyomma americanum ticks in a second infestation of guinea pigs.
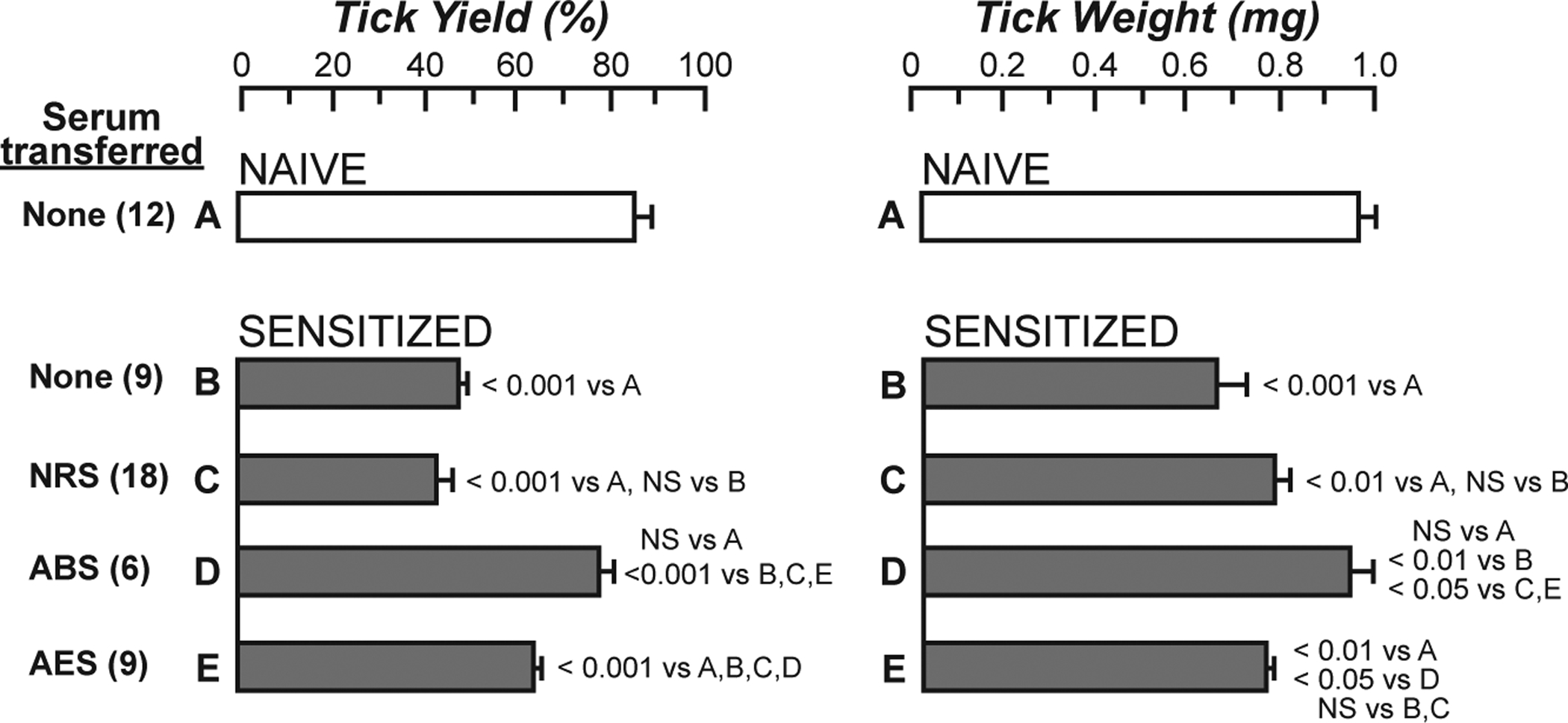
One hundred larval Amblyomma americanum ticks were placed on the flanks of “nonsensitized” (naïve) guinea pigs or guinea pigs that had been “sensitized” 26 days earlier by a primary infestation of A. americanum. The naïve guinea pigs (A) and one group of sensitized hosts (B) received no serum, other sensitized animals were treated with NRS (C), ABS (D), or AES (E), as described in [45]. The number (Tick Yield, left) and weight (Tick Weight, right) of engorged ticks was determined at 90 h of infestation. ABS completely ablated immunity; AES partially impaired resistance; NRS had no effect. Data (mean +/− SE) were pooled from three separate experiments, with the total number of animals in each group shown in parentheses. NS = not significant (P > 0.05). Differences among the experimental groups were analyzed by the Newman-Keuls multiple sample comparison test. [This is a modified version of Fig. 2 in Brown SJ, Galli SJ, Gleich GJ, Askenase PW: Ablation of immunity to Amblyomma americanum by anti-basophil serum: cooperation between basophils and eosinophils in expression of immunity to ectoparasites (ticks) in guinea pigs. J Immunol 1982;129:790–6 (ref. [45]) reprinted with the permission of the publisher. Copyright 1982. The American Association of Immunologists, Inc.]
To our knowledge, this was the first in vivo experimental evidence that went beyond correlative and observational studies (i.e., demonstrating the presence of basophils at tick feeding sites) indicating that basophils might represent one important component of acquired, antibody-dependent resistance to the feeding of an arthropod. Years later, it was possible to pursue similar experiments in mice genetically deficient in either mast cells or basophils. In convincing studies, Hiroshi Matsuda and Yukihiko Kitamura and colleagues (using mast cell-engrafted genetically mast cell deficient (WB/Re-W/+ X C57BL/6-Wv/+)F1-”W/Wv” mice) [51], and Hajime Karasuyama and colleagues (using mice genetically deficient in basophils [52]), provided evidence that both mast cells and basophils, as well as IgE, can contribute to acquired immunity to the feeding of Haemaphysalis longicornis ticks in mice. By contrast, it appears that basophils may have a more important role than mast cells in acquired resistance to the feeding of larval Dermacentor variabilis ticks in mice [53].
“Mast cell knock-in mice”.
Yukihiko Kitamura and colleagues discovered that (WB/Re-W/+ X C57BL/6-Wv/+)F1-”W/Wv” mice (now known as WBB6F1-KitW/KitW-v mice, since “W” later was shown to encode c-kit [54, 55]) not only had a moderate macrocytic anemia, a phenotype which had been reported decades earlier, but were profoundly deficient in tissue mast cells [56]. They also showed that mast cells developed in WBB6F1-W/Wv mice that had been engrafted with bone marrow cells from the WT (wild type) littermate WBB6F1-+/+ mice [56]. However, the recipient W/Wv mice also were cured of their anemia [56], as was initially shown by Elizabeth Russell [57]. Moreover, eventually, non-irradiated WBB6F1-W/Wv mice engrafted with sufficient (e.g., 1 × 107) WBB6F1-+/+ or other genetically-compatible WT whole bone marrow cells also undergo virtually complete replacement of multiple other hematopoietic lineages (including granulocytes and lymphocytes) with cells of donor origin [58–60].
Because transfer of WT bone marrow cells into W/Wv mice did not result in the selective engraftment of mast cells, due to the presence in bone marrow cells of hematopoietic stem and progenitor cell populations, an effort was undertaken to attempt to achieve a more selective “repair” of the mast cell deficiency of W/Wv mice by transferring in vitro-derived, “lineage-committed”, mast cells to the mice instead of whole bone marrow cells. This approach appeared plausible because it was clear that large populations of cells with features of “immature” mast cells could be generated in vitro from mouse hematopoietic progenitor cells [61] and that such cells exhibited features of additional mast cell maturation when exposed to sodium butyrate in vitro [62]. Nakano et al. showed that the transfer of WBB6F1-+/+ mouse bone marrow-derived cultured mast cells (BMCMCs) i.v., i.p. or i.d. into WBB6F1-W/Wv mice had no effect on the anemia of the recipient mice but resulted in the appearance of mast cells in their tissues, and that, over time, these mast cell populations came to exhibit certain phenotypic features similar to those present in the corresponding anatomical sites in WT mice [63].
Since that first study, many groups have used such mast cell-engrafted or, as we refer to them in the Galli lab, “mast cell knock-in mice” (Fig. 2), to analyze mast cell development, phenotype, heterogeneity and function in vivo [14, 71, 72]. An attractive aspect of this approach is that one can transfer into different genetically mast cell-deficient recipients either WT mast cells or mast cells that have been genetically manipulated or that are derived from various mutant or transgenic mice. This approach allows comparison of the function in vivo of mast cells that are normal or that lack (or have altered function of) various receptors, signaling molecules or mediators. Moreover, Tsai et al. showed that one also can generate such mast cells from embryonic stem cells, permitting the analysis in vivo of mast cells which lack products which, if absent in the germ line, would result in embryonic or perinatal lethality [64].
Fig. 2. Making “mast cell knock-in mice”.
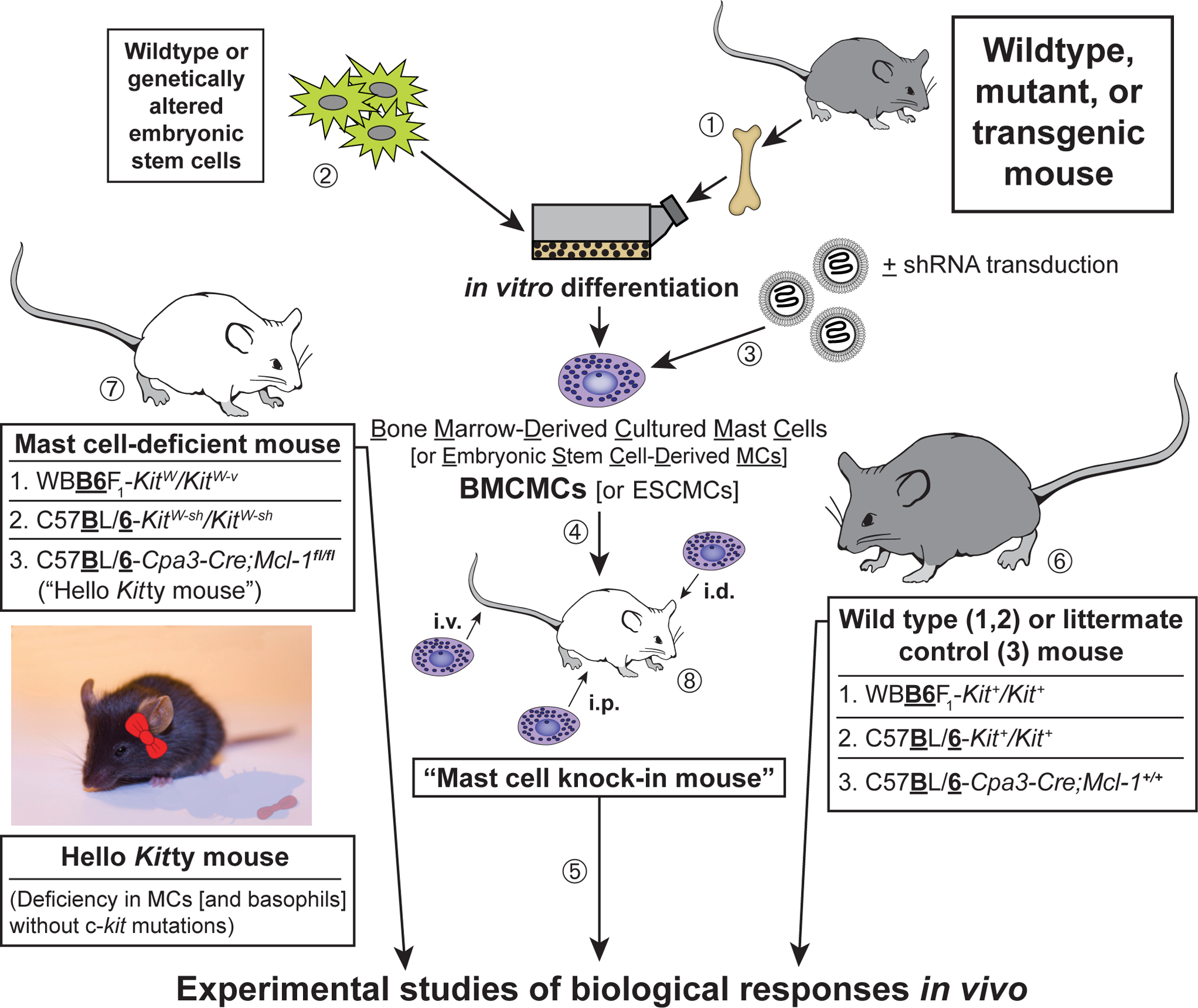
(1) Mast cells can be generated from bone marrow cells (or other hematopoietic cells; e.g., those in the fetal liver) from wild type mice or from mutant or transgenic mice with specific genetic alterations of interest [61–63]. Alternatively, (2) embryonic stem (ES) cell-derived cultured mast cells (ESCMCs) can be generated from wild type or genetically altered ES cells [64, 65], or (3) various mast cell populations can be transduced in vitro with shRNA to diminish expression of specific genes of interest [15, 66]. (4) Such bone marrow-, ES cell-, [or fetal liver-] derived cultured mast cells, or shRNA-transduced mast cells, can then be transplanted into mast cell-deficient c-kit mutant mice, such as WBB6F1-KitW/KitW-v mice [63, 67] or C57BL/6-KitW-sh/KitW-sh mice [68, 69], or into C57BL/6-Cpa3-Cre;Mcl-1fl/fl mice [70] (which we informally refer to as “Hello Kitty mice”, which have wild type c-kit), to produce mast cell knock-in mice. Note: BMCMCs can be injected into genetically mast cell-deficient mice intravenously (i.v.), intraperitoneally (i.p.), or intradermally (i.d.), or into the joints or meninges, etc., but there is a more limited experience with the engraftment of other types of MCs, such as EMCMCs, than with BMCMCs. (5) A suitable interval is then allowed for engraftment and phenotypic “maturation” of the adoptively-transferred mast cells (the length of this interval can be varied based on the route of mast cell transfer, the anatomical site of interest, the particular biological response being analyzed, etc.). The importance of mast cell function(s) in biological responses can be analyzed by comparison of the responses in the appropriate wild type or littermate control mice (6), the corresponding mutant mast cell-deficient mice (7), and selectively mast cell-engrafted mutant mice (mast cell knock-in mice) (8). The contributions of specific mast cell products (surface structures, signaling molecules, secreted products, and so on) to such biological responses can be analyzed by comparing the features of the responses of interest in mast cell knock-in mice engrafted with wild-type mast cells versus mast cells derived from mice or ES cells that lack or express genetically altered forms of such products or that have been transduced with shRNA to silence the specific genes that encode these products. An important part of the analysis of the mast cell knock-in mice used in particular experiments is to assess the numbers and anatomic distribution, and, for certain experiments, aspects of the phenotype, of the adoptively-transferred mast cells, as, depending on the type of in vitro-derived mast cells used, the route of administration, and other factors, these may differ from those of the corresponding native populations of mast cells in the corresponding wild type mice [14, 71, 72]. [This is a modified version of Fig. 2 in Metz M, Grimbaldeston MA, Nakae S, Piliponsky AM, Tsai M, Galli SJ. Mast cells in the promotion and limitation of chronic inflammation. Immunol Rev 2007; 217:304–28 (ref. [71]), reprinted with the permission of the publisher, John Wiley and Sons.]
In addition to using the original mast cell-deficient WBB6F1-KitW/KitW-v mice to prepare “mast cell knock-in mice”, this approach also can be employed using C57BL/6-KitW-sh/KitW-sh mice, which have the advantage of being inbred, fertile and not anemic [68, 69, 73]. We recently showed that this approach also can be pursued using C57BL/6-Cpa3-Cre+-Mcl-1fl/fl mice, which are mast cell-deficient (and also have substantially diminished numbers and function of basophils) due to the lineage-restricted ablation of the anti-apoptotic factor, myeloid cell leukemia 1 (Mcl-1), in lineages with sufficiently high expression of the Cpa3 gene [70]. Because the latter mice are mast cell deficient but, unlike WBB6F1-KitW/KitW-v or C57BL/6-KitW-sh/KitW-sh mice, have normal c-kit, we informally call them “Hello Kitty” mice [70] (Fig. 2).
New models for analyzing the functions of basophils and mast cells.
In addition to the models described above, those interested in the biology of mast cells, basophils or their mediators are now fortunate to have a large number of additional models to choose from. These include other lines of mice that exhibit constitutive or inducible deficiencies in populations of mast cells or in basophils, or which constitutively or inducibly lack various mast cell mediators or other molecules [72]. As reviewed in detail elsewhere [72, 74, 75], each of the various models currently available has features that must be kept in mind when interpreting the results of studies using such mice. Moreover,, the importance of particular mast cell (or basophil) roles in individual biological responses may vary both according to the details of the model used to study that biological response (e.g., whether one is studying a “weak” or “strong” model of that response) and based on the strain background of the mice. Accordingly, we have recommended that investigators consider using more than one type of genetic model to investigate the functions and importance of mast cells (or basophils) and/or their individual products in biological responses in vivo [72].
Identifying a beneficial role for mast cells in enhancing innate resistance to venoms.
Early work by Higginbotham and colleagues suggested that mast cells might be able to diminish the toxicity of certain venoms by degranulating and releasing heparin in response to venom exposure [76, 77]. However, this work was conducted before the description of mice deficient in mast cells or their individual mediators, so the importance of the roles of mast cell and their products in innate resistance to venoms could not be investigated more definitively in vivo. Even after Kitamura’s description of genetically mast cell-deficient mice, some time elapsed before any attention was paid to this question. One step in that direction was the finding that ET-1 can initiate a homeostatic mechanism whereby proteases released by mast cells activated by ET-1 can degrade that vasoactive peptide and thereby diminish its potential toxicity in vivo [65]. Using mast cell knock-in C57BL/6-KitW-sh/KitW-sh mice engrafted with ETA receptor-deficient or WT mast cells derived in vitro from ETA−/− or ETA−/+ embryonic stem cells, Maurer et al. found that mast cell activation by ET-1 via the ETA receptor contributed to this effect [65].
A homology search conducted by Martin Metz revealed that ET-1 was structurally similar to sarafotoxin 6b, one of the major toxins in the venom of the Israeli mole viper (Atractaspis engaddensis) [78]. Sarafotoxin 6b can induce activation of cells in the envenomated animals by binding to endothelin receptors [79]. Metz et al. showed that mast cells not only diminished the toxicity of sarafotoxin 6b and enhanced the survival of mice injected with that peptide, but also did so in mice injected with the whole venom of A. engaddensis [15]. Testing the ability of mast cells to influence responses to whole venoms is important, since snakes (and arthropods) don’t envenomate their prey with a single toxin but with a complex mixture of toxins that can induce pathology by different mechanisms [80].
Initial pharmacological studies suggested that chymase was the critical mast cell protease in this setting [65]. However, later work by our group, employing both pharmacological approaches and shRNA knock down of CPA3 in adoptively-transferred mast cells [15], as well as elegant studies by Schneider et al., who exploited a mouse they created which expressed only a catalytically inactive CPA3 [81], indicated that CPA3 is the key mast cell-derived protease that detoxifies both ET-1 and sarafotoxin 6b. Using mast cell knock-in mice, Metz et al. also provided evidence that mast cells were important in substantially enhancing the innate resistance of mice to honeybee (Apis mellifera) venom and to the venoms of two North American pit vipers, the western diamondback rattlesnake (Crotalus atrox) and the southern copperhead (Agkistrodon contortrix contortrix) [15].
A project led by Mitsuteru Akahoshi and Chang Ho Song then analyzed whether mast cells might enhance innate resistance to another pair of biologically active peptides, the endogenous mammalian peptide VIP and the structurally similar peptide helodermin (also known as exendin-2), which is one of the toxins present in the venom of the Gila monster (Heloderma suspectum) [18]. They tested both mast cell knock-in mice (including C57BL/6-KitW-sh/KitW-sh mice engrafted with WT versus chymase [mMCP4]-deficient mast cells) (Fig. 3, A and B) and mice which had mast cells but were genetically deficient in mMCP4 [82] or CPA3 [81] or produced a catalytically inactive CPA3 [81] (Fig. 3, C). This work showed that mast cells could diminish the toxicity of VIP, helodermin, and the whole venom of H. suspectum, and that this was largely or wholly dependent on mast cell-derived mMCP4 rather than CPA3 [18]. Similar approaches were used to provide evidence that mast cells and mMCP4 can contribute to enhanced innate resistance of mice to the venoms of two scorpions, one from the old world, the Deathstalker (Leiurus quiquestriatus hebraeus), and one from the new world, the Arizona bark scorpion (Centruroides exilicauda) [18].
Fig. 3. Mast cells can diminish Heloderma suspectum venom (H.s.v.)-induced hypothermia and mortality through MCP4-dependent mechanisms.
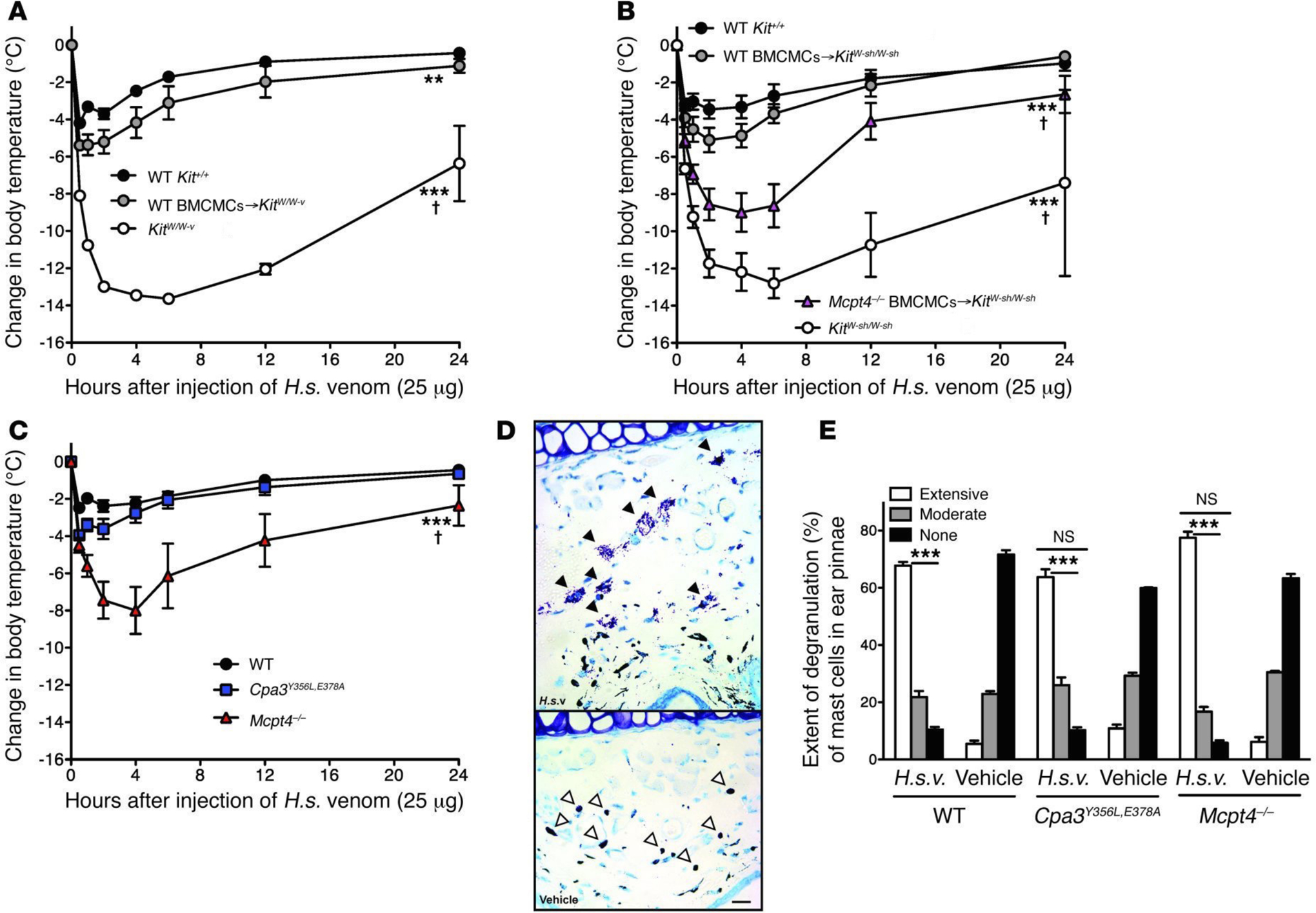
Changes in rectal temperatures after i.d. injection of H.s.v. (25 μg in 20 μl DMEM solution) into the ear pinnae (one ear pinna of each mouse) of: (A) WT WBB6F1-Kit+/+, mast cell-deficient WBB6F1-KitW/W-v, and WT BMCMCs→KitW/W-v mice (i.e., WBB6F1-KitW/W-v mice which had been engrafted, 6–8 weeks before venom challenge, in one ear pinna with 2 million BMCMCs derived from WT WBB6F1-Kit+/+ mice) (the death rates of Kit+/+, WT BMCMCs→KitW/W-v, and KitW/W-v mice within 24 h after H.s.v. injection were 0% [0/21], 7% [1/15, P = 0.42 vs. Kit+/+ mice], and 65% [13/20, P < 0.0001 vs. Kit+/+ mice], respectively); (B) WT C57BL/6-Kit+/+, mast cell-deficient C57BL/6-KitW-sh/W-sh, WT BMCMCs→KitW-sh/W-sh, and Mcpt4−/− BMCMCs→KitW-sh/W-sh mice (the death rates of Kit+/+, WT BMCMCs→KitW-sh/W-sh, Mcpt4−/− BMCMCs→KitW-sh/W-sh, and KitW-sh/W-sh mice within 24 h after H.s.v. injection were 5% [1/19], 11% [2/18, P = 0.48 vs. Kit+/+ mice], 43% [6/14, P = 0.01 vs. Kit+/+ mice], and 50% [10/20, P = 0.006 vs. Kit+/+ mice], respectively); or (C) WT C57BL/6-Kit+/+ mice, C57BL/6-Cpa3Y356L,E378A mice (which have a catalytically inactive CPA3) and C57BL/6-Mcpt4−/− mice (the death rates of Kit+/+, Cpa3Y356L,E378A, and Mcpt4−/− mice within 24 h after H.s.v. injection were 7% [1/15], 0% [0/14, P = 0.52 vs. Kit+/+ mice], 40% [6/15, P = 0.007 vs. Kit+/+ mice], respectively). Each figure shows data pooled from at least three independent experiments with each group of mice (n = 2–5 mice per group per each individual experiment). **P < 0.01, ***P < 0.001 versus WT WBB6F1-Kit+/+ or WT C57BL/6-Kit+/+ mice; †P < 0.01~0.001 versus each other group (A-C). (D) Extensive degranulation of mast cells (some indicated by closed arrowheads) 1 h after i.d. injection of H.s.v. (25 μg in 20 μl DMEM), but not vehicle (DMEM) alone (mast cells without evidence of degranulation are indicated by open arrowheads) in WT C57BL/6 mice (Toluidine blue stain; scale bar: 50 micrometers). (E) Degranulation of mast cells 60 min after i.d. injection of H.s.v. (25 μg in 20 μl DMEM) or vehicle (DMEM) alone in WT C57BL/6, Mcpt4−/−, or Cpa3Y356L,E378A mice (injection was into one ear pinna of each mouse). ***P < 0.001 versus corresponding vehicle-injected groups; NS = not significant (P > 0.05) versus values for WT mice. [This is a reproduction of Fig. 1 from Akahoshi M, Song CH, Piliponsky AM, Metz M, Guzzetta A, Abrink M, Schlenner SM, Feyerabend TB, Rodewald HR, Pejler G, Tsai M, Galli SJ. Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venom, and vasoactive intestinal polypeptide in mice. J Clin Invest 2011;121:4180–91 (ref. [18]), reprinted with the permission of the publisher, the American Society for Clinical Investigation.]
It is possible that future work will reveal that mast cell activation can increase, rather than decrease, the toxicity of some venoms. However, our initial evidence indicated that mast cells can increase the innate resistance of mice upon their first exposure to the venoms of 3 species of poisonous snakes, the Gila monster, the honeybee, or two especially dangerous scorpions. Mouse mast cells contain at least two different proteases, CPA3 and chymase (mMCP4), which permit such mast cells to respond, after their activation via cognate receptors that can bind either the endogenous or the structurally similar reptile-derived peptides, to high and potentially toxic levels of ET-1 and VIP, respectively. Such mast cells also can react when challenged with high levels of the similar peptides contained in the reptile venoms (Israeli mole viper sarafotoxin 6b and Gila monster helodermin, respectively) (Fig. 4). By undergoing degranulation and releasing proteases that can inactivate potentially toxic endogenous peptides or peptides in venoms, mouse mast cells can help to restore homeostasis, albeit while also enhancing features of the ensuing local and perhaps systemic inflammatory responses.
Fig. 4. Mast cells can enhance innate resistance to high levels of endogenous peptides and structurally similar peptides in reptile venoms.
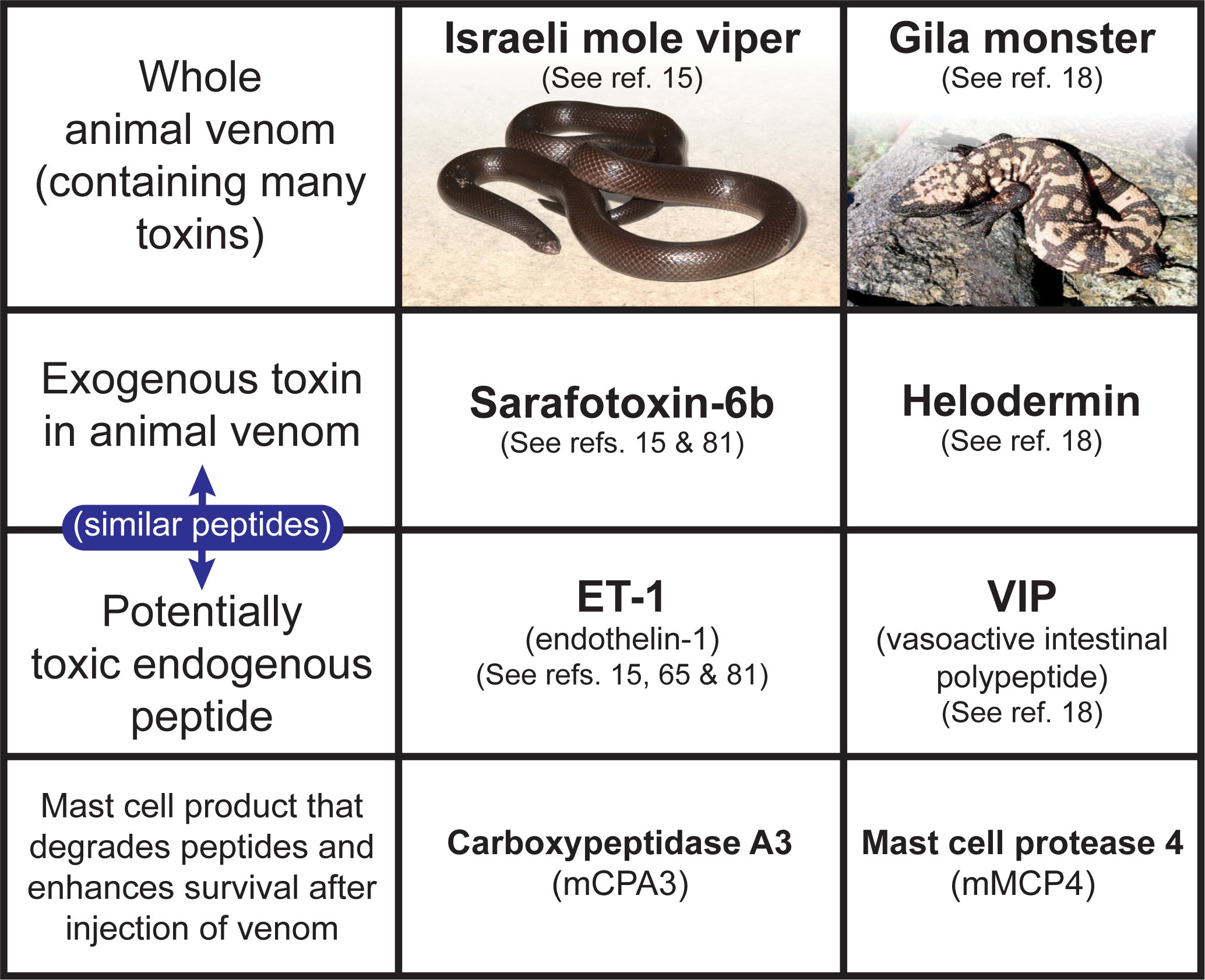
Mast cell cytoplasmic granules contain proteases such as carboxypeptidase A3 (CPA3 [mCPA3 = mouse CPA3]) and mast cell protease 4 (MCP4 [mMCP4 = mouse MCP4]) that, upon secretion by activated mast cells, can degrade certain endogenous peptides, such as endothelin-1 (ET-1) and vasoactive intestinal polypeptide (VIP), respectively, as well as structurally similar peptides contained in the venoms of poisonous reptiles, such as sarafotoxin 6b in the venom of the Israeli mole viper (Atractaspis engaddensis) and helodermin in the venom of the Gila monster (Heloderma suspectum). The ability of mast cells to be activated to degranulate by components of venoms such as these, which can act at the same receptors which recognize the corresponding structurally similar endogenous peptides, permits mast cells to release proteases that can reduce the toxicity of these peptides and which help to enhance the survival of mice injected with the whole venoms of these reptiles, that contain many toxins in addition to sarafotoxin 6b and helodermin. This mechanism may also permit mast cells to restore homeostasis in settings associated with markedly increase levels of the endogenous peptides. [This is a reproduction of Fig. 4 from Galli SJ. The 2014 Rous-Whipple Award Lecture. The mast cell-IgE paradox: From homeostasis to anaphylaxis. Am J Pathol 2016;186:212–24 (ref. [42]), reprinted with the permission of the publisher, Elsevier for the American Society for Investigative Pathology.]
Depending on the mammalian species, mast cells can contain several tryptases and chymases of distinct substrate specificity, as well as CPA3 [13, 72, 83]. This raises the possibility that mast cells of various species may employ differentproteases with the potential to degrade a variety of structurally distinct toxic compounds contained in animal venoms. Indeed, a recent study of human mast cell-derived proteases shows that tryptase (rather than chymase or CPA3) is more important at reducing the toxicity of the venoms of six different poisonous snakes, when embryonic zebra fish are used to assess such toxicity in vitro [84]. Mast cells might also contribute to innate resistance to venoms in other ways, such as by increasing local vascular permeability and thereby favoring the interstitial access of circulating molecules that can antagonize the effects of venom proteases [85] and other toxins.
IgE can contribute to host defense against arthropod and reptile venoms.
Many animals and some humans experience multiple episodes of envenomation by arthropods such as bees, wasps, and scorpions, or by various reptiles. Such envenomation not only provokes an innate inflammatory response and pathology related to the biological activities of the venoms’ toxins [86–88], but also can induce allergic sensitization associated with the development of specific IgE antibodies [89–94]. In some unfortunate people, such IgE responses to venoms put these individuals at risk to develop potentially fatal episodes of anaphylaxis [3, 7, 23, 94]. However, recent findings suggest, in accord with Profet’s “toxin hypothesis of allergy”, that this same “allergic” mechanism - involving IgE and mast cells - also can enhance host resistance to venoms.
Honeybee (Apis mellifera) venom consists of a mixture of cytolytic peptides (e.g., melittin), enzymes (e.g., phospholipase A2 [PLA2; considered the main allergen in bee venom]), hyaluronidase, neurotoxins and bioactive amines [86], and accounts for a large fraction of venom allergies in humans [94]. The venom of the Russell’s viper (Daboia russelii), one of the most dangerous snakes in the Indian subcontinent [95], is a complex mixture of growth factors and enzymes with pro-coagulant and neurotoxic activities [88]. We found that, in mice, IgE-associated type 2 immune responses against honeybee venom or Russell’s viper venom were able to increase significantly host resistance to challenge with potentially lethal doses of those venoms [96].
This was unexpected because both IgE and IgG1 antibodies produced during type 2 immune responses can orchestrate anaphylaxis and other allergic reactions in mice [7, 23, 97, 98] and because type 2 immune responses against venoms (that include the development of antivenom IgG1 [in mice] and IgE antibodies) are classically thought to exacerbate the outcome of subsequent venom exposure [90–94, 99]. By contrast, IgG class antibodies raised against animal venoms (or their F(ab’)2 fragments), are used to treat envenomated humans or animals [100].
So it was important to identify which antibodies contributed to the enhanced resistance to honeybee venom observed in mice with type 2 immune responses to that venom. Our evidence showed that IgE antibodies were the critical elements of the acquired host resistance to honeybee or Russell’s viper venom. We found that most or all of the acquired resistance induced in mice by a single exposure to honeybee venom was transferable to naïve mice with only 250 microliters of serum from honeybee venom-immunized mice [96]. Moreover, when such “immune serum” was depleted of IgE (either by adding a neutralizing antibody to IgE [35, 101] or by heating to 56° C for 1 hour, which eliminates the ability of IgE to bind to FcεRI and induce passive cutaneous anaphylaxis [102] while the function of other antibody classes, including IgG1, is not affected [103]), the immune serum’s ability to transfer enhanced resistance to naïve mice was essentially lost [96]. Finally, we showed that such “immune serum” failed to transfer enhanced venom resistance to mice lacking either the IgE antibody-binding α chain of the FcεRI or the γ chain of FcεRI that is necessary for signaling initiated by aggregation of the receptor [5, 6].
We also found that genetically IgE-deficient mice [97] could not develop acquired immunity to honeybee venom, even though they developed a robust IgG1 antibody response to the venom [96]. We also showed that “immune serum” from WT mice could passively transfer enhanced resistance to honeybee venom to naïve IgE-deficient mice, unless such “immune serum” was first treated to neutralize IgE or impair its ability to bind to FcεRI [96]. Finally, we revealed that naïve genetically mast cell-deficient C57BL/6-KitW-sh/KitW-sh or C57BL/6-Cpa3-Cre+-Mcl-1fl/fl mice which received immune serum from honeybee venom-immunized C57BL/6 WT mice actually exhibited worse survival after challenge with a high dose of honeybee venom than did mast cell-deficient mice which had received serum from PBS mock-immunized C57BL/6 WT mice [96]. The latter finding suggested that mast cells can contribute to IgE-mediated acquired resistance to honeybee venom, as well as enhance the innate resistance of mice to a first exposure to that venom [15]. Independently of our work, Palm et al. showed that mice immunized with the major allergen contained in honeybee venom, bee venom phospholipase A2 (bvPLA2), exhibited enhanced resistance to the ability of bvPLA2 to induce hypothermia upon its injection into mice, and also provided evidence that this enhanced immunity required B cells and was diminished significantly in mice which lacked the IgE-binding α chain of the FcεRI [104]. Taken together, these two initial studies [96, 104] support the hypothesis that one physiological function of IgE is to protect the host against noxious substances.
Subsequently, we found that the acquired enhanced resistance to Russell’s viper venom (RVV) which we observed in mice that had developed type 2 immune responses to that venom [96] also was highly dependent on IgE (Fig. 5) and FcεRI (see Fig. 4, A–E in ref. [105]), and could be effectively transferred by immune serum into normal mice (see Fig. 3, F–J in ref. [105]) but not into C57BL/6-Cpa3-Cre;Mcl-1fl/fl mice which were genetically markedly deficient in mast cells and which also had diminished numbers of basophils (see Fig. 4, G, H in ref. [105]) [105]. Notably, two different types of genetically mast cell-deficient mice also exhibited significantly diminished innate resistance to the toxicity and lethality of RVV (Fig. 6, A–C, E and F), supporting Higginbotham’s hypothesis that mast cells can contribute to enhanced innate resistance to this venom [76]. Compared to the corresponding mast cell-sufficient mice, such naïve mast cell-deficient mice also exhibited many fewer attempts to scratch sites of RVV injection (Fig. 6, D and G). The latter finding supports the idea proposed both by Stebbings [40] and Profet [39] that elements of allergic responses, in this case, mast cells, can confer benefit to hosts experiencing attacks by arthropods [40] or other sources of toxins [39] by altering the host’s behavior in ways that would help to eliminate, or at least permit the host to become aware of, the threat.
Fig. 5. Evidence that IgE antibodies contribute to acquired enhanced resistance to the toxicity and lethality of Russell’s viper venom.
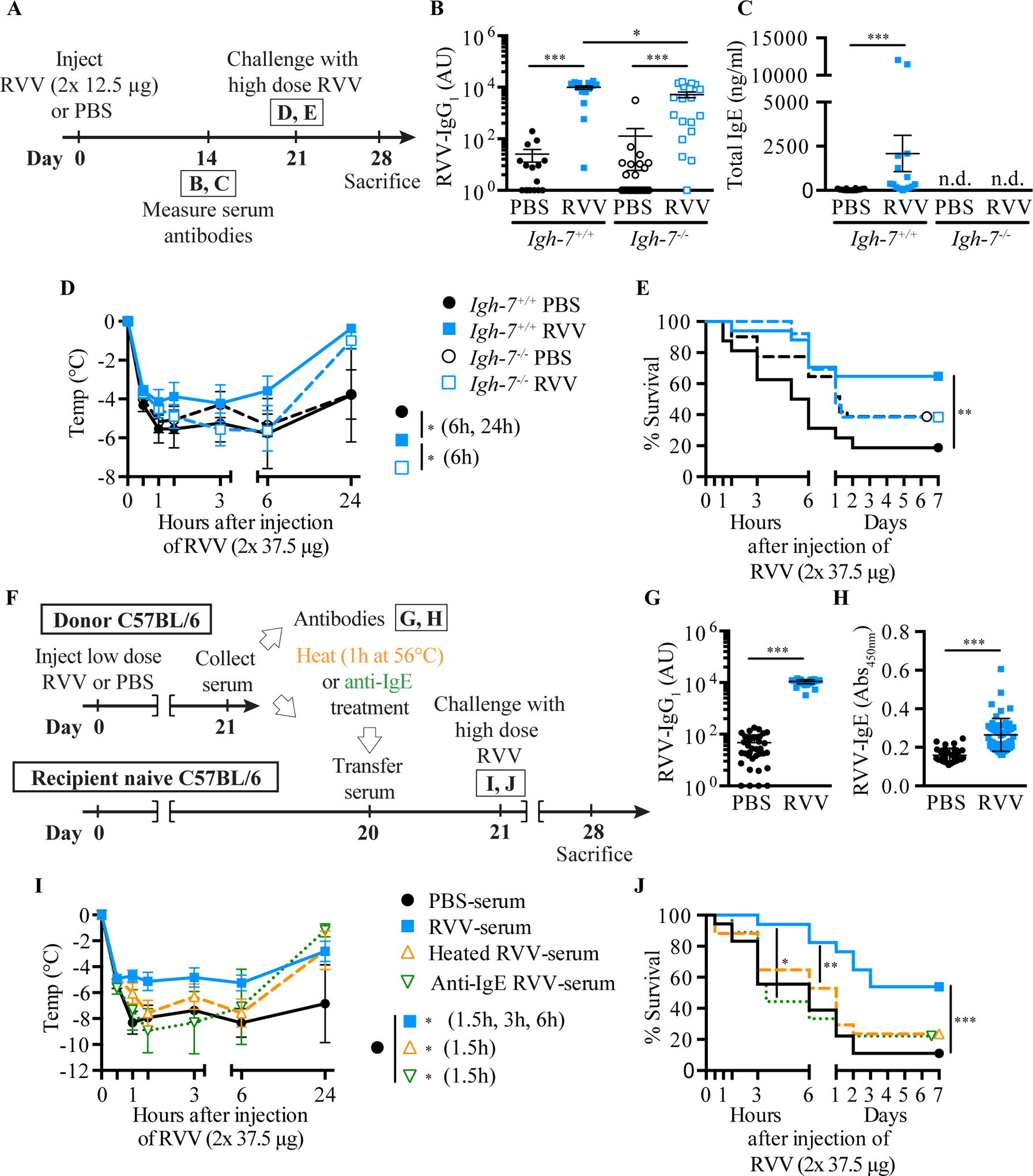
A. Outline of experiments with IgE-deficient (Igh-7−/−) and control (Igh-7+/+) C57BL/6 mice (B-E). B, C. Serum RVV-specific IgG1 (B) and total IgE (C). D, E. Body temperature (D) and survival (E). F. Outline of serum transfer experiments in C57BL/6 mice (G-J). G, H. Serum RVV-specific IgG1 (G) and total IgE (H). I, J. Body temperature (I) and survival (J). Data were pooled from 3–4 experiments (n= 9–25/group). P values: Mann-Whitney test (B, C, G, H), Student’s t test (D, I) and Mantel-Cox test (E, J). [This is a reproduction of Fig. 3 from Starkl P, Marichal T, Gaudenzio N, Reber LL, Sibilano R, Tsai M, Galli SJ. IgE antibodies, FcεRIα, and IgE-mediated local anaphylaxis can limit snake venom toxicity. J Allergy Clin Immunol 2016;137:246–57 (ref. [105]), reprinted with the permission of the publisher, Elsevier.]
Fig. 6. Evidence that mast cells contribute to innate resistance to the toxicity and lethality of Russell’s viper venom, as well as to behavioral responses to envenomation.
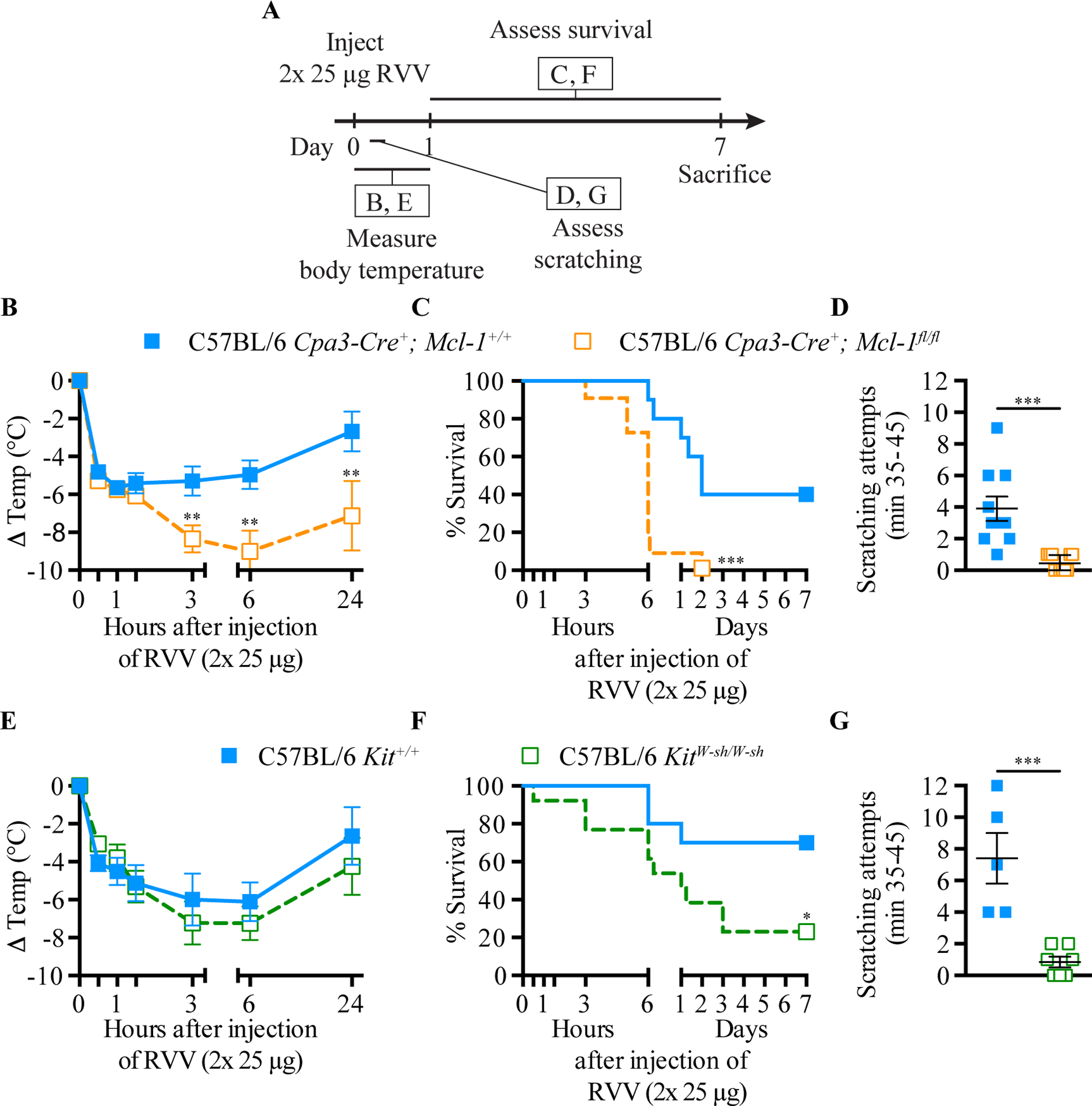
A. Experimental outline. B and E, body temperature; C and F, survival; D and G, scratching attempts, of mast cell-deficient Cpa3-Cre+; Mcl-1fl/fl (B-D) and KitW-sh/W-sh (E-G) mice and corresponding control mice after RVV injection. P values: (B, D, E, G) Student’s t test; (C, F) Mantel-Cox test. Data were pooled from 2–4 experiments (n=5–21/group). [This is a reproduction of Fig. 2 of Starkl P, Marichal T, Gaudenzio N, Reber LL, Sibilano R, Tsai M, Galli SJ. IgE antibodies, FcεRIα and IgE-mediated local anaphylaxis can limit snake venom toxicity. J Allergy Clin Immunol 2016;137:246–57 (ref. [105]), reprinted with the permission of the publisher, Elsevier.]
As noted above, Palm et al. reported that immunization of mice with honeybee venom-derived bvPLA2, which represents approximately 10% of the dry weight of whole BV [106], can reduce the toxicity-related hypothermia induced by subsequent challenge with a high dose of the same allergen in an antibody- and FcεRIα-dependent manner [104]. However, it was not clear whether an IgE response to a single constituent of an animal venom would be able to enhance resistance to the entire group of toxins contained in that venom. To investigate this, we passively sensitized WT mice locally against DNP-HSA by s.c. injections of anti-DNP IgE [107] (or with anti-DNP IgG1 or IgG2b as controls), or mock-sensitized the mice with saline, then challenged the animals s.c. at the same site 24 h later by injecting a mixture of RVV and DNP-HSA (Fig 7, A). We used amounts of anti-DNP IgE and DNP-HSA that were able to induce a local increase in vascular permeability at the DNP-HSA injection site without resulting in systemic hypothermia, and showed that the amount of DNP-HSA we used did not, by itself, influence the toxicity of RVV (see Fig E5 in the Online Repository of ref. [105]).
Fig. 7. IgE-dependent local mast cell activation induced by activation with a single antigen can enhance resistance to the lethality of Russell’s viper venom.
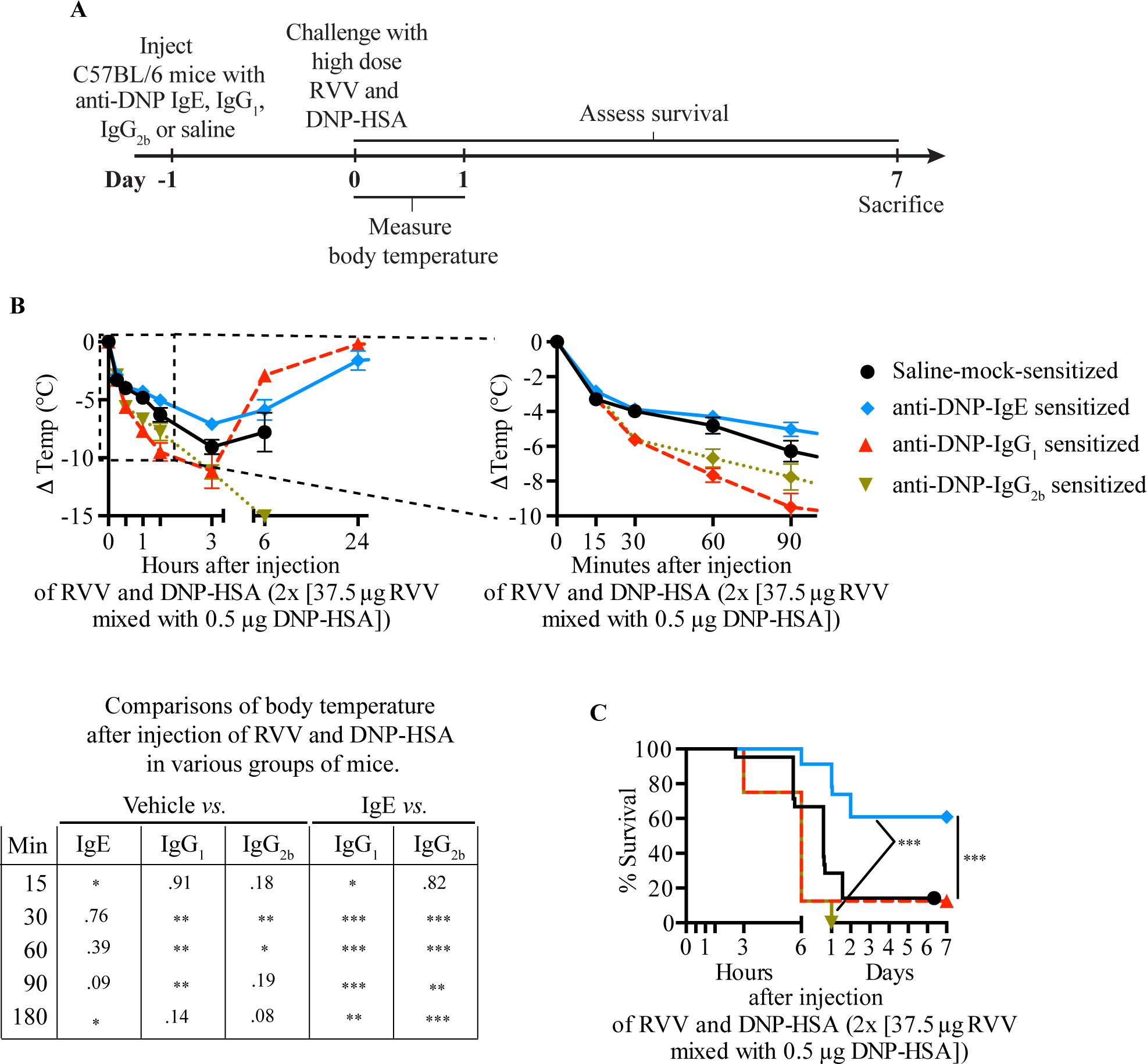
A. Experimental outline. B, C. Body temperature (B) and survival (C) of C57BL/6 mice treated with 3 s.c. injections of saline alone or containing 50 ng anti-DNP IgE, IgG1 or IgG2b antibody and challenged 18 h later with 2 s.c. injections, each containing 37.5 μg RVV and 0.5 μg DNP-HSA. Data were pooled from 2–5 independent experiments (n=10–25/group). P values: Student’s t test (B); Mantel-Cox test (C). [This is a reproduction of Fig. 5 from Starkl P, Marichal T, Gaudenzio N, Reber LL, Sibilano R, Tsai M, Galli SJ. IgE antibodies, FcεRIα and IgE-mediated local anaphylaxis can limit snake venom toxicity. J Allergy Clin Immunol 2016;137:246–57 (ref. [105]), reprinted with the permission of the publisher, Elsevier.]
We found that pre-sensitization with anti-DNP IgE significantly increased the resistance of C57BL/6 (Fig 7, B and C) or BALB/c (see Fig E5, H-I in the online repository of ref. [105]) mice to challenge with a potentially lethal amount of RVV admixed with DNP-HAS [105]. However, pre-sensitization of C57BL/6 mice with anti-DNP IgG1 or IgG2b, DNP-specific IgG isotypes with the capacity to activate effector cells via Fcγ receptors [108], not only failed to increase protection but also resulted in increased hypothermia at early time points compared to vehicle-treated or IgE-sensitized mice (Fig 7, B) [105]. These findings show that, in the tested mice, local tissue responses mediated by IgE and antigen can enhance host resistance against RVV even when that antigen is not a native constituent of the venom. These findings are consistent with the general idea that the host needs only to generate an IgE response against a limited number of the components of a complex venom (perhaps as few as one component) in order to manifest enhanced acquired resistance to that venom.
Conclusions.
Tissue resident cells with morphological, biochemical, and functional properties of mammalian mast cells, and which can produce histamine, heparin and serine proteases, are present in tunicates, whose ancestors appeared in evolution before the development of adaptive immunity [109, 110]. Such tunicates also have been reported to have cells resembling basophils [111]. After the appearance of acquired immunity and the development of antibodies, these ancient hematopoietic lineages acquired the ability to bind immunoglobulins such as IgE (in mammals) to their surface. This allowed such tissue-resident (or, in the case of basophils, circulating) cells to become “immunologically primed” or “sensitized” to undergo activation for mediator release upon encountering relatively small amounts of the antigen identified by their surface-bound IgE antibodies. The most extreme mal-adaptive example of an IgE-associated immune response resulting in the activation of mast cells (and basophils) is fatal anaphylaxis, in which the rapid, systemic and extensive release of mediators stored in these FcεRI-bearing effector cells results in a catastrophic and quickly lethal outcome.
Observational and epidemiological studies in humans, as well as studies in experimental animals (including those employing mice genetically deficient in mast cells, basophils or IgE), strongly suggest that one beneficial role of IgE, mast cells and basophils is to help to defend the host against ectoparasites such as ticks, and to diminish the numbers of parasites and burden of disease in mammals infected with certain helminths. However, in addition to parasites, vertebrates also have been subjected to evolutionary pressure through millions of years of co-evolution with venomous arthropods, reptiles, and other species. Evidence in mice indicates that mast cells can enhance innate resistance of mice to 4 species of poisonous snakes, the Gila monster, 2 species of scorpions, and the honeybee, and that mast cell proteases (specifically, CPA3 and the chymase MCP4) can contribute to such mast cell-dependent innate defenses by degrading toxins present in some of these venoms. Moreover, evidence derived from in vitro studies with embryonic zebra fish shows that human mast cell tryptase (but not human mast cell chymase or CPA3), can markedly reduce the toxicity of the venoms of six different poisonous snakes [84]. Finally, in mice, type 2 immune responses induced by a single exposure to honeybee venom or Russell’s viper venom, which “arm” mast cells with IgE antibodies that bear specificity for components of those venoms, can significantly increase the survival of such mice to challenge with doses of the venoms that would be lethal in naïve mice [96].
This evidence supports the notion [39–41] that key elements of “allergic reactivity”, including mast cells and IgE, indeed can importantly enhance innate and acquired host resistance to venoms. Yet much work remains to be done to answer several related, but unresolved, questions. These include the following. In addition to releasing proteases, are there other mechanisms by which mast cells can contribute to enhanced resistance to venoms during innate or acquired immune responses (e.g., ex vivo studies indicate that mast cell-derived heparin, which is highly anionic, can bind and thereby reduce the toxicity of cationic toxins in Russell’s viper venom [76]). Also, in what ways do venoms induce Th2 cell and IgE responses (for honeybee venom, this appears to involve a pathway by which products of bvPLA2 acting on host lipid membranes induce IL-33 production, which in turn can activate ILC2 cells to release cytokines that drive IgE production [104]). During vertebrate evolution, what has been the relative importance of exposure to ectoparasites (and the pathogens for which they serve as vectors), infection with helminths and other parasites, and interactions with venomous animals in shaping the features, function and immunological roles of mast cells, basophils, and IgE? Given that mast cells and basophils cooperate to enhance acquired resistance to the feeding of certain ticks, and that the hematophagous fluids produced by tick salivary glands can contain peptides similar to those in venoms [112], is there a role for basophils in enhancing resistance to some venoms? Finally, why do some subjects develop such severe IgE-dependent reactivity to venom that they are at risk for fatal anaphylaxis (an outcome far from a “protective” immune response)?
Our initial findings indicate that the propensity to develop protective vs. potentially harmful type 2 immune responses to venoms, at least in mice, can depend on the genetic background of the animal, the type and amount of venom to which the animal is exposed, and/or the frequency of such venom exposures [105]. But this is only the beginning of addressing this important issue. In considering this question, it should be noted that many people who develop type 2 immune responses to BV do not exhibit anaphylactic reactivity despite having venom-specific IgE antibodies [113]. Also, there is abundant evidence that Th2 cell-mediated responses are subject to immune regulation which can diminish pathology related to IgE-dependent reactivity to the inducing antigen, including honeybee venom [114–116]. One might speculate that such immune regulation of type 2 immune responses ideally would reduce the pathology associated with these responses while preserving their ability to confer enhanced protection when the elicited antigen is a toxin.
Acknowledgements
We thank the past and current members of the Galli lab and the many collaborators who have made important contributions to the work reviewed herein. The work reviewed herein was supported by grants to S.J.G. from the National Institutes of Health (e.g., R37 AI23990, R01 CA072074, R01 AR067145, and U19 AI104209) and the National Science Foundation, and from several other funding sources, including the Department of Pathology at Stanford University. M.M. was supported by grants from the German Research Foundation (DFG, ME 2668/3-2, ME 2668/2-1) and the Else Kröner-Fresenius-Foundation, P.S. was supported by the Austrian Science Fund (FWF): P31113-B30, a Max Kade Fellowship of the Max Kade Foundation and the Austrian Academy of Sciences, a Schroedinger Fellowship of the Austrian Science Fund (FWF): J3399-B21, and a Marie Curie fellowship of the European Commission (H2020-MSCAIF-2014), 655153. T.M. was supported by a Marie Curie International Outgoing Fellowship for Career Development: European Union’s Seventh Framework Programme (FP7-PEOPLE-2011-IOF), 299954, and a “Charge de recherches” fellowship of the Belgian National Fund for Scientific Research (F.R.S-FNRS).
Abbreviations
- ABS
rabbit anti-basophil serum
- AES
rabbit anti-eosinophil serum
- BMCMCs
bone marrow-derived cultured mast cells
- BV
honeybee venom
- bvPLA2
honeybee venom phospholipase A2
- CPA3
carboxypeptidase A3
- CBH
cutaneous basophil hypersensitivity
- DNP
dinitrophenol
- DNP-HSA
dinitrophenol-conjugated human serum albumin
- ESCMCs
embryonic stem cell-derived cultured mast cells
- ET-1
endothelin-1
- F(ab)
antigen-binding fragment of an immunoglobulin molecule
- FcεRI
the high affinity receptor for IgE
- IgE
immunoglobulin E (antibody)
- IgG
immunoglobulin G (antibody)
- IL
interleukin
- ILC2
innate lymphoid cells, type 2
- i.d.
intradermal
- i.p.
intraperitoneal
- i.v.
intravenous
- LPS
lipopolysaccharide
- MC(s)
mast cell(s)
- Mcl-1
myeloid cell leukemia 1
- MCP4
mast cell protease 4
- NRS
normal rabbit serum
- PAMPs
pathogen-associated molecular patterns
- RVV
Russell’s viper venom
- s.c.
subcutaneous
- shRNA
small hairpin RNA
- Th2
T helper cell type 2
- VIP
vasoactive intestinal polypeptide
- WT
wild type
Footnotes
All authors declare no conflict of interest.
This review is a modified and updated version of similar invited reviews that appeared in the American Journal of Pathology: Galli SJ: The 2014 Rous-Whipple Award Lecture. The mast cell-IgE paradox: From homeostasis to anaphylaxis. Am J Pathol, 2016; 186:212–224. PMID: 26776074 and in Allergology International: Galli SJ, Starkl P, Marichal T, Tsai M. Mast cells and IgE in defense against venoms: Possible “good side” of allergy. Allergol Int 2016; 65:3–15. PMID: 26666482. This article is based in part on a keynote lecture given by Stephen J. Galli on April 11, 2019 at the EAACI Allergy School on Insect Venom Allergy and Mastocytosis in Groningen, The Netherlands.
References
- 1.Pawankar R, Canonica GW, Holgate ST, Lockey RF. Allergic diseases and asthma: a major global health concern. Curr Opin Allergy Clin Immunol 2012; 12:39–41. [DOI] [PubMed] [Google Scholar]
- 2.Paul WE, Zhu J. How are TH2-type immune responses initiated and amplified? Nat Rev Immunol 2010;10:225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med 2012;18:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pulendran B, Artis D. New paradigms in type 2 immunity. Science 2012;337:431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinet JP. The high-affinity IgE receptor (FcƐRI): from physiology to pathology. Annu Rev Immunol 1999;17:931–72. [DOI] [PubMed] [Google Scholar]
- 6.Rivera J, Fierro NA, Olivera A, Suzuki R. New insights on mast cell activation via the high affinity receptor for IgE. Adv Immunol 2008;98:85–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oettgen HC, Geha RS. IgE in asthma and atopy: cellular and molecular connections. J Clin Invest 1999;104:829–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karasuyama H, Mukai K, Obata K, Tsujimura Y, Wada T. Nonredundant roles of basophils in immunity. Annu Rev Immunol 2011;29:45–69. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan BM, Liang HE, Bando JK, Wu D, Cheng LE, McKerrow JK, et al. Genetic analysis of basophil function in vivo. Nat Immunol 2011;12:527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Voehringer D. Protective and pathological roles of mast cells and basophils. Nat Rev Immunol 2013;13:362–75. [DOI] [PubMed] [Google Scholar]
- 11.Kawakami T, Kitaura J. Mast cell survival and activation by IgE in the absence of antigen: a consideration of the biologic mechanisms and relevance. J Immunol 2005;175:4167–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyce JA. Mast cells and eicosanoid mediators: a system of reciprocal paracrine and autocrine regulation. Immunol Rev 2007;217:168–85. [DOI] [PubMed] [Google Scholar]
- 13.Douaiher J, Succar J, Lancerotto L, Gurish MF, Orgill DP, Hamilton MJ, et al. Development of mast cells and importance of their tryptase and chymase serine proteases in inflammation and wound healing. Adv Immunol 2014;122:211–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol 2005;23:749–86. [DOI] [PubMed] [Google Scholar]
- 15.Metz M, Piliponsky AM, Chen CC, Lammel V, Abrink M, Pejler G, et al. Mast cells can enhance resistance to snake and honeybee venoms. Science 2006;313:526–30. [DOI] [PubMed] [Google Scholar]
- 16.Dawicki W, Marshall JS. New and emerging roles for mast cells in host defence. Curr Opin Immunol 2007;19:31–8. [DOI] [PubMed] [Google Scholar]
- 17.Abraham SN, St John AL: Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol 2010;10:440–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akahoshi M, Song CH, Piliponsky AM, Metz M, Guzzetta A, Abrink M, et al. Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venom, and vasoactive intestinal polypeptide in mice. J Clin Invest 2011;121:4180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol 2011;12:1035–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen BM, Frandsen PM, Raaby EM, Schiotz PO, Skov PS, Poulsen LK. Molecular and stimulus-response profiles illustrate heterogeneity between peripheral and cord blood-derived human mast cells. J Leukoc Biol 2014;95:893–901. [DOI] [PubMed] [Google Scholar]
- 21.McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015; 519:237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Portier MM, Richet C. De l’action anaphylactique de certains venims. C R Soc Biol 1902; 54:170–2. [Google Scholar]
- 23.Finkelman FD. Anaphylaxis: lessons from mouse models. J Allergy Clin Immunol 2007; 120:506–15.. [DOI] [PubMed] [Google Scholar]
- 24.Stetson DB, Voehringer D, Grogan JL, Xu M, Reinhardt RL, Scheu S, et al. Th2 cells: orchestrating barrier immunity. Adv Immunol 2004;83:163–89. [DOI] [PubMed] [Google Scholar]
- 25.Finkelman FD, Shea-Donohue T, Morris SC, Gildea L, Strait R, Madden KB, et al. Interleukin-4- and interleukin-13-mediated host protection against intestinal nematode parasites. Immunol Rev 2004;201:139–55. [DOI] [PubMed] [Google Scholar]
- 26.Fitzsimmons CM, Dunne DW. Survival of the fittest: allergology or parasitology? Trends Parasitol 2009;25:447–51. [DOI] [PubMed] [Google Scholar]
- 27.Spencer LA, Porte P, Zetoff C, Rajan TV. Mice genetically deficient in immunoglobulin E are more permissive hosts than wild-type mice to a primary, but not secondary, infection with the filarial nematode Brugia malayi. Infec Immun 2003;71:2462–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwartz C, Turqueti-Neves A, Hartmann S, Yu P, Nimmerjahn F, Voehringer D. Basophil-mediated protection against gastrointestinal helminths requires IgE-induced cytokine secretion. Proc Natl Acad Sci USA 2014;111:E5169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nawa Y, Kiyota M, Korenaga M, Kotani M. Defective protective capacity of W/Wv mice against Strongyloides ratti infection and its reconstitution with bone marrow cells. Parasite Immunol 1985;7:429–38. [DOI] [PubMed] [Google Scholar]
- 30.Knight PA, Wright SH, Lawrence CE, Paterson YY, Miller HR. Delayed expulsion of the nematode Trichinella spiralis in mice lacking the mucosal mast cell-specific granule chymase, mouse mast cell protease-1. J Exp Med 2000;192:1849–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furuta T, Kikuchi T, Iwakura Y, Watanabe N. Protective roles of mast cells and mast cell-derived TNF in murine malaria. J Immunol 2006;177:3294–302. [DOI] [PubMed] [Google Scholar]
- 32.Maurer M, Lopez Kostka S, Siebenhaar F, Moelle K, Metz M, Knop J, et al. Skin mast cells control T cell-dependent host defense in Leishmania major infections. FASEB J 2006; 20:2460–7. [DOI] [PubMed] [Google Scholar]
- 33.Ohnmacht C, Voehringer D. Basophils protect against reinfection with hookworms independently of mast cells and memory Th2 cells. J Immunol 2010;184:344–50. [DOI] [PubMed] [Google Scholar]
- 34.Arizono N, Kasugai T, Yamada M, Okada M, Morimoto M, Tei H, et al. Infection of Nippostrongylus brasiliensis induces development of mucosal-type but not connective tissue-type mast cells in genetically mast cell-deficient Ws/Ws rats. Blood 1993;81:2572–8. [PubMed] [Google Scholar]
- 35.Amiri P, Haak-Frendscho M, Robbins K, McKerrow JH, Stewart T, Jardieu P. Anti-immunoglobulin E treatment decreases worm burden and egg production in Schistosoma mansoni-infected normal and interferon gamma knockout mice. J Exp Med 1994;180:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newlands GF, Miller HR, MacKellar A, Galli SJ. Stem cell factor contributes to intestinal mucosal mast cell hyperplasia in rats infected with Nippostrongylus brasiliensis or Trichinella spiralis, but anti-stem cell factor treatment decreases parasite egg production during N brasiliensis infection. Blood 1995;86:1968–76. [PubMed] [Google Scholar]
- 37.Holgate ST, Polosa R. Treatment strategies for allergy and asthma. Nat Rev Immunol 2008;8:218–30. [DOI] [PubMed] [Google Scholar]
- 38.Artis D, Maizels RM, Finkelman FD. Forum: Immunology: Allergy challenged. Nature 2012;484:458–9. [DOI] [PubMed] [Google Scholar]
- 39.Profet M. The function of allergy: immunological defense against toxins. Q Rev Biol 1991; 66:23–62. [DOI] [PubMed] [Google Scholar]
- 40.Stebbings JH Jr. Immediate hypersensitivity: a defense against arthropods? Perspect Biol Med 1974;17:233–9. [DOI] [PubMed] [Google Scholar]
- 41.Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature 2012, 484:465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galli SJ: The 2014 Rous-Whipple Award Lecture. The mast cell-IgE paradox: From homeostasis to anaphylaxis. Am J Pathol 2016;186:212–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galli SJ, Colvin RB, Verderber E, Galli AS, Monahan R, Dvorak AM, et al. Preparation of a rabbit anti-guinea pig basophil serum: in vitro and in vivo characterization. J Immunol 1978;121:1157–66. [PubMed] [Google Scholar]
- 44.Dvorak HF. Cutaneous basophil hypersensitivity. J Allergy Clin Immunol 1976;58:229–40. [DOI] [PubMed] [Google Scholar]
- 45.Brown SJ, Galli SJ, Gleich GJ, Askenase PW. Ablation of immunity to Amblyomma americanum by anti-basophil serum: cooperation between basophils and eosinophils in expression of immunity to ectoparasites (ticks) in guinea pigs. J Immunol 1982;129:790–6. [PubMed] [Google Scholar]
- 46.Childs JE, Paddock CD. The ascendancy of Amblyomma americanum as a vector of pathogens affecting humans in the United States. Annu Rev Entomol 2003;48:307–37. [DOI] [PubMed] [Google Scholar]
- 47.Commins SP, James HR, Kelly LA, Pochan SL, Workman LJ, Perzanowski MS et al. The relevance of tick bites to the production of IgE antibodies to the mammalian oligosaccharide galactose-α−1,3-galactose. J Allergy Clin Immunol 2011;127:1286–93 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-α−1,3-galactose. N Engl J Med 2008;358:1109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown SJ, Askenase PW. Cutaneous basophil responses and immune resistance of guinea pigs to ticks: passive transfer with peritoneal exudate cells or serum. J Immunol 1981;127:2163–7. [PubMed] [Google Scholar]
- 50.Allen JR. Tick resistance: basophils in skin reactions of resistant guinea pigs. Int J Parasitol 1973;3:195–200. [DOI] [PubMed] [Google Scholar]
- 51.Matsuda H, Watanabe N, Kiso Y, Hirota S, Ushio H, Kannan Y, et al. Necessity of IgE antibodies and mast cells for manifestation of resistance against larval Haemaphysalis longicornis ticks in mice. J Immunol 1990;144:259–62. [PubMed] [Google Scholar]
- 52.Wada T, Ishiwata K, Koseki H, Ishikura T, Ugajin T, Ohnuma N, et al. Selective ablation of basophils in mice reveals their nonredundant role in acquired immunity against ticks. J Clin Invest 2010;120:2867–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steeves EB, Allen JR. Basophils in skin reactions of mast cell-deficient mice infested with Dermacentor variabilis. Int J Parasitol 1990;20:655–67. [DOI] [PubMed] [Google Scholar]
- 54.Chabot B, Stephenson DA, Chapman VM, Besmer P, Bernstein A. The proto-oncogene c-kit encoding a transmembrane tyrosine kinase receptor maps to the mouse W locus. Nature 1988;335:88–9. [DOI] [PubMed] [Google Scholar]
- 55.Geissler EN, Ryan MA, Housman DE. The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell 1988;55:185–92. [DOI] [PubMed] [Google Scholar]
- 56.Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood 1978;52:447–52. [PubMed] [Google Scholar]
- 57.Russell ES. Hereditary anemias of the mouse: a review for geneticists. Adv Genet 1979; 20:357–459. [PubMed] [Google Scholar]
- 58.Harrison DE, Astle CM. Population of lymphoid tissues in cured W-anemic mice by donor cells. Transplantation 1976;22:42–6. [DOI] [PubMed] [Google Scholar]
- 59.Nakano T, Waki N, Asai H, Kitamura Y. Lymphoid differentiation of the hematopoietic stem cell that reconstitutes total erythropoiesis of a genetically anemic W/Wv mouse. Blood 1989;73:1175–9. [PubMed] [Google Scholar]
- 60.Nakano T, Waki N, Asai H, Kitamura Y. Different repopulation profile between erythroid and nonerythroid progenitor cells in genetically anemic W/Wv mice after bone marrow transplantation. Blood 1989;74:1552–6. [PubMed] [Google Scholar]
- 61.Nabel G, Galli SJ, Dvorak AM, Dvorak HF, Cantor H. Inducer T lymphocytes synthesize a factor that stimulates proliferation of cloned mast cells. Nature 1981;291:332–4. [DOI] [PubMed] [Google Scholar]
- 62.Galli SJ, Dvorak AM, Marcum JA, Ishizaka T, Nabel G, Der Simonian H, et al. Mast cell clones: a model for the analysis of cellular maturation. J Cell Biol 1982;95:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakano T, Sonoda T, Hayashi C, Yamatodani A, Kanayama Y, Yamamura T, et al. Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice. Evidence that cultured mast cells can give rise to both connective tissue type and mucosal mast cells. J Exp Med 1985;162:1025–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsai M, Wedemeyer J, Ganiatsas S, Tam SY, Zon LI, Galli SJ. In vivo immunological function of mast cells derived from embryonic stem cells: an approach for the rapid analysis of even embryonic lethal mutations in adult mice in vivo. Proc Natl Acad Sci USA 2000;97:9186–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maurer M, Wedemeyer J, Metz M, Piliponsky AM, Weller K, Chatterjea D, et al. Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature 2004;432:512–6. [DOI] [PubMed] [Google Scholar]
- 66.Piliponsky AM, Chen CC, Nishimura T, Metz M, Rios EJ, Dobner PR, et al. Neurotensin increases mortality and mast cells reduce neurotensin levels in a mouse model of sepsis. Nat Med 2008;14:392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galli SJ, Kitamura Y. Genetically mast-cell-deficient W/Wv and Sl/Sld mice. Their value for the analysis of the roles of mast cells in biologic responses in vivo. Am J Pathol 1987;127:191–8. [PMC free article] [PubMed] [Google Scholar]
- 68.Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant KitW-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol 2005;167:835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wolters PJ, Mallen-St Clair J, Lewis CC, Villalta SA, Baluk P, Erle DJ, et al. Tissue-selective mast cell reconstitution and differential lung gene expression in mast cell-deficient KitW-sh/KitW-sh sash mice. Clin Exp Alergy 2005;35:82–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lilla JN, Chen CC, Mukai K, BenBarak MJ, Franco CB, Kalesnikoff J, et al. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 2011;118:6930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Metz M, Grimbaldeston MA, Nakae S, Piliponsky AM, Tsai M, Galli SJ. Mast cells in the promotion and limitation of chronic inflammation. Immunol Rev 2007;217:304–28. [DOI] [PubMed] [Google Scholar]
- 72.Galli SJ, Tsai M, Marichal T, Tchougounova E, Reber LL, Pejler G. Approaches for analyzing the roles of mast cells and their proteases in vivo. Adv Immunol 2015;126:45–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nigrovic PA, Gray DH, Jones T, Hallgren J, Kuo FC, Chaletzky B, et al. Genetic inversion in mast cell-deficient Wsh mice interrupts Corin and manifests as hematopoietic and cardiac aberrancy. Am J Pathol 2008;173:1693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brown MA, Hatfield JK. Mast cells are important modifiers of autoimmune disease: with so much evidence, why is there still controversy? Front Immunol 2012;3:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rodewald HR, Feyerabend TB. Widespread immunological functions of mast cells: fact or fiction? Immunity 2012;37:13–24. [DOI] [PubMed] [Google Scholar]
- 76.Higginbotham RD. Mast cells and local resistance to Russell’s viper venom. J Immunol 1965;95:867–75. [PubMed] [Google Scholar]
- 77.Higginbotham RD, Karnella S. The significance of the mast cell response to bee venom. J Immunol 1971;106:233–40. [PubMed] [Google Scholar]
- 78.Kloog Y, Ambar I, Sokolovsky M, Kochva E, Wollberg Z, Bdolah A. Sarafotoxin, a novel vasoconstrictor peptide: phosphoinositide hydrolysis in rat heart and brain. Science 1988;242:268–70. [DOI] [PubMed] [Google Scholar]
- 79.Kochva E, Bdolah A, Wollberg Z. Sarafotoxins and endothelins: evolution, structure and function. Toxicon 1993;31:541–68. [DOI] [PubMed] [Google Scholar]
- 80.Fry BG. From genome to “venome”: molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res 2005;15:403–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schneider LA, Schlenner SM, Feyerabend TB, Wunderlin M, Rodewald HR. Molecular mechanism of mast cell mediated innate defense against endothelin and snake venom sarafotoxin. J Exp Med 2007;204:2629–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tchougounova E, Pejler G, Abrink M. The chymase, mouse mast cell protease 4, constitutes the major chymotrypsin-like activity in peritoneum and ear tissue. A role for mouse mast cell protease 4 in thrombin regulation and fibronectin turnover. J Exp Med 2003;198:423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wernersson S, Pejler G. Mast cell secretory granules: armed for battle. Nat Rev Immunol 2014;14:478–94. [DOI] [PubMed] [Google Scholar]
- 84.Anderson E, Stavenhagen K, Kolarich D, Sommerhoff CP, Maurer M, Metz M. Human mast cell tryptase is a potential treatment for snakebite envenoming across multiple snake species. Front Immunol 2018;9:1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Neves-Ferreira AG, Perales J, Fox JW, Shannon JD, Makino DL, Garratt RC, et al. Structural and functional analyses of DM43, a snake venom metalloproteinase inhibitor from Didelphis marsupialis serum. J Biol Chem 2002;277:13129–37. [DOI] [PubMed] [Google Scholar]
- 86.Habermann E. Bee and wasp venoms. Science 1972;177:314–22. [DOI] [PubMed] [Google Scholar]
- 87.Mukherjee AK, Ghosal SK, Maity CR. Some biochemical properties of Russell’s viper (Daboia russelli) venom from Eastern India: correlation with clinico-pathological manifestation in Russell’s viper bite. Toxicon 2000;38:163–75. [DOI] [PubMed] [Google Scholar]
- 88.Risch M, Georgieva D, von Bergen M, Jehmlich N, Genov N, Arni RK, et al. Snake venomics of the Siamese Russell’s viper (Daboia russelli siamensis) -- relation to pharmacological activities. J Proteomics 2009;72:256–69. [DOI] [PubMed] [Google Scholar]
- 89.Saelinger CB, Higginbotham RD. Hypersensitivity responses to bee venom and the mellitin. Int Arch Allergy Appl Immunol 1974;46:28–37. [DOI] [PubMed] [Google Scholar]
- 90.Charavejasarn CC, Reisman RE, Arbesman CE. Reactions of anti-bee venom mouse reagins and other antibodies with related antigens. Int Arch Allergy Appl Immunol 1975;48:691–7. [DOI] [PubMed] [Google Scholar]
- 91.Jarisch R, Yman L, Boltz A, Sandor I, Janitsch A. IgE antibodies to bee venom, phospholipase A, melittin and wasp venom. Clin Allergy 1979;9:535–41. [DOI] [PubMed] [Google Scholar]
- 92.Wadee AA, Rabson AR. Development of specific IgE antibodies after repeated exposure to snake venom. J Allergy Clin Immunol 1987;80:695–8. [DOI] [PubMed] [Google Scholar]
- 93.Annila I. Bee venom allergy. Clin Exp Allergy 2000;30:1682–7. [DOI] [PubMed] [Google Scholar]
- 94.Bilo BM, Rueff F, Mosbech H, Bonifazi F, Oude-Elberink JN. Diagnosis of Hymenoptera venom allergy. Allergy 2005;60:1339–49. [DOI] [PubMed] [Google Scholar]
- 95.Simpson ID, Norris RL. Snakes of Medical Importance in India: Is the Concept of the “Big 4” Still Relevant and Useful? Wilderness Environ Med 2007;18:2–9. [DOI] [PubMed] [Google Scholar]
- 96.Marichal T, Starkl P, Reber LL, Kalesnikoff J, Oettgen HC, Tsai M, et al. A beneficial role for immunoglobulin E in host defense against honeybee venom. Immunity 2013;39:963–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Oettgen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, Leder P. Active anaphylaxis in IgE-deficient mice. Nature 1994;370:367–70. [DOI] [PubMed] [Google Scholar]
- 98.Miyajima I, Dombrowicz D, Martin TR, Ravetch JV, Kinet JP, Galli SJ. Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and FcγRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J Clin Invest 1997;99:901–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reimers AR, Weber M, Muller UR. Are anaphylactic reactions to snake bites immunoglobulin E-mediated? Clin Exp Allergy 2000;30:276–82. [DOI] [PubMed] [Google Scholar]
- 100.Meier J. Commercially available antivenoms (“hyperimmune sera”, “antivenins”, “antisera”) for antivenom therapy Handbook of Clinical Toxicology of Animal Venoms and Poisons. Edited by J Meier JW. Boca Raton, FL: CRC Press, 1995; 689–721. [Google Scholar]
- 101.Haak-Frendscho M, Saban R, Shields RL, Jardieu PM. Anti-immunoglobulin E antibody treatment blocks histamine release and tissue contraction in sensitized mice. Immunology 1998; 94:115–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Prouvost-Danon A, Binaghi RA, Abadie A. Effect of heating at 56 degrees C on mouse IgE antibodies. Immunochemistry 1977;14:81–4. [DOI] [PubMed] [Google Scholar]
- 103.Strait RT, Morris SC, Finkelman FD. IgG-blocking antibodies inhibit IgE-mediated anaphylaxis in vivo through both antigen interception and FcγRIIb cross-linking. J Clin Invest 2006;116:833–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Palm NW, Rosenstein RK, Yu S, Schenten DD, Florsheim E, Medzhitov R. Bee venom phospholipase A2 induces a primary type 2 response that is dependent on the receptor ST2 and confers protective immunity. Immunity 2013;39:976–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Starkl P, Marichal T, Gaudenzio N, Reber LL, Sibilano R, Tsai M, et al. IgE antibodies, FcεRIα and IgE-mediated local anaphylaxis can limit snake venom toxicity. J Allergy Clin Immunol 2016;137:246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Habermann E, Walsch P, Breithaupt H. Biochemistry and pharmacology of the cortoxin complex. II. Possible interrelationships between toxicity and organ distribution of phospholipase A, crotapotin and their combination. Naunyn Schmiedebergs Arch Pharmacol 1972;273:313–30. [DOI] [PubMed] [Google Scholar]
- 107.Liu FT, Bohn JW, Ferry EL, Yamamoto H, Molinaro CA, Sherman LA, et al. Monoclonal dinitrophenyl-specific murine IgE antibody: preparation, isolation, and characterization. J Immunol 1980;124:2728–37. [PubMed] [Google Scholar]
- 108.Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood 2012;119:5640–9. [DOI] [PubMed] [Google Scholar]
- 109.Cavalcante MC, de Andrade LR, Du Bocage Santos-Pinto C, Straus AH, Takahashi HK, Allodi S, et al. Colocalization of heparin and histamine in the intracellular granules of test cells from the invertebrate Styela plicata (Chordata-Tunicata). J Struct Biol 2002;137:313–21. [DOI] [PubMed] [Google Scholar]
- 110.Wong GW, Zhuo L, Kimata K, Lam BK, Satoh N, Stevens RL. Ancient origin of mast cells. Biochem Biophys Res Commun 2014;451:314–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.de Barros CM, Andrade LR, Allodi S, Viskov C, Mourier PA, Cavalcante MC, et al. The Hemolymph of the ascidian Styela plicata (Chordata-Tunicata) contains heparin inside basophil-like cells and a unique sulfated galactoglucan in the plasma. J Biol Chem 2007;282:1615–26. [DOI] [PubMed] [Google Scholar]
- 112.Fry BG, Roelants K, Champagne DE, Scheib H, Tyndall JDA, King GF, et al. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu Rev Genomics Hum Genetics 2009;10:483–511. [DOI] [PubMed] [Google Scholar]
- 113.Antonicelli L, Bilo MB, Bonifazi F. Epidemiology of Hymenoptera allergy. Curr Opin Allergy Clin Immunol 2002;2:341–6. [DOI] [PubMed] [Google Scholar]
- 114.Muller UR. Bee venom allergy in beekeepers and their family memberws. Curr Opin Allergy Clin Immunol 2005;5:343–7. [DOI] [PubMed] [Google Scholar]
- 115.Meiler F, Zumkehr J, Klunker S, Ruckert B, Akdis CA, Akdis M. In vivo switch to IL-10-secreting T regulatory cells in high dose allergen exposure. J Exp Med 2008;205:2887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ozdemir C, Kucuksezer UC, Akdis M, Akdis CA. Mechanisms of immunotherapy to wasp and bee venom. Clin Expl Allergy 2011;41:1226–34. [DOI] [PubMed] [Google Scholar]