

Abstract
Drug-like compounds are most of the time denied approval and use owing to the unexpected clinical side effects and cross-reactivity observed during clinical trials. These unexpected outcomes resulting in significant increase in attrition rate centralizes on the selected drug targets. These targets may be disease candidate proteins or genes, biological pathways, disease-associated microRNAs, disease-related biomarkers, abnormal molecular phenotypes, crucial nodes of biological network or molecular functions. This is generally linked to several factors, including incomplete knowledge on the drug targets and unpredicted pharmacokinetic expressions upon target interaction or off-target effects. A method used to identify targets, especially for polygenic diseases, is essential and constitutes a major bottleneck in drug development with the fundamental stage being the identification and validation of drug targets of interest for further downstream processes. Thus, various computational methods have been developed to complement experimental approaches in drug discovery. Here, we present an overview of various computational methods and tools applied in predicting or validating drug targets and drug-like molecules. We provide an overview on their advantages and compare these methods to identify effective methods which likely lead to optimal results. We also explore major sources of drug failure considering the challenges and opportunities involved. This review might guide researchers on selecting the most efficient approach or technique during the computational drug discovery process.
Keywords: Pharmacogenomics, genomics, machine learning, docking, drug targets
Introduction
Drug research and development pipeline entails the following steps: (a) target identification and validation, (b) hit to lead molecule generation, (c) lead molecule optimization and characterization, (d) drug formulation and delivery, (e) pharmacokinetics and drug disposition, (f) preclinical drug candidate identification and (g) bioanalytical testing and clinical trials [1]. Computational drug discovery has over the past few decades become very relevant mainly due to the reduced risks, time, cost effectiveness and resources as compared with the traditional experimental approaches [2]. This has been made possible due to the improved computational power and in silico methods. These complement experimental approaches by streamlining the research scope and guiding in vivo validation [3]. Discovery of sildenafil and thalidomide is one of the successes in the application of computational approaches to the drug design [4]. Traditional drug development from scratch to its availability in the market costs approximately $2.558 billion over a period of 10 to 15 years [1]. With this huge investment, the success rate of a drug progressing to the market is about 13% [1]. Rejection of potential drugs particularly during phase II and phase III clinical development is associated with unexpected clinical side effects and cross-reactivity. This result is significantly increasing attrition rate [5]. These unexpected outcomes centralize on drug targets which may be disease candidate proteins or genes, biological pathways, disease-associated microRNAs, biomarkers, crucial nodes of biological network or molecular functions [6]. This could be linked to inadequate knowledge on the drug targets, undesirable pharmacokinetic expressions upon target interaction or off-target effects. This challenge relies on the methods and population data used to identify targets especially for polygenic diseases, and this therefore serves as a major bottleneck in drug development. It is also due to the first fundamental stage of the drug development which is identifying and validating drug targets of interest for further downstream analysis [7]. This highlights the need for modulating drug targets to ameliorate the disease state observed and achieve the desired biological response by elucidating off-targets as observed in promiscuous kinase inhibitors [8, 9]. Experimental drug target identification approaches rely on the characterization of proteins of interest followed by the experimental validation using techniques, such as gene knockouts, animal studies and site-directed mutagenesis [10]. However, identifying drug targets through these methods is difficult as predicting off-targets which is almost impossible [11]. Off-target effects paved the way for the ‘magic-bullet paradigm’ characterized by maximally selective drug-like molecules [12].
In the post-genomic era where there is an exponential increase in open access biological data generated by bioinformatics pipelines, the drug discovery field has been revolutionized such that it involves the use of various biological datasets which enables scientists to understand comprehensively the biological system relevant to the disease in focus. Thus, the need arose to implement in silico methods that would facilitate designing and redesigning of drug-like molecules exhibiting desired bioactivity profiles as well as predicting and validating drug targets [13]. This is critical particularly considering the increased incidence of widespread drug-resistant strains threatening the efficacy of common drugs. Computational methods have brought about transformed rational and systematic approaches for exploring efficiently the space of drug combinations in combinatorial drug discovery.
In silico methods have strongly impacted on identifying new targets for old drugs [14, 15] as well as predicting side effects [16] and anatomical therapeutic indicators of approved drugs [17]. This implies that the inception of computational approaches has contributed immensely to a systematic rational guidance of the processes and in reducing the period required for the drug’s availability in the market [18]. This is possible based on the hypothesis that drug side effects would be minimized if the drug candidate is potent and highly selective [19].
The baseline criteria for selecting drug targets require the potential target(s) to be essential and indispensable to disease outcome. For instance, in genetic diseases, gene therapy involves identifying genes associated with mutations. However, with infectious diseases, the criteria require understanding the complex interplay between the host and the disease-causing organism or pathogen [20]. Host target(s) therefore must be unique and homologous to the microbe. Pathogen protein targets homologous to the host are eliminated in the computational drug discovery process primarily to avoid any drug reaction complications [21]. In addition to that, the effectiveness of a drug is highly dependent on the target protein(s) in the microbe or essential biological pathway(s) or process that is key to the survival and propagation of the pathogen in the host system.
Leveraging analytical platforms and omics databases containing biological information, computational approaches have become core components in the drug discovery pipeline [10]. For instance, analytical platforms help elucidate essential chemogenomic relationships between available target data and potential drug candidates or molecules, thereby facilitating the prospects of identifying novel druggable targets, possible off-targets, drug leads and potential repurposable drug candidates. So, it is expected that powerful computational models including but not limited to network-based and machine learning methods would lead to better prediction and understanding of drug target interactions and underlying disease molecular mechanisms.
The computational approach to drug discovery has helped to translate biological data into functional knowledge treatment interventions against diseases at a faster rate. This approach is characterized by providing a system view of the disease in relation to the biological system of interest. This helps elucidating important processes and molecular and cellular networks usually difficult to explore experimentally. The ability to reveal such patterns helps to design predictive models to identify disease biomarkers and potential drug targets [22]. Considering complex diseases which are distinguished by their ability to dysregulate biological functions and pathways, computational methods provide the means to understanding the regulatory mechanisms through gene regulatory network analysis [22]. Also, the development of a computational integrative framework using biological processes and functional datasets (protein–protein interactions between disease-causing pathogens and host) together with pharmaceutical datasets facilitates the extraction of drug targets and the identification of drugs possible for repositioning or repurposing against an infection [10, 11].
In the field of pharmacogenomics and pharmacomicrobiomics, computational techniques have facilitated the prediction of drug metabolism by elucidating inhibitors and substrates of specific enzymes involved in metabolism. This has led to an in-depth understanding of in silico evaluation of absorption, distribution, metabolism, excretion and toxicity (ADMET) properties though interactive optimization of leads, therefore mitigating the tendency of drug failure [2, 23]. For instance, in silico models have been proposed for cytochrome metabolism prediction [24].
Some review studies on in silico approaches to drug design exist, but they mainly focus on protein-associated methods in candidate drug target and drug-like molecule prediction [2, 3, 13]. Here we systematically dissect current computational approaches and tools applied in predicting or validating various drug targets and drug-like molecules. Furthermore, we present the strength of each approach and compare them to guide readers in their choice. We conclude by summarizing the sources of failure in the drug discovery process, taking into perspective the challenges and the opportunities they present to scientists.
Current computational approaches for drug target and potential drug candidate identification
Network-based analysis approach
The study of disease mechanisms to develop drugs or vaccines has evolved from single gene or protein analysis to an entire multiscale analysis of genomics, pharmacogenomics, metabolomics and proteomics relevant to the disease of interest. This approach consists of integrating these different large-scale datasets from heterogeneous sources to generate disease-specific networks, fostering a whole genome-based integrative approach to achieve a global view. The disease-specific network, which is a biological entity composed of sub-units connected as a whole, is used to elucidate essential nodes which could serve as targets due to their influence within the network [25]. A typical example is observed in the case of drugs, such as artemisinin combination therapies (ACTs) and clozapine for treating malaria and schizophrenia, respectively, which interact with multiple targets to deliver the required therapeutic response [26, 27]. This integrative approach presents a multiview perspective of elucidating causal genes, relevant pathways and novel drug targets. Also, it increases the reliability in predicting novel drugs and/or putative drugs as well as engineering drug targets to overcome drug resistance [28–30]. Integrating different biological datasets requires developing algorithms and systems biology tools together with the use of network analysis and functional genomic databases (Supplementary Table 1) to unify the dataset [30]. These tools (Supplementary Table 1) are used to interpret the interactions within the network by identifying sub-networks and regions of similarity and dissimilarity that best explains the disease of interest to narrow down the research scope for further enrichment and validation analysis to improve disease classification [31, 32], disease-associated gene prioritization [33, 34] and drug discovery [30].
The network-based approach is recommended when identifying targets and drug candidates for most complex diseases [30]. It allows uncovering biological mechanisms involved in development and differentiation of complex diseases [22]. The technique is implemented in analyzing nodes and edges in various types of networks including chemical structure and reaction networks, protein structure networks, protein–protein interaction networks, signal transduction networks, genetics interaction networks and metabolic networks. Supplementary Box 1 describes key terms and concepts in the network-based approach. Moreover, network-based approaches sometimes involve computational analysis of metabolisms during the life cycle of the pathogen. Network construction categorizes various metabolism processes into pathways and their reactions and enzymes [35]. This breakdown enables analysis of the entire network more conveniently. Flux balance analysis together with in silico knockout studies is implemented in studies during network analysis to identify vital reactions or biological processes essential for the pathogen’s survival, thus narrowing down the drug target search space [36]. There are evidences on the use of cellular networks to elucidate complex genotype-to-phenotype relationships among diseases and their associated genetic variants [37]. This technique has become an effective tool for predicting drug target associations.
Network-based approaches have been widely used to predict candidate targets and drug target interactions. Luo et al. [29] developed an integrative pipeline capable of integrating various data types as well as coping with the noise and the incomplete and high-dimensional nature of datasets by learning low-dimensional vector representations of essential features. They identified novel interactions between three drugs and cyclooxygenase which was experimentally verified and further showed to be potential for preventing inflammatory diseases. Also, various biological network pipelines and algorithms have been developed to predict essential molecular processes and pathways to enhance drug research, thus controlling pathway cross-talk and possible drug resistance [22, 28, 38].
Overall network-based approaches require a comprehensive understanding of the interaction network particularly regions where the potential drug target is located. This therefore requires pathway and enrichment analysis to accurately classify the potential drug target. Figure 1 describes the summarized workflow of the network-based approach.
Figure 1.
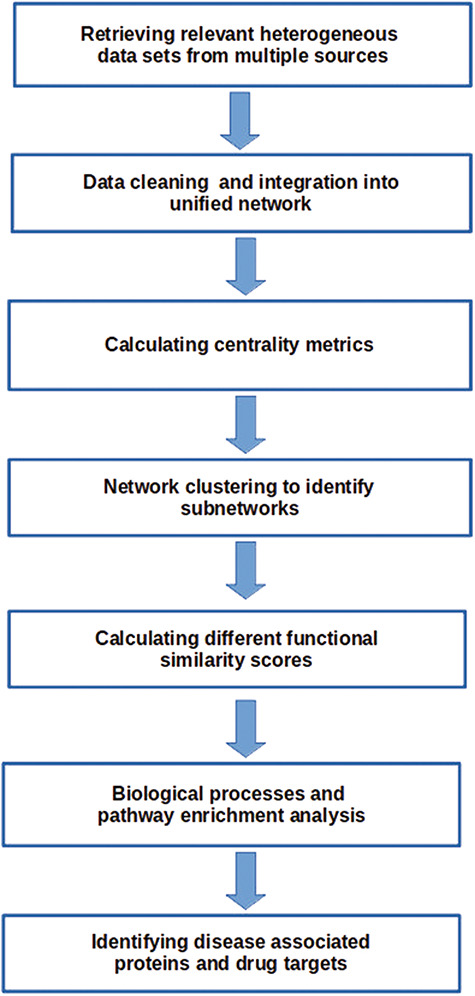
Generalized work flow of a network-based approach in predicting potential drug targets and drug candidates.
Data mining (DM)/machine learning (ML)
With the exponential increase in biological data from high-throughput and combinatorial synthesis, the technological and paradigm shift to data mining and machine learning-based methods have enhanced the extraction and processing of these datasets by combining both biological knowledge, computational tools and algorithms. These indispensable techniques are gaining most attention and credibility because of the reliability and accuracy in predicting key property values of compounds and its significant success rate [39]. This is attributed to their abilities to identify and map relationships between large number of compounds which is difficult to obtain using sub-structural similarities only [13, 40]. Also, machine learning techniques are implemented in both system and molecular methods to predict drug targets through proteomic, microarray and chemogenomic data mining and analysis [6].
Data mining approaches are primarily characterized by an automatic sub-setting of essential information from a pool of datasets. Data mining models ranging from simple parametric equations derived from linear methods to complex models derived from nonlinear methods [41] play a critical role in uncovering significant patterns in chemical and pharmacological property space essential for drug discovery. In addition to that, advanced machine learning models and algorithms such as support vector machines on databases [42], neural networks [39], logistic regression [43], naive Bayesian classification [39, 44], binary kernel discrimination [43], partial least squares [45] and random forest [39] as described in Supplementary Table 2 have been significantly instrumental in drug research. For instance, they have contributed to determining pattern recognition underlying the relationship between compounds and calculated molecular descriptors or experimental measurements within a large chemogenomic space [13, 41]. ML and DM attempt to find correlations between specific activities or classifications for a set of compounds and their features, thus enabling clustering similarities among drug-like compounds in multidimensional space [13, 41].
For example, Fatumo et al. [35] in their research to identify Plasmodium falciparum drug targets developed a machine learning-based metabolic network analysis tool that identifies essential reactions/enzymes as drug targets from the metabolic network of the pathogen. The authors identified 46 essential reactions of which 19 had been reported in literature. A study conducted by Sturm et al. [46] applied the neural network machine learning approach to develop an algorithm for microRNA target prediction. The algorithm developed can predict potential target sites with or without the presence of a seed match. The model was based on machine learning and automatic feature selection using a wide spectrum of compositional, structural and base pairing features covering current biological knowledge.
In relation to both structure-based and ligand-based virtual screening, the combination of DM approaches and a collection of selective pharmacological agents enables mapping of such chemogenomic libraries into biological activity space to predict potential targets [47]. Particularly, when training sets are available, ML methods are more effective in predicting the physical, chemical and biological properties of small molecules as compared with ab initio methods [48]. DM and ML methods are used to develop a quantitative structure–activity relationship (QSAR) or quantitative models for drug-like property predictions and chemical risk assessment [49]. Also, in silico in vitro absorption, distribution, metabolism, excretion and toxicity (ADMET) models and in vivo pharmacokinetic models for optimizing molecular properties and predicting pharmacokinetic parameters have been developed using ML and DM techniques [50, 51]. These models facilitate the selection of leads with improved strong binding affinity to targets.
Application of DM in target similarity search enables the identification of putative protein targets. This approach involves data mining of the pathogen’s sequence and querying against drug target databases to identify putative drug targets with a suitable druggability index [25]. In a study conducted by Mogire et al. [18] to identify putative drug targets against Plasmodium falciparum, a target similarity search of the parasite proteome against drug target databases such as Tropical Disease Research (TDR) target database [52], Therapeutic Target Database (TTD) [53] and Search Tool for Interaction of Chemicals (STITCH) database [54] was performed.
ML models are implemented in predicting sensitivity of drug candidates based on cell line response or the chemical properties of the drugs or a combination of both approaches. This improves the power of designing and systematically analyzing experimental screenings against panels of cell lines to identify potential drugs or repurposable drugs [55]. This approach is critical in personalized medicine in terms of leveraging genomic traits to drug sensitivity.
Menden and colleagues developed machine learning models that integrate chemical properties of drugs and genomic alterations such as copy number variation and sequence variation from cancer cell lines [55]. Their model predicts sensitivity of genomically characterized cancer cell lines to the drugs to ascertain the drug’s efficacy [55]. This model has the ability to optimize experimental design of drug cell screenings by estimating missing half maximal inhibitory concentration (IC50) values [55]. In addition, their model predicts essential target-specific association information between compounds and the target.
Nidhi and colleagues developed a multiple-category Laplacian-modified Bayesian model that works on the basis of chemical structures to predict targets for all MDL Drug Database Report (MDDR) database compounds [47]. The model generated was trained on extended-connectivity fingerprints of compounds from 964 target families characterized by various levels of annotation in the World Of Molecular BioAcTivity (WOMBAT) chemogenomics database. It was then used to predict the top three most likely targets for all MDDT database compounds. Nigsh et al. [56] compared the predictive power of the multiple-category Laplacian-modified Bayesian model and the Winnow algorithm, a linear threshold learning algorithm. The Winnow algorithm implements an additive machine learning rule to minimize ligand-target prediction-related errors [56]. It was observed that both algorithms predict slightly different targets due to compounds that are exclusively retrieved by each algorithm.
Recently, Polypharmacology Browser (PPB2), a new target predicting tool, has been reported [57]. This tool implements neural networks and Naive Bayesian classification models to classify ligands based on their molecular fingerprints or descriptors [57]. Figure 2 describes a summary of application of data mining and machine learning approaches in drug discovery whereas Supplementary Box 2 describes key terms and concepts in the ML and DM approaches.
Figure 2.
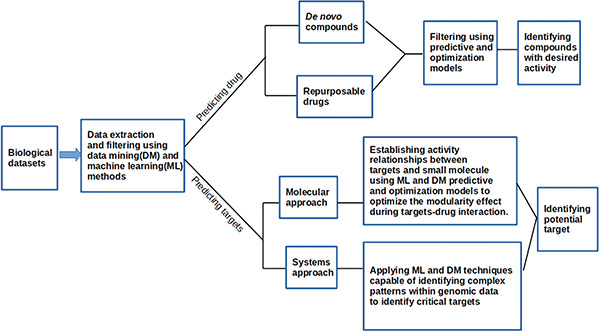
Generalized work flow of data mining and machine learning methods to biological data in predicting potential drug targets and drug candidates.
Reverse/inverse docking
This computational approach is used for identifying putative binding proteins from protein or genomic databases for small molecules with known biological activity [58]. Reverse or inverse virtual screening or inverse docking is a technique that facilitates developing hypothetical relationships among protein targets by chemical probing [59]. It is the structural-based approach of virtual screening unlike the ligand-based methods which require pharmacophores, two-dimensional (2D) fingerprints and three-dimensional (3D) similarity search [4]. It aims to identify drug targets by screening drug-like molecules against rightful protein databases [60]. Molecular docking simulation involves an optimization process of finding the most favorable 3D binding conformations of the ligand to the target [50]. The targets are assessed and scored using scoring function algorithms and ranked according to best binding modes and interaction [61]. Interestingly, reverse docking outputs could be used as a profile to characterize the druggability index or enzyme promiscuity of the protein structures [62]. The reverse docking approach remains a valuable computational technique for exploring alternative uses for existing drugs in terms of drug repurposing and drug rescue [4]. It therefore plays a vital role in the discovery of novel drugs, drug leads, natural products and other ligands for treating neglected diseases which most pharmaceutical industries are hesitant to invest in due to the fear of inadequate return on investment [63, 64].
Unlike conventional forward docking approaches wherein a variety of ligands is docked to a target, the reverse docking process involves screening to a set of different protein targets, a ligand or compound, to identify potential partners through statistical analysis of binding modes within the targets [58]. The framework of this technique is dependent on the knowledge of the distribution of nonhomogeneous proteins, their complexities due to the combination of domains and their conformational flexibility due to multiple folds [65, 66]. Due to that, a ligand fits or docks into the functional binding pocket of a specific protein fold based on its three-dimensional conformation and thus interacts with specific protein residues [59]. This technique enhances elucidation of mechanisms of action and control possible off-target effects. Various tools described in Supplementary Table 3 including TarFisDock [60], idTarget [67], INVDock [63] and AutoDock [68] have been proven to be very useful in drug leads and target prediction [69, 70]. These tools implement different scoring functions to approximate the standard chemical potentials of the system [68].
For instance, idTarget implements the AutoDock4 robust scoring functions [67, 71]. These functions have been shown to have better statistical performance in terms of binding mode prediction [71] and binding site searching efficiency even at the dimensionality of 30 [72]. TarFisDock, a valuable tool for target prediction, was developed from DOCK version 4. The tool however is still under improvement due to associated false positives because of inaccuracies in the scoring function for reverse docking [60]. These errors could be associated with less coverage due to limited target datasets and inability to incorporate protein flexibility during docking.
INVDock implements a scoring scheme capable of performing binding competitive analysis as well as evaluating the interaction energy between docked structures [63]. It is based on the concept of binding competitiveness such that a drug that binds to its target noncompetitively is likely to be less effective. AutoDock implements a machine learning-based scoring function that explores the Iterated Local Search global optimizer approach [68]. The scoring scheme is based on the advantages of knowledge-based potentials as well as extracting empirical information from conformations of receptor-ligand complexes and the experimental affinity measurements [68].
Inverse docking can predict off-targets for ligands aside facilitating predicting activity and selectivity of unknown ligands against known targets [73]. It has been applied in evaluating the binding energies (usually expressed in kcal/mol) and modes of libraries of compounds against a panel of proteins. This evaluation results in a defined group of protein-ligand complexes, thus enhancing the identification of lead compounds for subsequent biological tests. This application reduces cost involved in compound development and biological screening as well as reduces synthetic efforts and time required for de novo drug discovery [59].
Lauro et al. [73, 74] applied the above method to natural bioactive molecules to investigate their efficacy against a panel of cancer-associated proteins. The idea of polypharmacology led to the development of a selective optimization of side activities (SOSA) approach which enhances the generation of new biological activities [75]. The challenging part of this approach is the construction of a panel of target proteins taking into account careful selection of proteins not belonging to the same folds. Also, the accuracy and reliability of this approach are limited when 3D structures of the protein targets are not available.
Regardless of the advantages of reverse docking methods in in silico drug research, they are complex as compared with forward docking techniques such that larger target structure datasets are required to increase the coverage and predictive power [58, 62]. Also, aside its associated biases in inter-protein scoring yielding false positives [76], it requires high computational costs [62, 77].
Biological activity spectra (Biospectra) analysis
There are several reported evidences to the fact that most drugs establish therapeutic response through multiple-target modulation [78–81]. The ability to predict such functional consequences of biological perturbations between the genome or proteome of an organism and biologically profiled compounds is indispensable in drug research. In relation to that, analysis of the modularity effect drug-like molecules impact on the target’s function is a must to understanding the expressed phenotype or therapeutic response capacity of the molecule [82, 83]. Biospectra simply refers to the activities of compounds across potential targets which could enable investigating structure–property relationships [84]. Biospectra analysis is a probabilistic structure–activity relationship approach that complements experimental affinity-based studies [83]. The technique herein is used for measuring quantitatively the patterns and dynamics of the functional activity of a molecule across multiple potential targets [83, 84]. It is therefore a determinant of the inhibitory or stimulatory effect profiles of drug-like molecules on targets within a system. Studies have shown that the association between proteins, drug-like molecules and Biospectra serves as the building block for developing probabilistic approaches to drug discovery [79].
This method provides a firm foundation in determining quantitatively the correlation between molecular structures and biological effect profiles by providing estimates of the therapeutic effect of a molecule. Estimation is done by constructing a nonlinear multivariant model which provides an unbiased tool for investigating associations between structure and function similarities of molecules [83]. Such analysis is relevant for predicting drug targets for orphan compounds based on the concept of chemical structure similarity [84]. It provides the means to classifying molecules based on Biospectra similarity as well as predicting interacting capabilities of molecules with multiple targets. This classification mechanism allows for the identification of molecules with similar function with no prior information concerning the target which is difficult using experimental techniques. Biological activity spectra are an essential indicator of molecular property descriptor [85]. This method was implemented by Fliri and colleagues to identify agonist and antagonist effect profiles of medicinal agents on brain dopamine receptors belonging to the GPCR superfamily [83]. This technique facilitates the ability to conduct spectra similarity and hierarchical clustering methods through profile similarity measurements, thus establishing quantitative relationships between chemical structures and biological activity spectra [85]. Biospectra analysis has been shown to be critical in mining pharmacology datasets as well as predicting possible adverse drug effects based on profile similarity with drug-like molecules known for adverse reactions [84]. Similarity between molecules is measured using the Tanimoto similarity coefficient [86], cosine correlation [87], Euclidean distance [88] or city block distance [89].
Paolini and colleagues presented a comprehensive mapping of pharmacological space by applying a probabilistic model on integrated structure–activity relationship data [79]. They discovered 836 human genes discovering verified targets for small molecules. This integration enables the identification of unique molecular targets through construction of a ligand–target matrix. Since similar drug-like molecules express similar biospectra, this approach is useful for drug repurposing because it facilitates the translation of biological response data into chemical structure design [83]. This implies that the ability to correlate off-target effects with biological spectra would help map unto new targets where the response might be beneficial to address different diseases. For example, sildenafil initially developed to treat angina expressed a side effect of prolonged penile erection, and this resulted in a change of the treatment focus of the drug [90]. However, biospectra analysis is highly dependent on experimental data obtained from various ligand-binding assays or a matrix of targets which could be difficult.
Ligand-based in silico target prediction
A ligand-based computational approach is the framework for ligand-based drug discovery. It is based on the concept of chemical structure similarity, which states that similar ligands or compounds would bind to similar targets with almost the same binding affinity and express similar biological responses [65]. This concept of similarity has been extensively utilized in lead discovery and optimization primarily because it takes into account the polypharmacological nature of drugs [91]. Also, it is essential for quick investigation on primary and secondary targets as well as selectivity among target families [84]. The approach herein involves the interplay between characterized protein targets and characterized ligands with similar chemical structure, properties and pharmacophoric features to enable predicting biological targets. This is achieved by mapping the structures of compounds known to modulate cellular phenotypes (mostly natural products or orphan compounds) unto chemogenomics databases containing biologically profiled compounds with known targets [84].
In that regard, cheminformatics and bioinformatics have developed mapping models including but not limited to topological-based models, Bayesian classification models and atom pair-based models from available bioactivity data using machine learning and statistical methods [78]. These models are implemented in mapping compounds into the chemogenomical space or bioactivity database considering either 2D or 3D molecular descriptors [65, 92, 93] and chemical fingerprints [94, 95] for measuring similarity among structures to predict targets. An advantage of using chemical fingerprints in designing models is that it enables back-projections of correlation between characterized proteins and compounds unto orphan compounds with the knowledge that similar compound structures would exhibit similar affinity chemical fingerprints [84]. Molecular descriptors are numerical features extracted from the compounds based on their molecular properties [65, 92] whereas chemical fingerprints are high-dimensional vectors that encode the presence of sub-structural fragments [94, 95].
Ligand-based approaches to target prediction provide the platform to understanding the relationships between structurally dissimilar but functionally related proteins based on their ligand similarity, thus helping to form a hypothesis that can be verified using statistical methods. Similar to biospectra, the ligand-based approach is more informative for pharmacology, medicinal chemistry and biochemistry [78, 96]. Similarity among structures is measured using Minkowski distance metrics and the Tanimoto similarity coefficient and its complement, the Soergel distance [65]. The Tanimoto similarity coefficient can be applied to 3D structures [39]; however, this metric is susceptible to molecular size because it fails to account irrelevant features of a large molecule, thus resulting into odd size dependencies [65]. These measurable features of compounds have been implemented in developing tools such as Similarity Ensemble Approach (SEA) [78], Swiss Target Prediction (STP) [97], SpiDER [98], SuperPred [99], Polypharmacology Browser [100], HitPick [101], Prediction of Activity Spectra for Biologically Active Substance (PASS) [102], MOst-Similar ligand-based Target inference approach (MOST) [103], Candidate Ligand Identification Program (CLIP) [104] and Chemical Similarity Network Analysis Pulldown (CSNAP) [105]. These tools are described in Supplementary Table 4 which integrates fingerprints and/or structural similarity to predict ranked targets from ligand–target datasets in order of decreasing similarity score.
The ligand-based target prediction approach is not feasible in the cases of predicting targets with no or only a small number of bioactive ligands and ligands that exhibit activity cliffs characterized by high structural similarity but different activity [13, 106].
Derived or hybrid approaches
The derived approaches can be classified into two sub-groups: (a) target-based in silico prediction and (b) genomic analysis approach (see Supplementary File, Section 5).
The target-based approach involves the use of 3D structures of essential protein targets to identify potential ligands or drug-like compounds by investigating protein–ligand complex interactions and conformations. Genomic analysis involves the analysis of genetic data to explore their associations to specific phenotypes. This approach presents the ability to investigate the effect of genetic variants on a disease and functionally validate drug-targetable genes using genomics particularly in precision medicine.
Comparing different approaches in computational drug discovery
As described previously, several computational drug discovery approaches have been suggested, including genomic, biospectra, network-based, machine learning/data mining and virtual screening/molecular docking simulation approaches. Although these methods individually have their specific areas in drug discovery that best describes their usefulness, they have the ability to be integrated to understand complex biological systems in order to address challenges in computational drug discovery. This is because technological advancement has led to the generation of various dataset types describing biological systems from different dimensions, some of which are sequencing, gene expression activity and proteomics [107].
In predicting and assessing the pharmacological effects of a drug, the combination of these techniques has been instrumental in determining drug target interactions (DTIs) with high efficiency and low cost. In comparison to experimental techniques (in vitro and in vivo methods), computational methods have provided the technicalities to systematically determine all possible interactions to clearly elucidate the pharmacological patterns [108]. Higher-dimensional levels of predictions revolve around systematic analysis of biological complex networks and large integrated biomedical datasets, and as such, using a combinatorial approach is highly essential. Some approaches share similar concepts but applied in different forms in addressing similar issues; thus, combination helps to compensate for individual limitations. This in turn increases the accuracy of predicting and minimizing possible adverse effects [109]. For instance, molecular docking principles in elucidating DTIs require 3D structures. Due to that, there are associated biases and false positives when high-quality 3D structures are not available [108]. Unlike molecular docking approaches, ML drug target interaction-predictive models have the extended capacity of taking into consideration not only the 3D structures of targets but also molecular and protein sequence descriptors [108]. However, network-based methods in predicting DTIs to investigate pharmacological effects apply recommendation algorithms implemented in recommender systems [110] and link prediction algorithms [111] rather than 3D structures and molecular systems. Also, network-based methods are relatively faster compared with the other methods. This is because the DTI network of interest can be represented as a matrix on which calculations can be computed easily [108]. Aside that, they have the extended capacity of predicting drug effects through simple dynamic processes such as random walk, resource diffusion and collaborative filtering on biological networks [108].
For example, Paolini and colleagues studied the polypharmacology interaction network for human proteins by constructing the ligand-target matrix using a Laplacian-modified Bayesian probabilistic model to explore the relationships between chemical structure and targets by integrating diverse structure activity relationship data [79]. They observed 35% of 276,122 active compounds within their database to hit more than one target while 65% hit a target, thus indicating extensive promiscuity of drugs and leads across targets.
Data mining is highly essential in chemogenomics to mine chemogenomic datasets. This is critical in establishing the relationship between a set of potential drug targets and ligands. However, the interplay among a holistic picture of the biological system (network), molecular docking approaches and ML methods in chemogenomics presents a broader scope to investigate the effects of compounds on gene/protein expression. To efficiently identify and assess the effects of specific protein targets on specific drugs, robust molecular docking systems that implement ML and DM models have been developed to optimize the performance of predicting the drug’s effect across molecular networks [112]. These models provide the platform to avoiding unnecessary assumptions by specifically accounting for binding effects most often challenging to model without ML and DM techniques. Utilization of this approach in designing scoring functions has significantly enhanced the accuracy of establishing binding affinities of various protein-ligand complexes [113]. Ballester and colleagues developed a competitive high-performance scoring function that implements Random Forest to capture binding effects [113]. The flexibility of their scoring function compared with other rigid functions ensured that it has high predictive power when tested on trained datasets. Another application of this approach is developing machine learning-based scoring and binding affinity functions integrated with molecular docking tools to address difficulties involved in molecular docking. Hsin et al. [109] developed a computational screening approach using machine learning and docking packages to investigate the polypharmacological nature of compounds against potential targets within a biological network. The model developed can assess binding modes and predict the best binding mode to targets. This approach increases the reliability and confidence in assessing the binding conformations of compounds and predicting best modes [112]. It also helps to rate the performance of various docking packages as well as compensate for scoring function-associated errors [141, 115]. Advanced ML methods provide the technique to investigate drug effects in preclinical research and clinical trials. Also, they provide an efficient way to systematically and analytically extract meaningful biological information from clinical trial datasets. This therefore facilitates the ability to design the chemical structure of drugs to modulate drug target interactions. However, it is of importance in that the ability to interpret such datasets is challenging and as such requires experience and high technical skills.
Deep learning, a class of machine learning, has strong generalization ability and feature extraction capability. It has emerged as a powerful tool capable of identifying highly complex patterns in both homogeneous and heterogeneous datasets. In computational drug discovery, the deep learning method has enhanced prediction of bioactivity, de novo molecular design, virtual screening, activity scoring and synthesis prediction [42, 51, 114]. This is mainly because it has fewer generalization errors, thus yielding impressive results as compared with traditional machine learning. It has been extensively applied in functional genomics in discovering DNA-binding motifs and determining sequence specificity of DNA and RNA-binding proteins [115].
Data mining and machine learning models are implemented in computational drug discovery for unbiased mining and analysis of genetic datasets mostly in the focus of personalized medicine [116]. The aim of personalized medicine is to discover novel drugs and biomarkers for specific patient groups, most suffering from complex disorders. Developments in this field are applied most often in gene and immuno-oncology therapies for highly personalized and specific group treatments, respectively [117]. In view of that, the genomic approach together with ML methods provides the platform for identifying disease-associated genes (particularly rare disease variants) and their corresponding mutations from methods like DNA sequencing and genome-wide association studies [117]. This helps to translate functional results into treatment and strategic measures. Analyzing genetic functional networks with ML and DM methods enhances the chances of identifying novel biomarkers and drug targets. For example, the combination of a network-based approach and ML methods plays an increasingly significant role to predict novel mechanisms underlying disease-specific targetable genes or pathway associations. This in turn offers the opportunity for finding new applications for drugs as well as predicting potential adverse effects. Bari and colleagues developed a machine learning-assisted network inference algorithm capable of identifying class II cancer-associated genes in a cancer network generated from support vector machine models [118]. Also, this combination has been implemented in target fishing using chemical fingerprints [119].
Integration of ML and DM approaches together with network-based techniques is of noteworthy importance in analyzing biological networks to identify a potential set of genes or pathways that could serve as targets in combinatorial therapy. The rationale behind this combination strategy is not only to overcome resistance and limitations of monotherapy regimens but also to overcome the complexities of complex diseases such as cancer and HIV [120, 121]. In that regard, predictive models based on ML, DM, network-based and sometimes molecular docking approaches have been developed to investigate the synergistic effects of drug-like molecules on specified targets [122, 123]. These models incorporate heterogeneous datasets such as cell signaling pathway and transcriptomic and pharmacological datasets [123]. The models have the extended ability of providing insights into biological mechanisms underlying the synergistic combination.
The combination of the genomics approach with molecular docking simulations is mostly applied in discovering novel ligands or drug-like molecules to treat infectious diseases.
Computational assessment measures
Assessment measures from computational approaches in drug discovery field evaluate the performance of the model or algorithm used for prediction. These measures are very critical to ascertaining the model’s reliability, predictive power and ability to differentiate between positive and negative sets and exploring their relationships [128]. These include, but not limited to, precision, recall, classification accuracy, sensitivity, specificity and area under the curve (AUC) related to receiver operating characteristic (ROC) analysis of models or algorithms particularly in relation to DM and ML. Most of these measures, including those listed above, require the ground-truth sets of positives and negatives, which may be a problem [129] as these sets, especially the set of negatives, are not always available. Thus, models and algorithms which may produce the log-likelihood of the data, the Akaike or Bayesian ‘An Information Criterion’ (AIC or BIC) or the so-called Schwarz’s Bayesian criterion (SBC) can be used to assess model or algorithm performance. AIC, BIC and SBC are based on likelihood function, scoring how well the model or algorithm explains data while penalizing the number of estimated parameters.
Source of drug failure: challenges and opportunities
Incomplete knowledge on the biological mechanisms underlying certain diseases
A critical drawback in the success story of drug discovery is associated with poor understanding of the underlying mechanisms behind some diseases such as nervous system disorders, chronic kidney disease, idiopathic pulmonary fibrosis and other complex disorders [124]. Inability to elucidate genetic variants or biomarkers or pathways or proteins involved in the etiology of such diseases continues to be a challenge to drug research. Due to that, specific targeted drugs or vaccines have not yet been developed. Researchers have shown that in-depth knowledge of disease mechanisms and the elucidation of critical biomarkers would contribute significantly to drug development [124]. This could be associated with inadequate specific datasets available to help unravel the mystery behind a disorder. Due to that, there is an intensified scientific research into bridging the gap between disease mechanisms and drug development. Combination of omics, chemistry and clinical datasets together with advance ML techniques has been promising in exploring potential targets. A typical example is Alzheimer’s disease, in which various mechanisms are been identified through extensive research [125].
Drug resistance development
Drug resistance has been a major burden in drug use. More often, drugs, particularly, those targeting disease-causing pathogens in infectious diseases, lose potency with time primarily as a result of selective pressures resulting in drug-resistant strain development. This challenge contributes to disease resurgence and increased morbidity and mortality rates. In complex diseases, drugs targeting human cells develop resistance through factors like epigenetics, DNA damage repair and epithelial mesenchymal transition [126]. In general, drug efflux and drug inactivation are common factors linked to drug resistance. This phenomenon continuously necessitates further research and alternative treatment development. In addition, researchers are investigating the core biological associated activities resulting in resistance to identifying novel approaches to counter such effects.
Inability to reproduce generated disease-related datasets
Data reproducibility crisis remains a critical challenge in the post-genomic era. Data validation is a measure of the confidence and integrity of the datasets. It is of noteworthy that inconsistencies in results obtained from replicating experiments in different laboratories breed unsuccessful translation of discovery research because of the level of mistrust in the data [124]. This situation significantly slows the rate of translating biological data into functional knowledge and treatment interventions. However, it is argued that such differences in results could be attributed to confidence interval defined for the independent study as well as inadequate knowledge in essential statistical methods and tools used. Researchers have proposed that external validation and explicit reporting of experimental datasets could possibly increase reproducibility [124]. Also, this challenge presents the opportunity for researchers to develop standardized procedures tailored to each working environment to ensure reproducibility of results and continuity of scientific knowledge.
Complex unpredicted metabolism networks
Unpredicted interactions and mechanisms within a network due to associated kinetic interactions result in an incomplete picture of the cellular behavior [127]. However, overassumptions in modeling hinder the ability to develop accurate models to answer the biological hypothesis. As a result, algorithms developed for such models produce results that deviate from the true expectations. This therefore presents a challenge in modeling the system to overcome unknown associated metabolic fluxes. As such, there is a higher likelihood of missing essential information such as pathways and biological activities essential for drug research. Also, there are off-target metabolic interactions that models fail to account for due to modeling challenges. Off-target metabolic interactions can be responsible for expected and unexpected responses which most of the time are side effects. In overcoming these challenges and minimizing drug failures and associated adverse effects, predictive models for individual target networks that simultaneously detect metabolic similarity of associated metabolic pathways using joint learning algorithms can be implemented to investigate latent interactions.
Conclusion and Perspectives
In this review, we have presented various computational approaches and tools essential for in silico extraction of drug targets, predicting potential drug-like candidates, analyzing bioactivity profile and elucidating possible off-target effects in drug discovery. These approaches complement experimental techniques in drug development. Furthermore, we highlighted on the application of machine learning, data mining, genomics and network analysis techniques in investigating the dynamic patterns within integrated datasets from multiple sources to predict critical nodes, pathways and biological processes. These techniques are relevant in achieving a global perspective of the biological systems to investigate the interplay between multiple independent genes or proteins on disease etiology. This therefore provides the platform to elucidating a set of functional biological entities for drug and vaccine development. We discussed various molecular docking simulation techniques. We showed the specificity of each approach in terms of predicting potential drug-like molecules and protein targets in drug development. We emphasized on the application of these methods in drug repurposing and reuse particularly in addressing drug resistance and drug development for orphan diseases, thus contributing to limiting the risk of drug failure during trials. Also, we highlighted on the combination of machine learning and molecular docking techniques in designing various predictive models to investigate the structural and chemical properties of ligands or drug molecules and validate their efficacy in drug development. We have shown that these approaches can be combined to compensate for limitations of individual methods thereby increasing the predictive power. Finally, we presented sources of drug failure looking at the challenges and opportunities involved.
Due to the specificity of individual techniques, we recommend the use of multiple approaches, particularly integrative approaches in stages of computational drug research to compare and validate results prior to further experimental test, thus avoiding false positives and irreproducibility. Also, multiple performance assessment measures must be performed in validating results. We suggest the development of a comprehensive tool that produces systematic results based on individual approaches that give the user the opportunity to performance meta-analysis of results.
Supplementary Material
Francis E. Agamah is a master’s student at the University of Cape Town (UCT) Division of Human Genetics, Department of Pathology. Email: agmfra001@myuct.ac.za / francisagamahh@gmail.com.
Gaston K. Mazandu received his PhD in Bioinformatics at UCT. He holds a senior lecturer position at the Division of Human Genetics, Department of Pathology, and he is an honorary senior member of the Computational Biology Division at UCT and an Associate Researcher at the African Institute for Mathematical Sciences (AIMS) in South Africa. Email: gmazandu@gmail.com.
Radia Hassan is a PhD student at the University of Cape Town in The Division of Human Genetics, Department of Pathology. Email: ISMRAD001@myuct.ac.za
Christian D. Bope obtained his PhD in Biophysics at Nanyang Technological University, Singapore. He is currently a postdoctoral fellow at the Division of Human Genetics, Department of Pathology, UCT. Email: christian.bope@uct.ac.za
Nicholas E. Thomford obtained his PhD in Human Genetics from UCT. His research includes Pharmacogenomics and drug discovery. He is a researcher at UCT and a lecturer at UCCSMS. Email: nthomford@ucc.edu.gh
Anita Ghansah obtained her PhD in Genetic Epidemiology at the London School of Hygiene and Tropical Medicine. She is a Senior Researcher at Noguchi Memorial Institute for Medical Research, University of Ghana. Email: aghansah@noguchi.ug.edu.gh
Emile R. Chimusa received his PhD in Bioinformatics from University of Cape Town, South Africa. He is currently a Senior Lecturer/Researcher at the Division of Human Genetics, Department of Pathology, UCT. Email: emile.chimusa@uct.ac.za.
Acknowledgements
We acknowledge the staffs and colleagues from the Division of Human Genetics, University of Cape Town, and the Centre for High Performance Computing (CHPC), Cape Town, South Africa (www.chpc.ac.za/).
Funding
This work was supported through the DELTAS Africa Initiative [DELGEME grant 107740/Z/15/Z]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [DELGEME grant 107740/Z/15/Z] and the UK government. Some of the authors are supported in part by the National Institutes of Health (NIH) Common Fund under grant number U24HG006941 (H3ABioNet), NRF (RA171111285157/119056) and 1U01HG007459-01 (SADaCC). The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
References
- 1. Zhong F, Xing J, Liu X, et al. Artificial intelligence in drug design. Sci. China Life Sci 2018;1–14. [DOI] [PubMed] [Google Scholar]
- 2. Weilin Z, Jianfeng P, Luhua L. Computational multitarget drug design. J Chem Inf Model 2017;57(3):403–12. [DOI] [PubMed] [Google Scholar]
- 3. Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br J Pharmacol 2007;152:21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kharkar PS, Warrier S, Gaud RS. Reverse docking: a powerful tool for drug repositioning and drug rescue. Future Med Chem 2014;6:333–42. [DOI] [PubMed] [Google Scholar]
- 5. Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 2004;3:711. [DOI] [PubMed] [Google Scholar]
- 6. Yang Y, Adelstein SJ, Kassis AI. Target discovery from data mining approaches. Drug Discov Today 2012;17:S16–23. [DOI] [PubMed] [Google Scholar]
- 7. Dopazo J. Genomics and transcriptomics in drug discovery. Drug Discov Today 2014;19:126–32. [DOI] [PubMed] [Google Scholar]
- 8. Morphy R. Selectively nonselective kinase inhibition: striking the right balance. J Med Chem 2009;53:1413–37. [DOI] [PubMed] [Google Scholar]
- 9. Zhang X, Crespo A, Fernández A. Turning promiscuous kinase inhibitors into safer drugs. Trends Biotechnol 2008;26:295–301. [DOI] [PubMed] [Google Scholar]
- 10. Raman K, Yeturu K, Chandra N. targettb: a target identification pipeline for mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst Biol 2008;2:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mazandu GK, Chimusa ER, Rutherford K, et al. Large-scale data-driven integrative framework for extracting essential targets and processes from disease-associated gene data sets. Brief Bioinform 2017;19:1141–52. [DOI] [PubMed] [Google Scholar]
- 12. Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov 2004;3:353. [DOI] [PubMed] [Google Scholar]
- 13. Koutsoukas A, Simms B, Kirchmair J, et al. From in silico target prediction to multi-target drug design: current databases, methods and applications. J Proteomics 2011;74:2554–74. [DOI] [PubMed] [Google Scholar]
- 14. Keiser MJ, Setola V, Irwin JJ, et al. Predicting new molecular targets for known drugs. Nature 2009;462:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cameron RT, Coleman RG, Day JP, et al. Chemical informatics uncovers a new role for moexipril as a novel inhibitor of camp phosphodiesterase-4 (pde4). Biochem Pharmacol 2013;85:1297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lounkine E, Keiser MJ, Whitebread S, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012;486:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu L, Ai N, Liu Y, et al. Relating anatomical therapeutic indications by the ensemble similarity of drug sets. J Chem Inf Model 2013;53:2154–60. [DOI] [PubMed] [Google Scholar]
- 18. Mogire RM, Akala HM, Macharia RW, et al. Target-similarity search using plasmodium falciparum proteome identifies approved drugs with anti-malarial activity and their possible targets. PloS One 2017;12:e0186364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hopkins AL, Mason JS, Overington JP. Can we rationally design promiscuous drugs? Curr Opin Struct Biol 2006;16:127–36. [DOI] [PubMed] [Google Scholar]
- 20. Chen B, Butte A. Leveraging big data to transform target selection and drug discovery. Clin Pharmacol Ther 2016;99:285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hossain T, Kamruzzaman M, Choudhury TZ, et al. Application of the subtractive genomics and molecular docking analysis for the identification of novel putative drug targets against salmonella enterica subsp. enterica serovar poona. Biomed Res Int 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim YA, Wuchty S, Przytycka TM. Identifying causal genes and dysregulated pathways in complex diseases. PLoS Comput Biol 2011;7:e1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. H Andrade C, C Silva D, C Braga R. In silico prediction of drug metabolism by p450. Curr Drug Metab 2014;15:514–25. [DOI] [PubMed] [Google Scholar]
- 24. Arimoto R. Computational models for predicting interactions with cytochrome p450 enzyme. Curr Top Med Chem 2006;6:1609–18. [DOI] [PubMed] [Google Scholar]
- 25. Katsila T, Spyroulias GA, Patrinos GP, et al. Computational approaches in target identification and drug discovery. Comput Struct Biotechnol J 2016;14:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gobbi G, Janiri L. Clozapine blocks dopamine, 5-ht2 and 5-ht3 responses in the medial prefrontal cortex: an in vivo microiontophoretic study. Eur Neuropsychopharmacol 1999;10:43–9. [DOI] [PubMed] [Google Scholar]
- 27. Ashley EA, White NJ. Artemisinin-based combinations. Curr Opin Infect Dis 2005;18:531–6. [DOI] [PubMed] [Google Scholar]
- 28. Huthmacher C, Hoppe A, Bulik S, et al. Antimalarial drug targets in plasmodium falciparum predicted by stage-specific metabolic network analysis. BMC Syst Biol 2010;4:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Luo Y, Zhao X, Zhou J, et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat Commun 2017;8:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harrold J, Ramanathan M, Mager D. Network-based approaches in drug discovery and early development. Clin Pharmacol Ther 2013;94:651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee E, Chuang HY, Kim JW, et al. Inferring pathway activity toward precise disease classification. PLoS Computational Biol 2008;4:e1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chuang HY, Lee E, Liu YT, et al. Network-based classification of breast cancer metastasis. Mol Syst Biol 2007;3:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Köhler S, Bauer S, Horn D, et al. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet 2008;82:949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu X, Jiang R, Zhang MQ, et al. Network-based global inference of human disease genes. Mol Syst Biol 2008;4:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fatumo S, Adebiyi E, Schramm G, et al. An in silico approach to detect efficient malaria drug targets to combat the malaria resistance problem. In: 2009 International Association of Computer Science and Information Technology-Spring Conference. IEEE, 2009, 576–80. [Google Scholar]
- 36. Rout S, Patra NP, Mahapatra RK. An in silico strategy for identification of novel drug targets against plasmodium falciparum. Parasitol Res 2017;116:2539–59. [DOI] [PubMed] [Google Scholar]
- 37. Wang X, Wei X, Thijssen B, et al. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat Biotechnol 2012;30:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cava C, Bertoli G, Castiglioni I. In silico identification of drug target pathways in breast cancer subtypes using pathway cross-talk inhibition. J Transl Med 2018;16:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lo YC, Rensi SE, Torng W, et al. Machine learning in chemoinformatics and drug discovery. Drug Discov Today 2018;23:1538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen R, Liu X, Jin S, et al. Machine learning for drug-target interaction prediction. Molecules 2018;23:2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weaver DC. Applying data mining techniques to library design, lead generation and lead optimization. Curr Opin Chem Biol 2004;8:264–70. [DOI] [PubMed] [Google Scholar]
- 42. Burbidge R, Trotter M, Buxton B, et al. Drug design by machine learning: support vector machines for pharmaceutical data analysis. Comput Chem 2001;26:5–14. [DOI] [PubMed] [Google Scholar]
- 43. Nasrabadi NM. Pattern recognition and machine learning. J Electron Imaging 2007;16: 049901. [Google Scholar]
- 44. Flach PA, Lachiche N. Naive Bayesian classification of structured data. Mach Learn 2004;57:233–69. [Google Scholar]
- 45. Mazandu GK, Opap K, Mulder NJ. Contribution of microarray data to the advancement of knowledge on the mycobacterium tuberculosis interactome: use of the random partial least squares approach. Infect Genet Evol 2011;11:725–33. [DOI] [PubMed] [Google Scholar]
- 46. Sturm M, Hackenberg M, Langenberger D, et al. Targetspy: a supervised machine learning approach for microrna target prediction. BMC Bioinformatics 2010;11:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nidhi a GM, Davies JW, et al. Prediction of biological targets for compounds using multiple-category bayesian models trained on chemogenomics databases. J Chem Inf Model 2006;46:1124–33. [DOI] [PubMed] [Google Scholar]
- 48. Azencott CA, Ksikes A, Swamidass SJ, et al. One-to four-dimensional kernels for virtual screening and the prediction of physical, chemical, and biological properties. J Chem Inf Model 2007;47:965–74. [DOI] [PubMed] [Google Scholar]
- 49. Golbraikh A, Wang XS, Zhu H, et al. Predictive QSAR modeling: methods and applications in drug discovery and chemical risk assessment. Handb Comput Chem 2016;1–48. [Google Scholar]
- 50. Lavecchia A. Machine-learning approaches in drug discovery: methods and applications. Drug Discov Today 2015;20:318–31. [DOI] [PubMed] [Google Scholar]
- 51. Panteleev J, Gao H, Jia L. Recent applications of machine learning in medicinal chemistry. Bioorg Med Chem Lett 2018. [DOI] [PubMed] [Google Scholar]
- 52. Magariños MP, Carmona SJ, Crowther GJ, et al. TDR Targets: a chemogenomics resource for neglected diseases. Nucleic Acids Res 2011;40:D1118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen X, Ji ZL, Chen YZ. Ttd: therapeutic target database. Nucleic Acids Res 2002;30:412–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuhn M, von Mering C, Campillos M, et al. Stitch: interaction networks of chemicals and proteins. Nucleic Acids Res 2007;36:D684–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Menden MP, Iorio F, Garnett M, et al. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS One 2013;8:e61318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nigsch F, Bender A, Jenkins JL, et al. Ligand-target prediction using winnow and naive bayesian algorithms and the implications of overall performance statistics. J Chem Inf Model 2008;48:2313–25. [DOI] [PubMed] [Google Scholar]
- 57. Awale M, Reymond JL. The polypharmacology browser ppb2: target prediction combining nearest neighbors with machine learning. J Chem Inf Model 2018;59:10–7. [DOI] [PubMed] [Google Scholar]
- 58. Lee M, Kim D. Large-scale reverse docking profiles and their applications. BMC Bioinformatics. BioMed Central 2012;13:S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sarnpitak P, Mujumdar P, Taylor P, et al. Panel docking of smallmolecule libraries—prospects to improve efficiency of lead compound discovery. Biotechnol Adv 2015;33:941–7. [DOI] [PubMed] [Google Scholar]
- 60. Li H, Gao Z, Kang L, et al. Tarfisdock: a web server for identifying drug targets with docking approach. Nucleic Acids Res 2006;34:W219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Warren GL, Andrews CW, Capelli AM, et al. A critical assessment of docking programs and scoring functions. J Med Chem 2006;49:5912–31.17004707 [Google Scholar]
- 62. Lee A, Lee K, Kim D. Using reverse docking for target identification and its applications for drug discovery. Expert Opin Drug Discov 2016;11:707–15. [DOI] [PubMed] [Google Scholar]
- 63. Chen Y, Zhi D. Ligand–protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins 2001;43:217–26. [DOI] [PubMed] [Google Scholar]
- 64. Cai J, Han C, Hu T, et al. Peptide deformylase is a potential target for anti-helicobacter pylori drugs: reverse docking, enzymatic assay, and x-ray crystallography validation. Protein Sci 2006;15:2071–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Byrne R, Schneider G. In silico target prediction for small molecules. Sys Chem Biol, 2019; Springer. pp. 273–309. [DOI] [PubMed] [Google Scholar]
- 66. Koonin EV, Wolf YI, Karev GP. The structure of the protein universe and genome evolution. Nature 2002;420:218. [DOI] [PubMed] [Google Scholar]
- 67. Wang JC, Chu PY, Chen CM, et al. idtarget: a web server for identifying protein targets of small chemical molecules with robust scoring functions and a divide-and-conquer docking approach. Nucleic Acids Res 2012;40:W393–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Trott O, Olson AJ. Autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ma C, Tang K, Liu Q, et al. Calmodulin as a potential target by which berberine induces cell cycle arrest in human hepatoma Bel7402 cells. Chem Biol Drug Des 2013;81:775–83. [DOI] [PubMed] [Google Scholar]
- 70. Scafuri B, Marabotti A, Carbone V, et al. A theoretical study on predicted protein targets of apple polyphenols and possible mechanisms of chemoprevention in colorectal cancer. Sci Rep 2016;6: 32516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang JC, Lin JH, Chen CM, et al. Robust scoring functions for protein–ligand interactions with quantum chemical charge models. J Chem Inf Model 2011;51:2528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chang DTH, Lin JH, Hsieh CH, et al. On the design of optimization algorithms for prediction of molecular interactions. In: 2009 Ninth IEEE International Conference on Bioinformatics and BioEngineering. IEEE, 2009, 208–15. [Google Scholar]
- 73. Lauro G, Romano A, Riccio R, et al. Inverse virtual screening of antitumor targets: pilot study on a small database of natural bioactive compounds. J Nat Prod 2011;74:1401–7. [DOI] [PubMed] [Google Scholar]
- 74. Lauro G, Masullo M, Piacente S, et al. Inverse virtual screening allows the discovery of the biological activity of natural compounds. Bioorg Med Chem 2012;20:3596–602. [DOI] [PubMed] [Google Scholar]
- 75. Wermuth CG. Selective optimization of side activities: the SOSA approach. Drug Discov Today 2006;11:160–4. [DOI] [PubMed] [Google Scholar]
- 76. Wang W, Zhou X, He W, et al. The interprotein scoring noises in glide docking scores. Proteins 2012;80:169–83. [DOI] [PubMed] [Google Scholar]
- 77. Yuriev E, Holien J, Ramsland PA. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J Mol Recognit 2015;28:581–604. [DOI] [PubMed] [Google Scholar]
- 78. Wang Z, Liang L, Yin Z, et al. Improving chemical similarity ensemble approach in target prediction. J Cheminform 2016;8:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Paolini GV, Shapland RH, van Hoorn WP, et al. Global mapping of pharmacological space. Nat Biotechnol 2006;24:805. [DOI] [PubMed] [Google Scholar]
- 80. Keiser MJ, Roth BL, Armbruster BN, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol 2007;25:197. [DOI] [PubMed] [Google Scholar]
- 81. Morphy R, Kay C, Rankovic Z. From magic bullets to designed multiple ligands. Drug Discov Today 2004;9:641–51. [DOI] [PubMed] [Google Scholar]
- 82. Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol 2008;4:682. [DOI] [PubMed] [Google Scholar]
- 83. Fliri AF, Loging WT, Thadeio PF, et al. Biospectra analysis: model proteome characterizations for linking molecular structure and biological response. J Med Chem 2005;48:6918–25. [DOI] [PubMed] [Google Scholar]
- 84. Jenkins JL, Bender A, Davies JW. In silico target fishing: predicting biological targets from chemical structure. Drug Discov Today Technol 2006;3:413–21. [Google Scholar]
- 85. Fliri AF, Loging WT, Thadeio PF, et al. Biological spectra analysis: linking biological activity profiles to molecular structure. Proc Natl Acad Sci U S A 2005;102:261–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Godden JW, Xue L, Bajorath J. Combinatorial preferences affect molecular similarity/diversity calculations using binary fingerprints and Tanimoto coefficients. J Chem Inf Comput Sci 2000;40:163–6. [DOI] [PubMed] [Google Scholar]
- 87. Downs GM, Willett P, Fisanick W. Similarity searching and clustering of chemical structure databases using molecular property data. J Chem Inf Comput Sci 1994;34:1094–102. [Google Scholar]
- 88. Breu H, Gil J, Kirkpatrick D, et al. Linear time Euclidean distance transform algorithms. IEEE Trans Pattern Anal Mach Intell 1995;17:529–33. [Google Scholar]
- 89. de Souza RM, De CFA. Clustering of interval data based on city–block distances. Pattern Recognit Lett 2004;25:353–65. [Google Scholar]
- 90. Campillos M, Kuhn M, Gavin AC, et al. Drug target identification using side-effect similarity. Science 2008;321:263–6. [DOI] [PubMed] [Google Scholar]
- 91. Bender A, Glen RC. Molecular similarity: a key technique in molecular informatics. Org Biomol Chem 2004;2:3204–18. [DOI] [PubMed] [Google Scholar]
- 92. Khan AU. Danishuddin. Descriptors and their selection methods in qsar analysis: paradigm for drug design. Drug Discov Today 2016;21:1291–302. [DOI] [PubMed] [Google Scholar]
- 93. Nettles JH, Jenkins JL, Bender A, et al. Bridging chemical and biological space:“target fishing” using 2d and 3d molecular descriptors. J Med Chem 2006;49:6802–10. [DOI] [PubMed] [Google Scholar]
- 94. Hert J, sWillett P, Wilton DJ, et al. Comparison of topological descriptors for similarity-based virtual screening using multiple bioactive reference structures. Org Biomol Chem 2004;2:3256–66. [DOI] [PubMed] [Google Scholar]
- 95. Raymond JW, Willett P. Effectiveness of graph-based and fingerprint-based similarity measures for virtual screening of 2d chemical structure databases. J Comput Aided Mol Des 2002;16:59–71. [DOI] [PubMed] [Google Scholar]
- 96. Hert J, Keiser MJ, Irwin JJ, et al. Quantifying the relationships among drug classes. J Chem Inf Model 2008;48:755–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gfeller D, Grosdidier A, Wirth M, et al. Swisstargetprediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res 2014;42:W32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Reker D, Rodrigues T, Schneider P, et al. Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc Natl Acad Sci U S A 2014; 201320001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dunkel M, Günther S, Ahmed J, et al. Superpred: drug classification and target prediction. Nucleic Acids Res 2008;36:W55–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Awale M, Reymond JL. The polypharmacology browser: a web-based multi-fingerprint target prediction tool using chembl bioactivity data. J Cheminform 2017;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Liu X, Vogt I, Haque T, et al. Hitpick: a web server for hit identification and target prediction of chemical screenings. Bioinformatics 2013;29:1910–2. [DOI] [PubMed] [Google Scholar]
- 102. Lagunin A, Stepanchikova A, Filimonov D, et al. Pass: prediction of activity spectra for biologically active substances. Bioinformatics 2000;16:747–8. [DOI] [PubMed] [Google Scholar]
- 103. Huang T, Mi H, Cy L, et al. Most: most-similar ligand based approach to target prediction. BMC Bioinform 2017;18:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Rhodes N, Willett P, Calvet A, et al. Clip: similarity searching of 3d databases using clique detection. J Chem Inf Comput Sci 2003;43:443–8. [DOI] [PubMed] [Google Scholar]
- 105. Lo YC, Senese S, Li CM, et al. Large-scale chemical similarity networks for target profiling of compounds identified in cell-based chemical screens. PLoS Comput Biol 2015;11:e1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cruz-Monteagudo M, Medina-Franco JL, Perez-Castillo Y, et al. Activity cliffs in drug discovery: Dr jekyll or mr hyde? Drug Discov Today 2014;19:1069–80. [DOI] [PubMed] [Google Scholar]
- 107. Zitnik M, Nguyen F, Wang B, et al. Machine learning for integrating data in biology and medicine: Principles, practice, and opportunities. Inf Fusion 2019;50:71–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wu Z, Li W, Liu G, et al. Network-based methods for prediction of drug-target interactions. Front Pharmacol 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hsin KY, Kitano H, Matsuoka Y, et al. Application of machine learning approaches in drug target identification and network pharmacology, International Conference on Intelligent Informatics and Biomedical Sciences (ICIIBMS). IEEE, 2015, 219–9. [Google Scholar]
- 110. Lu L, Medo M, Yeung CH, et al. Recommender systems. In: 2012;CoRR abs/1202, 1112.
- 111. Zhou T. Link prediction in complex networks: a survey. Physica A 2011;390:1150–70. [Google Scholar]
- 112. Hsin KY, Ghosh S, Kitano H. Combining machine learning systems and multiple docking simulation packages to improve docking prediction reliability for network pharmacology. PloS One 2013;8:e83922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ballester PJ, Mitchell JB. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics 2010;26:1169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Chen H, Engkvist O, Wang Y, et al. The rise of deep learning in drug discovery. Drug Discov Today 2018;23:1241–50. [DOI] [PubMed] [Google Scholar]
- 115. Zou J, Huss M, Abid A, et al. A primer on deep learning in genomics. Nat Genet 2018;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Pan Y, Zhang Y, Liu J. Text mining-based drug discovery in cutaneous squamous cell carcinoma. Oncol Rep 2018;40:3830–42. [DOI] [PubMed] [Google Scholar]
- 117. Cardon LR, Harris T. Precision medicine, genomics and drug discovery. Hum Mol Genet 2016;25:R166–72. [DOI] [PubMed] [Google Scholar]
- 118. Bari MG, Ung CY, Zhang C, et al. Machine learning-assisted network inference approach to identify a new class of genes that coordinate the functionality of cancer networks. Sci Rep 2017;7:6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wale N, Karypis G. Target fishing for chemical compounds using target-ligand activity data and ranking based methods. J Chem Inf Model 2009;49:2190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Madani Tonekaboni SA, Soltan Ghoraie L, Manem VSK, et al. Predictive approaches for drug combination discovery in cancer. Brief Bioinform 2016;19:263–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Csermely P, Agoston V, Pongor S. The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci 2005;26:178–82. [DOI] [PubMed] [Google Scholar]
- 122. Li P, Huang C, Fu Y, et al. Large-scale exploration and analysis of drug combinations. Bioinformatics 2015;31:2007–16. [DOI] [PubMed] [Google Scholar]
- 123. Zhao XM, Iskar M, Zeller G, et al. Prediction of drug combinations by integrating molecular and pharmacological data. PLoS Comput Biol 2011;7:e1002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Altevogt BM, Davis M, Pankevich DE, et al. Improving and accelerating therapeutic development for nervous system disorders: workshop summary. National Academies Press, 2014. [PubMed] [Google Scholar]
- 125. Magalingam KB, Radhakrishnan A, Ping NS, et al. Current concepts of neurodegenerative mechanisms in Alzheimer’s disease. Biomed Res Int 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Housman G, Byler S, Heerboth S, et al. Drug resistance in cancer: an overview. Cancers 2014;6:1769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Vasilakou E, Machado D, Theorell A, et al. Current state and challenges for dynamic metabolic modeling. Current Opin Microbiol 2016;33:97–104. [DOI] [PubMed] [Google Scholar]
- 128. Flach P. Performance evaluation in machine learning: the good, the bad, the ugly and the way forward. In: 33rd AAAI Conference on Artificial Intelligence, 2019.
- 129. Mazandu GK, Geza E, Seuneu M, et al. Orienting future trends in local ancestry deconvolution models to optimally decipher admixed individual genome variations. IntechOpen, Bioinformatics Tools for Detection and Clinical Interpretation of Genomic Variations: 2019. DOI: 10.5772/intechopen.82764. Available from: https://www.intechopen.com/online-first/orienting-future-trends-in-local-ancestry-deconvolution-models-to-optimally-decipher-admixed-individ. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.