

Abstract
Identifying new gene functions and pathways underlying diseases and biological processes are major challenges in genomics research. Particularly, most methods for interpreting the pathways characteristic of an experimental gene list defined by genomic data are limited by their dependence on assessing the overlapping genes or their interactome topology, which cannot account for the variety of functional relations. This is particularly problematic for pathway discovery from single-cell genomics with low gene coverage or interpreting complex pathway changes such as during change of cell states. Here, we exploited the comprehensive sets of molecular concepts that combine ontologies, pathways, interactions and domains to help inform the functional relations. We first developed a universal concept signature (uniConSig) analysis for genome-wide quantification of new gene functions underlying biological or pathological processes based on the signature molecular concepts computed from known functional gene lists. We then further developed a novel concept signature enrichment analysis (CSEA) for deep functional assessment of the pathways enriched in an experimental gene list. This method is grounded on the framework of shared concept signatures between gene sets at multiple functional levels, thus overcoming the limitations of the current methods. Through meta-analysis of transcriptomic data sets of cancer cell line models and single hematopoietic stem cells, we demonstrate the broad applications of CSEA on pathway discovery from gene expression and single-cell transcriptomic data sets for genetic perturbations and change of cell states, which complements the current modalities. The R modules for uniConSig analysis and CSEA are available through https://github.com/wangxlab/uniConSig.
Keywords: quantification of genome function, disease gene discovery, causal pathway discovery
Background
The identification of causal genes and pathways underlying diseases (such as cancer) based on proteomic, genomic, transcriptomic, or single-cell profiling/sequencing data sets is a daunting task, yet critical to translational research. The first challenge is that genomics data can provide molecular evidence for the pathological factors contributing to certain diseases; however, it is often challenging for biologists to interpret the possible functions of an overwhelming number of aberrant genes cataloged by genomics, and prioritize those genes based on existing human knowledge of their biological functions. This often happens in cancer genomics studies where hundreds of candidate pathological genetic alterations are identified, making it exceedingly difficult to pinpoint the key causal genes and prioritize them for experimental validation. This calls for innovative algorithms to compute novel gene–disease or gene–function associations based on genome knowledge databases. Here, we define ‘gene set’ as a group of genes that are functionally related, such as a set of genes that function in certain diseases (disease gene set) or pathways (pathway gene set). Thus, this type of associations can be generalized as ‘gene to gene set associations’.
Another major challenge is to interpret the pathways characteristic of a list of aberrant genes, such as differentially expressed or mutated genes. Here, we define a nominal gene list of interest cataloged by genomics data as an experimental gene list. Pathway gene set analysis, originally derived from analyzing gene expression data, has progressively been applied to genetic data. The current gene set analysis methods have been outlined in a recent review [1]. While pathway tools are vastly available for gene expression data of continuous variables, only handful of tools are available for genetic data of nominal variables (Table 1). A majority of these tools rely on overrepresentation analysis (ORA) that performs statistical test to assess the statistical overrepresentation of experimental gene list compared to the pathway gene sets and are thus severely limited by their dependence on the genes included in the experimental gene list and the pathways. To illustrate, suppose two gene sets A and B do not share any common genes. Even if gene sets A and B each consist entirely of genes involved in DNA repair, they will not be found to have functional relationship by current approaches. While recent studies has attempted to resolve this issue by quantifying the interactome network topology between gene sets [2–4], these methods cannot take advantage of the vast molecular concept data to analyze the variety of ways that genes can be functionally related. Thus, an algorithm that can better compute the functional relations between gene sets based on the framework of the vast knowledge databases will be of utmost importance.
Table 1.
Pathway enrichment methods for interpreting pathways characteristic of an experimental gene list
| Tools | Statistical approach | Pathway database | PMID |
|---|---|---|---|
| GO-Elite | Hypergeometric distribution and Fisher’s exact test | Gene Ontology, WikiPathways, KEGG, microRNA, user defined | 22743224 |
| GeneTrail | Hypergeometric distribution and Fisher’s exact test | KEGG, TRANSPATH, Gene Ontology, DIP | 17526521 |
| ConceptGen | Modified Fisher’s exact test | Gene Ontology, MiMI, KEGG, Panther, BioCarta | 21715386 |
| KOBAS 2.0 | Binomial test, chi-square test, Fisher’s exact test and hypergeometric test | KEGG, PID curated, PID BioCarta, PID Reactome, BioCyc, Panther | 21715386 |
| DAVID | Kappa statistics | Gene Ontology, PANTHER, BIND, MINT, DIP | 17576678 |
| Enrichr | Fisher’s exact test and z score of the deviation from the expected rank by the Fisher’s exact test | NCI-Nature, PANTHER, metabolic pathway, Gene Ontology, BioCarta, user defined | 23586463 27141961 |
| NEA | Use z score to compute the enrichment statistics based on the interactome network topology | KEGG, Gene Ontology, user defined | 28361684 |
| TopoGSA | Target genes are mapped to an interaction network to compute topological properties and are compared with pathway genes | PPI network, KEGG, BioCarta, Gene Ontology | 20335277 |
| TPEA | TPEA measures topological properties of pathways of the genes and calculates the area under the enrichment curve | KEGG | 28968630 |
| EnrichNet | Target genes are mapped to a network, and random walk procedure scores the functional associations (distance) between target and pathway genes | KEGG, BioCarta, Reactome, WikiPathways, Gene Ontolgy, NCI Pathway | 22962466 |
Molecular concepts, also known as gene sets, are sets of biologically related genes, such as gene ontologies, pathways, molecular interactions, and protein domains [5]. The sum of these concepts represents the current human knowledge about the biological functions of genes. Here, we will take the quantification of new gene functions underlying cancer as an example. We previously developed a concept signature (ConSig) algorithm to compute the functional relevance of genes underlying cancer by assessing their associations with the cancer gene signature concepts (i.e. oncogenic pathways, interactions with key oncogenic proteins, characteristic protein domains, specific gene ontologies). This algorithm provides a unique quantitative estimation of new gene functions underlying cancer, which have been successfully applied by our group to identify new cancer genes from genomics data sets [6–10]. Other algorithms may utilize network analysis, text mining, or similarity profiling to rank candidate genes [11–24], which generally fail to account for the variety of ways in which genes can be related to each other.
It bears notable that all gene set-based algorithms including ConSig require merging of a wide array of molecular concept databases thanks to their outgrowth in recent years [25–33]. Such data fusion, however, carries the inherent challenge of data redundancy because different data sources follow different rules for the nomenclature of concepts and different categories of concepts possess levels of inherent redundancy. Hence, the ability of the gene set-based algorithms to identify the redundancy and sieve only the effective or unique signature concepts for further calculation is critical. Here, we developed a powerful algorithm called universal ConSig (uniConSig) analysis, which has substantially improved performance for genome-wide quantification of gene functions based on redundant molecular concept databases (Figure 1A). Based on this algorithm, we further developed a ConSig enrichment analysis (CSEA) for the quantification of precise functional associations between molecular concepts (Figure 1B) by deep interpreting their shared signature concepts rather than assessing shared gene numbers, which cannot be carried out by any of the previous methods. The uniConSig and CSEA algorithms directly measure the functional interconnectedness of genes and gene sets, which will have wide applications in genomics studies, such as discovering new gene functions underlying certain disease or identifying the pathways underlying experimentally defined gene lists.
Figure 1.
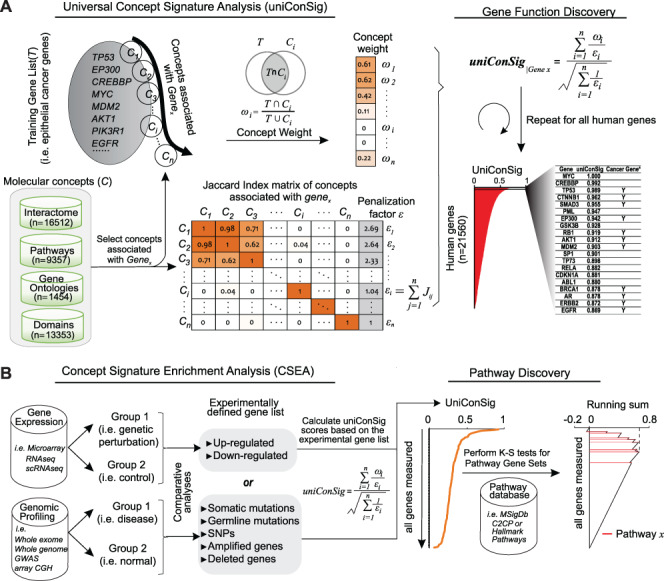
The schematic of the uniConSig and CSEA algorithms. (A) The uniConSig algorithm computes new gene functions underlying a certain disease or a biological process based on a training gene list and a gene knowledge database, which we termed molecular concept database. The training gene list is a collection of genes that carry out a specific biological function or drive certain diseases such as cancer. The molecular concept database integrates diverse sources of gene annotation data sets, including gene ontologies, pathways, interactions, domains, etc. Here, we define the concept weight (ωi) as the Jaccard index between a molecular concept of Genex and the training gene list; a given gene’s uniConSig score is calculated as the average concept weight of all concepts of Genex. The uniConSig algorithm eliminates the redundancies between the concepts of Genex through an innovative mathematical scheme penalizing their overlaps (Jij). A realistic example of molecular concept redundancy is shown in the middle–lower panel, in which the overlap (Jij) between concepts varies from 0 to 1. (B) The CSEA facilitates pathway discovery from an experimental gene list defined from genomics data. Using an experimentally defined gene set (i.e. differentially expressed genes, mutated genes, amplified or deleted genes, etc.) as training gene list, a uniConSig score can be calculated for each gene in the genome (red line), which is used to sort all genes in the genome. For gene expression data, differentially expressed genes can be identified, and the top up- or downregulated genes can then be used as a training gene list to calculate uniConSig scores for all human genes. To identify the pathways characteristic of the experimental gene set, the enrichment of all pathways (i.e. in the MSigdb c2cp or hallmark pathways) in this sorted gene list can be assessed by K-S tests. The resulting ES can be used as a quantitative measure of the functional association between these pathways with the experimentally defined gene set.
Materials and methods
Compiling the molecular concept database and training gene lists
To generate a comprehensive and reliable knowledge base for the calculation of ConSig and uniConSig scores, we compiled 40,676 molecular concepts (Supplementary Table S1) from the Molecular Signatures Database (MSigdB) [34] (C2 and C5 gene sets from http://software.broadinstitute.org/gsea/msigdb), the Pathway commons database [35] (http://www.pathwaycommons.org), the NCBI EntrezGene interactome database and conserved domain database [27] (https://www.ncbi.nlm.nih.gov/gene) and the VisAnt interactome database [36] (http://visant.bu.edu/). Here, we only included these human-curated gene sets in the compiled molecular concept knowledge base to increase the reliability of the calculations.
To calculate ConSig and uniConSig scores for cancer, type 2 diabetes and nucleotide excision repair pathway, we have compiled three training gene lists. The known cancer causal gene set was downloaded from the Cancer Gene Census (CGC) [37] (http://cancer.sanger.ac.uk/census/). The cancer genes were then classified to different cancer entities via mapping the cancer type annotations of CGC cancer genes to TCGA cancer types. The ‘DIABETES MELLITUS, TYPE II’ gene set was obtained from OMIM [38] (https://www.omim.org/, OMIM number: 125853) and the ‘KEGG Nucleotide Excision Repair’ gene set was obtained from MSigdb C2CP database [34].
The uniConSig algorithm
For a given Genex, we define the basic uniConSig score to be the average of the concept weights of each Concepti associated with Genex, which indicates the average similarities between the Genex-associated concepts and the training gene list. The concept weight (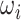 ) for each Concepti is defined as the Jaccard index between the Concepti and the training gene list, which measures the similarities between each concept and the training gene list (Figure 1A). One key problem in the concept analysis is that many concepts are similar to each other, which will cause the over estimation of uniConSig scores for most studied genes (Supplementary Figure S1). To remove the effect of redundancy in the knowledge base, we introduced a penalization factor, ε, which is given by
) for each Concepti is defined as the Jaccard index between the Concepti and the training gene list, which measures the similarities between each concept and the training gene list (Figure 1A). One key problem in the concept analysis is that many concepts are similar to each other, which will cause the over estimation of uniConSig scores for most studied genes (Supplementary Figure S1). To remove the effect of redundancy in the knowledge base, we introduced a penalization factor, ε, which is given by
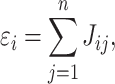 |
(1) |
where Jij is the Jaccard index between each pair of the molecular concepts associated with Genex and n is the total number of concepts associated with Genex. Thus, εi is an estimator of redundancy among the molecular concepts associated with Genex, which ranges from 1 (all the concepts are completely different from each other) to n (all the concepts are exactly the same) (Figure 1A; Supplementary Figure S2). During the development of uniConSig, we found that the algorithm calculating the penalization parameter ε tends to aggravate the impact of small overlaps between the molecular concepts (Figure 2A). To remove the negative effect introduced by the small overlaps between the concepts associated with Genex, we introduced a cutoff to remove the small Jaccard scores caused by random overlaps. We tested different cutoffs from 0.01 to 0.1 (Supplementary Figure S3). The enrichment scores (ESs) and the SDs of the ES indicate a cutoff of 0.05 for Jij can achieve a good ES with less variations. Therefore, the new penalization factor ε′ for a given concept Ci of Genex is calculated as
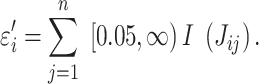 |
(2) |
Figure 2.
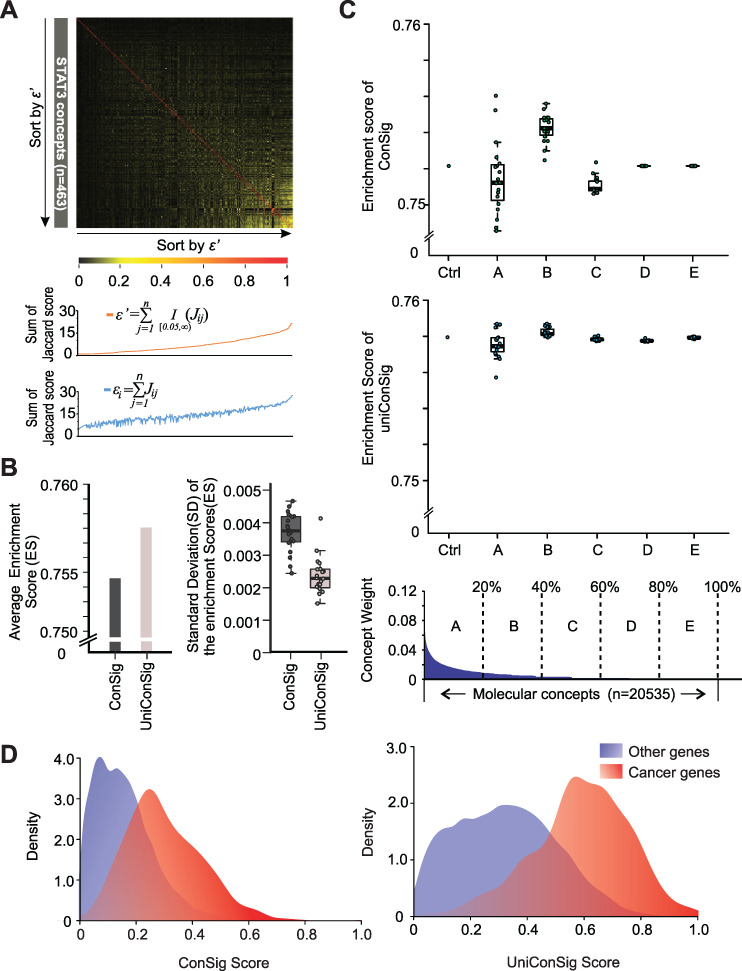
Benchmarking the performance of the uniConSig algorithm over simulated concept redundancy. (A) The effect of minor overlaps between molecular concepts on calculating the penalizing factor εi for concept redundancy. The Jaccard indexes between the 463 molecular concepts associated with the STAT3 gene are plotted in the upper heatmap. The concepts are sorted by the adjusted penalizing factor εi’ calculated with a Jaccard index cutoff of 0.05 (orange line, middle plot). In contrast, without adjustment, the minor overlaps (which are presumably biologically irrelevant) significantly inflate εi, which leads to the underestimation of ECW and ECN (blue line, lower plot). (B) The uniConSig algorithm outperforms the ConSig algorithm over simulated concept redundancy. Here, we randomly selected two-thirds of the CGC cancer genes as training gene list and the remaining one-third as testing gene set and repeated this process for 20 times to generate 20 pairs of training/testing gene sets. We then randomly duplicated 50% of the molecular concept database and repeated this for 20 times and calculated the ConSig and uniConSig scores based on each of the training gene list, as well as ES based on the paired testing gene set. The average ESs generated using ConSig and uniConSig are shown on the left panel, and the average SDs from 20 database duplications using each of the 20 training gene lists are shown on the right panel. (C) The uniConSig algorithm showed substantially improved performance over random duplications of selected concepts with different levels of overlaps with the cancer gene list. Here, we used the known cancer gene set generated by CGC as the training gene list and calculated its Jaccard index with each of the molecular concepts in the database as concept weights. Then the 20,535 all molecular concepts are divided into five groups (A–E) based on their different levels of overlaps with the CGC cancer gene list. We then randomly duplicated 50% of the concepts in each of these five groups for 20 times and calculated the ConSig or uniConSig scores. The performance of the resulting scores on prioritizing the known cancer genes are benchmarked with K-S tests. ‘Ctrl’ is the result of the original molecular concept database without concept duplications. (D) The uniConSig algorithm better separates known cancer genes from other human genes. ConSig scores and uniConSig scores were calculated based on the known cancer genes collected from CGC. Density plots of the known cancer genes (red) and other human genes (blue) are shown for ConSig scores (left) and uniConSig scores (right).
The Jaccard score Jij will be adjusted to 0 if Jij < 0.05. Based on εi′, we then penalized the concept weight ωi by εi′, resulting in the effective concept weight (ECW):
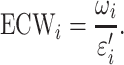 |
(3) |
The harmonious sum of εi′ was then used to calculate the effective concept number (ECN):
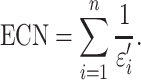 |
(4) |
We also tested different transformation of ECN to improve the ESs using cancer gene list as training gene list (Supplementary Figure S4). We chose square root as the final form for ECN to be consistent with former ConSig algorithm. Taken together, the uniConSig score of a given Genex is then calculated as
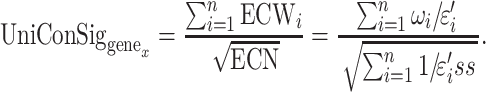 |
(5) |
For a given training gene list, this calculation is applied to each of the genes in genome; thus, all the genes are assigned with a uniConSig score. If a gene is included in the training gene list, then the number of overlapping genes between a concept and the training gene list will be subtracted by 1 to avoid the inflation of the concept weight (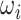 ). The final uniConSig score is scaled to [0, 1]. To avoid bias, we excluded the molecular concepts with fewer than five genes during calculation of uniConSig scores.
). The final uniConSig score is scaled to [0, 1]. To avoid bias, we excluded the molecular concepts with fewer than five genes during calculation of uniConSig scores.
Calculating duniConSig scores
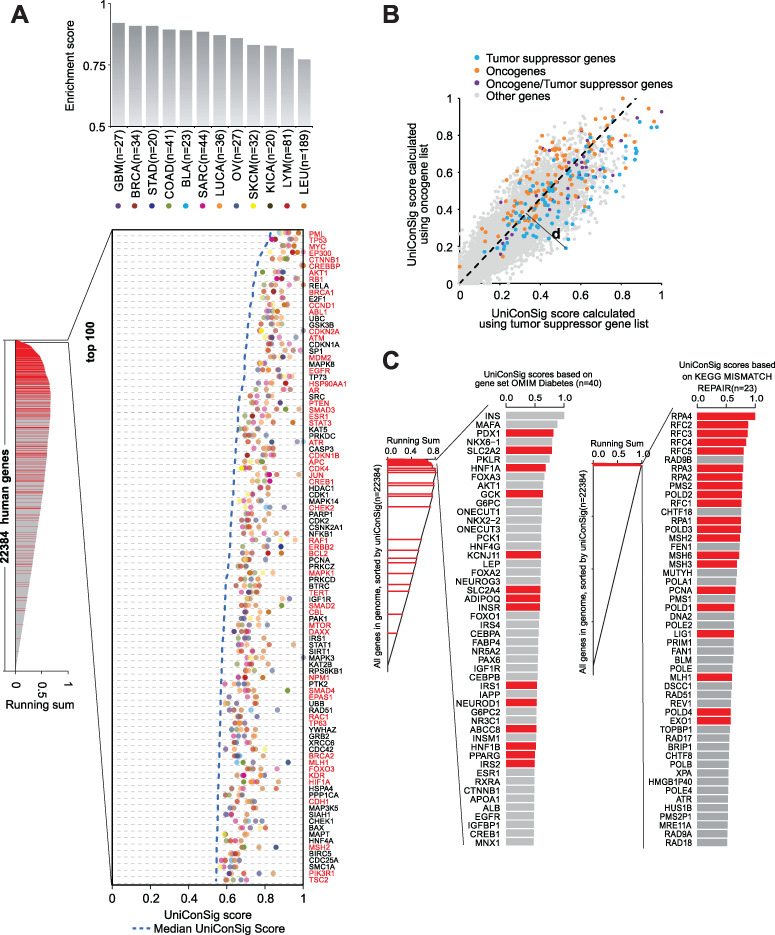
Here, we take comparing the oncogene and tumor suppressor gene sets as example to illustrate the duniConSig algorithm. Based on the oncogene and tumor suppressor gene sets compiled from the CGC [37], we first calculated the oncogene and tumor suppressor uniConSig scores for each Genei in the genome (Figure 3B). Here, we define uniConSigi|Onco to be the oncogene uniConSig score of Genei and uniConSigi|TSG to be the tumor suppressor uniConSig score of Genei. Then we sorted the genes on either oncogene set or tumor suppressor gene set by (uniConSigi|Onco)/(uniConSigi|TSG) in descending order. Scanning from the top to the bottom of this sorted gene list, the percentage of correctly classified genes in the oncogene gene set is calculated by
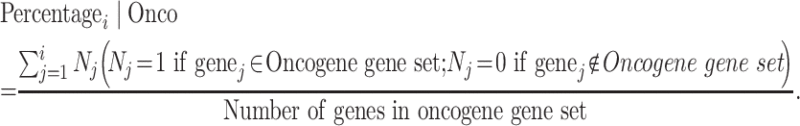 |
(6) |
Figure 3.
The applications of the uniConSig algorithm in the discovery of new gene functions. (A) Quantifying the role of human genes underlying different cancer entities based on the lists of known cancer genes for different cancers. Gene sets for different cancer types were collected from CGC. The resulting cancer gene ESs based on K-S tests for different cancer entities are shown at the top (gray columns). We calculated the cancer type-specific uniConSig scores for the human genome using these 12 cancer gene sets as training gene lists and then sorted all human genes by the median of their uniConSig scores (dashed blue line). The top five uniConSig scores for each gene are shown in the chart, which represent five different cancer types (different colored dots). Circles outlined in black indicate that the gene is included in the CGC database for that specific cancer type. Known cancer gene names are highlighted in red. The running sum of the random walk K-S test of CGC cancer gene set is shown to the left. Red lines are the genes that are on the CGC known cancer gene list. (B) uniConSig scores for Oncogenes and tumor suppressors. In this dot plot, the uniConSig scores calculated based on the oncogene gene set and tumor suppressor gene set from CGC are shown in y- and x-axis, respectively. The dashed line indicates the distinction line (D-line), which was calculated based on the ROC-like curve shown in Supplementary Figure S7, where we selected the D-line’s slope based on the maximum of the Youden index. Here, we define the distance of a gene to the D-line as ‘dConSig’ score. (C) Quantifying the functional relevance of human genes underlying diabetes or mismatch repair pathway based on the OMIM diabetes gene set or the KEGG mismatch repair gene set. uniConSig scores of each human gene were calculated based on OMIM diabetes gene set (left) or KEGG’s mismatch repair pathway gene set (right). Top 50 genes are shown in the plots. Red bars are the genes that are on the OMIM diabetes gene set (left) or KEGG mismatch repair pathway gene set (right).
Similarly, the percentage of correctly classified genes in the tumor suppressor gene set is calculated by
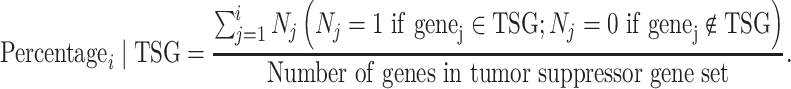 |
(7) |
Next, we plotted the above percentages in a receiver operating characteristic (ROC)-like curve (Supplementary Figure S7). The optimal separation line (D-line) for oncogenes and tumor suppressors can be determined using the ROC Youden index:
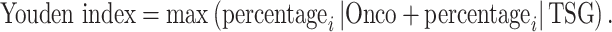 |
With this computation, if a gene has high probability to be oncogenes or tumor suppressors, the uniConSigi|Onco or uniConSigi|TSG will be high, respectively. Thus, the distance of each gene to the D-line in the two-dimensional plot for oncogene and tumor suppressor uniConSig scores will correlate with the role of tested genes in cancer, which we termed duniConSig score.
ConSig enrichment analysis
To assess the association between two gene sets A and B, we used one gene set A (i.e. experimentally defined gene set) to calculate the uniConSig score for each gene in the genome and sorted all human genes by this score (Figure 1B). Then, the ES for the other gene set B (i.e. pathway gene set) in this sorted list is calculated by the weighted step-up method. This method assesses the enrichment of the genes in gene set B with high uniConSig scores calculated based on gene set A. The steps of calculations are similar to the equal step-up method, except the step-up for a given Genei is calculated by
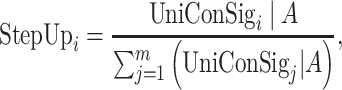 |
(8) |
where m is the number of genes in gene set B. The step-down for a given Genei is calculated by
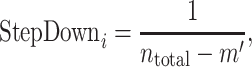 |
(9) |
where ntotal is the total number of genes in genome. To assess the functional relations of multiple gene sets with a given gene set (i.e. multiple pathways with an experimental defined gene set), the ES needs to be normalized, since the number of genes in gene sets are different. The normalization can be carried out by randomly picking the same number of genes as the pathway gene set from the total genome and then calculating the ES using the above method. Repeat this step until we get 1000 positive ESs, and then, the normalized ES (NES) can be calculated by
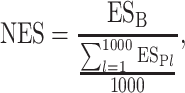 |
(10) |
where ESB is the ES of gene set B and ESPl is the ES of a permutation l. This is the same as calculating the NES score in the Kolmogorov–Smirnov (K-S) test used in Gene Set Enrichment Analysis (GSEA) methods, and we used 1000 permutations as recommended by GSEA.
Benchmarking the ConSig and uniConSig algorithms
To assess the performance of the different algorithms in prioritizing known cancer genes, we randomly selected two-thirds of the genes from 567 known cancer genes from the CGC as the training gene list for the calculation of uniConSig scores and the remaining one-third of genes as a testing gene set to benchmark the performance of the uniConSig scores. Such random selection was repeated 20 times (Supplementary Figure S5). To benchmark the performance of the algorithms, we used a random walk K-S test to assess the enrichment of testing gene set in the top-ranked genes by the ConSig/uniConSig scores calculated using the training gene list. This is similar to the equal step-up K-S test option used in the GSEA analysis [34]. To scale the ES, we used the ratio of the ES divided by the maximum possible ES, given by
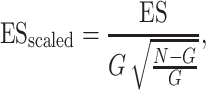 |
(11) |
where G is the total number of genes in CGC cancer gene list and N is the total number of all human genes included in this calculation. 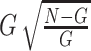 gives the maximum possible ES (if all of the CGC cancer genes were enriched at the top). Here, we have used equal step-up in this analysis to take more account of the effect of known cancer genes with low ConSig scores that would be overlooked by the weighted step-up K-S test.
gives the maximum possible ES (if all of the CGC cancer genes were enriched at the top). Here, we have used equal step-up in this analysis to take more account of the effect of known cancer genes with low ConSig scores that would be overlooked by the weighted step-up K-S test.
CSEAs of gene expression and scRNA-seq data sets
The gene expression data sets were downloaded from Gene Expression Omnibus: GSE31812 by Freed-Pastor et al. [39] and GSE84970 by Zhao et al. [40]. Because the two data sets used different microarray platforms, including Affymetrix Human Gene 1.0 ST Array and Affymetrix Human Genome U133 Plus 2.0 Array, respectively, the corresponding R packages Oligo and Affy were used for normalization. The data sets were normalized by Robust Multiarray Averaging (RMA) and GeneChip RMA (GC-RMA), respectively, and the gene expression differences are compared by the R package Limma [41]. Specifically, for the data from Freed-Pastor et al. [39], the downregulated genes were identified by comparing the expression data of MDA-468.shp53 cells with or without mutant TP53 inhibition (DOX+ versus DOX−). For the data from Zhao et al. [40], the downregulated genes were identified by comparing the expression data of LnCap cells following CHD1 knockout with the control. To capture the most significantly up- or downregulated pathways, the top 50 downregulated genes were used as input for the following CSEAs. After calculating uniConSig scores, all the human genes were sorted by their uniConSig scores, and the ESs of each of the MSigdb C2CP pathways (for data from Freed-Pastor et al. [39]) or hallmark pathways (for data from Zhao et al. [40]) were calculated by the weighted step-up of the random walk K-S test, as described above.
GSEA analysis of gene expression data sets
In GSEA analysis, the parameters for processing the two data sets are identical to the CSEA. Gene expression data were loaded into R by Affy for Zhao et al [40] and by Oligo for Freed-Pastor et al. [39] and normalized by RMA and GCRMA, respectively; analyzed by Limma; and then exported to GSEA 3.0. Since GSEA takes all the sorted genes into account, there is no cutoff required following Limma analysis. The MSigdb C2CP pathways (for data from Freed-Pastor et al. [39]) or hallmark pathways (for data from Zhao et al. [40]) were used in the GSEA analyses. The minimum number of genes in a concept was set to 5 as in CSEA (default 15). Other parameters were default.
Analysis of single-cell transcriptomic data
For pathway discovery from single-cell transcriptomic data of hematopoietic stem cells (HSCs), the up- or downregulated gene lists in quiescent HSCs compared to active HSCs were obtained from Table S6 of the scRNA-seq study by Yang et al. [42]. The genes marked as ‘Active’ in the table were the downregulated genes in quiescent HSCs, while the genes marked as ‘Quiescent’ were the upregulated genes in quiescent HSCs. Mouse gene IDs were converted to human gene IDs by R package ‘biomaRt’. CSEA was performed using these gene lists as input and the default parameters against the merged MSigdb C2CP and Hallmark pathways, which were also used for the following comparative pathway enrichment analyses. The ORA and network enrichment analysis (NEA) used the same up- or downregulated gene lists in quiescent HSCs as CSEA. The ORA enrichment analysis was carried out by an R function ‘enricher’ from package ‘clusterProfiler’ [43], which used hypergeometric test. The NEA analysis was performed using NEA render (https://cran.r-project.org/web/packages/NEArender/index.html) [3] and the recommended merged network provided by Merid et al. [44]. For GSEA, we first reconstruct the t-Distributed Stochastic Neighbor Embedding (t-SNE) plot using the same parameters as described in the article by Yang et al. [42], to obtain the mapping list of cells to quiescent/active groups. The log2(FPKM + 1) of the expression data were then fed into GSEA desktop software using default parameters.
Results
The impact of concept redundancy on the ConSig algorithm
In ConSig analysis, the quantification of the function of human genes underlying a certain disease requires a list of known disease genes as a training gene list and a molecular concept database compiled from multiple sources (Figure 1A). For example, to quantify the function of human genes underlying cancer, we leveraged the cancer causal genes collected by the CGC [37] as a training gene list (n = 567) and compiled an integrated molecular concept database from different resources [27, 34–36, 45], which represents the sum of current human knowledge about the functions of the human genome (Supplementary Table S1). The compilation of molecular concept databases greatly enriches the scope of the knowledge base; however, unavoidable data redundancy was introduced into this database by the database merging process (Supplementary Figure S1). This is attributable to the different nomenclature of molecular concepts from different data sources or even from the same source (i.e. ‘PI-3K cascade’ and ‘PI3K signaling’) and the different levels of overlaps between functionally similar molecular concepts (i.e. EGFR pathway and ERBB pathway; Supplementary Table S2).
To demonstrate the impact of data redundancy on the ConSig algorithm, we performed random duplications of selective subsets of molecular concepts in the database. The resulting duplicated concept databases were used to calculate ConSig scores based on CGC cancer genes. To examine the performance of the ConSig scores in prioritizing the CGC cancer genes, we applied the random walk K-S test, which estimates the enrichment of the genes in the testing gene set among all of the genes ranked by a set of ConSig scores. We found that the performance of the ConSig algorithm was most greatly affected by duplication of concepts with a higher degree of overlap to the training gene list (CGC cancer genes; Figure 2C, upper panel). Interestingly, we found that the database duplication can lead to both increase and decrease of its performance, and the deviation of the K-S scores can be considered as an indicator of the algorithm’s performance under random database duplication. The general solutions to remove the concept redundancy include removing one of the overlapping gene sets or taking the union of overlapping gene sets. These approaches, however, will lead to substantial loss of information, because many gene sets have partial overlaps with multiple other gene sets (Supplementary Figure S1). For example, we cannot take the union of the overlapping gene sets in Supplementary Figure 1 indicated by yellow color, which are mostly ‘interrelated’. Here, we developed an innovative penalty algorithm to minimize the effect of data redundancy.
The uniConSig algorithm
To develop a uniConSig algorithm that was unaffected by concept redundancy, we first analyzed a simple scenario to calculate the functional association of a given Genex with a disease gene list based on redundant molecular concepts (Supplementary Figure S2). Assume there are five molecular concepts (Ci) associated with Genex, the association of Genex with the training gene list (T) can be simply calculated as the average of the Jaccard similarity coefficients between each Genex concept (Ci) and the training gene list, which is calculated as the intersection over the union of the two comparing gene lists. Here, we term this Jaccard index as the weight ωί for Concepti. If among the five molecular concepts, concepts 1 (C1) and 2 (C2) are identical, then the association score should be calculated as if there were only four unique molecular concepts, which we term as effective concepts (by taking the average of the ECWs, 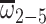 ).
).
Molecular concepts have varying degrees of overlap with each other (i.e. the EGFR pathway has 64.3% overlap with the ERBB2 pathway). In this case, although the two concepts are highly redundant, they still introduce different information to the calculation. Thus, using a simple cutoff to remove the redundant information in the knowledge database is not optimal. To overcome this problem, we introduced a penalization parameter ε, which is the sum of the Jaccard index of a given Concepti compared to each of the other concepts associated with Genex. The εi indicates the degree of overlap between Concepti with other Genex-associated concepts. The effective concepts can then be calculated as the division of the concept weight ωί by the penalization parameter εi. In Supplementary Figure S2, concept C1 and C2 are identical, and the sum of the ECWs is equivalent to the sum of the concept weights without the concept C1. Theoretically, this algorithm will effectively remove the impact of the overlapping concepts on concept weights. However, we discovered that the algorithm calculating the penalization parameter ε tends to aggravate the impact of small overlaps between the molecular concepts associated with Genex (Figure 2A). Such low-level overlaps, albeit individually insignificant, can result in a large sum (εi) of Jaccard indexes when a large number of concepts are associated with Genex. This problem was resolved by setting a cutoff (α) for the Jaccard index (Jij) when calculating the εi, and a cutoff of 0.05 achieved the relative highest ESs and lowest SDs of the ESs in the presence of random duplications of 50% of molecular concepts (Supplementary Figure S3). Therefore, in the following calculation of uniConSig scores, we set the minimal Jaccard index between overlapping concepts to 0.05 when calculating εi.
Next, we take the sum of the reciprocals of εi to calculate the ECN based on different degrees of concept overlaps. If there is no overlap between the concepts associated with Genex, then 1/εi will be 1. If there are two out of a total of five concepts of Genex that 100% overlap with each other, the ECN will be calculated as ½ + ½ + 1 + 1 + 1 = 4. Thus, using the ECN has the same effect as removing duplicated concepts. Incorporating ε in the calculation of concept weight and ECN not only generates a similar result as simply removing the duplicated concept C1 but also avoids setting an arbitrary cutoff to remove duplicated concepts (Figure 1A). We then tested the performance of different transformations of the ECN, including square, square root, log10 and linear algorithms. We speculate that the attenuating transformations such as square root can accentuate the weights more on the final scores, similar to a sample test statistic where the sample size is now the number of concepts. Indeed, the square root and log10 algorithms achieved the best performance as shown by K-S tests (Supplementary Figure S4). To be consistent with other statistics, we applied square root to transform the ECN in the final uniConSig algorithm. Finally using the ECWs, ECN and square root normalization, we define a uniConSig algorithm that can more effectively assess the gene to concept associations using redundant molecular concept databases:
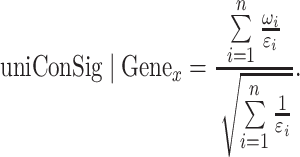 |
(12) |
Benchmarking the performance of the uniConSig algorithm
To determine the performance of the new uniConSig algorithm, we randomly selected two-thirds of the known cancer genes as training gene lists and the other one-third as testing gene sets, for 20 times (Supplementary Figure S5). To determine the variation of uniConSig scores caused by data redundancy, we randomly duplicated 50% of the molecular concept database for each of the permutated testing gene sets. Such random duplication was repeated 20 times for each permutation of the training/testing gene sets. This generated a total of 400 sets of uniConSig scores. Using K-S tests, we examined the performance of uniConSig versus the original ConSig in prioritizing the known cancer genes in the testing gene set among all of the human genes ranked by a set of uniConSig scores. The uniConSig algorithm showed a better performance, as indicated by a higher average ES (Figure 2B, left), and smaller deviations resulting from artificial database duplications, as indicated by a lower SD of the ESs (Figure 2B, right).
Next, we further tested the performance of the ConSig and uniConSig algorithms under selected duplications of molecular concepts with different levels of overlap with the training gene list. All molecular concepts are divided into five groups based on their different levels of overlaps with the cancer causal gene list. We then randomly duplicated 50% of the concepts in each group and calculated the cancer gene uniConSig scores. The performance of the resulting scores on prioritizing these known cancer genes are benchmarked with K-S tests. The results show that uniConSig algorithm shows the much lower deviations compared to the original ConSig algorithm across different levels of database duplications (Figure 2C), supporting its outstanding performance against concept redundancy.
We next assessed the performance of the uniConSig algorithms in computing functional relevance of human genes underlying cancer. The density plots of the ConSig and uniConSig scores calculated based on CGC cancer genes show significant improvement of the latter in separating known cancer genes from other human genes (Figure 2D). In contrast, the uniConSig algorithm did not produce enrichment of the random training gene lists generated by randomly selecting the same number of genes as CGC cancer gene list (Supplementary Figure S6), indicating that such separation is generated by the shared functional traits of cancer genes.
The application of the uniConSig algorithm on gene function discovery
Next, we sought to demonstrate the application of the uniConSig algorithm on gene function discovery using cancer gene discovery as example. We first explored the possibility of calculating cancer type-specific uniConSig scores by compiling gene sets for different cancer entities using the CGC database. The uniConSig scores calculated using these gene sets showed the best prioritization results for glioblastoma and worst for lymphoma and leukemia, even though these liquid tumors have many more known cancer genes than the solid tumors (Figure 3A). This may be attributed to the many distinct subtypes of lymphoma and leukemia. Notably, the uniConSig scores for different cancers not only prioritized known cancer genes in these cancers but also nominated the cancer genes that are not on the CGC database, including those well-known cancer genes that are missed by the CGC, such as CDKN1A, SRC, HDAC1 and PAK1. Next, we examined the capability of uniConSig to distinguish oncogenes from tumor suppressors. We calculated uniConSig scores based on the CGC oncogene and tumor suppressor gene sets, which were plotted against each other for all human genes (Figure 3B). The optimal separation line for oncogenes and tumor suppressors was determined using an ROC curve (Supplementary Figure S7). At this cutoff, 63.7% of oncogenes can be separated from 77.8% of tumor suppressors. The area under the ROC curve, which we termed aucConSig, is indicative of the functional difference between oncogenes and tumor suppressors (aucConSig = 0.77).
To demonstrate the wide applications of uniConSig algorithm in discovering novel disease causal genes or pathway genes, we tested its utility in prioritizing genes involved in diabetes or nucleotide excision repair. We used the ‘OMIM Diabetes Mellitus, Type II’ gene set or the ‘KEGG Nucleotide Excision Repair’ gene set as training gene lists (see Methods) to calculate the uniConSig scores (Figure 3C). As expected, the genes included by these two gene sets are highly enriched in the genes with top uniConSig scores. In addition, a number of genes not included in the two gene sets were assigned high uniConSig scores, respectively. Interestingly, the gene with the highest uniConSig score in the results for type 2 diabetes was INS, which encodes the precursor of insulin. It is common knowledge that insulin plays a major role in diabetes, even when it is not the causal gene. The gene with the second highest uniConSig score was MAFA, which is known to bind RIPE3b and regulate the expression of INS [46]. Other top-ranked genes have also been shown to be functionally associated with glucose metabolism, including NKX6-1 [47], PKLR [48], FOXA3 [49] and AKT1 [50]. Similarly, the highest ranked genes not included in nucleotide excision repair were RAD9B and CHTF18. RAD9B is associated with the activation of DNA damage checkpoint and DNA repair pathways [51], while CHTF18 is known to be a component of the replication factor C complex, which is a positive regulator of the replication stress response [52]. These results demonstrate that uniConSig can identify novel candidate genes associated with a wide range of diseases or pathways based on existing gene sets for the disease or pathway.
ConSig enrichment analysis
One of the common goals of genomics studies is to identify the pathways that are enriched in the experimental gene lists (i.e. differentially expressed, mutated, amplified or deleted genes) defined from genomics data sets. This is commonly done by Fisher’s exact test or similar approaches, which, however, are all on assessing overlapping genes. We hypothesized that this problem could be overcome by leveraging the functional quantification of genes by the uniConSig algorithm to assess the functional similarity between gene sets. We thus developed a CSEA, which uses the experimentally defined gene list to calculate the uniConSig scores and then tests the enrichment of the pathway genes in the top genes ranked by uniConSig scores using K-S tests, which will bypass the limitations of calculating pathway uniConSig scores (Figure 1B). If the two gene sets are functionally similar, the uniConSig scores calculated based on one gene set (i.e. experimental gene list) will be high for the genes of the other gene set (i.e. pathway); therefore, the ES based on the K-S test can be used as an indicator of the functional similarity between the two gene sets.
To test the performance of CSEA, we identified nine growth factor signaling pathways and nine DNA damage-related pathways. The similarities between each pair of these pathways were calculated using the Fisher’s exact test or CSEA algorithm, and the pathways were then clustered based on the resulting scores (Figure 4). Fisher’s exact test was able to identify similarities between growth factor signaling pathways due to the high number of overlapping genes, but it was unable to identify similarities between DNA repair pathways, which are functionally similar but distinct in terms of the specific genes they contain. The CSEA result shows a much better separation and clustering of growth factor signaling pathways from DNA repair pathways compared to the Fisher’s exact test.
Figure 4.
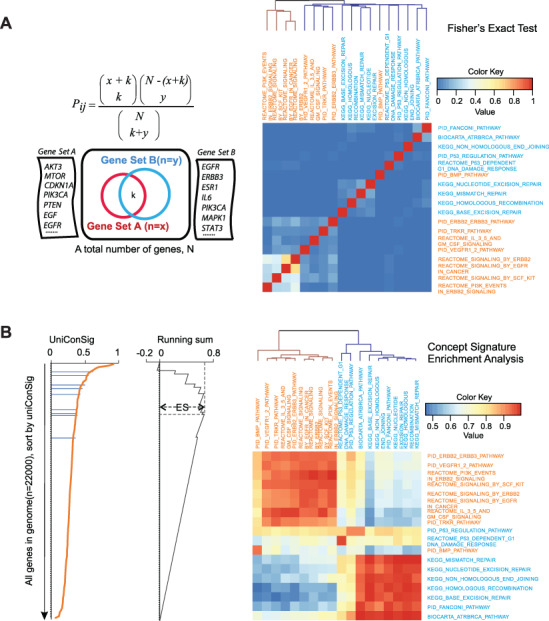
Quantification of the functional associations between different gene sets based on uniConSig scores. (A) Quantification of the functional associations between the selected growth factor pathways (orange) and DNA damage repair pathways (blue) by Fisher’s exact tests. P-values were −log 10 transformed and normalized to 1. (B) Quantification of functional associations by CSEA for the selected growth factor pathways (orange) and DNA damage repair pathways (blue). Pathways on the horizontal axis were used to calculate uniConSig scores; pathways on the vertical axis were used to calculate ESs. The clustering tree shows the CSEA was able to clearly distinguish the two groups of pathways. The matrix resulting from CSEA analysis is asymmetric as we used one pathway to calculate uniConSig scores and then performed enrichment analysis for the other pathway, so the results for each pair of pathways are slightly different when the order of the analysis is reversed.
CSEA facilitates pathway discovery from experiment gene sets defined by genomic data sets
Next, we sought to assess the application of the CSEA algorithm on pathway discovery from experiment gene sets defined by genomic data sets. We first determined the minimal number of genes required to calculate effective uniConSig scores from experimental gene sets by randomly selecting varying numbers of cancer genes to calculate uniConSig scores (Supplementary Figure S8). While a minimal of 10 genes in the training gene list can produce significant enrichment, the performance of the uniConSig algorithm dropped significantly when the training gene lists had less than 30 genes. As the size of 50 genes in the training gene lists started to show optimal and stable performance, in the following analyses, we performed the pathway enrichment analyses based on the 50 most up- or downregulated genes defined by the gene expression data sets. This will also help prevent the small denominators in the Jaccard calculations that may introduce noise when assessing the concept weights based on the experimental gene sets.
To demonstrate the application of the CSEA algorithm in de novo pathway discovery, we compiled the expression data sets of two genetic perturbation studies. One data set (GSE31812) compared the breast cancer cell line MDA-468.shP53 inducibly expressing mutant TP53 shRNA in the presence or absence of doxycycline induction (DOX+/−), and the mevalonate pathway (labeled ‘biosynthesis of steroids’ in C2CP data set) was known to downregulate following mutant TP53 depletion [39]. The other data set (GSE84970) compared the CRISPR edited CHD1 knockout LNCap cells with control LNCap cells, and the NF-κB pathway is known to downregulate following CHD1 knockout [40]. Gene expression data were first analyzed for differentially expressed genes using Limma package [41]. Then uniConSig scores were calculated based on the top 50 downregulated genes following mut-TP53 knockdown or CHD1 knockout, and the pathways enriched in these top-scored genes were assessed using K-S tests. To be consistent with the original studies, for the TP53 data set, we analyzed the enrichment of the C2CP pathways [53], and for the CHD1 knockout data set, we used the hallmark pathways [54], all of which from the MSigDb. For comparison, we also performed GSEA analysis using same gene expression data processed by Limma. As a result, CSEA identified the ‘steroid biosynthesis’ pathway and the ‘TNFα signaling via NF-κB pathway’ as one of the most downregulated pathways in MDA-468.shP53 cells following mutant TP53 depletion and in LNCap cells following CHD1 knockout, respectively, which were lower ranked by GSEA analysis (Figure 5A–B, D–E).
Figure 5.
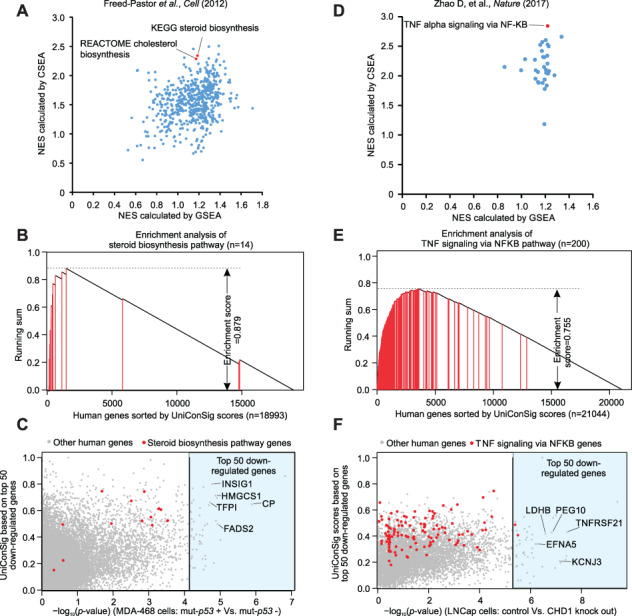
CSEA identifies the pathways characteristic of genetic perturbations from the experimental gene sets defined from genomics data. (A) The NESs of C2CP pathways downregulated following mutant-p53 knockdown, which are calculated by CSEA and GSEA using data from Freed-Pastor et al. [39]. (B) The running sum of the ‘steroid biosynthesis’ pathway in human genes sorted by their uniConSig scores of top 50 downregulated genes following mutant p53 knockdown. (C) The dot plot showing the Limma P-values comparing the control with mutant p53 knockdown and uniConSig scores calculated using the top 50 down genes (highlighted in the light blue area) for all the genes included in the microarray data of Freed-Pastor et al. [39]. (D) The NES scores calculated by CSEA and GSEA for the hallmark pathways downregulated following CHD1 knockout based on the data of Zhao et al. [40]. (E) The running sum of the ‘TNF signaling via NFKB pathway’ in human genes sorted by their uniConSig scores of the top 50 downregulated genes following CHD1 knockout. (F) The dot plot showing the Limma P-values comparing the control with CHD1 knockout and uniConSig scores calculated using the top 50 downregulated genes (light blue area) for all the genes included in the microarray data of Zhao et al. [40].
Of note, while the top 50 downregulated gene lists contained no genes in the steroid biosynthesis pathway or only two genes in TNFα signaling via NF-κB pathway, their uniConSig scores still ranked these pathway genes to the top (Figure 5C, F). This suggests that many of the top 50 genes are functionally related to these pathways but are not included in their original gene sets. This hypothesis is supported by subsequent literature investigations. For example, CP is the second most downregulated gene following TP53 depletion in MDA-468 cells and is involved in the peroxidation of Fe (II) to Fe (III), a common mechanism engaged by cytochrome P450 genes during the steroid biosynthesis [55]. Other downregulated genes in this study are also shown to be functionally related to steroid biosynthesis, such as FADS2 [56], HMGCS1 [57], INSIG1 [58] and TFPI [59]. Similarly, the most downregulated genes following CHD1 knockout in LNCap cells are also more or less related to the NF-κB pathway. Among these, TNFRSF21 (DR6) has been shown to activate the NF-κb and JNK pathways [60, 61]; KCNJ3 (Kir3.1) has been implicated in NF-κB activation in THP-1 cells [62]; PEG10 has been shown to interact with SIAH2 [63], which decreases TNFα-dependent induction of JNK activity and transcriptional activation of NF-κB [64]. LDHB can be upregulated by a natural NF-κB inhibitor, panepoxydone [65], and DFNA5 was reported to be a target gene of NF- κB [66]. These results demonstrate the power of CSEA in identifying the pathways enriched in experimentally defined gene lists through deep assessment of the functional relations.
CSEA outperforms other methods on interpreting complex pathway changes during change of cell state from single-cell transcriptomics
Single-cell RNA sequencing (scRNAseq) is a rapidly growing technology that is becoming more and more popular. However, single-cell transcriptomics usually have much lower coverage than bulk sequencing, which limits the detection of differentially expressed genes and thus challenges the pathway analysis. We speculate that the capability of CSEA to deep interpret the functions of the differentially expressed genes may greatly enhance pathway discovery from the limited experimental gene list cataloged by single-cell transcriptomics. To test this, we performed CSEA on the differentially expressed gene lists detected by scRNAseq comparing the active and quiescent populations of HSCs from a recent study [42]. The single-cell RNA-seq was performed using the fluidigm C1 system and the Illumina HiSeq2500 with 101 bp paired-end sequencing strategy. A total of 112 HSC and 109 MPP1 single cells are included in the data set.
Change of cellular states usually involve more complicated changes of transcriptional programs compared to single genetic perturbations, which makes the pathway analyses more challenging. In particular, quiescent HSCs have several hallmark pathway changes, including cellular quiescence, repressed DNA replication and repair, repressed differentiation, low RNA content, and quick response to hematopoietic cytokines. Interestingly, CSEA interpreted a comprehensive picture of pathway alterations characteristic of quiescent HSCs consistent with current knowledge of their hallmarks, which are not reported in the original study (Figure 6): (1) downregulation of E2F targets indicating cellular quiescence [67]; (2) downregulation of MYC targets indicating reduced differentiation [68]; (3) DNA replication, cell cycle progression and mitotic pathways are repressed, which are hallmarks of quiescent HSCs [42]; (4) RNA transcription pathways are repressed, which is consistent with the reduced total RNA amount in quiescent HSCs [69]; (5) upregulation of cytokine signaling (i.e. interferons, interleukins, GM-CSF and TNFα), which are required for the quiescent HSCs to respond to hematopoietic cytokines [70]; (6) downregulation of DNA repair pathways. It is known that quiescent HSCs accumulate DNA damage, which is repaired upon entry into active state [71].
Figure 6.
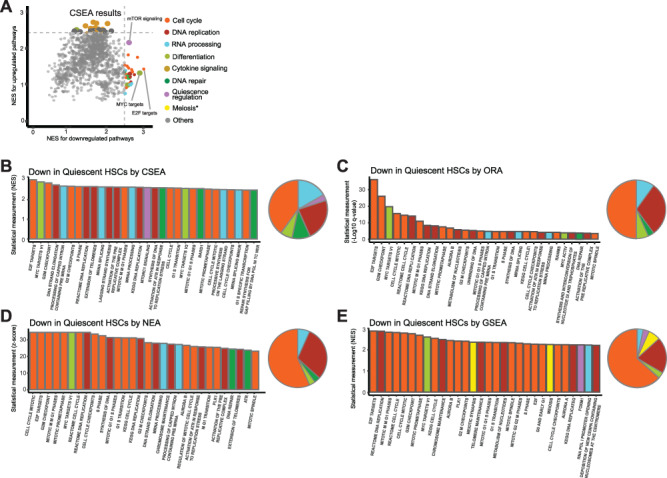
Differentially expressed pathways characteristic of quiescent HSCs revealed by CSEA and comparative analyses with ORA, NEA and GSEA methods. (A) Dot plot showing NESs of the pathways calculated based on the up- or downregulated gene lists in quiescent HSCs compared to active HSCs. Top 30 up- and downregulated genes-related pathways were painted with colors based on their classifications. (B) Bar chart showing the NES of top 30 downregulated pathways in quiescent HSCs compared to active HSCs revealed by CSEA. (C) Bar chart showing the −log 10 of q values for top 30 downregulated pathways in quiescent HSCs compared to active HSCs revealed by ORA. (D) Bar chart showing the z scores of top 30 downregulated pathways in quiescent HSCs compared to active HSCs revealed by NEA. (E) Bar chart showing the NES of top 30 downregulated pathways in quiescent HSCs compared to active HSCs revealed by GSEA. The pathways illustrated in B–E were painted with same colors as in A based on their classifications. The pie charts in B–E show the distribution of top 30 enriched pathways in different pathway classifications.
Next, we compared CSEA with the two most common classes of pathway enrichment analyses for nominal experimental gene list—ORA [43] and NEA [3]. We also compared CSEA with the GSEA method for pathway interpretation from gene expression data of continuous variables. Our result showed that CSEA achieved more balanced detection of different levels of pathway alterations discussed above than the other three methods while avoiding biased pathways such as meiosis-related pathways. ORA, NEA and GSEA tends to detect the pathways whose gene expressions are mostly altered, such as cell cycle, but less sensitive to multiple levels of pathway changes. This may be attributed to the capability of CSEA in deep functional assessment of the experimental gene list. Together, this result supports the utility of CSEA in pathway discovery from single-cell transcriptomics and its advantage over other popular methods on interpreting complex pathway changes.
Discussion
As our main motivation, we sought to develop methods for quantifying new gene and gene set functions that took a broader approach, not solely relying on intersecting genes or interactome network topology. To achieve this, we exploited other types of molecular knowledge to help inform the strength of functional relationships. This novel approach allows the functional assessments of genes and gene sets at a much deeper level, and the general framework can be integrated into other integrative gene prioritization methods that are based on complementary genomic information, such as mutations and other molecular data. We begin by demonstrating how knowledge database redundancy affects the performance of the ConSig algorithm and then introduce the new uniConSig algorithm, illustrating its improvement with gene function discovery. This innovation allows this algorithm to take advantage of the molecular concept data sets compiled from varying sources without bias from data redundancy.
We then move beyond gene prioritization to show how our general approach can be used to assess the functional relations between gene sets as well and developed the CSEA. Our analysis of the mutant TP53 inhibition and CHD1 knockout data sets exemplifies how CSEA uses the genes that were indirectly related to pathways but nonetheless caused those pathways to be enriched. Such deep interpretation cannot be achieved by other algorithms, such as Fisher’s exact test or GSEA, which purport to do the similar analysis but are limited by their reliance on known genes in the pathways. We have shown that the CSEA algorithm is able to reliably assess the functional relationships between pathways and experimental genes to identify the critical pathways altered following genetic perturbation. The preponderance of highly downregulated genes, which are functionally associated with the top-ranked pathway but not on the pathway gene list, highlights the need for the algorithms that can identify the actual functional relationships between gene sets, rather than relying upon assessing the levels of numerical overlaps between the gene sets as in the current modalities, which are severely limited in discovering functionally related pathways. Furthermore, a major advantage of CSEA over the approach based on interactome network is that CSEA is grounded on the framework of the vast knowledge databases and thus can comprehensively assess all functional aspects when computing the functional relations. Considering these advantages, CSEA will have broad applications on the discovery of pathways that are enriched in experimental defined gene sets.
More important, through its capability of deep functional assessment of experimental gene lists, CSEA will be particularly useful for pathway discovery from single-cell transcriptomics, for which pathway analysis is severely limited by the low coverage of the current single-cell sequencing technology. Through analysis of a single-cell transcriptomic data set comparing the active and quiescent populations of HSCs, we demonstrate the excellent performance of CSEA in identifying the signature pathways characteristic of quiescent HSCs, providing the pathway insights not previously reported. Through deep functional assessment of the experimental gene set, CSEA achieved more balanced detection of different levels of pathway changes during HSC quiescence than ORA, NEA and GSEA methods. This suggests that CSEA could be particularly useful for interpreting complex pathway changes such as during change of cell state.
Taken together, the uniConSig algorithm can be used to investigate the causal genes of any disease or the functional genes in any pathways, provided that an initial list of known functional genes can be curated. The CSEA algorithm can be used to investigate the pathways enriched in an experimentally defined gene list, such as over- or underexpressed genes as well as mutated, amplified, deleted or polymorphism genes related to disease predisposition, causation, progression, therapeutic resistance, etc. This tool kit provides a framework for investigating the function of genomes and generating the hypotheses that link individual genes to functions, pathways and diseases and link pathways to gene expression alterations, genetic aberrations, diseases, etc. As more knowledge about the genomes are acquired, the compendium of molecular concepts will become more thorough and robust, which in turn will allow these algorithms to provide ever more powerful calculations and predictions.
Authors’ contributions
X.C. performed the bioinformatics analysis, optimized the algorithms, analyzed the data and cowrote the manuscript. M.A.S. helped algorithm development, provided statistical support to the bioinformatics analysis, analyzed the data and revised the manuscript. M.A. performed the initial bioinformatics analysis. P.H. and S.P. wrote the original R script of the uniConSig algorithm. S.L. and M.W. revised the manuscript. X-S.W. designed the study, conceived the algorithms, supervised and performed bioinformatics analysis, analyzed the data and wrote the manuscript.
Data availability
The uniConSig and CSEA methods are embodied in R packages (https://github.com/wangxlab/uniConSig). The input files and the result files of our analyses are available from https://github.com/wangxlab/uniConSig/tree/master/AdditionalData.
Key Points
Robust algorithms that can quantify the novel functional relations between genes, gene sets and pathways will be of utmost importance for interpreting the wealth of genomic data sets.
uniConSig analysis is a novel algorithm for computing the new functions of genes underlying any biological or pathological process based on their association with the signature molecular concepts.
CSEA computes the functional relationship between genomics-defined gene sets and pathways grounded on the framework of shared concept signatures, which enables deep assessments of the functional relations.
These algorithms will offer powerful new tools for investigating the genome functions by taking a much deeper approach for functional assessments than currently available methods.
Through its capability of deep functional assessment of experimental gene lists, CSEA will be particularly useful for pathway discovery from single-cell transcriptomics, for which pathway analysis is severely limited by the low coverage of the current single-cell sequencing technology.
Supplementary Material
Acknowledgments
This research was supported in part by the University of Pittsburgh Center for Research Computing (CRC) through the resources provided. We specifically acknowledge the assistance of Fangping Mu and Kim F. Wong from CRC. This work also used the Extreme Science and Engineering Discovery Environment, which is supported by National Science Foundation Grant Number OCI-1053575. Specifically, it used the Bridges system, which is supported by NSF Award Number ACI-1445606 at the Pittsburgh Supercomputing Center.
Xu Chi is a post-doctoral researcher in the Department of Pathology, School of Medicine, at the University of Pittsburgh. He has been working in the field of bioinformatics and genomics. Current address: CAS Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, China.
Maureen A. Sartor is an associate professor in the Department of Computational Medicine & Bioinformatics, Medical School, at the University of Michigan. She has been working in the field of bioinformatics and biostatistics.
Sanghoon Lee is a graduate student in the Department of Biomedical Informatics at the University of Pittsburgh. His research area is bioinformatics and machine learning.
Meenakshi Anurag is an assistant professor in the Department of Medicine at the Baylor College of Medicine. Her research specialties are cancer multiomics analysis and next-gen sequencing (NGS) data analysis.
Snehal Patil is an application programmer in the Department of Computational Medicine & Bioinformatics at the University of Michigan. She is an expert in information science.
Pelle Hall is a researcher in the Department in Computational Medicine & Bioinformatics at the University of Michigan. His research area is bioinformatics and statistics.
Matthew Wexler is a graduate student in the Department of Pathology, School of Medicine at the University of Pittsburgh. He has been researching in the field of cancer genomics.
Xiaosong Wang is an associate professor in the Department of Pathology and Biomedical Informatics, School of Medicine, at the University of Pittsburgh. He has been researching in the field of cancer genomics, bioinformatics, and Molecular Cancer Biology.
Funding
This study was supported by National Institutes of Health Grants 1R01CA181368 (XS.W.) and 1R01CA183976 (X-S.W.), the Michigan Lifestage Environmental Exposures and Disease National Institute of Environmental Health Sciences (NIEHS) Center of Excellence (P30 ES017885) (M.A.S.), the Commonwealth of PA Tobacco Phase 15 Formula Fund (X-S.W.), the Shear Family Foundation and the Hillman Foundation.
REFERENCES
- 1. de CA, Neale BM, Heskes T, et al. The statistical properties of gene-set analysis. Nat Rev Genet 2016;17:353–64. [DOI] [PubMed] [Google Scholar]
- 2. Wang Q, Sun J, Zhou M, et al. A novel network-based method for measuring the functional relationship between gene sets. Bioinformatics 2011;27:1521–8. [DOI] [PubMed] [Google Scholar]
- 3. Jeggari A, Alekseenko Z, Petrov I, et al. EviNet: a web platform for network enrichment analysis with flexible definition of gene sets. Nucleic Acids Res 2018;46:W163–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Glaab E, Baudot A, Krasnogor N, et al. TopoGSA: network topological gene set analysis. Bioinformatics 2010;26:1271–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tomlins SA, Mehra R, Rhodes DR, et al. Integrative molecular concept modeling of prostate cancer progression. Nat Genet 2007;39:41–51. [DOI] [PubMed] [Google Scholar]
- 6. Wang XS, Prensner JR, Chen G, et al. An integrative approach to reveal driver gene fusions from paired-end sequencing data in cancer. Nat Biotechnol 2009;27:1005–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Veeraraghavan J, Tan Y, Cao XX, et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun 2014;5:4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim JA, Tan Y, Wang X, et al. Comprehensive functional analysis of the tousled-like kinase 2 frequently amplified in aggressive luminal breast cancers. Nat Commun 2016;7:12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu L, Liang Y, Cao X, et al. Identification of MYST3 as a novel epigenetic activator of ERalpha frequently amplified in breast cancer. Oncogene 2017;36:2910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan Y, Ge N, Wang X, et al. Amplification and over-expression of MAP 3K3 gene in human breast cancer promotes formation and survival of breast cancer cells. J Pathol 2014;232:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kohler S, Bauer S, Horn D, et al. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet 2008;82:949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nitsch D, Tranchevent LC, Goncalves JP, et al. PINTA: a web server for network-based gene prioritization from expression data. Nucleic Acids Res 2011;39:W334–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen J, Bardes EE, Aronow BJ, et al. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009;37:W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fontaine JF, Priller F, Barbosa-Silva A, et al. Genie: literature-based gene prioritization at multi genomic scale. Nucleic Acids Res 2011;39:W455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. James RA, Campbell IM, Chen ES, et al. A visual and curatorial approach to clinical variant prioritization and disease gene discovery in genome-wide diagnostics. Genome Med 2016;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adie EA, Adams RR, Evans KL, et al. SUSPECTS: enabling fast and effective prioritization of positional candidates. Bioinformatics 2006;22:773–4. [DOI] [PubMed] [Google Scholar]
- 17. JMS DS, Markus S. GeneDistiller—distilling candidate genes from linkage intervals. PLoS One 2008;3:e3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Makita Y, Kobayashi N, Yoshida Y, et al. PosMed: ranking genes and bioresources based on semantic web association study. Nucleic Acids Res 2013;41:W109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pers TH, Dworzynski P, Thomas CE, et al. MetaRanker 2.0: a web server for prioritization of genetic variation data. Nucleic Acids Res 2013;41:W104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tranchevent LC, Ardeshirdavani A, ElShal S, et al. Candidate gene prioritization with Endeavour. Nucleic Acids Res 2016;44:W117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van S, Thienpont B, Menten B, et al. Mapping biomedical concepts onto the human genome by mining literature on chromosomal aberrations. Nucleic Acids Res 2007;35:2533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu W, Wulf A, Liu T, et al. Gene prospector: an evidence gateway for evaluating potential susceptibility genes and interacting risk factors for human diseases. BMC Bioinformatics 2008;9:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van MA, Cuelenaere K, Kemmeren PP, et al. GeneSeeker: extraction and integration of human disease-related information from web-based genetic databases. Nucleic Acids Res 2005;33:W758–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie B, Agam G, Balasubramanian S, et al. Disease gene prioritization using network and feature. J Comput Biol 2015;22:313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Radivojac P, Clark WT, Oron TR, et al. A large-scale evaluation of computational protein function prediction. Nat Methods 2013;10:221–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zou D, Ma L, Yu J, et al. Biological databases for human research. Genom Proteom Bioinf 2015;13:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brown GR, Hem V, Katz KS, et al. Gene: a gene-centered information resource at NCBI. Nucleic Acids Res 2015;43:D36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sayers EW, Barrett T, Benson DA, et al. Database resources of the National Center for biotechnology information. Nucleic Acids Res 2012;40:D13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sayers EW, Barrett T, Benson DA, et al. Database resources of the National Center for biotechnology information. Nucleic Acids Res 2011;39:D38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sayers EW, Barrett T, Benson DA, et al. Database resources of the National Center for biotechnology information. Nucleic Acids Res 2010;38:D5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2015;44:D457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kutmon M, Riutta A, Nunes N, et al. WikiPathways: capturing the full diversity of pathway knowledge. Nucleic Acids Res 2015;44:D488–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, et al. The BioGRID interaction database: 2015 update. Nucleic Acids Res 2015;43:D470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cerami EG, Gross BE, Demir E, et al. Pathway commons, a web resource for biological pathway data. Nucleic Acids Res 2011;39:D685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hu Z, Chang YC, Wang Y, et al. VisANT 4.0: integrative network platform to connect genes, drugs, diseases and therapies. Nucleic Acids Res 2013;41:W225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Futreal PA, Coin L, Marshall M, et al. A census of human cancer genes. Nat Rev Cancer 2004;4:177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hamosh A, Scott AF, Amberger JS, et al. Online Mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005;33:D514–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Freed-Pastor WA, Mizuno H, Zhao X, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012;148:244–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao D, Lu X, Wang G, et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature 2017;542:484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang J, Tanaka Y, Seay M, et al. Single cell transcriptomics reveals unanticipated features of early hematopoietic precursors. Nucleic Acids Res 2017;45:1281–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Merid SK, Goranskaya D, Alexeyenko A. Distinguishing between driver and passenger mutations in individual cancer genomes by network enrichment analysis. BMC Bioinformatics 2014;15:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Marchler-Bauer A, Derbyshire MK, Gonzales NR, et al. CDD: NCBI's conserved domain database. Nucleic Acids Res 2015;43:D222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Olbrot M, Rud J, Moss LG, et al. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A 2002;99:6737–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tessem JS, Moss LG, Chao LC, et al. Nkx6.1 regulates islet β-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc Natl Acad Sci U S A 2014;111:5242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang H, Chu W, Das SK, et al. Liver pyruvate kinase polymorphisms are associated with type 2 diabetes in northern European Caucasians. Diabetes 2002;51:2861–5. [DOI] [PubMed] [Google Scholar]
- 49. Ma X, Xu L, Gavrilova O, et al. Role of forkhead box protein A3 in age-associated metabolic decline. Proc Natl Acad Sci U S A 2014;111:14289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu S, Sun F, Li W, et al. Apelin stimulates glucose uptake through the PI3K/Akt pathway and improves insulin resistance in 3T3-L1 adipocytes. Mol Cell Biochem 2011;353:305–13. [DOI] [PubMed] [Google Scholar]
- 51. Perez-Castro AJ, Freire R. Rad9B responds to nucleolar stress through ATR and JNK signalling, and delays the G1-S transition. J Cell Sci 2012;125:1152–64. [DOI] [PubMed] [Google Scholar]
- 52. Kaneko Y, Daitoku H, Komeno C, et al. CTF18 interacts with replication protein a in response to replication stress. Mol Med Rep 2016;14:367–72. [DOI] [PubMed] [Google Scholar]
- 53. Liberzon A. A description of the molecular signatures database (MSigDB) web site. Methods Mol Biol 2014;1150:153–60. [DOI] [PubMed] [Google Scholar]
- 54. Liberzon A, Birger C, Thorvaldsdottir H, et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meunier B, de SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem Rev 2004;104:3947–80. [DOI] [PubMed] [Google Scholar]
- 56. Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 2016;16:732–49. [DOI] [PubMed] [Google Scholar]
- 57. Clendening JW, Pandyra A, Boutros PC, et al. Dysregulation of the mevalonate pathway promotes transformation. Proc Natl Acad Sci U S A 2010;107:15051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jo Y, Debose-Boyd RA. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit Rev Biochem Mol Biol 2010;45:185–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sekiya A, Morishita E, Maruyama K, et al. Fluvastatin upregulates the expression of tissue factor pathway inhibitor in human umbilical vein endothelial cells. J Atheroscler Thromb 2015;22:660–8. [DOI] [PubMed] [Google Scholar]
- 60. Pan G, Bauer JH, Haridas V, et al. Identification and functional characterization of DR6, a novel death domain-containing TNF receptor. FEBS Lett 1998;431:351–6. [DOI] [PubMed] [Google Scholar]
- 61. Dell'Accio F, De C, El NM, et al. Activation of WNT and BMP signaling in adult human articular cartilage following mechanical injury. Arthritis Res Ther 2006;8:R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jo HY, Kim SY, Lee S, et al. Kir3.1 channel is functionally involved in TLR4-mediated signaling. Biochem Biophys Res Commun 2011;407:687–91. [DOI] [PubMed] [Google Scholar]
- 63. Okabe H, Satoh S, Furukawa Y, et al. Involvement of PEG10 in human hepatocellular carcinogenesis through interaction with SIAH1. Cancer Res 2003;63:3043. [PubMed] [Google Scholar]
- 64. Habelhah H, Frew IJ, Laine A, et al. Stress-induced decrease in TRAF2 stability is mediated by Siah2. EMBO J 2002;21:5756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Arora R, Schmitt D, Karanam B, et al. Inhibition of the Warburg effect with a natural compound reveals a novel measurement for determining the metastatic potential of breast cancers. Oncotarget 2015;6:662–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gu JM, Wang DJ, Peterson JM, et al. An NF-kappaB—EphrinA5-dependent communication between NG2(+) interstitial cells and myoblasts promotes muscle growth in neonates. Dev Cell 2016;36:215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kwon JS, Everetts NJ, Wang X, et al. Controlling depth of cellular quiescence by an Rb-E2F network switch. Cell Rep 2017;20:3223–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wilson A, Murphy MJ, Oskarsson T, et al. C-myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 2004;18:2747–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Huttmann A, Liu SL, Boyd AW, et al. Functional heterogeneity within rhodamine123(lo) Hoechst33342(lo/sp) primitive hemopoietic stem cells revealed by pyronin Y. Exp Hematol 2001;29:1109–16. [DOI] [PubMed] [Google Scholar]
- 70. Robb L. Cytokine receptors and hematopoietic differentiation. Oncogene 2007;26:6715–23. [DOI] [PubMed] [Google Scholar]
- 71. Beerman I, Seita J, Inlay MA, et al. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014;15:37–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The uniConSig and CSEA methods are embodied in R packages (https://github.com/wangxlab/uniConSig). The input files and the result files of our analyses are available from https://github.com/wangxlab/uniConSig/tree/master/AdditionalData.
Key Points
Robust algorithms that can quantify the novel functional relations between genes, gene sets and pathways will be of utmost importance for interpreting the wealth of genomic data sets.
uniConSig analysis is a novel algorithm for computing the new functions of genes underlying any biological or pathological process based on their association with the signature molecular concepts.
CSEA computes the functional relationship between genomics-defined gene sets and pathways grounded on the framework of shared concept signatures, which enables deep assessments of the functional relations.
These algorithms will offer powerful new tools for investigating the genome functions by taking a much deeper approach for functional assessments than currently available methods.
Through its capability of deep functional assessment of experimental gene lists, CSEA will be particularly useful for pathway discovery from single-cell transcriptomics, for which pathway analysis is severely limited by the low coverage of the current single-cell sequencing technology.