

Abstract
Genome editing to correct a defective β-globin gene or induce fetal globin (HbF) for patients with beta-hemoglobinopathies has the potential to be a curative strategy available to all. HbF reactivation has long been an area of intense interest given the HbF inhibition of sickle hemoglobin (HbS) polymerization. Patients with HbS who also have high HbF tend to have less severe or even minimal clinical manifestations. Approaches to genetically engineer high HbF include de novo generation of naturally occurring hereditary persistence of fetal hemoglobin (HPFH) mutations, editing of transcriptional HbF repressors or their binding sites and/or regulating epigenetic intermediates controlling HbF expression. Recent preclinical and early clinical trial data show encouraging results; however, long-term follow-up is lacking, and the safety and efficacy concerns of genome editing remain.
Introduction
Beta-globin disorders, including sickle cell disease (SCD) and beta-thalassemia, are the most common monogenic disorders worldwide, with more than 600 000 newborns affected each year (1,2). Supportive care, vaccines and antibiotic treatment in the developed world have improved life expectance and quality of life for SCD; however, the majority of the disease burden is in the developing world, particularly sub-Saharan Africa and the Middle East, where mortality remains high. Currently, the only curative option is allogeneic hematopoietic stem cell (HSC) transplantation, and it is mostly utilized for patients with severe disease and a matched-sibling donor. The scarce availability of having a human leukocyte antigen (HLA)-matched donor (<15%) limits the wide use of this approach (3,4). Research to expand donor options is underway; however, allogeneic transplantation carries the risk for graft-versus-host disease (GVHD), graft rejection, and transplant-related morbidity and mortality. Genetic modification of autologous HSCs is, therefore, an appealing option that would be available to all patients and would avoid the morbidity and mortality associated with GVHD. Such systems include gene addition versus gene editing strategies, with the former currently having more available outcome data. Recent advances in viral gene delivery systems have demonstrated the efficient transfer of healthy adult β-globin, γ-globin or modified version of β-globin (βT87Q-globin) with enhanced anti-sickling properties (5,6). Although gene addition approaches are beyond the scope of the review, clinical trials with βT87Q-globin gene addition show encouraging results, demonstrating circumvention of the disease phenotype and transition to a transfusion-independent stage (7,8).
The field of genome editing has grown quickly given technological advances, specifically with the introduction of the safer, more efficient and relatively inexpensive clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system. The CRISPR/Cas system is, therefore, attractive for ex vivo correction of defective β-globin genes, allowing for autologous HSC correction and transplantation (Fig. 1). However, this process is challenging as homology-directed repair (HDR) is less efficient compared to non-homologous end joining (NHEJ), and therefore, requires more optimization in ex vivo culture conditions and animal models before transitioning into clinical trials (9).
Figure 1.
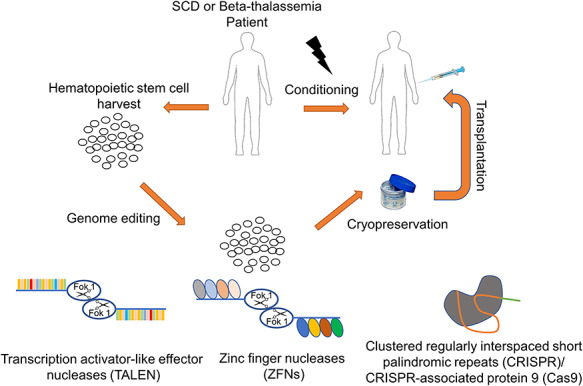
Ex vivo genome editing with TALENs, ZFNs and CRISPR/Cas9 for autologous hematopoietic stem cell transplantation for beta-hemoglobinopathy patients.
Naturally occurring elevated fetal hemoglobin (α2γ2, HbF) termed hereditary persistence of fetal hemoglobin (HPFH) occurs rarely in some individuals. When co-inherited with a hemoglobinopathy (i.e. SCD) the disease phenotype is significantly less severe (10). While beta-hemoglobinopathies have been studied for more than a century, there are only four FDA-approved drugs for the treatment of SCD, three of which were only recently approved between 2017 and 2019. The most utilized therapy, hydroxyurea, induces partial benefit due to HbF induction; however, no drug fully eliminates the complications of the disease, and there is significant patient-dependent variability (11). Genetic strategies aimed at reactivating HbF, recapitulating the phenotype seen in patients with naturally occurring HFPF, offer a potential definitive solution for disease- and treatment-free survival.
Globin switching and the importance of HbF for beta-hemoglobinopathies
HbF is the most abundant hemoglobin produced by the fetus in utero and is gradually replaced by adult globin (α2β2, HbA) after birth. By approximately 6 months of life, the contribution of HbF is generally less than 1%. The concentration of HbF is not pancellular; HbF is not evenly distributed among red blood cells (RBCs); rather it is concentrated in particular cells (termed F-cells). The protective effect of high HbF was first noted in 1945 when a delay in RBC sickling was noted in newborns with SCD compared to their affected mothers (12) and was confirmed when milder clinical outcomes were noted in patients with HPFH (13). High HbF offsets defective β-globin production in hemoglobinopathies and additionally exerts a protective anti-sickling effect in SCD. γ-globin reduces sickling in RBCs of SCD patients by reducing the overall concentration of sickle hemoglobin (α2βS2, HbS) and by forming either non-polymerizing HbF tetramers (α2γ2) or hybrid tetramers (α2γβS), which enter sickle polymerization with a much lesser extent than HbS (14).
Given the protective effects of high HbF in hemoglobinopathies, research efforts have focused on approaches that induce HbF. Multiple strategies are being explored and include the use of chemical inducers or small molecules, silencing transcriptional γ-globin repressors, de novo generation of elevated HbF-related mutations or reactivation γ-globin expression using forced chromatin looping vectors (5). Here we discuss the current status of the research on HbF induction through genome editing applications.
Genome editing for HbF induction
Genome editing relies on the creation of a double-strand break (DSB) in a sequence-specific manner followed by repair by either NHEJ or HDR in the presence of a donor template. NHEJ changes the genomic sequence through small insertions/deletions (INDEL), resulting in interrupted expression of the targeted gene or by changing the sequence motif required for protein binding. Given cell preference for NHEJ over HDR, and that the creation of INDELs is sufficient for the various approaches to induce HbF, derepressing HbF has been a natural first choice for gene editing in hemoglobinopathies. The most commonly used genome editing tools are zinc finger nucleases (ZFNs), transcription-activator like effector nucleases (TALENs) and the CRISPR/Cas9 system. While all three systems have their own limitations, the biggest advantage of the CRISPR/Cas9 system is its ease of use, low relative cost and general efficiency (11). Approaches to induce HbF in adult RBCs using these tools include creating natural mutations related with high HbF, knocking out HbF repressors and regulating epigenetic intermediates controlling HbF expression.
Generation of HPFH mutations
The human β-globin locus is composed of five globin genes (β-, Aγ-,Gγ-, ε- and δ-globin) located on a short region of chromosome 11. Naturally occurring HPFH mutations present with either small or large deletions within the human β-globin locus, leading to pancellular expression of elevated HbF levels (~30%) (Fig. 2). Early attempts to mimic the naturally occurring Sicilian HPFH mutation in human CD34+ cells demonstrated significant HbF induction by two mechanisms: generation of the ~13 kb deletion of the β-globin locus and creation of a 13 nt deletion in the γ-globin promoter sequence (15,16). A more detailed investigation deleted a 3.5 kb γ-δ intergenic region containing the putative HbF repressor protein (BCL11A) binding site, a 7.2 kb Corfu region and a larger 13.6 kb deletion, starting from the 5′ breakpoint of the Corfu deletion and extending further to 3′ to include the promoter and first exon of the β-globin gene (17). In patients with SCD, this 13.6 kb deletion would simultaneously upregulate γ-globin synthesis and inactivate the βS-globin gene. Disruption of just the putative HbF silencer resulted in a mild increase in γ-globin expression, whereas deletion or inversion of the 13.6 kb region caused a robust reactivation of HbF synthesis in adult erythroblasts. This work confirmed that gene competition between fetal and adult globins may limit the degree of HbF induction and that chromosomal configuration plays a critical role given that the inversion of the 13.6 kb area was as effective as the deletion of the site. Further mechanistic dissection studies revealed that point mutations at −115 (BCL11A binding site) and −200 bp (ZBTB7A, also known as LRF, binding site) upstream of the transcription start site of γ-globin gene enhanced robust HbF induction in the immortalized RBC progenitor cell line, HUDEP-2 (18). As a different strategy, a −115 and −114 bp sequence alteration was created using a base editor (hA3A-BE3) to avoid the deleterious effect of DSBs and reported potent HbF induction from 6.8–13.7% to 44.2–58.1% in differentiated edited cells (19).
Figure 2.
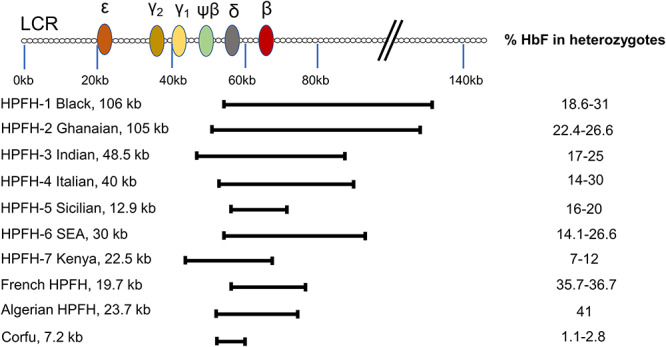
Common hereditary persistence of fetal hemoglobin (HPFH) mutations and relative fetal hemoglobin (HbF) levels in adults. The figure is adapted from (15,70).
To test the feasibility of the de novo HPFH mutation in progenitor cells, NBSGW mice were transplanted with CD34+ cells with a 13 nt BCL11A binding motif deleted (20). There was durable CD34+ engraftment (60%), HbF activation to potentially therapeutic levels (~30%) in RBC progeny, with no detectable genotoxicity reported. In a recent study, disruption of the LRF binding site led to γ-globin induction, correction of the sickling phenotype in SCD patient-derived CD34+ cells and persistence of high editing efficiency in NSG mice (21). In a rhesus-macaque model utilizing a 13 nt deletion of rhesus CD34+ cells, engraftment of 18% of edited cells (>1-year follow-up) with 1–5% γ-globin expression in vivo was recently shown (22). Stable engraftment of edited cells and detectable levels of HbF in rhesus macaques is encouraging; however, optimization is needed to improve engraftment of edited cells, and higher HbF expression is needed to achieve therapeutically meaningful levels.
Editing of transcriptional repressors
Globin expression is controlled by a developmentally regulated mechanism known as globin switching and includes the interaction of transcriptional factors with the upstream enhancer cluster and gene promoters (23). In the last decade, significant research has been devoted to elucidating the underlining mechanism for globin switching. Specific globin expression is thought to be the result of chromosomal looping created through complex interactions of transcriptional factors. Genome-wide association studies (GWAS) identified crucial genomic locations regulating globin expression, for example, that HBS1L-MYB intergenic region and BCL11A are associated with elevated HbF levels (24,25).
In targeting BCL11A as an important repressor for HbF expression, various transcription factors including GATA1, SOX6, KLF1 and MYB have been identified as forming a multiprotein complex interacting with BCL11A in the chromosomal reorganization of globin synthesis (26). Among these, the erythroid-specific transcription factor, KLF1, is proven to exert a dual effect on HbF expression by both direct involvement in the looping construct (27) and acting as an indirect silencer through regulating BCL11A expression (28). Significant HbF activation has been reported in ex vivo models (primary human erythroid cells, K562 cells) for these transcription factors either through direct gene editing (29–31) or disruption of their binding sites (18,32–34). KLF1 and MYB have critical roles in erythropoiesis and hematopoiesis, respectively; however, strategies aimed at most of these transcription factors have not been evaluated in vivo in mice or large-animal models and do not appear to be as strong as BCL11A for the robust HbF induction needed for clinical application (35).
Many studies validate BCL11A as a master HbF regulator (28,36,37). Inactivation of BCL11A prevented clinical complications in a mouse model of SCD through high-level pancellular expression of HbF (28.3 versus 1.3% of total β-like human globins in adult SCD/Bcl11a−/− mice compared with SCD mice) (38). A non-human primate model investigating BCL11A knockout using TALEN mRNA reported a much lower level of indel persistence (~0.3%) and negligible γ-globin induction (~0.05%) at 1-year post-transplantation (39). As rare BCL11A haploinsufficiency in humans is associated with a mild clinical course due to elevated HbF levels (40,41), optimized and efficient BCL11A editing has become an attractive and promising clinical approach. Importantly, however, BCL11A expression is essential for HSC function and B-cell development (42–44); therefore, focus is directed toward knocking down erythroid-specific expression. Identification of the erythroid-specific enhancer for BCL11A expression, intron-2, as sufficient to specify developmentally restricted, erythroid-specific gene expression made such an approach feasible (45). A follow-up study determined pivotal minimal features of the enhancer by targeting a series of gRNAs. Here, the investigators demonstrated that editing of the enhancer using specific gRNA targets induced substantial HbF expression in RBCs without affecting erythropoiesis and the expression levels of BCL11A in other lineages (46). Encouragingly, CD34+ cells edited at the GATA1 binding site of the enhancer by either CRISPR/Cas9 or ZFN systems displayed multilineage engraftment potential (≤95%) in the immunocompromised mouse model (47–49). Ribonucleoprotein (RNP) base editing of the BCL11A enhancer in human HSCs as an alternative to nuclease editing (and therefore the destructive effects of DSB) was validated using an A3A (N57Q)-BE3 base editor and provided comparable levels of HbF induction compared to standard Cas9 application (50).
To test the feasibility and safety of erythroid-specific enhancer editing, we have conducted a rhesus macaque model verifying the persistence of high-level edited cells (up to 80%) with robust γ-globin induction (up to 29%) (51). Consistent with the clinical trials performing ZFN-mediated BCL11A enhancer editing (NCT03653247) (52), and erythroid-specific knockdown of BCL11A expression using a lentiviral vector shmiR (NCT03282656) (53), we have observed a peak in the HbF levels in rhesus macaques at early time points of transplantation followed by a significant drop, possibly due to stress erythropoiesis HbF induction overestimating editing effectiveness at early time points. Another clinical trial evaluating CRISPR/Cas9-based BCL11A enhancer editing has released early data (NCT03745287), demonstrating 10.1 g/dL HbF at 9 months post-transplantation for a patient with β-thalassemia and a total hemoglobin level of 11.3 g/dL with 46.6% HbF at 4 months post-transplantation for a patient with SCD (54). Given concerns for the long-term persistence of HbF induction, long-term follow up data from preclinical and clinical trials are essential to determine the feasibility, safety and efficacy of enhancer editing.
Epigenetic factors are another protein family involved in the regulation of globin expression through chromatin structure reorganization, histone modifications and DNA methylation (55). The majority of this approach uses pharmacological inhibitors to induce HbF by targeting epigenetic mediators (i.e. NuRD/HDAC, DNMT, EHMT and KDM1A) rather than creating permanent gene editing. Oral drugs including decitabine as a DNMT1 inhibitor (NCT01375608), INCB059872 as LSD1 inhibitor (NCT03132324), and panobinostat (NCT01245179) and pomalidomide (NCT01522547) as histone deacetylase inhibitors have been tested for SCD patients in clinical trials with some clinical benefit reported (56). However, long-term safety and efficacy of these drugs are needed. Genome editing tools, in fact, have helped elucidate the HbF potential of these various genes. It was shown that CRISPR/Cas9 based knockout of MBD2 gene of the NuRD complex reactivates HbF in HUDEP-2 cells but not MBD3 (57). A recent study reported a more detailed investigation of the NuRD family protein members for their HbF activation potential using CRISP/Cas9 comprehensive mutagenesis in HUDEP-2 cells (58). The results revealed that only 5 members of the complex (CHD4, GATAD2A, HDAC2, MBD2 and MTA2) were required for HbF silencing. The major hurdle in epigenetic editing, however, seems to be cell specificity as various side effects including neutropenia, thrombocytopenia or thrombophilia have been reported (59) in addition to the various contributions of these intermediates in hematopoiesis (60). Though inhibition of LSD1 has been associated with elevated HbF levels in cell culture conditions, erythroid-specific inhibition could be biologically problematic, for instance, in mouse and large animal models where LSD1 is important for erythroid maturation (61,62).
Challenges and Prospects
Given the hurdles in gene correction efficiency, including a preference for NHEJ over HDR, genome editing by knockout appears to be as effective for HbF induction (63) and is a potential clinically applicable treatment modality for beta-hemoglobinopathies. The erythroid-specific BCL11A enhancer is currently the most sought after candidate for clinical translation; however, long-term data and continued research may identify new targets or combination approaches with higher HbF induction, safer editing or improved engraftment. Cell culture models with reporter globin genes (64), CRISPR/Cas9-based genome-wide screening protocols (29) or nuclease-dead mutants of Cas9 that either block (CRISPRi) or activate (CRISPRa) gene expression (65) may uncover new, improved or potentially synergistic targets.
The safety and specificity of genome editing approaches are critical parameters that will ultimately determine how and when these therapeutic tools become widely clinically available. Recently, two studies demonstrated a significant portion of adults with a pre-existing adaptive immune response to Cas9 (66,67). While these reports did not address the effects of these antibodies on the efficiency of CRISPR/Cas9-based treatments and host safety, these concerns should be addressed in large-animal models and clinical trials. Another critical consideration regarding genome editing is off-target activity. The majority of reports demonstrate no off-target effects in their experimental models; however, off-target detection sensitivities and false-negatives of in vitro off-target assays remain a significant hurdle that must be improved to avoid unwanted phenotypic outcomes.
For most of the editing applications, electroporation of the RNP complex is preferred due to its superior editing efficiency and faster kinetics. However, electroporation-induced HSC toxicity may limit the application for patients with SCD whose HSC quality and quantity are of concern (68). One potential solution to this problem would be the development of toxicity-reducing electroporation solutions (49). Another definitive solution would be the establishment of targeted in vivo delivery of RNPs that does not require stem cell harvest or transplantation. Given ex vivo manipulation and culture of HSCs reduces stem cell property, function and engraftment potential, successful in vivo genome editing would increase the impact of the application (69). An in vivo model may also expand access of these curative strategies to developing countries where resources as less widely available to carry out transplantation approaches.
Recent improvements in genome editing approaches demonstrate HbF induction as a feasible and potentially clinically meaningful treatment modality for beta-hemoglobinopathies. Long-term data showing long-term engraftment with high gene expression is currently lacking and limits the broad application of this therapy. Whether these expensive applications will be successful is yet to be determined, as is the ability to implement such modalities in resource-limited regions such as sub-Saharan Africa where the burden of disease is highest. To be applicable for the largest number of patients, editing systems must be proven safe and effective prior to a transition into a large-scale, cost-effective system that is needed to provide this disease-modifying and potentially curative strategy to patients.
Conflict of Interest Statement. None declared.
References
- 1. Khosravi M.A., Abbasalipour M., Concordet J.-P., Vom Berg J., Zeinali S., Arashkia A., Azadmanesh K., Buch T. and Karimipoor M. (2019) Targeted deletion of BCL11A gene by CRISPR-Cas9 system for fetal hemoglobin reactivation: a promising approach for gene therapy of beta thalassemia disease. Eur. J. Pharmacol., 854, 398–405. [DOI] [PubMed] [Google Scholar]
- 2. Piel F.B., Hay S.I., Gupta S., Weatherall D.J. and Williams T.N. (2013) Global burden of sickle cell anaemia in children under five, 2010–2050: modelling based on demographics, excess mortality, and interventions. PLoS Med., 10, e1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guilcher G.M., Truong T.H., Saraf S.L., Joseph J.J., Rondelli D. and Hsieh M.M. (2018) Curative therapies: allogeneic hematopoietic cell transplantation from matched related donors using myeloablative, reduced intensity, and nonmyeloablative conditioning in sickle cell disease. Semin. Hematol., 55, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walters M., Patience M., Leisenring W., Rogers Z., Aquino V., Buchanan G., Roberts I., Yeager A.M., Hsu L. and Adamkiewicz T. (2001) Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol. Blood Marrow Transplant., 7, 665–673. [DOI] [PubMed] [Google Scholar]
- 5. Demirci S., Uchida N. and Tisdale J.F. (2018) Gene therapy for sickle cell disease: an update. Cytotherapy, 20, 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Orkin S.H. and Bauer D.E. (2019) Emerging genetic therapy for sickle cell disease. Annu. Rev. Med., 70, 257–271. [DOI] [PubMed] [Google Scholar]
- 7. Bonner M., Kanter J., Macari E., Lane R., Lewis G., Coles P., Kassenaar S., Mynampati S., Schulze R. and Hebert M. (2019) The relationships between target gene transduction, engraftment of HSCs and RBC physiology in sickle cell disease gene therapy. Blood, 134, 206. [Google Scholar]
- 8. Lal A., Locatelli F., Kwiatkowski J.L., Kulozik A.E., Yannaki E., Porter J.B., Thuret I., Sauer M.G., Elliot H. and Chen Y. (2019) Northstar-3: interim results from a phase 3 study evaluating Lentiglobin gene therapy in patients with transfusion-dependent β-thalassemia and either a β0 or IVS-I-110 mutation at both alleles of the HBB gene. Blood, 134, 815. [Google Scholar]
- 9. Ghiaccio V., Chappell M., Rivella S. and Breda L. (2019) Gene therapy for beta-hemoglobinopathies: milestones, new therapies and challenges. Mol. Diagn. Ther., 23, 173–186. [DOI] [PubMed] [Google Scholar]
- 10. Stamatoyannopoulos G., Wood W., Papayannopoulou T. and Nute P. (1975) A new form of hereditary persistence of fetal hemoglobin in blacks and its association with sickle cell trait. Blood, 46, 683–692. [PubMed] [Google Scholar]
- 11. Demirci S., Leonard A., Haro-Mora J.J., Uchida N. and Tisdale J.F. (2019) CRISPR/Cas9 for sickle cell disease: applications, future possibilities, and challenges. Adv. Exp. Med. Biol., 5, 37–52. [DOI] [PubMed] [Google Scholar]
- 12. Watson J. (1948) A study of sickling of young erythrocytes in sickle cell anemia. Blood, 3, 465–469. [PubMed] [Google Scholar]
- 13. Conley C.L., Weatherall D.J., Richardson S.N., Shepard M.K. and Charache S. (1963) Hereditary persistence of fetal hemoglobin: a study of 79 affected persons in 15 negro families in Baltimore. Blood, 21, 261–281. [PubMed] [Google Scholar]
- 14. Eaton W.A. and Bunn H.F. (2017) Treating sickle cell disease by targeting HbS polymerization. Blood, 129, 2719–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ye L., Wang J., Tan Y., Beyer A.I., Xie F., Muench M.O. and Kan Y.W. (2016) Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and β-thalassemia. Proc. Natl. Acad. Sci., 113, 10661–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Traxler E.A., Yao Y., Wang Y.-D., Woodard K.J., Kurita R., Nakamura Y., Hughes J.R., Hardison R.C., Blobel G.A. and Li C. (2016) A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med., 22, 987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Antoniani C., Meneghini V., Lattanzi A., Felix T., Romano O., Magrin E., Weber L., Pavani G., El Hoss S. and Kurita R. (2018) Induction of fetal hemoglobin synthesis by CRISPR/Cas9-mediated editing of the human β-globin locus. Blood, 131, 1960–1973. [DOI] [PubMed] [Google Scholar]
- 18. Martyn G.E., Wienert B., Yang L., Shah M., Norton L.J., Burdach J., Kurita R., Nakamura Y., Pearson R.C. and Funnell A.P. (2018) Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet., 50, 498–503. [DOI] [PubMed] [Google Scholar]
- 19. Wang L., Li L., Ma Y., Hu H., Li Q., Yang Y., Liu W., Yin S., Li W. and Fu B. (2020) Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies. Cell Res., 30, 276–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Métais J.-Y., Doerfler P.A., Mayuranathan T., Bauer D.E., Fowler S.C., Hsieh M.M., Katta V., Keriwala S., Lazzarotto C.R. and Luk K. (2019) Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv., 3, 3379–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weber L., Frati G., Felix T., Hardouin G., Casini A., Wollenschlaeger C., Meneghini V., Masson C., De Cian A. and Chalumeau A. (2020) Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci. Adv., 6, eaay9392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E. and Negre O. (2019) Therapeutically relevant engraftment of a CRISPR-Cas9–edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med., 11, eaaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartman C.R., Hsu S.C., Hsiung C.C.-S., Raj A. and Blobel G.A. (2016) Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol. Cell, 62, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Menzel S., Garner C., Gut I., Matsuda F., Yamaguchi M., Heath S., Foglio M., Zelenika D., Boland A. and Rooks H. (2007) A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet., 39, 1197–1199. [DOI] [PubMed] [Google Scholar]
- 25. Uda M., Galanello R., Sanna S., Lettre G., Sankaran V.G., Chen W., Usala G., Busonero F., Maschio A. and Albai G. (2008) Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc. Natl. Acad. Sci., 105, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wienert B., Martyn G.E., Funnell A.P., Quinlan K.G. and Crossley M. (2018) Wake-up sleepy gene: reactivating fetal globin for β-hemoglobinopathies. Trends Genet., 34, 927–940. [DOI] [PubMed] [Google Scholar]
- 27. Deng W., Lee J., Wang H., Miller J., Reik A., Gregory P.D., Dean A. and Blobel G.A. (2012) Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell, 149, 1233–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou D., Liu K., Sun C.-W., Pawlik K.M. and Townes T.M. (2010) KLF1 regulates BCL11A expression and γ-to β-globin gene switching. Nat. Genet., 42, 742–744. [DOI] [PubMed] [Google Scholar]
- 29. Grevet J.D., Lan X., Hamagami N., Edwards C.R., Sankaranarayanan L., Ji X., Bhardwaj S.K., Face C.J., Posocco D.F. and Abdulmalik O. (2018) Domain-focused CRISPR screen identifies HRI as a fetal hemoglobin regulator in human erythroid cells. Science, 361, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shariati L., Khanahmad H., Salehi M., Hejazi Z., Rahimmanesh I., Tabatabaiefar M.A. and Modarressi M.H. (2016) Genetic disruption of the KLF1 gene to overexpress the γ-globin gene using the CRISPR/Cas9 system. J. Gene Med., 18, 294–301. [DOI] [PubMed] [Google Scholar]
- 31. Shariati L., Rohani F., Heidari Hafshejani N., Kouhpayeh S., Boshtam M., Mirian M., Rahimmanesh I., Hejazi Z., Modarres M. and Pieper I.L. (2018) Disruption of SOX6 gene using CRISPR/Cas9 technology for gamma-globin reactivation: an approach towards gene therapy of β-thalassemia. J. Cell. Biochem., 119, 9357–9363. [DOI] [PubMed] [Google Scholar]
- 32. Martyn G.E., Wienert B., Kurita R., Nakamura Y., Quinlan K.G. and Crossley M. (2019) A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site. Blood, 133, 852–856. [DOI] [PubMed] [Google Scholar]
- 33. Fanis P., Kousiappa I., Phylactides M., Kyrri A., Hadjigavriel M., Christou S., Sitarou M. and Kleanthous M. (2019) A novel mutation in the erythroid transcription factor KLF1 is likely responsible for ameliorating β-thalassemia major. Hum. Mutat., 40, 1768–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li B., Zhu X., Hossain M.A., Guy C.R., Xu H., Bungert J. and Pace B.S. (2018) Fetal hemoglobin induction in sickle erythroid progenitors using a synthetic zinc finger DNA-binding domain. Haematologica, 103, e384–e387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Martino M., Sessa R., Storino M.R., Giuliano M., Trombetti S., Catapano R., Bianco A.L., Izzo P. and Grosso M. (2020) Transcriptional repressors of fetal globin genes as novel therapeutic targets in Beta-thalassemia. Beta Thalassemia, 1–9. doi: 10.5772/intechopen.90762. [DOI] [Google Scholar]
- 36. Sankaran V.G., Xu J. and Orkin S.H. (2010) Transcriptional silencing of fetal hemoglobin by BCL11A. Ann. N. Y. Acad. Sci., 1202, 64–68. [DOI] [PubMed] [Google Scholar]
- 37. Sankaran V.G., Xu J., Ragoczy T., Ippolito G.C., Walkley C.R., Maika S.D., Fujiwara Y., Ito M., Groudine M. and Bender M. (2009) Developmental and species-divergent globin switching are driven by BCL11A. Nature, 460, 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu J., Peng C., Sankaran V.G., Shao Z., Esrick E.B., Chong B.G., Ippolito G.C., Fujiwara Y., Ebert B.L. and Tucker P.W. (2011) Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science, 334, 993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Humbert O., Peterson C.W., Norgaard Z.K., Radtke S. and Kiem H.-P. (2018) A nonhuman primate transplantation model to evaluate hematopoietic stem cell gene editing strategies for β-hemoglobinopathies. Mol. Ther. Methods Clin. Dev., 8, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dias C., Estruch S.B., Graham S.A., McRae J., Sawiak S.J., Hurst J.A., Joss S.K., Holder S.E., Morton J.E. and Turner C. (2016) BCL11A haploinsufficiency causes an intellectual disability syndrome and dysregulates transcription. Am. J. Hum. Genet., 99, 253–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Funnell A.P., Prontera P., Ottaviani V., Piccione M., Giambona A., Maggio A., Ciaffoni F., Stehling-Sun S., Marra M. and Masiello F. (2015) 2p15-p16. 1 microdeletions encompassing and proximal to BCL11A are associated with elevated HbF in addition to neurologic impairment. Blood, 126, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu P., Keller J.R., Ortiz M., Tessarollo L., Rachel R.A., Nakamura T., Jenkins N.A. and Copeland N.G. (2003) Bcl11a is essential for normal lymphoid development. Nat. Immunol., 4, 525–532. [DOI] [PubMed] [Google Scholar]
- 43. Luc S., Huang J., McEldoon J.L., Somuncular E., Li D., Rhodes C., Mamoor S., Hou S., Xu J. and Orkin S.H. (2016) Bcl11a deficiency leads to hematopoietic stem cell defects with an aging-like phenotype. Cell Rep., 16, 3181–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsang J.C., Yu Y., Burke S., Buettner F., Wang C., Kolodziejczyk A.A., Teichmann S.A., Lu L. and Liu P. (2015) Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells. Genome Biol., 16, 178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bauer D.E., Kamran S.C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M.C., Smith E.C. and Pinello L. (2013) An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science, 342, 253–257.24115442 [Google Scholar]
- 46. Canver M.C., Smith E.C., Sher F., Pinello L., Sanjana N.E., Shalem O., Chen D.D., Schupp P.G., Vinjamur D.S. and Garcia S.P. (2015) BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature, 527, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chang K.-H., Smith S.E., Sullivan T., Chen K., Zhou Q., West J.A., Liu M., Liu Y., Vieira B.F. and Sun C. (2017) Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Mol. Ther. Methods Clin. Dev., 4, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Psatha N., Reik A., Phelps S., Zhou Y., Dalas D., Yannaki E., Levasseur D.N., Urnov F.D., Holmes M.C. and Papayannopoulou T. (2018) Disruption of the BCL11A erythroid enhancer reactivates fetal hemoglobin in erythroid cells of patients with β-thalassemia major. Mol. Ther. Methods Clin. Dev., 10, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K. and Baricordi C. (2019) Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med., 25, 776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zeng J., Wu Y., Ren C., Bonanno J., Shen A.H., Shea D., Gehrke J.M., Clement K., Luk K. and Yao Q. (2020) Therapeutic base editing of human hematopoietic stem cells. Nat. Med., 26, 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Demirci S., Zeng J., Wu Y., Uchida N., Gamer J., Yapundich M., Drysdale C., Bonifacino A.C., Krouse A.E. and Linde N.S. (2019) Durable and robust fetal globin induction without Anemia in rhesus monkeys following autologous hematopoietic stem cell transplant with BCL11A Erythroid enhancer editing. Blood, 134, 4632. [Google Scholar]
- 52. Smith A.R., Schiller G.J., Vercellotti G.M., Kwiatkowski J.L., Krishnamurti L., Esrick E.B., Williams D.A., Miller W.P., Woolfson A. and Walters M.C. (2019) Preliminary results of a phase 1/2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent Beta thalassemia. Blood, 134, 3544. [Google Scholar]
- 53. Esrick E.B., Achebe M., Armant M., Bartolucci P., Ciuculescu M.F., Daley H., Dansereau C., Di Caprio G., Goncalves B. and Hebert N. (2019) Validation of BCL11A as therapeutic target in sickle cell disease: results from the adult cohort of a pilot/feasibility gene therapy trial inducing sustained expression of fetal hemoglobin using post-transcriptional gene silencing. Blood, 134, LBA-5. [Google Scholar]
- 54. CRISPR (2019) Therapeutics and Vertex Announce Positive Safety and Efficacy Data From First Two Patients Treated with Investigational CRISPR/Cas9 Gene-Editing Therapy CTX001® for Severe Hemoglobinopathies. Zug, Switzerland, CRISPR Therapeutics. [Google Scholar]
- 55. Paikari A. and Sheehan V.A. (2018) Fetal haemoglobin induction in sickle cell disease. Br. J. Haematol., 180, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carden M.A. and Little J. (2019) Emerging disease-modifying therapies for sickle cell disease. Haematologica, 104, 1710–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu X., Azzo A., Bilinovich S.M., Li X., Dozmorov M., Kurita R., Nakamura Y., Williams D.C. and Ginder G.D. (2019) Disruption of the MBD2-NuRD complex but not MBD3-NuRD induces high level HbF expression in human adult erythroid cells. Haematologica, 104, 2361–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sher F., Hossain M., Seruggia D., Schoonenberg V.A., Yao Q., Cifani P., Dassama L.M., Cole M.A., Ren C. and Vinjamur D.S. (2019) Rational targeting of a NuRD subcomplex guided by comprehensive in situ mutagenesis. Nat. Genet., 51, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rivers A., Molokie R. and Lavelle D. (2019) A new target for fetal hemoglobin reactivation. Haematologica, 104, 2325–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goyama S. and Kitamura T. (2017) Epigenetics in normal and malignant hematopoiesis: an overview and update 2017. Cancer Sci., 108, 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kerenyi M.A., Shao Z., Hsu Y.-J., Guo G., Luc S., O'Brien K., Fujiwara Y., Peng C., Nguyen M. and Orkin S.H. (2013) Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. elife, 2, e00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rivers A., Vaitkus K., Ibanez V., Ruiz M.A., Jagadeeswaran R., Saunthararajah Y., Cui S., Engel J.D., DeSimone J. and Lavelle D. (2016) The LSD1 inhibitor RN-1 recapitulates the fetal pattern of hemoglobin synthesis in baboons (P. anubis). Haematologica, 101, 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lomova A., Clark D.N., Campo-Fernandez B., Flores-Bjurström C., Kaufman M.L., Fitz-Gibbon S., Wang X., Miyahira E.Y., Brown D. and DeWitt M.A. (2019) Improving gene editing outcomes in human hematopoietic stem and progenitor cells by temporal control of DNA repair. Stem Cells, 37, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Breveglieri G., Salvatori F., Finotti A., Cosenza L.C., Zuccato C., Bianchi N., Breda L., Rivella S., Bresciani A. and Bisbocci M. (2019) Development and characterization of cellular biosensors for HTS of erythroid differentiation inducers targeting the transcriptional activity of γ-globin and β-globin gene promoters. Anal. Bioanal. Chem., 411, 7669–7680. [DOI] [PubMed] [Google Scholar]
- 65. Kampmann M. (2018) CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem. Biol., 13, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L. and Bode N.M. (2019) Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med., 25, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Simhadri V.L., McGill J., McMahon S., Wang J., Jiang H. and Sauna Z.E. (2018) Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Mol. Ther. Methods Clin. Dev., 10, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Leonard A., Bonifacino A., Dominical V.M., Luo M., Haro-Mora J.J., Demirci S., Uchida N., Pierciey F.J. Jr. and Tisdale J.F. (2019) Bone marrow characterization in sickle cell disease: inflammation and stress erythropoiesis lead to suboptimal CD34 recovery. Br. J. Haematol., 186, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cox D.B.T., Platt R.J. and Zhang F. (2015) Therapeutic genome editing: prospects and challenges. Nat. Med., 21, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Joly P., Lacan P., Garcia C., Couprie N. and Francina A. (2009) Identification and molecular characterization of four new large deletions in the β-globin gene cluster. Blood Cells Mol. Dis., 43, 53–57. [DOI] [PubMed] [Google Scholar]