

Abstract
Nanobodies (nAbs) are recombinant antigen binding variable domain fragments obtained from the heavy chain only immunoglobulins. Among mammals these are unique to camelids (camels, llamas, alpacas, etc.). Nanobodies are of great use in biomedical research due to their efficient folding and stability under a variety of conditions, as well as their small size. The latter characteristic is particularly important for nAbs used as immunolabeling reagents since this can improve penetration of cell and tissue samples compared to conventional antibodies, as well as reduce the gap distance between signal and target thereby improving imaging resolution. In addition, their recombinant nature allows for unambiguous definition and permanent archiving in the form of DNA sequence, enhanced distribution in the form of sequences or plasmids, and easy and inexpensive production using well-established bacterial expression systems, such as the IPTG induction method described here. This unit will review the basic workflow and process for developing, screening and validating novel nAbs against neuronal target proteins. The protocols described make use of the most common nAb development method, wherein an immune repertoire from an immunized llama is screened via phage display technology. Selected nAbs can then be taken through validation assays for use as immunolabels or as intrabodies in neurons.
Keywords: nanobodies, recombinant antibodies, phage display, ELISA, immunocytochemistry, immunohistochemistry, intrabodies
INTRODUCTION
Nanobodies (nAbs) are recombinant antigen binding fragments derived from heavy chain only immunoglobulins and as such are also termed VHH fragments (Muyldermans, 2013) versus the VH fragments obtained from conventional heavy chains. nAbs are of particular interest due to their small size as compared to Fab, Fv or scFv derived from conventional heavy and light chain antibodies, and are the smallest functional antibody-derived fragment (MW ≈ 15 kD or 1/10 the molecular mass of a conventional IgG antibody). The small size of nAbs is especially useful in immunolabeling, allowing for improved sample penetration and image resolution over conventional antibodies. In addition, they are well suited for use as intracellular antibodies, or intrabodies, since they are stable in the range of conditions present in the cell, including the reducing cytoplasmic environment of mammalian neurons (Dong et al., 2019). Other advantages of nAbs are 1) that only one polypeptide domain needs to be cloned and expressed, 2) expression in bacteria is typically higher than that of antigen-binding fragments from conventional antibodies, and 3) nAbs exhibit a high degree of stability as compared to other miniaturized antibody forms. As recombinant antibodies they also have unambiguous molecular definition, effective archiving as plasmids or as DNA sequence, and the potential for engineering to enhance their antigen binding properties, incorporate epitope tags, site-specific labeling sequences, or other fusion partners to enhance detection of, and or, confer biological function to the nAb.
While there are a variety of methods to identify and isolate target-specific nanobodies, the one most commonly cited in the literature is based on the technique known as phage display (Smith, 1985). Phage display is a method based on the presentation of molecular libraries on the surface of certain bacteriophages (Bradbury, 2010). The molecule to be displayed, in this case a nAb, is expressed as a fusion with a phage coat protein. The bacteriophages most commonly used for this technology are E. coli filamentous bacteriophages (f1, fd, M13), which are specific to F plasmid-containing E. coli bacteria and which do not kill their host during infection. Displayed peptides (in this case nAbs) are typically fused with the coding region of the minor phage coat protein (pIII), causing loss of coat protein functionality which can be compensated by use of hybrid phages. These consist of a complete wild-type genome and a copy of the fusion gene, commonly in a phagemid vector. Phagemids are plasmids that contain origins of replication for phage and bacteria, the pIII gene with appropriate cloning sites, and antibiotic resistance genes. However, they require the assistance of “helper phage” for packing into M13 particles. These helper phage (such as M13KO7) supply all the remaining structural proteins for generation of the virion while containing a slightly defective origin of replication. This allows for preferential packaging of the phagemid genome, which will be important during selection experiments.
The protocols described here detail the steps involved in development of novel nAbs for use as immunolabels and intrabodies. The immune repertoire isolated from llamas immunized with the target antigen is turned into a phage display library, using the pComb3XSS phagemid (Andris-Widhopf, Rader, Steinberger, Fuller, & Barbas, 2000). This vector is designed to express the nAb (the VHH antibody fragments) fused to the carboxy-terminal domain of pIII, with insertion of the gene being aided by the unique SfiI restriction enzyme sites. The fusion junction also includes an amber stop codon between the 3’ SfiI site and the 5’ end of gene III, thus allowing for expression of the nAb without the pIII fusion partner in non-suppressor bacterial strains. The junction region also includes 6XHis and HA tags C-terminal to the nAb but prior to the amber stop codon for ease of detection and purification. Once constructed, the phage library undergoes repeated cycles of panning against the target protein to serially increase the representation of phage displaying target-specific nAbs, which are then subjected to further screening and validation assays.
The pComb3XSS phagemid vector allows use of the lac operator-repressor inducible expression system (Andris-Widhopf et al., 2000). This results in secretion of the expressed nAbs into the periplasm, due to the presence of the OmpA leader sequence fused to the N-terminus of the nAb coding sequence (Choi & Lee, 2004). It is a common procedure to then extract the periplasmic proteins for recovery of the nAb prior to testing (Pardon et al., 2014). However, in an effort to work in a more high-throughput manner, and parallel to the use of conditioned media from hybridomas or COS-1 (i.e., tissue culture supernatants) for antibody screening (Andrews et al., 2019; Gong, Murray, & Trimmer, 2016), this protocol makes use of bacterial culture supernatants (BCS) for screening of the nAbs. The use of BCS eliminates the need to perform periplasmic protein extraction and nAb purification during the screening and validation phases, thus simplifying the workflow and increasing the number of nAbs that can be screened simultaneously. Typical screening assays include ELISA against the target protein.
Here we describe several validation assays for nAbs intended to be used in brain neurons as immunolabels or as intrabodies. Screening employs transiently transfected mammalian non-neuronal cell lines. Expressing the target proteins in heterologous cells yields a mosaic population of transfected and non-transfected cells in the same sample, allowing for direct comparison of nAb labeling in the expressing and null cells by immunolabeling. In the case of screening nAbs for use as intrabodies, the OmpA leader sequence is removed to allow for expression in the cytoplasm of transfected mammalian cells. Plasmids encoding nAbs are then co-transfected with target encoding plasmids and nAbs evaluated for colocalization with target protein in co-transfected cells. Candidates are then tested for target-specific immunolabeling in mammalian brain sections and on immunoblots against crude brain membrane fractions, and as intrabodies via transfection of cultured hippocampal neurons.
STRATEGIC PLANNING
Immunogen selection
There are several important considerations for immunogen selection. The protocol will need to be modified depending on the nature of the target protein, whether this is a soluble protein, membrane protein or multi-protein complex (Pardon et al., 2014). For many proteins it is not practical to produce the full-length protein at sufficient levels and quality for immunizations. In this case it is critical to select the appropriate fragment. There are many considerations that have been extensively documented, including that the fragment used be as unique to the proteins of interest as is feasible. Other considerations (hydropathy, charge, surface probability, effective folding, etc.) are typically taken into consideration. It is also important to consider the intended use of the nAbs. For example, for nAbs to be used as intrabodies, a fundamental consideration is that the immunogen comprises or contains intracellular domains of the target protein.
Llama immunization
Procedures involving llamas (commercial or in-house) need to be performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and adhere to the institution’s or vendor’s Institutional Animal Care and Use Committee. There are many different protocols that can be used, including employing purified recombinant proteins (either full-length or protein fragments), membrane proteins immobilized on supports such as nanodiscs, transfected cells expressing high levels of target protein, etc. These may differ in the immunization vehicle (e.g., whether and which type of adjuvant is used), and the route and frequency of injections. Detailed example protocols have been published previously [e.g., (Chow et al., 2019; Pardon et al., 2014).
We have employed recombinant fragments of neuronal target proteins in our immunizations (Dong et al., 2019). These fragments were obtained via expression and purification from E. coli bacteria. A set of five target protein-derived fragments were then combined into a cocktail containing 0.2 mg of each protein and used to subcutaneously inject llamas five times at biweekly intervals. Initial test bleeds were obtained after the fifth immunization and an antiserum fraction prepared. The presence of target-specific llama antibodies was evaluated by ELISA (See Support Protocol 1), where a titer of a 1:100,000 dilution of antiserum yielding ≥3X difference in ELISA signal over the no primary antibody negative control as our standard for proceeding with library construction. It is possible to purify a whole IgG fraction from the antiserum, and to selectively purify the heavy chain only IgG2 and IgG3 antibody fractions away from the conventional IgG1 antibodies also present in camelids, should one wish to perform analyses of the respective contributions of these antibodies to the overall ELISA signal. Note that additional rounds of immunizations and test bleeds/antiserum evaluation can be performed if sufficient antibody titer is not obtained after this conventional immunization schedule.
BASIC PROTOCOL 1: Total RNA isolation from camelid leukocytes
Once a sufficient titer is obtained, a whole blood sample is collected from the llama and used as the source of leukocytes for RNA extraction (Figure 1). The blood sample contains the antibody secreting B lymphocytes and is used to subsequently isolate total RNA for conversion to cDNA, and then PCR employing custom llama-specific primers used to prepare a nAb library. This protocol is based on the use of the LeukoLOCK™ Total RNA Isolation System (Life Technologies Catalog# AM1293). Reagents and materials provided in the kit are also listed below.
Figure 1.

Flowchart of the llama leukocyte RNA isolation procedure performed using the LeukoLok™ Total RNA isolation system from Thermo Fisher (Catalog #AM1923).
Materials
RNase decontamination solution (Fisher Catalog# AM9780)
RNase-free pipette tips (Fisher Catalog# 2707450)
1X PBS, pH 7.4
RNAlater® Solution
LeukoLOCK™ Filter
LeukoLOCK™ Elution Solution
LeukoLOCK™ Lysis/Binding Solution
LeukoLOCK™ pH Adjustment Solution
Processing Tubes (RNase and DNase free)
Wash Solution 1 Concentrate
Wash Solution 2/3 Concentrate
Nuclease-free Water
RNA Binding Beads, stored at 4°C
1X LeukoLOCK™ DNase Buffer
Proteinase K
TURBO™ DNase (20 U/μL)
100% isopropanol
Whole llama blood, provided by llama immunization service
Orbital ≈
Vortex
Centrifuge
Prior preparation and considerations
RNA is an unstable molecule to work with, due to the high number of RNases that are present in the laboratory environment. Due to this it is imperative that surfaces and pipettors are previously cleaned with an RNase decontamination solution, laboratory gloves and masks are used, and the method is done with RNase-free pipette tips.
Method
Remove 10 mL of whole blood as provided by the llama immunization service with a syringe. Attach the syringe to the LeukoLOCK™ Filter and pass the blood through the filter to isolate the leukocytes, which will be captured by the filter.
-
Collect 3 mL of 1XPBS with a syringe and flush the filter to remove any residual red blood cells. Then, using a syringe, flush the filter with 3 mL of RNAlater solution.
The filter can be capped and sealed for storage at −80°C at this stage if needed. If stored frozen, thaw at RT ≈5 min before proceeding.
Prepare the pH-adjusted Lysis/Binding Solution by mixing 2.5 mL of Lysis/Binding Solution Concentrate and 70 μL of pH Adjustment Buffer per reaction.
Attach a 1 mL or 3 mL syringe, with the plunger retracted, to the inlet port of the LeukoLOCK Filter, then depress the plunger and flush the filter with air to expel residual RNAlater from the filter and ports.
Load a 3 mL syringe with 2.5 mL of pH-adjusted Lysis/Binding Solution and pass through the LeukoLOCK Filter. Collect the lysate in a 15 mL conical tube (Tube A). The process should take 5–10 seconds. Disconnect syringe, retract plunger, and then reconnect it to the LeukoLOCK Filter. Depress plunger to expel any residual drops of cell lysate into the same tube.
Add 2.5 mL of Nuclease-free Water to the collected lysate (Tube A) and mix thoroughly by vortexing or inversion.
Add 25 μL of Proteinase K solution (Tube A).
Incubate the tube at moderate speed (≈250 rpm) on an orbital shaker for 5 min at RT. Alternatively, the sample can be mixed manually by intermittent inversion.
Resuspend the RNA Binding Beads by vortexing, then remove 50 μL of RNA Binding Beads and add these to Tube A. Vortex briefly to mix. Add 2.5 mL of 100% isopropanol to Tube A and vortex briefly to mix. Incubate with mixing for 5 min at RT to allow the RNA to bind to the beads.
Pellet the beads by centrifugation at 2000 × g for 3 min.
Carefully decant or aspirate supernatant without disturbing RNA Binding Beads. Discard the supernatant.
Add 600 μL of Wash Solution-1 to the RNA Binding Beads (Tube A), vortexing or pipetting up and down to disperse the beads sufficiently to transfer the mixture to a 1.5 mL Processing Tube. The beads may not fully disperse during this step; this will not affect RNA purity or yield.
Rinse the 15 mL tube (Tube A) with a second 600 μL aliquot of Wash Solution-1, and transfer to the same 1.5 mL (Tube B) Processing Tube.
Centrifuge the sample (Tube B) for 30 sec in a microcentrifuge set at ≈16,000 × g to pellet the beads.
Carefully decant or aspirate and discard the supernatant without disturbing the beads.
Add 750 μL Wash Solution-2/3 to Tube B and vortex vigorously for 15–30 sec to dislodge the pellet.
Recover the RNA Binding Beads by centrifugation for 15–30 sec at ≈16,000 × g.
-
Carefully aspirate and discard the supernatant without disturbing the RNA Binding Beads.
At this stage you can proceed to the optional treatment with TURBO™ DNAse, or skip to step 21.
Prepare the TURBO™ DNase master mix by adding 4 μL of TURBO™ DNase (20 U/μL) into 296 μL of 1× LeukoLOCK DNase Buffer per reaction. Add 300 μL of the prepared TURBO DNase master mix to each sample. Vortex briefly or pipet up and down to disperse the bead pellet, then agitate gently (1000 rpm on an orbital shaker) for 10 min at RT.
Add 300 μL of Lysis/Binding Solution to each sample.
Add 300 μL of 100% isopropanol, mix, and briefly centrifuge (≈2 sec) at low speed (<1000 g) to remove any liquid from the lid of the tube.
Incubate at RT for 3 min.
Recover the RNA Binding Beads by centrifugation for 30 sec at ≈16,000 × g. Carefully aspirate and discard the supernatant without disturbing the beads.
Add 750 μL Wash Solution-2/3 and vortex vigorously for 15–30 sec.
Recover the RNA Binding Beads by centrifugation for 30 sec at ≈16,000 × g. Carefully aspirate and discard the supernatant without disturbing the beads.
Centrifuge the tube briefly and remove any remaining liquid from the tube, then leave the tube open at RT for ≈3 min to allow any residual liquid to evaporate.
Add 50 μL Elution Solution and vortex vigorously for 30 seconds.
Thoroughly pellet the RNA Binding Beads by centrifugation for 2 min at ≈16,000 g. Transfer the RNA-containing supernatant to a new Processing Tube.
Store the purified RNA at −20°C.
BASIC PROTOCOL 2: First strand cDNA synthesis; VHH and VH repertoire PCR
After total RNA has been isolated from the immunized llama’s leukocytes, the phage library is constructed. This includes making cDNA from the isolated RNA, isolating the VHH and VH repertoire through PCR and using these fragments to prepare a library of nAb expressing plasmids.
A). First strand cDNA synthesis
This protocol is designed for use with the SuperScript ® III First-Strand Synthesis System for RT-PCR (Invitrogen Catalog# 18080–051). This contains the materials and reagents needed for the process, as listed below.
Reagents and materials
Oligo(dT) 20 (50 μM)
Random hexamers (50 ng/μL)
10X RT buffer
0.1 M DTT
25 mM MgCl2
10 mM dNTP mix
SuperScript III RT (200 U/μL)
RNaseOUT (40 U/μL)
E. coli RNase H (2 U/μL)
DEPC-treated water, 1.2 mL
Total HeLa RNA (10 ng/μL)
Sense Control Primer (10 μM)
Antisense Control Primer (10 μM)
Source RNA, isolated from immunized llama’s leukocytes
Heat block or water bath for temperature-controlled incubation
Centrifuge
Method
Prepare two sets of mixture with two primers. Prepare one tube with 8 μL RNA, 1 μL Oligo dT and 1 μL dNTP per reaction. Prepare another with 8 μL RNA, 1 μL random hexamers and 1 μL dNTP per reaction.
Incubate the tubes at 65°C for 5 min, then place on ice for at least 1 min.
- Prepare the following cDNA Synthesis Mix:
Component 1 rxn (μL) 10X RT buffer 2 25 mM MgCl2 4 0.1 M DTT 2 RNaseOUT™ (40 U/μL) 1 SuperScript® III RT (200 U/μL) 1 Add 10 μL of cDNA Synthesis Mix to each RNA/primer mixture, mix gently, and collect by brief centrifugation. Incubate the tube primed with Oligo(dT) 20 for 50 min at 50°C. Incubate the tube primed with random hexamers for 10 min at 25°C, followed by 50 min at 50°C.
Terminate the reactions incubating at 85°C for 5 min. Chill on ice.
Collect the reactions by brief centrifugation. Add 1 μL of RNase H to each tube and incubate the tubes for 20 min at 37°C.
Store the synthesized cDNA at −20°C or use it directly for PCR
B). VHH and VH repertoire PCR
This protocol employs llama-specific custom primers and is designed for use with the Platinum Taq DNA Polymerase (Invitrogen Catalog# 10966–018). This kit contains the materials and reagents needed for the process, as listed below, except the custom primers.
Reagents and materials
Platinum Taq DNA Polymerase (5U/μL)
10X PCR buffer (no Mg)
50 mM MgCl2
10 mM dNTP mix
50 mM primers (see sequences below)
QiaQuick Gel Extraction kit (Qiagen Catalog# 28704)
PCR tubes (Genemate Catalog# T-3035–2)
Forward and reverse primers, 50mM (see sequences below)
cDNA (OligodT primed and hexamer primed), see Basic Protocol 2A
Thermocycler
Primers
| Forward primers | |
|---|---|
| VH1 (SfiI) | 5’-CATGCCATGACTCGCGGCCCAGGCGGCCATGGCCCAGGTGCAGCTGGTGCAGTCTGG-3’ |
| VH3 (SfiI) | 5’-CATGCCATGACTCGCGGCCCAGGCGGCCATGGCCGAGGTGCAGCTGGTGGAGTCTGG-3’ |
| VH4 (SfiI) | 5’-CATGCCATGACTCGCGGCCCAGGCGGCCATGGCCCAGGTGCAGCTGCAGGAGTCGGG-3’ |
| F1 (SfiI) | 5’-CATGCCATGACTGTGGCCCAGGCGGCCATGCAGKTGCAGCTCGTGGAGTC-3’ |
| F2 (SfiI) | 5’-CATGACTGTGGCCCAGGCGGCCATGCAGGTGCAGCTCGTGGASWCHGGNGGAGGMTTGGT-3’ |
| VHH1BACK (SfiI) | 5’-GTTATTACTCGCGGCCCAGGCGGCCATGGCCCAGGTSMARCTGCAGSAGTCWGG-3’ |
| VHH6BACK (SfiI) | 5’-T GTTATTATCTGCGGCCCAGGCGGCCATGGCCGATGTGCAGCTGCAGGCGTCTGGRGGAGG-3’ |
| Reverse primer | |
| JH | 5’-CCACGATTCTGGCCGGCCTGGCCTGAGGAGACRGTGACCTGGGTCC-3’ |
Method
Set up a PCR tube for each combination of forward and reverse primers.
- Prepare the PCR reactions as follows:
Component 1 rxn (μL) Nuclease free water 37. 8 10×PCR buffer, -Mg 5 50 mM MgCl2 2 10 mM dNTP 1 cDNA (Oligo dT primed) 1 cDNA (hexamers primed) 1 Forward primer (50 mM) 1 Reverse primer (50 mM) 1 Taq 0.2 -
Incubate the reactions in a thermocycler with the following PCR cycles:
1 cycle: 94°C for 2 min;
30 cycles: 94°C for 30 s, 55°C for 30 s, 72°C for 1 min;
1 cycle: 72°C for 10 min;
Purify the ≈400 bp PCR products on an agarose gel using the Qiagen Gel Extraction Kit.
BASIC PROTOCOL 3: Preparation of the phage display library
Once the VHH and VH repertoire has been amplified via PCR, the amplified fragments are used to prepare a library of nAb expressing plasmids. Restriction enzyme digested fragments are cloned into the pComb3XSS phagemid thus constructing a multi-target recognizing phage library ready for further screening against the individual target proteins. Successful library preparation should yield a complexity of >108 (the number of transformants when transforming the library into E. coli bacteria) and a phage titer of >1012 cfu/mL.
A). Restriction enzyme SfiI digestion of VHH and VH fragments and pComb3XSS
This protocol is designed for use with the NEB SfiI enzyme (New England Biolabs Catalog# R0123S). This comes with the materials and reagents needed for the process, included in the list below.
Reagents and materials
Nuclease free water
SfiI enzyme (2 U/μL)
1× CutSmart buffer: 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 100 μg/mL BSA, pH 7.9
pComb3XSS plasmid (Addgene Plasmid# 63890)
QIAquick PCR Purification kit (Qiagen Catalog# 28104)
QIAquick Gel Extraction kit (Qiagen Catalog# 28704)
1.5 mL microcentrifuge tubes (Fisher Catalog# 5408129)
VHH and VH PCR fragments, see Basic Protocol 2B
Agarose powder (Fisher Catalog# BP1356–100)
1X TAE buffer (1 mM EDTA, 40 mM Tris base, 20 mM glacial acetic acid)
Heat block or water bath for temperature controlled incubations
Agarose gel electrophoresis rig
- Prepare two tubes with the digestion reactions as follows.
Reagents 1 rxn (μL) 16 rxns (μL) Nuclease free water To bring to 50 To bring to 800 Cut Smart buffer 5 80 pComb3XSS 1 μg 16 μg SfiI 2 32 Reagents 1 rxn (μL) 12 rxns (μL) Nuclease free water To bring to 50 To bring to 600 Cut Smart buffer 5 60 VHH and VH PCR fragments 1 μg 12 μg SfiI 2 24 Incubate for 16 h at 50°C.
Use QIAGEN’s QIAquick PCR Purification Kit to purify the VHH and VH PCR fragment after digestion.
Electrophorese the digested pComb3XSS on an agarose gel. The SfiI digestion of pComb3XSS should produce 2 bands: 1,600 and 3,400 bp in size, respectively. Excise the 3400 bp band and extract the DNA, using QIAGEN’s Gel Extraction kit.
B). Ligation and transformation of VHH and backbone
The VHH and VH fragments and pComb3XSS plasmid are now ligated to generate the phage display library. The ligations are then transformed into electrocompetent ER2738 E. coli bacteria using electroporation. A negative control of plasmid without the VHH and VH fragments is used to compare the transformation efficiencies of ligated and self-ligated reactions. The background efficiency of self-ligated plasmid should be less than 10% of that with an insert.
Reagents and materials
Nuclease free water
10X Buffer
Purified VHH and VH fragments
Purified pComb3XSS plasmid
Ligase (1 U/μL)
Lucigen electrocompetent ER2738 cells (Catalog# 60522)
Lucigen Recovery Medium
LB+ 1.5% w/v agar + 100 μg/mL ampicillin bacterial culture plates
5X PEG/NaCl solution: 100 g of PEG8000, 75 g of NaCl in 500 mL of MilliQ water, sterilized
Suspension buffer: PBS containing 1× protease inhibitor cocktail, 0.02% NaN3, and 0.5% BSA
3 M Sodium acetate, pH 5.2
95% ethanol
70% ethanol
Super Broth (SB): 30 g/L tryptone, 20 g/L yeast extract, 10 g/L MOPS, pH 7.2
100 mg/mL ampicillin stock
20 mg/mL tetracycline stock
70 mg/mL kanamycin stock
1.5 mL microcentrifuge tubes (Fisher Catalog# 5408129)
Electroporation cuvettes (BioRad Catalog# 165–2089)
2 L Erlenmeyer flask
Centrifuge bottles
0.22 μm syringe filter (Millex Catalog# SLGV013VL)
Temperature-controlled orbital shaking incubator
Bacterial electroporator (such as BioRad Micropulser, Catalog# 1652100)
- Prepare separate tubes for the transformation reactions as follows.
Each experimental reaction Negative control reaction ddH2O To bring to 20 μL To bring to 20 μL 10X buffer 2 2 VHH and VH fragments ≈ 53 μg 0 μg pComb3XSS ≈ 150 μg ≈ 150 μg Ligase 1 1 Carry out electroporation using 3 μL of ligation mixture per 30 μL of ER2738. Follow electroporation conditions as described in the Lucigen manual.
Have Recovery Medium and 17 mm × 100 mm sterile culture tubes readily available at RT (one tube for each transformation reaction).
Place electroporation cuvettes (0.1 cm gap) and microcentrifuge tubes on ice (one cuvette and one microfuge tube for each transformation reaction).
Remove electrocompetent cells from the −80°C freezer and place on ice until they thaw completely (10–15 min). When the cells are thawed, mix them by tapping gently. Aliquot 30 μL of cells into the chilled microcentrifuge tubes on ice.
Add 3 μL of heat-denatured ligation reaction to the aliquoted cells. Stir briefly with a pipet tip; do not pipet up and down to mix, which can introduce air bubbles and warm the cells.
Carefully pipet 25 μL of the cell/DNA mixture into a chilled electroporation cuvette without introducing bubbles. Quickly flick the cuvette downward with your wrist to deposit the cells across the bottom of the well. Electroporate according to the selected conditions, determined by the Lucigen manual.
Within 10 seconds of the pulse, add 975 μL of Recovery Medium to the cuvette and pipet up and down three times to resuspend the cells. Transfer the cells and Recovery Medium to a culture tube.
Incubate the tube for 1 h at 37°C with orbital shaking at 250 rpm.
Spread up to 10 μL of transformed cells from each ligation on LB + agar + ampicillin bacterial culture plates.
Incubate the plates overnight at 37°C.
Compare the ligation reaction plates to the negative control plates to see transformation efficiency and library diversity (number of clones minus number of clones for pComb3XSS self-ligation estimated from small scale experiment).
If the ratio of (number of clones / background number of clones for pComb3XSS self-ligation) is greater than 10 prepare 20 more ligation reactions as described in steps 1 to 12.
- Scale up the ligation as above to 20×rxn, then purify and concentrate the ligated library with ethanol precipitation of DNA.
- Add 1/10 volume of Sodium Acetate
- Add 2.5–3.0× volume (calculated after addition of sodium acetate) of at least 95% ethanol.
- Incubate at −20°C overnight.
- Centrifuge at ≥ 14,000 × g for 30 min at 4°C. Discard supernatant carefully, making sure the DNA pellet (which may not be visible) is not discarded
- Rinse with 70% ethanol.
- Centrifuge again for 15 min. Discard supernatant and dissolve pellet in 60 μl of sterilized water. Make sure that the water comes in contact with the whole surface of the tube since a significant portion of DNA may be deposited on the walls instead of in the pellet.
After the 1 h recovery, pool all bacterial cultures into a 2000 mL flask containing 400 mL SB medium containing ampicillin (100 μg/mL) and tetracycline (20 μg/mL).
Incubate the flask for 2 h at 37°C with orbital shaking at 250 rpm.
Add 1 mL of M13KO7 helper phage (1×1012 cfu/mL; see Support Protocol 2) and incubate the flask at 37°C for 30 min without shaking.
Incubate the flask at 37°C for 2 h with orbital shaking at 250 rpm.
Add kanamycin to 70 μg/mL and incubate at 37°C overnight with orbital shaking at 250 rpm.
The following day centrifuge the culture at 10,000 rpm for 15 min at 4°C.
Transfer the clear supernatants to new centrifuge bottles and add 100 mL of 5× PEG/NaCl solution into each bottle, followed by a 2 h incubation on ice.
Centrifuge at 12,000 rpm for 20 min at 4°C, resuspend the white phage pellet in 100 mL of sterile PBS. Add 25 mL of 5× PEG/NaCl into each bottle and incubate another 2 h on ice.
Centrifuge the bottles at 12,000 rpm for 20 min at 4°C and re-suspend with 10 mL of suspension buffer
Filter the phage suspension through a 0.22 μm syringe filter.
Store the phage library aliquots at −80°C.
BASIC PROTOCOL 4: Panning of the phage display library against individual target proteins
It is necessary to pan the library against the desired target protein. This will increase the representation of phage expressing target specific nAbs. Typically, the panning is performed sequentially using decreasing concentrations of target protein and phage clones selected from the last round. To maximize sequence diversity, clones can also be selected from all panning rounds. Below we describe conventional and sandwich panning against purified protein (Figure 2). These two variations have the potential to yield distinct pools of selected nAbs due to the distinct manner in which the target protein is presented to the phage display library.
Figure 2.
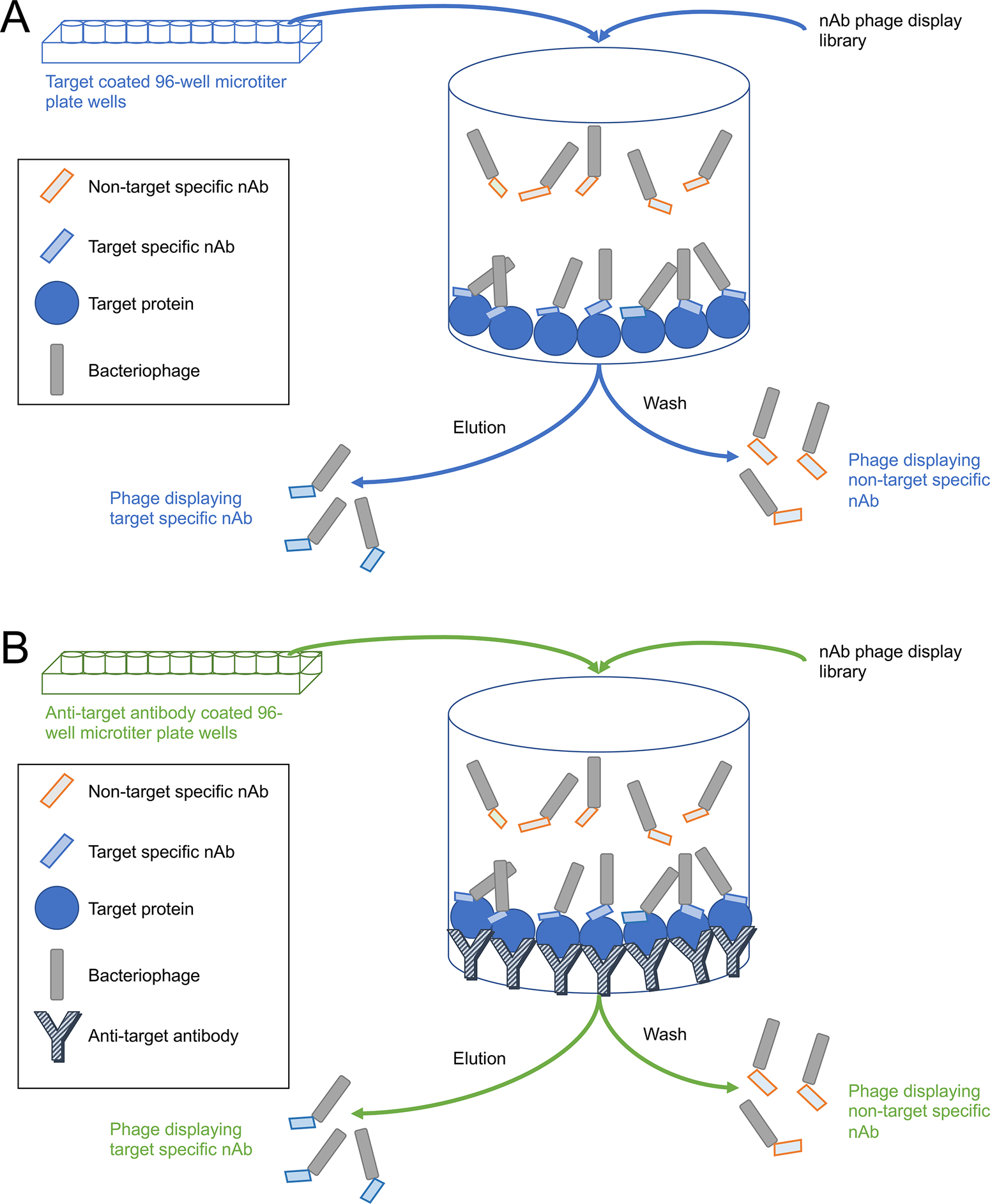
Schematic diagrams of the conventional and sandwich panning procedures. A) Conventional panning. Wells of a 96-well microtiter plate are coated directly with the target protein. B) Sandwich panning. Wells of a 96-well plate are first coated with an antibody against the target protein, then incubated with the target protein which binds to the antibody. In both cases the target-containing wells are then incubated with the nAb phage display library. Phage displaying target-specific nAbs on their pili will bind to the attached target protein whereas those displaying non-target specific nAbs will not and remain in solution. After washing away the non-target specific nAbs, the bound target-specific nAbs are eluted and retained for further analysis and use. Steps common to both procedures are in black font.
Reagents and materials
1X Phosphate Buffered Saline (PBS): 8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, 0.24 g/L KH2PO4
1X PBST: 0.1% v/v Tween-20 in PBS
Luria Broth (LB): 10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl
Super Broth (SB): 30 g/L tryptone, 20 g/L yeast extract, 10 g/L MOPS, pH 7.2
0.5% Bovine Serum Albumin (BSA) in PBS
1X TBSC: 137 mM NaCl, 1 mM CaCl2, 10 mM Tris, pH 7.4
5X PEG/NaCl: 200 g/L PEG8000, 150 g/L NaCl, sterilized
Protease inhibitor cocktail II (RPI Catalog# P50800–1)
20 mg/mL tetracycline stock
100 mg/mL ampicillin stock
70 mg/mL kanamycin stock
LB+ 1.5% w/v agar+ 20 μg/mL tetracycline bacterial culture plates
LB+ 1.5% w/v agar + 100 μg/mL ampicillin+20 μg/mL tetracycline bacterial culture plates
E. coli K12 ER2738 bacteria (Lucigen Catalog# 60522, NEB Catalog# E4104)
96-well high-binding microplates (Greiner Bio-One Catalog# 655061)
15 mL round bottom tubes (Falcon Catalog# 352059)
(For Protocol 4B, see below) Anti-target antibody, the purified whole IgG fraction from the llama immunization process or another available target-specific antibody can be used for this step
0.1 μg/μL desired target protein in PBS
(Optional, see below) 0.1 μg/μL protein tag (e.g., GST) in PBS
1012 cfu/mL M13KO7 helper phage (NEB Catalog# N0315S)
Phage library aliquots, see Basic Protocol 3
1.5 mL microcentrifuge tubes (Fisher Catalog# 5408129)
A). Conventional panning
In this method the wells of a microtiter plate are directly coated with target protein (Figure 2A). An aliquot of the library is incubated in these wells and non-binding phage are washed away. If the target protein used for immunizations includes tags, such as GST or 6×His tags, the library can be pre-depleted against these tags.
Day 1
Streak the LB + agar + tetracycline bacterial culture plate with E. coli ER2738 bacteria and incubate overnight at 37 ℃.
Day 2
Inoculate a single colony of E. coli ER2738 into 3 mL of LB medium containing 20 μg/mL tetracycline and incubate overnight at 37°C with orbital shaking at 250 rpm.
Coat 2–3 wells with 100 μL per well of target protein at 10 μg per well in PBS. Incubate at 4°C overnight.
Coat 2–3 wells for pre-depletion. If a GST fusion protein is used, use wells coated with GST to perform depletion panning under the same conditions as for target-specific panning. If using an untagged protein, use wells coated with 0.5% BSA in PBS. Incubate at 4°C overnight.
Day 3
Inoculate 30 μl of the overnight E. coli ER2738 culture into 3 mL of LB medium containing 20 μg/mL tetracycline. Incubate the culture ≈3 h at 37°C with orbital shaking at 250 rpm, until OD600 =0.6–0.8. Continue with steps 2 through 6 while waiting for the culture to grow.
Block the pre-coated wells with 150 μl of 0.5% BSA in PBS for 1 h at 37°C.
Add 100 μL of the phage library (≈1011 particles) into the wells coated for pre-depletion. Incubate 1 h at RT.
Transfer the unbound phage to the target protein coated wells. Incubate 1 h at RT.
Remove the unbound phage. Rinse the wells 8 times with PBST followed by another 8 times with PBS.
Elute the target-specific bound phage with 100 μL per well of 10 mg/mL trypsin in TBSC buffer. Incubate 30 min at 37°C, then pipette the eluate into a microcentrifuge tube and add protease inhibitor to inactivate the trypsin.
- Titer the eluted phage:
- Prepare 10-fold serial dilutions of elute phage in LB medium.
- Infect 100 μl of the E. coli ER2738 log-phase culture from step 1 with 10 μL of each dilution of the eluted phage.
- Incubate 10 min at 37°C with orbital shaking at 250 rpm.
- Plate infected E. coli ER2738 on LB + agar + ampicillin + tetracycline plates. Incubate overnight at 37°C.
- Assess the titer of these output phage the following day.
Inoculate a single colony of E. coli ER2738 into 3 mL of LB medium containing 20 μg/mL tetracycline for library amplification. Incubate the culture overnight at 37°C with orbital shaking at 250 rpm.
Day 4
Inoculate 5 mL of SB medium containing 20 μg/mL tetracycline with 50 μL of the overnight E. coli ER2738 culture. Incubate the culture ≈3 h at 37°C with orbital shaking at 250 rpm, until OD600 =0.6–0.8.
Infect the log-phase E. coli ER2738 culture with 100 μL of the eluted phage from day 3. Incubate at 37°C for 10 min.
Add 3 mL of SB medium containing 1.6 μL of 100 mg/mL ampicillin and 3 μL of 20 mg/mL tetracycline. Incubate 1 h at 37°C with orbital shaking at 250 rpm.
Add 2.4 μL of 100 mg/mL ampicillin and incubate for an additional 1 h at 37°C.
Add 100 μL of M13KO7 helper phage (1012 cfu/mL; see Supplementary Protocols) and incubate 10 min at 37°C.
Transfer the mixture to 32 mL of SB medium containing 36 μL of 100 mg/mL ampicillin and 32 μL of 20 mg/mL tetracycline. Incubate 2 h at 37°C with orbital shaking at 250 rpm.
Add 40 μL 70 mg/mL kanamycin to the culture and incubate overnight at 37°C with orbital shaking at 250 rpm.
Day 5
Transfer the overnight culture to a centrifuge tube and centrifuge10 min at 12,000×g at 4°C.
Transfer the supernatant to a fresh centrifuge tube and add 10 mL of 5× PEG/NaCl solution. Precipitate phage for 2h at 4℃.
Centrifuge the PEG precipitation 15 min at 12,000 g at 4 ℃.
Discard the supernatant and suspend the pellet in 2 mL 0.5% BSA in PBS plus protease inhibitor.
Filter the suspension with a sterilized 0.22 μm syringe filter to remove residual bacterial cells. Store the flow through containing amplified phage at 4°C and use as input for the next round of panning.
Repeat the process using decreasing concentrations of target protein (i.e., 5 μg per well then 1 μg per well) for subsequent rounds of panning.
B). Sandwich panning
In this method the wells of a microtiter plate are coated first with anti-target antibody, and then with target protein, to capture and display the target protein in a different orientation than when it is used to directly coat the wells of microtiter plate (Figure 2B). The nAb phage library is then panned against these wells and non-binding phage are washed away, essentially as detailed above. If the target protein used for immunizations includes tags such as GST or 6×His tags, the library can be pre-depleted against these tags.
Day 1
Streak an LB + agar + tetracycline bacterial culture plate with E. coli ER2738 bacteria and incubate overnight at 37 ℃.
Day 2
Inoculate a single colony of E. coli ER2738 into 3 mL 20 μg/mL tetracycline in LB, and incubate overnight at 37°C with orbital shaking at 250 rpm.
Coat 2–3 wells with 100 μL per well of the appropriate pure antibody at 10 μg per well in PBS. Incubate at 4°C overnight.
Coat 2–3 wells for pre-depletion. Use 100 μL per well of the appropriate pure antibody at 10 μg per well in PBS. Incubate at 4°C overnight.
Day 3
Inoculate 30 μL of the overnight E. coli ER2738 culture into 3 mL 20 μg/mL tetracycline in LB. Incubate the culture ≈3 h at 37°C with orbital shaking at 250 rpm, until OD600 =0.6–0.8. During this incubation continue with steps 2 through 6.
Block the pre-coated wells with 150 μL of 0.5% BSA in PBS for 1 h at 37°C.
Add 100 μL of the phage library (≈1011 particles) into the wells coated for pre-depletion. Add 100 μL per well of 10 μg/mL target protein into the antibody-coated capture wells. Incubate 1 h at RT.
Wash the antigen captured wells 2 times with PBS to remove unbound target protein. Transfer the unbound phage to the target protein captured wells. Incubate 1 h at RT.
Remove the nonbinding phage. Rinse the wells 8 times with PBST followed by another 8 times with PBS.
Elute the target-specific bound phage with 100 μL per well of 10 mg/mL trypsin in TBSC buffer. Incubate 30min at 37°C, then pipette the eluate into a microcentrifuge tube and add protease inhibitor to inactivate the trypsin.
- Titer the elute phage:
- Prepare 10-fold serial dilutions of elute phage in LB medium.
- Infect 100 μL of the E. coli ER2738 log-phase culture from step 1 with 10 μL of each dilution of the eluted phage.
- Incubate 10 min at 37°C.
- Plate infected E. coli ER2738 on LB + agar + ampicillin + tetracycline bacterial culture plates. Incubate overnight at 37°C.
- Assess the titer of these output phage the following day.
Inoculate a single colony of E. coli ER2738 into 3 mL LB medium containing 20 μg/mL tetracycline for library amplification. Incubate the culture overnight at 37°C with orbital shaking at 250 rpm.
Day 4
Inoculate 5 mL SB medium containing 20 μg/mL tetracycline with 50 μL of the overnight E. coli ER2738 culture. Incubate the culture ≈3 h at 37°C with orbital shaking at 250 rpm, until OD600 =0.6–0.8.
Infect the log-phase E. coli ER2738 culture with 100 μL of the eluted phage from day 3. Incubate at 37°C for 10 min.
Add 3 mL of SB medium containing 1.6 μL of 100 mg/mL ampicillin and 3 μL of 20 mg/mL tetracycline. Incubate 1 h at 37°C with orbital shaking at 250 rpm.
Add 2.4 μL of 100 mg/mL ampicillin and incubate for an additional 1 h at 37°C with orbital shaking at 250 rpm.
Add 100 μL of M13KO7 helper phage (1012 cfu/mL; see Supplementary Protocols) and incubate 10 min at 37°C with orbital shaking at 250 rpm.
Transfer the mixture to 32 mL of SB medium containing 36 μl of 100 mg/mL ampicillin and 32 μL of 20 mg/mL tetracycline. Incubate 2 h at 37°C with orbital shaking at 250 rpm.
Add 40 μL 70 mg/mL kanamycin to the culture and incubate overnight at 37°C with orbital shaking at 250 rpm.
Day 5
Transfer the overnight culture to a centrifuge tube and centrifuge 10 min at 12,000 × g at 4°C.
Transfer the supernatant to a fresh centrifuge tube and add 10 mL of 5× PEG/NaCl solution. Precipitate phage for 2h at 4℃.
Centrifuge the PEG precipitation 15 min at 12,000 × g at 4℃.
Discard the supernatant and suspend the pellet in 2 mL 0.5% BSA in PBS plus protease inhibitor.
Filter the suspension with a sterilized 0.22 μm syringe filter to remove residual bacterial cells. Store the flow through containing amplified phage at 4°C and use as input for the next round of panning.
Repeat the process using decreasing concentrations of target protein (i.e., 5 μg per well then 1 μg per well) for subsequent rounds of panning.
BASIC PROTOCOL 5: Small scale nAb expression
Once the phage library has been enriched for target-specific nAbs we can express the antibodies for screening purposes. This is done on a small scale so as to select the most promising nAbs in a high-throughput manner.
Reagents and materials
2XYT medium: 16 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl
Super Broth (SB): 30 g/L tryptone, 20 g/L yeast extract, 10 g/L MOPS, pH 7 2
20 mg/mL tetracycline stock
100 mg/mL ampicillin stock
100 mM IPTG stock, prepared fresh
50% glycerol
96 well 2 mL deep well plates (ThermoFisher Catalog# 278743)
14 mL round bottom tubes (Falcon Catalog# 352059)
Output phage titer plates from biopanning process, see Basic Protocol 4
Temperature-controlled orbital shaking incubator
Centrifuge
Method
Day 1
Pick individual colonies from the titer plates of output phage obtained during the biopanning. Inoculate individual colonies into wells of a 96 well deep well plate with 200 μL of 100 μg/mL ampicillin and 20 μg/mL tetracycline in 2×YT medium.
Incubate the plate overnight at 37°C with orbital shaking at 250 rpm.
Day 2
Transfer 50 μL of the overnight culture to 2 mL of SB medium containing 100 μg/mL ampicillin and 20 μg/mL tetracycline in 14 mL round bottom tubes. Incubate ≈4 h at 37°C with orbital shaking at 250 rpm, until OD600 = 0.5–1.
Prepare frozen glycerol stocks of the selected clones by adding 150 μL of 50% glycerol to the remainder of the overnight culture in the 96 well deep well plates. Freeze the stocks at −80°C for possible later use.
Once the cultures have reached OD600 = 0.5–1, add IPTG at 0.5 mM-1 mM into each tube to induce the expression of the nanobody-pIII fusion.
Incubate the cultures overnight at 37°C with orbital shaking at 250 rpm.
Day 3
Centrifuge the tubes at 3000 × g for 15min.
Collect the supernatant and save for testing.
BASIC PROTOCOL 6: Analysis and initial validation of selected clones
The small-scale preparations of nAb can be evaluated in a protein ELISA against the target protein (See Support Protocol 1). We have primarily selected candidates exhibiting a three-fold difference in ELISA signal over the no phage negative control as our standard for proceeding with sequencing and further validation. The plasmids encoding ELISA-positive nAbs are then purified and subjected to sequencing to identify unique versus redundant nAb clones. The plasmid DNA is also used to transform a non-amber suppressor strain of E. coli bacteria to generate nAb BCS for subsequent analyses.
Reagents and materials
Qiagen QIAprep Spin Miniprep Kit (Qiagen Catalog# 27104)
14 mL round bottom tubes (Falcon Catalog# 352059)
20 mg/mL tetracycline stock
100 mg/mL ampicillin stock
LB + 1.5% w/v agar + 100 μg/mL ampicillin + 20 μg/mL tetracycline bacterial culture plates
Glycerol stocks of ELISA positive nAb clones, see Basic Protocol 5
Sequencing Primers
| Forward primer | |
|---|---|
| pCombF | 5’-TTAGGCACCCCAGGCTTTACACT-3’ |
| Reverse primer | |
| gback | 5’-GCCCCCTTATTAGCGTTTGCCATC-3’ |
Method
Day 1
Streak the selected ELISA positive nAb clones on LB + agar + ampicillin + tetracycline plates from the glycerol stocks made during the small-scale expression. Incubate overnight at 37°C.
Day 2
Inoculate 3 mL LB medium containing 100 μg/mL ampicillin and 20 μg/mL tetracycline in 14 mL round bottom tubes with a single colony for each nAb selected for further analysis. Incubate overnight at 37°C with orbital shaking at 250 rpm. If possible, grow the culture for no more than 14 h.
Day 3
Following the QIAprep miniprep kit protocol, miniprep the plasmids for all the ELISA positive nanobodies. The typical DNA yield from the pComb3XSS nAb plasmids in ER2738 was 8 μg (40 μL at 0.2 μg/μL).
Send prepared plasmids for sequencing with the sequencing primers pCombF and gback (2 reactions per plasmid).
After sequencing results are returned
Trim the sequences between the sites for the enzyme SfiI (GGCCCAGGCGGCC) and the 6×His tag (CACCATCACCATCACCAT).
Align trimmed sequences and select nAb clones with unique sequences for further validation.
BASIC PROTOCOL 7: Validation of nanobodies as immunolabels
nAbs that will recognize full-length target protein in fixed and permeabilized mammalian neurons, are initially evaluated by performing immunocytochemistry on transiently transfected mammalian cells expressing the full-length target protein. When performed in 96 well microtiter plates, this assay offers a robust and sufficiently high-throughput assay to test dozens of candidate nAbs (Gong et al., 2016). For these assays it is recommended to prepare new BCS samples, using an amber suppressor cell line, as this increases nAb concentration in the BCS and also reduces background arising from the presence of the PIII protein (See Support Protocol 3).
A). Immunofluorescence immunocytochemistry on transiently transfected COS-1 cells
This assay makes use of transient transfection to directly compare nAb binding to a mosaic sample of cells in the same well with and without target protein expression. Using this approach, it is simple to determine whether a particular nAb exhibits specific immunolabeling signal to cells expressing the target protein versus nonspecific labeling of fixed and permeabilized mammalian cells as visualized as labeling of non-transfected cells in the same sample
Prior considerations and experimental design
Typically, testing of each nAb against a target requires a double labeling with a distinct previously validated antibody against the same target (positive control). If available, it is also useful to include separate samples of a previously validated nAb against cells expressing its target to serve as a positive control for nAb detection. This nAb can also serve as a negative control against the new target being evaluated. A media only negative control (the same media in which the nAbs were produced but without nAb) can also be employed. Nanobodies are considered to be positive in this assay when the nAb signal colocalizes with that of the positive control antibody at the cellular and subcellular level.
Reagents and materials
COS-1 mammalian cells (ATCC# CRL-1650), to be thawed and maintained according to ATCC protocols including BSL-2 considerations.
Lipofectamine 2000 (Invitrogen Catalog# 52881)
1X Optimem (Gibco Catalog# 31985–062)
COS cell media: 10% BCS, 100 μg/mL penicillin-streptomycin in DMEM
DMEM (Gibco Catalog# 11965–092)
Penicillin-streptomycin (Gibco Catalog# 15140–122)
Bovine Calf Serum (BCS) (Hyclone Catalog# SH3007303)
0.05% Trypsin-EDTA (Gibco Catalog# 25300–054)
10 cm Tissue Culture dishes (Falcon Catalog# 353003)
96 well plates (Greiner Bio-One Catalog# 655090)
1X DPBS: 2.67 mM KCl, 1.47 mM KH2PO4, 136.9 mM NaCl, 8.10 mM Na2HPO4
1X DPBS + 1% NaN3
Fixative: 4% formaldehyde (FA) prepared fresh from paraformaldehyde (PFA), 1 mM CaCl2, 1 mM MgCl2, 0.1% Triton X-100, 1X DPBS
Blocking solution (BLOTTO-T): 20 mM Tris-HCl, 0.15 M NaCl, 4% dry milk powder + 0.1 % Triton X-100
Alexa Fluor 488 anti-HA mouse IgG1 mAb 16B12 (ThermoFisher Catalog# A21287)
Alexa Fluor 555 anti-mouse IgG subclass-specific antibodies, dependent on the controls used
Hoechst 33258 (ThermoFisher Catalog# H21491)
Candidate nAb BCS, see Support Protocol 3
1.5 mL microcentrifuge tubes (Fisher Catalog# 5408129)
Fluorescent microscope
Method
Day 1
- The stock culture plates of COS-1 cells should not exceed 80% confluence before starting. Determine the number of 10 cm dishes of cells (assumed to have 10 mL of media) needed by using the following formula:
Remove media from the dishes and wash with 10 mL DPBS to remove dead cells. Use 1 mL trypsin to detach cells from the dish, swirling and incubating the dish up to 5 min. Add 9 mL of media to deactivate the enzyme and using a cell scraper gently detach the cells from the surface of the dish.
Remove the amount of media needed, as calculated in step 1. Bring this volume up with cell media, to a suitable final volume for pipetting 100 μL per microtiter plate well.
Using a multichannel pipettor, pipette 100 μL per well of microtiter plate. Make sure to swirl the cell suspension prior to pipetting, to avoid cells settling in the reservoir and to ensure they are evenly dispensed.
Incubate plates of cells overnight at 37°C in a 5% CO2 incubator.
Day 2
In a tissue culture hood, prepare sterile 1.5 mL microcentrifuge tubes with the necessary amount of target DNA. Use 0.42 μg total DNA for every 12 wells. Add 25 μL Optimem to each tube.
Prepare a stock of Lipofectamine 2000 in Optimem. Add (0.42*number of tubes prepared in step 1) μL of Lipofectamine 2000 to (25* number of tubes prepared in step 1) μL of Optimem.
Incubate at RT for 5 min.
Add 25 μL of the mix prepared in step 2 to each of the tubes prepared in step 1. Mix well and incubate at RT for 20 min.
Bring each tube up to a final volume of 300 μL per 12 wells with cell media.
Add 25 μL of transfection mixture to each well of plated cells, as prepared on day 1.
Incubate at 37°C in a 5% CO2 incubator for 48 h.
Day 3
(Optional) Check cells are growing and are healthy. Incubate in a 37°C incubator overnight.
Day 4
Pipette off the cell media. Working in a fume hood, add 50 μL of FA fixative to each well. Incubate 30 min at 4°C.
Pipette off fixative and dispose of as per institutional guidelines for formaldehyde waste.
Rinse plates three times with 100 μL per well of ice-cold DPBS.
Block plates with 100 μL per well blocking solution. Incubate 1 h at RT.
Dilute target-specific positive control antibody to the appropriate concentration in blocking solution in a volume sufficient to use 25 μL for each well. Add 25 μL to each well using a multichannel pipettor. Add 25 μL of each candidate nAb BCS undiluted to each well. Incubate 1 h at RT with gentle mixing.
Wash plates three times for 10 min each with 100 μL per well blocking solution.
Dilute Alexa Fluor anti-HA antibody and positive control antibody-specific secondary antibodies at 1:2000 in blocking solution in a volume sufficient to use 50 μL for each well. Add a nuclear stain such as Hoechst 33258 (200 ng/mL final). Incubate 45 min at RT with gentle mixing, keep away from light.
Wash plates three times for 10 min each with 100 μL per well of DPBS.
Remove the final wash. Add 50 μL per well of DPBS + 1% NaN3. Store plates at 4°C until visual analysis using a fluorescent microscope. For example images of results from this type of analysis, please refer to previously published work (Dong et al., 2019).
B). Immunoperoxidase staining of free floating brain sections
Validation of the nAbs for whether they immunolabel endogenous targets in brain neurons is performed using immunohistochemistry against brain sections. The regional, cellular and subcellular pattern of labeling is then evaluated versus neuroanatomical hallmarks of target protein expression, and if possible in a side-by-side comparison to a positive control antibody. In our work, we evaluate this using conventional fixation and perform the immunolabeling on free-floating sections. The initial evaluation is typically performed using immunoperoxidase -DAB/NAS immunohistochemistry including avidin-biotin complex signal amplification due to the enhanced sensitivity of this procedure.
Prior considerations and experimental design
As with the other assays it is important to choose a correct series of controls that will accompany the nAbs being evaluated. In this case positive controls could include previously validated antibodies against the target protein, allowing for a direct side-by-side comparison of patterns of correct immunolabeling. A critical negative control is the bacterial media as a no primary control.
This protocol makes use of brain sections from a rat subjected to transcardial perfusion with formaldehyde. The perfusion and preparation of brain sections was performed as described in a previously published protocol (Paletzki & Gerfen, 2019) with the following modifications: 4% formaldehyde fixative is prepared directly from paraformaldehyde the same day it is to be used and does not include NaCl, heparin sulfate (1 mL/kg) and sodium nitrite (0.5 mL of a 3% solution) is injected into the left ventricle prior to saline perfusion, and there is an additional cryoprotection step in 30% sucrose.
Reagents and materials
30 μm-thick sagittal rat brain sections. Sections should be collected in 0.1 M PB with 10 mM NaN3.
1X TBS: 0.15 M NaCl, 50 mM Tris, pH 7.5
1X TB: 50 mM Tris, pH 7.5
0.5% TX-100 in TBS
NGS: normal goat serum (GeneTex Catalog# GTX73249)
Vehicle solution: 5% normal goat serum, 0.1% TX-100 in TBS
Avidin, egg white (ThermoFisher Catalog# A887)
Biotin (Millipore/Sigma Catalog# B4501)
Anti-HA tag monoclonal antibody (2–2.2.14) (ThermoFisher Catalog# 26183)
Biotin SP conjugated goat anti-mouse IgG1 (Jackson Immunochemicals Catalog# 115-065-205)
VECTASTAIN® Elite ABC-HRP Kit, Peroxidase (Standard) (Vector Laboratories Catalog# PK-6100).
3,3’ diaminobenzidine tetrahydro-chloride DAB tablet (0.01 g, Millipore/Sigma Catalog# D5905)
NAS: Nickel ammonium sulfate hexahydride (ThermoFisher Catalog# N48)
Hydrogen Peroxide (H2O2): 30% solution (Millipore/Sigma Catalog# HX0635)
DAB/NAS solution, freshly prepared and filtered before use: For 25 mL use 1 DAB tablet, and 75 mg NAS in 25 mL TB, add 1.5 μL H2O2 immediately before use
Citrus Clearing Solvent (Richard-Allan Scientific/ThermoFisher Catalog# 8301)
DEPEX mountant (Electron Microscopy Sciences Catalog# 13515)
70%, 95%, 100% ethanol
Gelatin coated microscope slides
6 well plates, with net inserts (Corning Catalog# 351146)
15 cm Petri dishes (Corning Catalog# 351029)
Candidate nAb BCS, see Support Protocol 3
Brushes,
Plate rocker
Method
Day 1
-
Wash sections twice for 5 min each with TBS at RT with gentle rocking/agitation.
Use a 6-well plate with net insert for accelerated washing. Make sure not to overflood the well with TBS since floating sections may be caught at the rims of the inserts. Be gentle lifting and lowering the inserts while transferring to prevent sections from breaking due to surface tension of the buffer.
Permeabilize sections by adding 800 μL per well TBS with 0.5% Triton X-100, 10 μg/mL avidin and incubating for 45 min at RT with gentle rocking/agitation.
Wash sections two times for 5 min each in TBS at RT with gentle rocking/agitation.
Block sections by adding 800 μL per well vehicle (TBS, 0.1% Triton-X-100, 5% NGS) plus 18.75 μg/mL biotin and incubating for 45 min at RT with gentle rocking/agitation.
Add 800 μL per well primary antibodies (nAb BCS and controls). Control antibodies are diluted in ice-cold vehicle as needed. nAb BCS are used undiluted, adding Triton X-100 to 0.1% and NGS to 5% in each well. Incubate sections overnight at 4°C with gentle rocking/agitation.
Day 2
Wash sections five times for 5 min each in TBS at RT with gentle rocking/agitation.
Add 800 μL per well anti-HA 2–2.2.14 IgG1 mAb diluted 1:2,000 in vehicle and incubate for 1 h at RT with gentle rocking/agitation.
Wash sections five times for 5 min each TBS at RT with gentle rocking/agitation.
Add 800 μL per well Biotin SP conjugated goat anti-mouse IgG1 secondary antibody at 0.5 μg/mL in vehicle and incubate for 1 h at RT with gentle rocking/agitation.
Just before the end of this incubation, make ABC reagent. For each 5 mL of TBS, add 2 drops of ABC kit Solution A and 2 drops of Solution B. The ABC solution requires a 30 min pre-incubation period at 4°C to work properly. Make sufficient volume for 800 μL per well.
While the ABC complex is being formed, wash sections five times for 5 min each TBS at RT with gentle rocking/agitation.
Incubate sections in ABC solution for 1 h at RT with gentle rocking/agitation.
Wash sections three times for 5 min each in TBS at RT with gentle rocking/agitation.
-
3.
Wash sections two times for 5 min each in TB (50 mM Tris, pH 7.5) at RT with gentle rocking/agitation.
-
4.
During these washes, prepare and filter 100 mL DAB/NAS developing solution. Make sufficient volume for 800 μL per well. Just prior to use add H2O2. DAB is a suspected carcinogen so it should be handled under a hood with appropriate protective gear, and all glassware used during preparation of the solution should be cleaned with bleach.
-
5.
Develop sections in DAB/NAS solution. The developing duration will vary so it is necessary to visually monitor the intensity of the reaction constantly so as to avoid over developing. The optimal development time is dependent on the specific nAb. Develop the negative control for a time equal to the longest nAb development time.
-
6.
To stop development, transfer the developed sections to TB and rinse two times with TB.
-
7.
Mount sections on gelatin-coated microscope slides: Use a 15 cm Petri dish filled with TB, submerge a slide in the buffer. Using a brush, position a section right above the desired mounting area and while holding the section with the brush, gently lift the slide out of the TB. Tilt the slide to remove excessive buffer and let it air dry for at least 2 h or overnight at RT.
-
8.
Dehydrate mounted sections successively in 70%, 95%, and 100% (two times) ethanol, each step for 5 min each at RT. Clear in Citrus Clearing Solvent, two times for 10 min each at RT. Perform all steps under a chemical fume hood. Coverslip with Electron Microscopy Sciences DEPEX mountant. The slides should lay flat in the fume hood for 3–5 days prior to analysis.
C). Multiplexed immunofluorescence immunohistochemistry on brain sections
Enhanced validation of nAbs for immunohistochemistry is performed using multiplexed immunofluorescence labeling of brain sections. This allows for direct comparison of nAb labeling to that of controls antibodies against the same target or anatomical markers on the same section.
Prior considerations and experimental design
As with the other assays it is important to choose a correct series of controls that will accompany the nAbs to be evaluated. In this case positive controls include previously validated antibodies against the same target protein, and established markers for specific cells or subcellular structures.
This protocol makes use of brain sections from a rat subjected to transcardial perfusion with formaldehyde. The perfusion and preparation of brain sections was performed as described in a previously published protocol (Paletzki & Gerfen, 2019) with the following modifications: 4% formaldehyde fixative is prepared directly from paraformaldehyde the same day it is to be used and does not include NaCl, heparin sulfate (1 mL/kg) and sodium nitrite (0.5 mL of a 3% solution) is injected into the left ventricle prior to saline perfusion, and there is an additional cryoprotection step in 30% sucrose.
Reagents and materials
30 μm-thick sagittal rat brain sections. Sections should be collected in 0.1 M PB with 10 mM NaN3.
0.1 M PB: 0.1 M Na2PO4, pH 7.4
NGS: normal goat serum (GeneTex Catalog# GTX73249)
Vehicle solution: 10% NGS in 0.1 M PB+ 0.3% Triton X-100
Alexa Fluor 488 anti-HA mouse IgG1 mAb 16B12 (ThermoFisher Catalog# A21287)
Alexa Fluor 555 anti-mouse IgG subclass-specific or anti-rabbit secondary antibodies, dependent on the controls used
Hoechst 33258 Pentahydrate (bis-Benzimide)-FluoroPure™ Grade (ThermoFisher Catalog# H21491)
Prolong™ Gold antifade mountant (ThermoFisher Catalog# P36930)
15 cm Petri dishes (Corning Catalog# 351029)
Plate rocker at 4°C
Fluorescent microscope
Method
Day 1
Wash sections four times 5 for min each with gentle rocking/agitation at 4°C in ice-cold 0.1 M PB (0.1 M Na Phosphate, pH 7.4).
Block sections in vehicle solution for 1 h with gentle rocking/agitation at 4°C.
Incubate sections in designated immunolabels, nAbs and controls, diluted in ice-cold vehicle. nAb BC supes are to be used with minimal dilution. Incubate sections overnight at 4°C with gentle rocking/agitation.
Day 2
Wash sections four times for 5 min each with gentle rocking/agitation at 4°C in ice-cold vehicle solution.
Incubate sections for 1 h with gentle rocking/agitation at 4°C in appropriate secondary antibody cocktail. From this step on the sections should be protected from room light.
Wash sections for 5 min in vehicle with gentle rocking/agitation at 4°C.
Wash sections for 5 min in 0.1 M PB with gentle rocking/agitation at 4°C.
Wash sections for 5 min in 0.05 M PB with gentle rocking/agitation at 4°C.
Mount on gelatin-coated microscope slides (floating in 0.05 M PB in a 15 cm Petri dish).
Air dry slides. Add 15 μL mounting media to each section, coverslip and cure per manufacturer’s recommendations.
After curing
Analyze and image slides using a fluorescent microscope with the appropriate fluorescence filter sets. For example images of results from this type of analysis, please refer to previously published work (Dong et al., 2019).
BASIC PROTOCOL 8: Generation of nAb-pEGFP mammalian expression constructs
When evaluating nAbs for intrabody function it is necessary to express them in mammalian cells. To accomplish this, the coding sequences of previously sequenced nAbs need to be transferred from pComb3XSS to a mammalian expression plasmid. This is performed using Gibson assembly (Gibson et al., 2009). This protocol is designed for use with the Phusion DNA Polymerase (ThermoFisher Catalog# F530S). This contains the materials and reagents needed for the process, included in the list below. A key consideration is that the OmpA leader sequence needs to be removed from the N-terminus of the encoded nAbs to obtain cytoplasmic expression in mammalian cells. This will also remove the start codon at the beginning of the OmpA sequence. It is crucial to ensure that a natural start codon exists at the 5’ end of the nAb coding region, or to add one during cloning. We have generated expression plasmids in which the nAb is expressed with an appended GFP, retaining the 6XHis and HA tags at the junction between the nAb and GFP.
Reagents and materials
5 U/μL Phusion DNA Polymerase (ThermoFisher Catalog# F530S)
Nuclease free water
10X PCR buffer (GC or HF)
50 mM MgCl2
10 mM dNTP mix
100 mM primers (see sequences below)
SmaI Restriction enzyme (NEB Catalog# R0141)
QIAquick Gel Extraction kit (Qiagen Catalog# 28704)
Chemically competent cells (e.g., XL1-Blue, DH5alpha, DH10beta)
QIAprep Spin Miniprep Kit (Qiagen Catalog# 27104)
14 mL round bottom tubes (Falcon Catalog# 352059)
PCR tubes (Genemate Catalog#T-3035–2)
LB+ 1.5% w/v agar+ 50 μg/mL kanamycin bacterial culture plates
nAb plasmids, see Basic Protocol 6
Thermocycler
Primers
| Forward primer | |
|---|---|
| F1 | 5’-CTTCGAATTCTGCAGTCGACGGTACCGCGGGGCCATGCAGKTGCAGCTCGTGGAGTC-3’ |
| Reverse primer | |
| R1 | 5’-CTCACCATGGTGGCGACCGGTGGATCCCTAGCGTAGTCCGGAACGTCGTACGGGTA-3’ |
Method
Day 1
- Prepare the PCR reactions as follows:
1 rxn (μL) Nuclease free water 36 10X Buffer (either GC or HF) 10 dNTPs 2 Phusion Polymerase 1 Forward primer 1 Reverse primer 1 Template (nAb plasmid) 1 -
Incubate the reactions in a thermocycler with the following PCR cycles:
1 cycle: 98 °C for 30 s;
31 cycles: 98°C for 10s, 70°C for 30 s, 72°C for 2 min;
1 cycle: 72°C for 10 min;
- Linearize the pEGFP-N1 plasmid by digestion with the SmaI restriction enzyme. Prepare the restriction enzyme reactions as follows:
1 rxn (μL) pEGFP-N1 vector 1 μg 10X CutSmart Buffer 5 SmaI 1 Nuclease free water To bring to 50 μL total Incubate the reaction at 25°C for 15 min. Heat inactivate at 65°C for 10 min.
Run the PCR product and linearized plasmid on a 0.8% agarose gel and confirm sizes. Linearized pEGFP-N1 should be ≈ 4.7 kb and the PCR product should be ≈ 0.5 kb.
Excise the bands and purify the linearized plasmid and individual PCR products using the Qiagen Gel Extraction Kit per the manufacturer’s guidelines.
- Assemble the Gibson assembly reaction. Prepare the following reaction on ice:
Gel purified insert (≈ 100 ng/μL) 4 μL SmaI digested vector (≈ 20 ng/μL) 1 μL 2X Gibson Assembly Master Mix 5 μL Incubate the samples in a thermocycler at 50°C for 60 min. After the incubation store samples on ice or at −20°C for subsequent transformation. Transform the reaction into chemically competent cells per the manufacturer’s protocol.
Plate transformations on LB + agar + kanamycin plates and incubate overnight at 37°C.
Day 2
To confirm the assembly worked and the construct is correct, colonies can be picked from the plate, miniprepped per the manufacturer’s protocol, and sequenced. Sequencing is done using the CMV Fwd primer.
BASIC PROTOCOL 9: Validation of nanobodies as intrabodies
Once the mammalian expression constructs have been generated (see Basic Protocol 8), nAbs can be evaluated for intrabody function. This is initially done by co-expression of the GFP-tagged nAb and the target protein in heterologous cells by transient transfection. This can be performed in 96 well microtiter plates to allow for simultaneous evaluation of dozens of candidate nAbs. This is followed by expression of the GFP-tagged nAb in primary cultures of neurons expressing endogenous target protein. Analyses of expression and subcellular localization of the nAb and target by immunocytochemistry allow for a determination of intrabody function of each nAb by determining whether the two signals colocalize, and whether the subcellular localization of either protein is impacted by co-expression.
A). nAb-EGFP expression +/− target co-expression in heterologous COS-1 cells
This assay makes use of COS-1 cells that do not endogenously express detectable levels of target protein. This allows for a controlled comparison of the subcellular localization of nAb and target expressed singly and then co-expressed.
Prior considerations and experimental design
The cells are transiently co-transfected to express the GFP-tagged nAb and the target. Separate wells are transfected singly with either each of the GFP-tagged nAbs or the target protein. Control wells are transfected with target protein co-expressed with GFP alone. Nanobodies are considered to be positive in this assay when co-transfected cells show colocalization of the nAb and the target. An additional evaluative criterion is whether the subcellular localization of the GFP-tagged nAb or the target protein is altered by co-expression, which would be indicative of a direct interaction impacting localization. For example, we have found that GFP-tagged nAbs expressed alone have nuclear localization that is reduced by co-expression with and presumably binding to cytoplasmically or plasma membrane localized target proteins.
Reagents and materials
COS-1 (ATCC# CRL-1650) cells, to be thawed and maintained according to ATCC protocols and institutional BSL-2 guidelines.
Lipofectamine 2000 (Invitrogen Catalog# 52881)
1X Optimem (Gibco Catalog# 31985–062)
COS cell media: 10% BCS, 100 μg/mL penicillin-streptomycin in DMEM
DMEM (Gibco Catalog# 11965–092)
Penicillin-streptomycin (Gibco Catalog# 15140–122)
Bovine Calf Serum (BCS) (Hyclone Catalog# SH3007303)
0.05% Trypsin-EDTA (Gibco Catalog# 25300–054)
96 well plates (Greiner Bio-One Catalog# 655090)
10 cm Tissue Culture dishes (Falcon Catalog# 353003)
Sterile reagent reservoir (Corning Catalog#4872)
1X DPBS: 2.67 mM KCl, 1.47 mM KH2PO4, 136.9 mM NaCl, 8.10 mM Na2HPO4
1X DPBS + 1% NaN3
Fixative: 4% formaldehyde (FA) prepared fresh from paraformaldehyde (PFA), 1 mM CaCl2, 1 mM MgCl2, 0.1% Triton X-100, 1X DPBS
Blocking solution (BLOTTO-T): 20 mM Tris-HCl, 0.15 M NaCl, 4% dry milk powder + 0.1 % Triton X-100
Alexa Fluor 555 anti-mouse IgG subclass-specific or anti-rabbit secondary antibodies, dependent on the controls used
Hoechst 33258 (ThermoFisher Catalog# H21491)
10 mL stripette (Falcon Catalog# 357530)
1.5 mL microcentrifuge tubes (Fisher Catalog# 5408129)
nAb candidates in mammalian expression plasmids, see Basic Protocol 8
Fluorescent microscope
Method
Day 1
Stock culture plates of COS-1 cells should be ≈80% confluent before starting. Determine the number of 10 cm dishes of cells needed, so as to have 2,500 cells/well.
Remove media from the dishes and wash with 10 mL DPBS to remove dead cells. Use 1 mL trypsin to detach cells from the dish, swirling and incubating the dish up to 5 min. Add 9 mL of media to deactivate the enzyme and using a stripette gently detach the cells from the surface of the dish.
Remove the amount of media needed, as calculated in step 1. Bring this volume up with cell media, to a suitable final volume for pipetting 100 μL/well. Transfer to a sterile reagent reservoir.
Using a multichannel pipettor, pipette 100 μL/well. Make sure to swirl reservoir prior to pipetting, to avoid cells settling in the reservoir to ensure they are evenly dispensed.
Incubate plates overnight at 37°C in a 5% CO2 incubator.
Day 2
In a tissue culture hood, prepare sterile 1.5 mL microcentrifuge tubes with the necessary amount of DNA. Use 0.1 μg total DNA per well. In wells where target and nAb plasmids are co-transfected, 0.05 μg of each plasmid should be used. Add 25 μL Optimem to each tube.
Prepare a stock of lipofectamine 2000 in Optimem. Add (0.42*number of tubes prepared in step 1) μL of Lipofectamine 2000 to (25* number of tubes prepared in step 1) μL of Optimem.
Incubate at RT for 5 min.
Add 25 μL of the mix prepared in step 2 to each of the tubes prepared in step 1. Mix well and incubate at RT for 20 min.
Bring each tube up to a final volume of 300 μL per 12 wells with cell media.
Add 25 μL of transfection mixture to each well of plated cells, as prepared on day 1.
Incubate at 37°C in a 5% CO2 incubator for 48 h.
Day 3
(Optional) Check cells are growing and are healthy. Incubate in a 37°C incubator overnight.
Day 4
-
10.
Pipette off the cell media. Working in a fume hood, add 50 μL of FA fixative to each well. Incubate 30 min at 4°C.
-
11.
Pipette off fixative and dispose of according to institutional guidelines for formaldehyde disposal.
-
12.
Rinse plates three times with 100 μL per well of ice-cold DPBS.
-
13.
Block plates with 100 μL per well blocking solution. Incubate 1 h at RT.
-
14.
Dilute selected anti-target antibodies in blocking solution to the appropriate concentrations. Add 50 μL per well and incubate 1 h at RT.
-
15.
Wash plates three times for 10 min each with 100 μL per well blocking solution.
-
16.
Dilute Alexa Fluor secondary antibodies to 1:2000 in blocking solution. Include a nuclear stain such as Hoechst 33258. Incubate 45 min at RT, keep away from light.
-
17.
Wash plates three times for 10 min each with 100 μL per well of DPBS.
-
18.
Remove the final wash. Add 50 μL per well of DPBS + 1% NaN3. Store plates at 4°C until analysis with a fluorescent microscope. For example images of results from this type of analysis, please refer to previously published work (Dong et al., 2019).
B). nAb-EGFP expression in cultured neurons
Once GFP-tagged nAbs are selected as being positive for intrabody function in transfected heterologous cells, they are next evaluated for intrabody function in an appropriate neuronal culture system in which the target protein is endogenously expressed. This involves exogenous expression of the GFP-tagged nAbs in the cultured neurons and evaluation of whether they localize to the same sites as the endogenous target protein as visualized by immunocytochemistry.
Prior considerations and experimental design
It is critical to select a neuronal culture system that endogenously expresses the target protein at levels sufficient to detect with immunocytochemistry. For our purposes, cultured hippocampal neurons have been used. The GFP-tagged nAbs are expressed by transient transfection, followed by immunocytochemistry with target-specific antibodies. The GFP-tagged nAbs are considered to be positive in this assay when transfected cells show colocalization with target. Cells transfected with GFP alone are used as a negative control for colocalization with target.
Reagents and materials
Embryonic hippocampal neurons, to be cultured following established protocol (Kaech & Banker, 2006)
Lipofectamine 2000 (Invitrogen Catalog# 52881)
1X Optimem (Gibco Catalog# 31985–062)
DMEM (Gibco Catalog# 11965–092)
Penicillin-streptomycin (Gibco Catalog# 15140–122)
Bovine Calf Serum (BCS) (Hyclone Catalog# SH3007303)
0.05% Trypsin-EDTA (Gibco Catalog# 25300–054)
0.25% (w/v) trypsin (ThermoFisher Cat# 15050065) in HBSS
Plating medium: Neurobasal medium (ThermoFisher Catalog# 21103049) with 10% FBS (ThermoFisher Catalog# 16140071), 2% B27 (ThermoFisher Catalog# 17504044), 2% GlutaMAX (ThermoFisher Catalog# 35050061), and 0.001% gentamycin (ThermoFisher Catalog# 15710064)
0.5 mg/mL poly-L-lysine (Millipore/Sigma Catalog# P2636)
Cytosine-D-arabinofuranoside (Millipore/Sigma Catalog# 251010)
10 cm Tissue Culture dishes (Falcon Catalog# 353003)
Glass bottom 35 mm dishes (MatTek Catalog# P35G-1.5–14-C)
1X DPBS: 2.67 mM KCl, 1.47 mM KH2PO4, 136.9 mM NaCl, 8.1 mM Na2HPO4
1X DPBS + 1% NaN3
Fixative: 4% formaldehyde (FA) prepared fresh from paraformaldehyde (PFA), 1 mM CaCl2, 1 mM MgCl2, 4% w/v sucrose, 1X DPBS
Blocking solution (BLOTTO-T): 20 mM Tris-HCl, 0.15 M NaCl, 4% dry milk powder + 0.1 % Triton X-100
Alexa Fluor 555 anti-mouse IgG subclass-specific or anti-rabbit secondary antibodies, dependent on the controls used
Hoechst 33258 (ThermoFisher Catalog# H21491)
1.5 mL microcentrifuge tubes (Fisher Catalog# 5408129), sterile
nAb candidates in mammalian expression plasmids, see Basic Protocol 8
Method
Days 1 to 7
Embryonic hippocampal neurons (cultured hippocampal neurons or CHNs) were cultured following an established protocol (Kaech & Banker, 2006). Briefly, hippocampi from embryonic day 18 rat embryos are dissected and dissociated enzymatically for 20 min at 37°C in 0.25% trypsin in HBSS. They are then dissociated mechanically by trituration with glass polished Pasteur pipettes. Dissociated cells are suspended in plating medium and plated at 60,000 cells per dish in poly-L-lysine coated glass bottom 35 mm dishes. Transfections are performed essentially as described previously (Lim, Antonucci, Scannevin, & Trimmer, 2000).
Day 7
Add cytosine-D-arabinofuranoside to a final concentration of 5 μM to inhibit non-neuronal cell growth.
- Transiently transfect neurons with 1 μg of each GFP-nAb mammalian expression plasmid using Lipofectamine 2000 for 1 h. Transfections are as follows:
- Dilute 3 μL of Lipofectamine 2000 into 50 μL of unsupplemented Neurobasal medium in a sterile microcentrifuge tube. Let stand for 5 min at RT.
- In a separate sterile microcentrifuge tube, dilute 1 μg of plasmid DNA into 50 μL unsupplemented Neurobasal medium.
- Pipette the Lipofectamine/Neurobasal solution into the DNA/Neurobasal solution and let stand at RT for 20 min. During this incubation warm sufficient volume (1.1 mL medium per transfection) of unsupplemented Neurobasal medium to 37°C in a water bath.
- Immediately prior to transfection, add 1 mL of warmed unsupplemented medium to plated neurons.
- Working quickly, to minimize temperature and pH fluctuations of the neuronal culture medium, gently pipette the Lipofectamine/DNA/Neurobasal solution drop-wise onto the neurons. Return dishes to the incubator.
- After incubating 1 h, change to fresh supplemented media. Leave the culture for 48h in the incubator before proceeding.
Day 9
Gently pipette off the cell media. Fix CHNs in ice-cold FA/sucrose fixative for 15 min at 4°C.
In a fume hood, pipette off fixative and dispose of according to institutional guidelines for formaldehyde disposal. Wash three times for 10 min each with 1 mL of ice-cold DPBS with gentle agitation. The second and third washes can be done outside of the fume hood.
Add 1 mL of blocking solution. Incubate 1 h at RT.
Dilute selected anti-target antibodies in blocking solution to the appropriate concentrations. Add 500 μL per well and incubate 1 h at RT.
Wash three times for 10 min each with 1 mL per well blocking solution.
Dilute Alexa Fluor secondaries at 1:2000 in blocking solution. Incubate 45 min at RT, keep away from light.
Wash three times for 10 min each with 1 mL per well of DPBS.
Remove the final wash. Mount coverslips using Prolong Gold mounting medium. Leave to cure per manufacturer’s protocol prior to analysis with the fluorescent microscope. For example images of results from this type of analysis, please refer to previously published work (Dong et al., 2019).
SUPPORT PROTOCOL 1: ELISA for llama serum testing, phage titer, and screening of selected clones
Throughout the protocols different samples (antiserum, phage, BC supes) will require ELISA testing before proceeding with the next step. The initial serum obtained from immunized llamas will be tested on multiple occasions, to ensure there is sufficient signal against the desired target protein before construction of the phage library. During panning of the phage library, it can be useful to screen the enriched sub-library by ELISA to confirm the panning is enriching for positive nAb-expressing phage as expected. Finally, ELISA is used to inform which of the secreted nAbs retain binding to the selected target protein, justifying further characterization. For this last use, if the immunization was against a tagged protein it is important to account for nAbs that might bind the tag and not the specific protein, in these cases nAbs are also screened in parallel against the tag, either by itself for large tags such as GST, or in the context of an unrelated protein for smaller tags such as 6XHis.
The different uses for this protocol relate to differences in the input sample, the choice of coating protein, and the detection proteins, as indicated in the following table.
| Function of ELISA | Coating protein | Input Sample, Primary incubation | Secondary incubation |
|---|---|---|---|
| a. Llama serum testing | Target protein | Immunized llama serum dilutions | anti-llama-HRP |
| a. Antigen-specific phage titer | Target protein | Panned phage library | 12CA5 mAb and goat anti-mouse IgG2b-HRP |
| b. Clone screening | Target protein, tag/non target protein (e.g., GST) | Bacterial culture supernatants | 12CA5 mAb and goat anti-mouse IgG2b-HRP |
Reagents and materials
Sample to be tested depending on objective of assay
96-well high-binding microplates (Greiner Bio-One Catalog# 655081)
0.6 % TMB substrate (Millipore/Sigma Catalog# T2885, keep this away from light)
Citrate buffer: 14.71 g/L sodium citrate, pH 5.5
1% (v/v) H2O2
1 N H2SO4
1X Phosphate Buffered Saline (PBS): 8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, 0.24 g/L KH2PO4
1–20 μg/mL target protein in PBS
(Optional) 10 μg/mL protein tag (e.g., GST) in PBS
BLOTTO: 20 mM Tris-HCl, 0.15 M NaCl, 4% dry milk powder
(a.) anti-llama IgG-HRP conjugated antibody (Abcam Catalog# AB112786)
(b. and c.) Anti-HA-HRP conjugated antibody (Invitrogen Catalog# PA1–29751)
Plate rocker
450 nm wavelength microplate reader
Method
Day 1
Coat high binding plates with 50 μL per well of target protein solution. The number of plates will depend on the number of samples to screen, plus wells for designated positive and negative controls. Incubate at 4°C overnight.
Optional: Coat an equal number of plates with 50 μL per well of tag or tagged protein using the same concentration as the tagged target protein. Incubate at 4°C overnight.
Day 2
Block plates with 200 μL per well BLOTTO for 1 h at RT. This can also be left overnight at 4°C.
Remove blocking solution. Add 50 μL per well of antibody test sample. For llama antiserum run a two-fold serial dilution series (e.g., 1:4,000–1:128,000) for each sample. For enriched phage sublibraries run a dilution series of 1:10–1:640 for each sample. For nAb BCS use undiluted. For control antibodies use at their designated dilutions. Incubate in primary antibody 1 h at RT. This incubation can also be performed overnight at 4°C.
Wash the plates three times for 10 min each with 150 μL per well of BLOTTO with gentle agitation.
Add 50 μL per well of HRP-conjugated secondary antibody at a 1:10,000 dilution. Incubate 1 h at RT.
Wash the plates three times for 5 min each with 150 μL per well of PBS with gentle agitation. During the last wash prepare the developing solution by combining 6.25 mL citrate buffer, 100 μL 0.6% TMB, and 25 μL 1% H2O2 per plate. Keep this solution away from the light.
Add 50 μL per well of developing solution. Incubate at RT for 10 to 20 min in the dark.
Stop the developing by adding 50 μL per well of 1 N H2SO4.
Read the plates in a plate reader at 450 nm.
- To analyze the ELISA results, compare OD450 values for to appropriate controls, as follows:
- For antiserum use preimmune serum as the negative control. Antiserum samples are scored as positives when exhibiting OD450 values ≥ 3X above the preimmune serum control at a 1:100,000 dilution.
- For phage use a no phage control. Phage samples are scored as positives when exhibiting OD450 values ≥ 3X above no phage controls.
- For nAb BCS use wells with culture medium alone as the negative control. Samples are scored as positives when exhibiting OD450 values ≥ 3X above the culture medium alone controls.
- In all cases, when comparing tagged target proteins against the tag-coated plates, samples that exhibit OD450 values ≥ 3X against the tagged target protein versus the tag alone are selected for further analysis.
SUPPORT PROTOCOL 2: Amplification of M13KO7 helper phage stock
pComb3XSS is a phagemid vector engineered for phage display. It is employed in conjunction with a helper phage such as M13KO7, in which case single-stranded phagemid DNA is preferentially packaged into phage that are then secreted into the culture medium. In the case of pComb3XSS, the protein encoded by the phagemid insert is expressed on the phage surface as a fusion with pIII coat-protein. It is necessary to prepare stocks of M13KO7 helper phage for this purpose.
Reagents and materials
E. coli ER2738 bacteria (NEB Catalog#E4104)
Super Broth (SB): 30 g/L tryptone, 20 g/L yeast extract, 10 g/L MOPS, pH 7.2
1X Phosphate Buffered Saline (PBS): 8 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, 0.24 g/L KH2PO4
5X PEG/NaCl: 200 g/L of PEG8000, 150 g/L NaCl, sterilized
Phage suspension buffer: 13.29 mL PBS, 1X protease inhibitor cocktail, 0.02% NaN3, 0.5% BSA)
20 mg/mL tetracycline stock
100 mg/mL ampicillin stock
70 mg/mL kanamycin stock
Protease inhibitor cocktail II (RPI Catalog# P50800–1)
1012 cfu/mL M13KO7 helper phage (NEB Catalog# N0315S)
LB + 1.5% w/v agar + 20 μg/mL tetracycline bacterial culture plates
14 mL round bottom tubes (Falcon Catalog# 352059)
2 L Erlenmeyer flask
0.22 μm syringe filter (Millex Catalog# SLGV013SL)
Temperature-controlled orbital shaking incubator
Centrifuge
Method
Day 1
Streak the LB + agar + tetracycline plate with E. coli ER2738 bacteria and incubate overnight at 37℃.
Day 2
Inoculate 5 mL in SB medium containing 20 μg/mL tetracycline with a single E. coli ER2738 colony. Incubate culture overnight at 37°C with orbital shaking at 250 rpm.
Day 3
Inoculate 400 mL of SB medium in a 2 L flask with 4 mL of the overnight culture. Incubate the culture ≈2–3 h, until the OD600= 0.5–0.6.
Add 800 μL of helper phage stock and incubate 30 min at 37°C without shaking.
Incubate the infected culture 1 h at 37°C with orbital shaking at 250 rpm.
Add 400 μL of 70 mg/mL kanamycin stock. Incubate the flask overnight at 37°C with orbital shaking at 250 rpm.
Day 4
Centrifuge the overnight culture for 15 min at 10,000 rpm. Transfer the supernatant to clean centrifuge bottles.
Add a fifth of the volume (≈100 mL total) of 5× PEG/NaCl and incubate the bottles for 2 h on ice.
Centrifuge at 12,000 rpm for 20 min and resuspend the phage pellet in 100 mL of PBS.
Centrifuge at 12,000 rpm for 20 min and resuspend the pellet in 15 mL of phage suspension buffer.
Filter the suspension with a 0.22 μm filter and store the aliquots at −80°C.
SUPPORT PROTOCOL 3: nAb expression in non-amber suppressor E. coli strains
To express the nAbs in E. coli bacteria without being fused to the pIII protein it is necessary to transform the phagemid DNA into a non-amber suppressor strain of E. coli, such as TOP10F’. This results in increased expression of the nAbs, and increases the signal in subsequent assays. The pIII protein can also confer higher background in certain assays.
Reagents and materials
Super Broth (SB): 30 g/L tryptone, 20 g/L yeast extract, 10 g/L MOPS, pH 7.2
SOC medium (optional): 2% Tryptone, 0.5% Yeast Extract, 0.4% glucose, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2 and 10 mM MgSO4.
20 mg/mL tetracycline stock
100 mg/mL ampicillin stock
100 mM IPTG stock, prepared fresh
14 mL round bottom tubes (Falcon Catalog# 352059)
Top10F’ chemically competent cells (ThermoFisher Catalog# C303003)
1 M NaN3 in PBS
1.5 mL microcentrifuge tubes (Fisher Catalog# 5408129)
nAb candidates in bacterial expression plasmids
Heat block or water bath, for temperature controlled incubation
Temperature-controlled orbital shaking incubator
Centrifuge
Method
Day 1
Warm a water bath or heat block to 42°C.
Remove aliquots of competent cells from −80°C and thaw on ice for 10 min. The number of cells will depend on the number of transformations to be done. Typically, 25–50 μL of cells will be used per transformation.
Add 1–5 μL of DNA to cells in pre-labeled tubes. Tap tube to mix, do not vortex the tube. Incubate on ice for 30 min.
Place tubes in pre-warmed 42°C water bath or heat block for 30 seconds. Return the tubes to ice for 2 min.
Add 200 μL of recovery medium (SB or SOC) without antibiotics to each tube. Incubate the tubes for 1 h at 37°C with orbital shaking at 250 rpm.
Plate cells by spreading 50–200 μL on LB + agar + ampicillin + tetracycline bacterial culture plates. Incubate overnight at 37°C.
Day 2
Prepare 14 mL round bottom tubes with 2 mL of SB medium containing 100 μg/mL ampicillin and 20 μg/mL tetracycline.
Pick single colonies from the plates of transformed cells and inoculate prepared tubes.
Incubate the tubes overnight at 37°C with orbital shaking at 250 rpm.
Day 3
Prepare 14 mL round bottom tubes with 2 mL of SB medium containing 100 μg/mL ampicillin and 20 μg/mL tetracycline.
Transfer 50 μL of the overnight culture these tubes. Incubate ≈4 h at 37°C with orbital shaking at 250 rpm, until the OD600 = 0.5–1.
Add IPTG to 0.5 mM-1 mM into each tube to induce the expression of the nAb.
Incubate the tubes overnight at 37°C with orbital shaking at 250 rpm.
Day 4
Centrifuge the tubes at 3000×g for 15 min.
Collect the supernatant, add NaN3 to 1 mM, and save for testing as immunolabels (See Basic Protocol 7).
COMMENTARY
General considerations when developing nAbs for diverse uses.
Similar to other antibodies, there are a wide variety of uses for nAbs in neuroscience research. We have focused on two of those here, namely the use of nAbs as immunolabels and as intrabodies. Additional uses are varied but include using nAbs to capture and purify targets proteins, and as chaperones to stabilize specific target proteins conformational states for structural analyses (Pardon et al., 2014). Each of these intended uses may rely on the availability of distinct nAbs, each of which is specific for the target in different states. For example, using nAbs as immunolabels of the target protein in fixed cells and tissues in immunocyto- and immunohisto-chemistry likely involves the nAb recognizing the protein in a different state than it would in a living cell when the same nAb is expressed as an intrabody. We found this to hold for our collection of anti-Homer1 nAbs, where a different set of nAbs was judged to be the most effective as immunolabels on fixed samples than those deemed best as intrabodies in living cells (Dong et al., 2019). As such, it is advantageous to begin with a substantial set of unique nAbs from the phage display and ELISA steps prior to their evaluation in the diverse applications in which they will be used. There exist many alternatives to customize these early steps, for example by performing the panning against a form of the protein more similar to the intended use (e.g., expressed in cells and used live or fixed). However, most protocols employ panning against purified recombinant protein as a reliable and sensitive method to isolate a diverse enough repertoire of target-specific nAbs from which application-specific nAbs can subsequently be identified.
Evaluation of nAbs as immunolabels
We describe above a two-step approach for evaluating nAbs for use as immunolabels. The first step is assaying for efficacy and specificity in immunocytochemistry in heterologous cells exogenously expressing the target protein. While the ultimate use of the nAbs is in native preparations, this proxy assay has numerous advantages. It allows for a relatively high throughput when performed in a 96 well microtiter plate format. The use of transient transfection also provides a sample that is a mosaic of cells with and without detectable target protein expression. Moreover, the expressing cells typically have high levels of target protein expression increasing sensitivity. When used in conjunction with double immunofluorescence labeling for the target protein, this represents a powerful system for evaluating a large collection of candidate nAbs. The visual evaluation of such samples is also straightforward, in determining whether the nAb labels the same subset of cells as the control antibody, and whether in these cells the pattern matches at the subcellular level. We note that it is not necessary to double label with an anti-target antibody, should one have available plasmid for expressing the target protein tagged with GFP or another fluorescent protein. We have found when developing mAbs for use in neuroscience research that this robust and simple assay is predictive, in that it is unlikely that samples negative in this assay will be positive in immunocytochemistry or immunohistochemistry against samples endogenously expressing the target protein (Gong et al., 2016). However, not all antibodies that are positive in this assay will exhibit effective and specific labeling in native samples as discussed in prior publications [e.g., (Gong et al., 2016)]. However, it serves as a useful filter to reduce the number of samples for immunohistochemistry on brain sections, which is more labor intensive, less sensitive, and more difficult to evaluate in terms of target labeling specificity. However, using the protocols above and applying the considerations detailed in prior publications for antibody validation [e.g., (Gong et al., 2016)], as well as those approaches consistent with general guidelines for antibody validation [e.g., (Taussig, Fonseca, & Trimmer, 2018; Uhlen et al., 2016)] this is an effective approach to identify nAbs that can be employed for specific labeling of the target protein in brain sections.
Purification of nAbs
The immunolabeling screening protocols above (ELISA, ICC, IHC) make use of nAbs in BCS preparations, that is nAbs that are secreted into the culture medium. This yields nAb preparations with sufficiently high concentrations of nAbs for detection in these assays, and the use of BCS is time and labor effective, allowing for screening of many more candidates and increasing the likelihood of obtaining nAbs for each intended purpose. Alternative protocols make use of periplasmic extracts of the expressing bacteria. The bacterial periplasm contains substantial amounts of nAbs that have been secreted from the bacterial cells but that do not cross the cell wall into the culture media. There are many protocols for making crude periplasmic extracts that are also commonly used for early stage evaluation of nAbs [e.g., (Pardon et al., 2014)]. There are also experimental needs for purified nAbs. The use of the pComb3XSS phagemid generates nAbs with C-terminal 6XHis tags that allow for one-step chromatographic purification over Ni-NTA columns. This approach can be used to purify nAbs from BCS and/or periplasmic extracts. Yields of nAbs can approach 10 mgs or more from a 1 L culture, at concentrations of 1 mg/mL or higher. It is important to note that as nAbs are 1/10 the size of conventional IgG antibodies, these values represent 10X higher molar equivalents than conventional antibodies.
Critical Parameters
The quality and characteristics of protein used for immunization, panning and ELISA
The protocols described above rely heavily on the protein preparation that is used to immunize the llama, and to coat the wells of the microtiter plates used for panning and ELISA. These steps define the pool of nAbs available for evaluation in experimental applications. As discussed above, use of a full-length protein or a specific fragment representing a particular domain or region will fundamentally impact the uses of the nAb. However, there are other parameters related to the proteins used in these procedures that are also critical. Using higher purity preparations of the target protein will allow for a more focused representation of target-specific nAbs that emerge from these steps. This is in contrast to less pure preparations, for example those contaminated with proteins from the bacterial expression system, which will generate contaminate-specific nAbs that will be carried through the panning, ELISA and sequencing step and lead to time, labor and effort being spent on spurious nAbs. A somewhat analogous situation can also occur with highly purified preparations of tagged target proteins. In general, an affinity tag (GST, 6XHis, etc.) is needed to purify the recombinant target protein after it is expressed. These tags differ in size (e.g., GST is 26 kD while a 6XHis tag is only six sequential His residues). It is advantageous to use a smaller tag, or have high quality protein preparations with different tags, and use a different preparation for immunization than for panning and ELISA. However, larger tags such as GST can play a crucial role in stabilizing the partner proteins during expression and purification (Esposito & Chatterjee, 2006), such that producing proteins without this tag may not be practical. While methods exist to remove large tags after purification, this can introduce additional problems (Harper & Speicher, 2011). As such, we describe approaches employed at the early stages of panning and ELISA to eliminate anti-tag nAbs from the candidate pool.
Expression in amber suppressor versus non-suppressor strains
During the initial panning of the nAbs it is important to use an amber suppressor strain, the protocols described here make use of E. coli ER2738 bacteria. In the nAb phagemid construct there is an amber stop codon between the nAb gene and the coat protein pIII gene, which in amber suppressor strains of E. coli bacteria is ignored allowing for complete translation of the nAb-pIII fusion protein. Since only proteins where this fusion has worked correctly can be expressed on the phage surface, this is a critical step for phage display. However, once the phage panning has been completed and it is necessary to test the nAb, this fusion is of no use. At this point it is recommended to switch to use of non-suppressor bacterial strains, such as E. coli Top10F’. In these bacterial strains, translation will stop at the amber stop codon and only the nAb protein will be expressed. As a general rule, this results in higher concentrations of nAb, which could be an important component for success in later nAb validation assays.
Anti-HA epitope tag detection antibodies for brain immunohistochemistry
This protocol makes extensive use of anti-HA epitope tag detection antibodies to detect the nAbs used as immunolabels, based on the HA tag that is present at the C-terminus of nAbs expressed from the pComb3XSS plasmid. We have made use of the anti-HA mouse IgG2b mAb 12CA5 for immunocytochemical detection of HA-tagged proteins expressed in transfected heterologous cells and primary neurons [e.g., (Lim et al., 2000)]. We also use this in antiserum and phage ELISAs as detailed above. However, in our hands 12CA5 gives unacceptable levels of background labeling in mammalian brain sections, as observed in no-nAb control sections. We evaluated a number of anti-HA commercial antibodies and found that Alexa 488-conjugated anti-HA mouse IgG1 mAb 16B12 yielded the highest signal to noise for immunofluorescence immunohistochemistry on brain sections. The best signal for immunoperoxidase immunohistochemistry on brain sections is obtained by including avidin in the permeabilization step, and biotin in the blocking step, and by using unconjugated anti-HA mouse IgG1 mAb 2–2.2.14 as the anti-HA antibody for nAb detection.
Time Considerations
The total time from the start of immunizations until specific clones have been selected and sequenced can vary. This is particularly reliant on how long is needed for immunized llama antiserum to show sufficient immunoreactivity against each target protein. However, since immunization of a llama with up to five targets can be performed simultaneously, the overall time investment per target is reduced. The initial steps in the process involve construction of a phage library that includes nAbs for all the targets used in the immunization. It is only at the biopanning phase that target-specific selection takes place. As a general rule, immunizations will take approximately three months, with the remainder of the process requiring approximately one additional month from start to finish for each target.
SIGNIFICANCE STATEMENT.
Numerous forms of basic neuroscience research employ antibodies as key reagents. Recombinant camelid nanobodies offer many advantages over conventional antibodies due to their stability and small size. As such, they are emerging as valuable biomedical research reagents. We describe developing, screening and validating nanobodies for basic neuroscience research applications, including their use as immunolabels and as intrabodies.
Acknowledgements
Our work developing nAbs for neuroscience research was supported by National Institutes of Health BRAIN Initiative Grants U01NS099714 and U24NS109113 to J. S. Trimmer. We thank Yongam Lee, Leon Palao III, Stephanie Palacio, Camelia Dumitras, Belvin Gong, Karl Murray and Deborah van der List for their contributions to this work.
Footnotes
Conflict of Interest
The authors have no conflict of interest to declare.
Literature Cited
- Andrews NP, Boeckman JX, Manning CF, Nguyen JT, Bechtold H, Dumitras C, … Trimmer JS (2019). A toolbox of IgG subclass-switched recombinant monoclonal antibodies for enhanced multiplex immunolabeling of brain. eLife, 8, e43322. doi: 10.7554/eLife.43322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andris-Widhopf J, Rader C, Steinberger P, Fuller R, & Barbas CF 3rd. (2000). Methods for the generation of chicken monoclonal antibody fragments by phage display. J Immunol Methods, 242(1–2), 159–181. [DOI] [PubMed] [Google Scholar]
- Bradbury ARM (2010). The use of phage display in neurobiology. Curr Protoc Neurosci, 51, 5.12.11–15.12.27. doi:DOI: 10.1002/0471142301.ns0512s51 [DOI] [PubMed] [Google Scholar]
- Choi JH, & Lee SY (2004). Secretory and extracellular production of recombinant proteins using Escherichia coli. Appl Microbiol Biotechnol, 64(5), 625–635. doi: 10.1007/s00253-004-1559-9 [DOI] [PubMed] [Google Scholar]
- Chow KM, Whiteheart SW, Smiley JR, Sharma S, Boaz K, Coleman MJ, … Vander Kooi CW (2019). Immunization of alpacas (Lama pacos) with protein antigens and production of antigen-specific single domain antibodies. J Vis Exp(143). doi: 10.3791/58471 [DOI] [PubMed] [Google Scholar]
- Dong JX, Lee Y, Kirmiz M, Palacio S, Dumitras C, Moreno CM, … Trimmer JS (2019). A toolbox of nanobodies developed and validated for use as intrabodies and nanoscale immunolabels in mammalian brain neurons. eLife, 8, e48750. doi: 10.7554/eLife.48750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D, & Chatterjee DK (2006). Enhancement of soluble protein expression through the use of fusion tags. Curr Opin Biotechnol, 17(4), 353–358. doi: 10.1016/j.copbio.2006.06.003 [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, & Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods, 6(5), 343–345. doi: 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- Gong B, Murray KD, & Trimmer JS (2016). Developing high-quality mouse monoclonal antibodies for neuroscience research - approaches, perspectives and opportunities. N Biotechnol, 33(5 Pt A), 551–564. doi: 10.1016/j.nbt.2015.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper S, & Speicher DW (2011). Purification of proteins fused to glutathione S-transferase. Methods Mol Biol, 681, 259–280. doi: 10.1007/978-1-60761-913-0_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, & Banker G (2006). Culturing hippocampal neurons. Nat Protoc, 1(5), 2406–2415. doi: 10.1038/nprot.2006.356 [DOI] [PubMed] [Google Scholar]
- Lim ST, Antonucci DE, Scannevin RH, & Trimmer JS (2000). A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron, 25(2), 385–397. doi:S0896–6273(00)80902–2 [pii] [DOI] [PubMed] [Google Scholar]
- Muyldermans S (2013). Nanobodies: natural single-domain antibodies. Annu Rev Biochem, 82, 775–797. doi: 10.1146/annurev-biochem-063011-092449 [DOI] [PubMed] [Google Scholar]
- Paletzki RF, & Gerfen CR (2019). Basic neuroanatomical methods. Curr Protoc Neurosci, 90(1), e84. doi: 10.1002/cpns.84 [DOI] [PubMed] [Google Scholar]
- Pardon E, Laeremans T, Triest S, Rasmussen SG, Wohlkonig A, Ruf A, … Steyaert J (2014). A general protocol for the generation of nanobodies for structural biology. Nat Protoc, 9(3), 674–693. doi: 10.1038/nprot.2014.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GP (1985). Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science, 228(4705), 1315–1317. doi: 10.1126/science.4001944 [DOI] [PubMed] [Google Scholar]
- Taussig MJ, Fonseca C, & Trimmer JS (2018). Antibody validation: a view from the mountains. N Biotechnol, 45, 1–8. doi: 10.1016/j.nbt.2018.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Bandrowski A, Carr S, Edwards A, Ellenberg J, Lundberg E, … Yamamoto T (2016). A proposal for validation of antibodies. Nat Methods, 13(10), 823–827. doi: 10.1038/nmeth.3995 [DOI] [PMC free article] [PubMed] [Google Scholar]