

The crucial role of CD8+ T cells in vaccine-mediated protection is a valuable evidence for new insights in vaccine development.
Abstract
Zika virus (ZIKV) is associated with congenital malformations in infants born to infected mothers, and with Guillain-Barré syndrome in infected adults. Development of ZIKV vaccines has focused predominantly on the induction of neutralizing antibodies, although a suboptimal antibody response may theoretically enhance disease severity through antibody-dependent enhancement (ADE). Here, we report induction of a protective anti-ZIKV CD8+ T cell response in the HLA-B*0702 Ifnar1−/− transgenic mice using an alphavirus-based replicon RNA vaccine expressing ZIKV nonstructural protein NS3, a potent T cell antigen. The NS3 vaccine did not induce a neutralizing antibody response but elicited polyfunctional CD8+ T cells that were necessary and sufficient for preventing death in lethally infected adult mice and fetal growth restriction in infected pregnant mice. These data identify CD8+ T cells as the major mediators of ZIKV NS3 vaccine–induced protection and suggest a new strategy to develop safe and effective anti-flavivirus vaccines.
INTRODUCTION
Zika virus (ZIKV) is a reemerging member of the flaviviridae family, which includes dengue (DENV), yellow fever, Japanese encephalitis, and West Nile viruses. ZIKV was first isolated in Uganda in 1947, and since then, outbreaks have occurred in several regions of the globe. The major route of ZIKV transmission is through the bite of Aedes spp. mosquitoes, but it is also transmitted through sexual contact and blood transfusions, as well as transplacentally. ZIKV has been found to persist in the semen, testes, and female reproductive tract of humans and animal models for up to 6 months after infection (1, 2). The consequences of vertical transmission of ZIKV are particularly devastating, often resulting in developmental malformations, known as congenital Zika syndrome, in the fetuses of infected mothers. Women infected during the first trimester of pregnancy have a high risk of miscarriage or of fetuses with microcephaly and other brain abnormalities (3). During the 2015–2016 outbreak in Brazil, about 2000 cases of microcephaly per 200,000 ZIKV infections were reported (3). Viral RNA and whole virus have been detected in the brains of fetuses with central nervous system abnormalities, confirming this link (4, 5). ZIKV has also been detected in the central nervous system of adult patients and is linked to development of various neurological complications, including Guillain-Barré syndrome (6). Moreover, ZIKV infection of memory-related regions in adult human and mouse brains suggests that memory and cognitive deficits may be important neurologic sequelae to ZIKV infection in adults (7). These observations underscore the urgent need to develop a vaccine that is safe for use during pregnancy and in infants, children, and adults. Despite intense efforts, there are no currently approved ZIKV vaccines.
In common with other flaviviruses, the ZIKV genome encodes a polyprotein that is cleaved into three structural proteins [capsid (C), premembrane (prM), and envelope (E)] and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). Of these, prM, E, NS1, NS3, and NS5 proteins have been shown to be the predominant targets of the adaptive anti-ZIKV immune response in humans (8–10) and mice (11–16). Apart from whole-virus vaccines, prM and E (prM-E) proteins are the major constituents of the more than 40 anti-ZIKV vaccine candidates developed since 2016, most of which are based on DNA, conventional nonreplicating mRNA, inactivated virus, attenuated virus, or viral vectors (17). These anti-ZIKV vaccines have been designed to elicit neutralizing antibody (nAb) responses, which are assumed to be the key mechanism of protection against infection. However, there is a severe and potentially lethal drawback to eliciting a suboptimal Ab response, because weak or poorly nAb responses may enhance the severity of a subsequent infection with a heterologous flavivirus or serotype. This phenomenon, known as Ab-dependent enhancement (ADE), occurs when subneutralizing Abs promote virus infection by facilitating uptake into Fcγ receptor–bearing cells (18–20). A growing body of evidence from long-term cohort studies in DENV-endemic countries (18, 20) and from clinical trials of the first DENV vaccine (Dengvaxia) to be licensed for human use (19) implicates a major role for ADE in human DENV pathogenesis. Although ZIKV exists as a single serotype, it is closely related to and cocirculates with the four DENV serotypes. Because of the genetic and antigenic similarities between ZIKV and DENV, Ab cross-reactivity between the two viruses is extensive (21, 22). Abs to DENV enhance ZIKV infection and disease severity in mice (21, 23, 24) and human placental explants (25), although there is limited epidemiologic evidence demonstrating ZIKV-ADE in humans (26). Conversely, studies in mice and rhesus macaques suggest that ZIKV Ab can mediate ADE of DENV infection. For example, cross-reactive monoclonal Abs generated against ZIKV mediate DENV-ADE in mice (22); previous ZIKV infection results in increased peak DENV viremia in rhesus macaques (27); and maternally acquired ZIKV Abs enhance DENV disease severity in mice (28). These studies of ZIKV and DENV pathogenesis highlight the need for anti-flaviviral vaccines that avoid the possibility of facilitating ADE.
For these reasons, we focused our efforts on developing anti-flaviviral vaccines that elicit protective T cell–mediated responses to ZIKV infection. Work by our group and others have shown that CD8+ T cells are key mediators of protection against primary ZIKV infection (11, 12, 15) and sequential DENV infection followed by ZIKV infection in nonpregnant (16, 29) and pregnant (30) mice. In line with our finding that previous DENV immunity confers cross-protection against subsequent ZIKV infection via CD8+ T cells in mice (29, 30), recent studies have revealed that previous DENV exposure also provides protection against subsequent ZIKV infection in humans (31–33). Although the immunological basis for the cross-protection in humans remains unclear, one study suggested that nAbs are unlikely to be responsible (33). In addition, our study using human leukocyte antigen (HLA) transgenic mice revealed that the immunodominance pattern of the CD8+ T cell response to ZIKV is altered by previous DENV infection (16). This is also consistent with human studies reporting that DENV-exposed individuals harbor T cells with cross-reactivity to ZIKV (8–10, 34) and that DENV exposure before ZIKV infection influences the magnitude and kinetics of the CD8+ T cell response to subsequent ZIKV infection (8). Thus, both mouse and human studies demonstrate that DENV immunity affords cross-protection against ZIKV and shapes the anti-ZIKV CD8+ T cell response, and mouse studies provide direct evidence that CD8+ T cells protect against ZIKV infection, thereby providing the rationale to develop a protective CD8+ T cell–directed ZIKV vaccine.
To this end, we developed a vaccine based on a synthetic alphavirus-derived RNA replicon to express ZIKV NS3, which is the dominant antigenic target of T cell responses to natural ZIKV infection in humans (8, 14). We also created a vaccine expressing ZIKV prM-E, which combines the major ZIKV Ab-eliciting proteins in humans (17), as a control in the immunogenicity and efficacy experiments. We examined the immunogenicity of the vaccines in immunocompetent C57BL/6 wild-type mice and additionally investigated both the immunogenicity and protective capabilities of the vaccines in transgenic HLA-B*0702 Ifnar1−/− mice. These mice express a broadly representative HLA molecule that presents epitopes relevant to the human T cell response to ZIKV, but they lack interferon (IFN) α/β receptors, rendering the mice more susceptible to viral infection. We show that ZIKV NS3 and prM-E vaccines both induce strong immunity that protects against lethal ZIKV challenge and prevents transplacental transmission. Notably, however, whereas both vaccines elicit an antiviral T cell response, the NS3 vaccine does not induce neutralizing Abs. Mechanistically, CD8+ T cells are shown to be necessary and sufficient for NS3 vaccine–induced protection. These data provide proof of concept that an NS3-based, T cell–centric vaccine can effectively protect against ZIKV infection and suggest that induction of a T cell response should be included as a major design factor in the development of safe and effective anti-flaviviral vaccines that avoid the potential for ADE.
RESULTS
Construction and characterization of RNA replicon–based NS3 and prM-E vaccines
Alphavirus-based self-replicating RNA replicons are ideal vaccine platforms capable of high levels of transient heterologous gene expression and induction of strong cellular, humoral, and mucosal immune responses (35). Replicons are positive strand RNAs consisting of the alphavirus 5′ and 3′ untranslated regions, replicase (nonstructural proteins 1 to 4), and 26S promoter to drive expression of a heterologous gene of interest. All viral structural genes are deleted and can be replaced by a gene or antigen of interest. To express ZIKV NS3 and prM-E proteins, we used the Synthetically Modified Alpha RNA Replicon Technology (SMARRT) platform (Synthetic Genomics Inc.), which is an alphavirus (Venezuelan equine encephalitis virus)–based replicon engineered to evade the antiviral immune response (Fig. 1A). A ZIKV prM-E–based vaccine lacking the immunodominant EDII fusion loop (FL) epitope has previously been shown to induce protection against ZIKV while minimizing the production of Abs that mediate DENV-ADE (36). We therefore introduced four mutations (T76R, Q77E, W101R, and L107R) within the E protein to eliminate the production of EDII FL–specific Abs that could result in DENV-ADE.
Fig. 1. Generation of ZIKV vaccines using SMARRT.
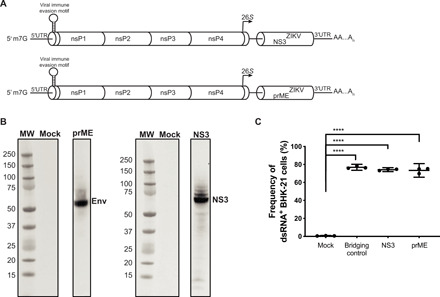
(A) Schematic of the ZIKV prM-E and ZIKV NS3 vaccines. All structural genes were removed from the Venezuelan equine encephalitis virus (strain TC-83) while retaining the four nonstructural proteins (nsP1 to nsP4) that encode the replicase. prM-E or NS3 genes from ZIKV strain SPH2015 replaced the structural genes downstream of the viral 26S promoter. BHK-21 cells were electroporated with water (Mock), an irrelevant RNA (bridging control), or RNA encoding either ZIKV prM-E or NS3 and then analyzed 20 hours after electroporation. UTR, untranslated region. (B) Western blot analysis of whole-cell lysates probed with anti-ZIKV E or NS3 Abs. MW, molecular weight. (C) Quantification of launch efficiency by intracellular staining of BHK-21 cells with an anti-dsRNA (J2) Ab. Data are presented as the mean ± SD of triplicates from one experiment, representative of two independent experiments. The nonparametric Mann-Whitney test was used to compare Mock versus each group; ****P < 0.0001.
To confirm the ability of these constructs to launch and produce protein efficiently, baby hamster kidney (BHK)–21 cells were electroporated with the replicon RNA and analyzed 20 hours later. Western blot analysis of cell lysates with Abs against ZIKV NS3 or E revealed expression of proteins with the predicted molecular weights (Fig. 1B), and flow cytometry of cells stained with an anti–double-stranded RNA (dsRNA) Ab indicated that approximately 80% of cells were dsRNA positive (Fig. 1C). These data confirm efficient launch and protein production by the alphavirus-derived replicon in mammalian cells. For the mouse experiments, the replicon constructs formulated in lipid nanoparticles are referred to as vaccines.
Immunogenicity of ZIKV NS3 and prM-E vaccines in C57BL/6 mice
To assess the immunogenicity of the ZIKV vaccines, we first examined T and B cell responses in wild-type C57BL/6 mice. Age- and gender-matched groups of naïve 4- to 5-week-old female and male mice were intramuscularly injected with 10 μg of NS3 or prM-E vaccine or saline alone (control) and boosted 28 days later in the same manner (Fig. 2A). Three weeks later (day 49), splenocytes were prepared and stimulated for 20 hours in vitro with a pool of H-2b–restricted peptides derived from ZIKV NS3 or prM-E, which were previously identified as epitopes for CD8+ and CD4+ T cells in C57BL/6 mice (Table 1) (11, 13). After stimulation, IFNγ-producing T cells [spot-forming cells (SFCs)] were enumerated using enzyme-linked immunospot (ELISpot) assays. IFNγ-producing CD8+ T cells (Fig. 2B) and CD4+ T cells (Fig. 2C) were more abundant in the spleens of NS3- and prM-E–vaccinated mice compared with control mice, and they were also more abundant in the spleens of prM-E–vaccinated mice compared with NS3-vaccinated mice (Fig. 2, B and C). The latter result may reflect the higher number of immunodominant T cell epitopes located in the prM-E protein compared with the NS3 protein.
Fig. 2. Immunogenicity of NS3 and prM-E vaccines in immunocompetent C57BL/6 mice.
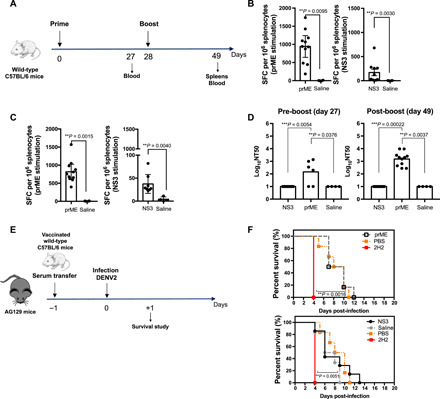
(A) C57BL/6 wild-type mice were intramuscularly injected with 10 μg of NS3 (n = 10) and prM-E (n = 11) vaccines or with saline (n = 4) and boosted in the same manner on day 28. (B and C) On day 49, splenocytes were stimulated with pooled ZIKV prM-E– or NS3-derived peptides. IFNγ-producing CD8+ T cells (B) and CD4+ T cells (C) were quantified as SFCs per 106 of splenocytes. (D) U937-DC-SIGN cell-based flow cytometric assay of ZIKV neutralizing activity (NT50) of sera collected on days 27 and 49. (E and F) Sera from ZIKV NS3- and prM-E–vaccinated C57BL/6 wild-type mice were collected on day 49, and 1 to 10 μl were intraperitoneally injected into 5- to 6-week-old AG129 mice (n = 6). Mice (n = 6) were injected with 15 μg of anti–DENV-prM Ab (2H2) or PBS as positive and negative controls, respectively. One day later, all AG129 mice were intravenously infected with 105 focus-forming units (FFU) of DENV2 S221. (F) Survival of mice treated as in (E). Data are presented as the mean ± SEM pooled from two independent experiments. The nonparametric Kruskal-Wallis and Mann-Whitney tests were used to compare three groups (D) and two groups (B and C), respectively. The Gehan-Breslow-Wilcoxon test was used to compare survival data.
Table 1. Summary of ZIKV-derived CD8+ and CD4+ T cell epitopes.
| Mouse model | Sequence position | Sequence | Peptides per pool | T cell specificity |
| Wild-type C57BL/6 | NS31656–1664 | VVIKNGSYV | 2 | CD8 |
| NS31866–1874 | PSVRNGNEI | CD8 | ||
| NS31740–1754 | GLPVRYMTTAVNVTH | 1 | CD4 | |
| PrM169–177 | ATMSYECPM | 3 | CD8 | |
| E294–302 | IGVSNRDFV | CD8 | ||
| E297–305 | SNRDFVEGM | CD8 | ||
| E644–658 | PVGRLITANPVITES | 2 | CD4 | |
| E346–360 | VRSYCYEASISDMAS | CD4 | ||
| HLA-B*0702 Ifnar1−/− | NS3206–215 | APTRVVAAEM | 5 | CD8 |
| NS3427–436 | GPMPVTHASA | CD8 | ||
| NS3574–582 | KPRWMDARV | CD8 | ||
| NS3405–413 | RVIDSRRCL | CD8 | ||
| NS3309–317 | FPDSNSPIM | CD8 | ||
| prM4–12 | LPSHSTRKL | 6 | CD8 | |
| E38–45 | KPTVDIEL | CD8 | ||
| E170–178 | TPNSPRAEA | CD8 | ||
| E173–180 | SPRAEATL | CD8 | ||
| E233–242 | TPHWNNKEAL | CD8 | ||
| E337–347 | GPCKVPAQMAV | CD8 |
To evaluate the neutralization capacity of the anti-ZIKV Ab response induced by NS3 and prM-E vaccines, we performed in vitro neutralization assays with serum samples collected on days 27 and 49 after vaccination using a cell-based flow cytometric assay. Sera prepared from mice on day 27 after vaccination with prM-E already contained Abs capable of blocking ZIKV infection of U937-DC-SIGN cells in vitro, and the neutralization titer (NT50) was further increased in the sera prepared after boosting (Fig. 2D). In contrast, sera prepared from NS3-vaccinated and control mice both lacked neutralizing activity (Fig. 2D). Quantification of anti-ZIKV immunoglobulin G (IgG) titers by enzyme-linked immunosorbent assay (ELISA) revealed the presence of anti-ZIKV E protein Abs in sera from prM-E–vaccinated mice (fig. S1A). These results demonstrate the presence of ZIKV E protein–specific IgG and anti-ZIKV nAbs in prM-E–vaccinated mice and indicate that the NS3 vaccine failed to elicit neutralizing anti-ZIKV Abs. This finding is consistent with previous work demonstrating that the dominant nAb response in humans is to ZIKV prM and E proteins.
Because the modified EDII FL epitope used in the prM-E vaccine is engineered to reduce the production of cross-reactive poorly neutralizing Abs that are known to increase ADE of infection (36), we investigated whether prM-E or NS3 vaccine–induced Abs could induce ADE in vivo. For these experiments, we used AG129 mice, which lack type I and II IFN receptors (IFNα/β/γR−/−) and are thus highly susceptible to lethal infection with flaviviruses. Groups of AG129 mice were injected with 1 or 10 μl of serum pooled from C57BL/6 mice that had been primed and boosted with NS3 or prM-E vaccines or saline (Fig. 2E). Additional groups of mice were injected with the anti-DENV prM Ab 2H2 as a positive control inducer of ADE, as previously described (37), or phosphate-buffered saline (PBS) as negative control. One day later, the AG129 mice were intravenously injected with DENV2 S221 and the mice were monitored for survival. As expected, 100% of the mice injected with 2H2 died 4 days after DENV2 infection, while 50% of the mice injected with PBS and then infected with DENV2 remained alive on day 10 (Fig. 2F). Mice receiving 1 μl (Fig. 2F) or 10 μl (fig. S1, B and C) of serum from control, NS3-vaccinated, or prM-E–vaccinated mice showed a similar pattern of survival to that of PBS-injected mice, indicating that the transferred serum did not mediate ADE. Collectively, these results demonstrate that the NS3 and prM-E vaccines are both highly immunogenic for CD4+ and CD8+ T cells, whereas only the prM-E vaccine elicits anti-ZIKV nAbs, and these Abs induced by the prM-E vaccine do not enhance DENV2 pathogenesis under the conditions tested.
Immunogenicity of ZIKV NS3 and prM-E vaccines in HLA-B*0702 Ifnar1−/− mice
HLA-B*0702 is one of the most common major histocompatibility complex (MHC) class I allele supertypes in humans and provides good representation of populations from diverse ethnic and geographic backgrounds worldwide. We previously showed that DENV epitopes identified as immunogenic in transgenic HLA-B*0702 Ifnar1−/− mice reflect the T cell repertoire in humans following natural exposure to DENV (38) and that changes in the immunodominance pattern of the T cell response to heterotypic secondary infections mirror those occurring in humans (39). Additional work has proven the utility of these mice in identifying immunodominant T cell epitopes in ZIKV and in characterizing the CD8+ T cell response to ZIKV infection (16). For these reasons, we used HLA-B*0702 Ifnar1−/− mice to further evaluate the CD8+ T cell response to ZIKV NS3 vaccine. Using a similar protocol to that used for the wild-type C57BL/6 experiments described above, we intramuscularly injected 4- to 5-week-old HLA-B*0702 Ifnar1−/−mice with 1 or 10 μg NS3 vaccine and boosted them in the same manner 28 days later (Fig. 3A). To examine the class I–restricted CD8+ T cell response in NS3-vaccinated mice, splenocytes were isolated on day 49 and stimulated in vitro with pooled NS3 peptides (Table 1). As expected, the NS3 vaccine induced a dose-dependent increase in the number of IFNγ-producing cells compared with the saline-injected mice, as measured using ELISpot assays (Fig. 3B). Based on these results, we used HLA-B*0702 Ifnar1−/−mice vaccinated with 10 μg of NS3 or prM-E vaccines for the rest of this study.
Fig. 3. Induction of anti-ZIKV immune response by NS3 and prM-E vaccines in HLA-B*0702 transgenic mice.
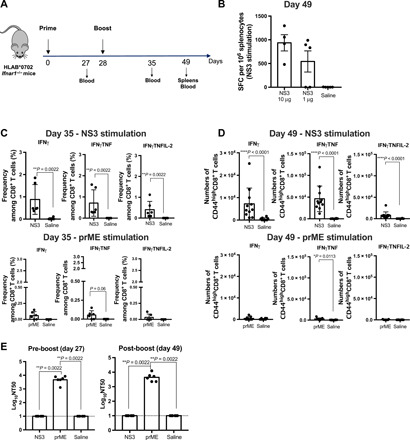
(A) Experimental protocol. (B) HLA-B*0702 Ifnar1−/− mice were intramuscularly injected with 10 μg (n = 4) or 1 μg (n = 5) of NS3 vaccine or with saline (n = 5) and boosted in the same manner on day 28. On day 49, splenocytes were stimulated with pooled ZIKV NS3 peptides and IFNγ-producing SFCs were evaluated by ELISpot. This experiment was performed once. (C and D) HLA-B*0702 Ifnar1−/− mice were intramuscularly injected with 10 μg (n = 6 and n = 11 for days 35 and 49) of NS3 or prM-E (n = 6 and n = 9 for days 35 and 49) vaccines or with saline (n = 5 and n = 10 for days 35 and 49) and boosted on day 28. (C) Peripheral blood mononuclear cells (day 35) and (D) splenocytes (day 49) were stimulated with pooled ZIKV NS3 or prM-E peptides. Cells producing IFNγ, IFNγ and TNF, or IFNγ, TNF, and IL-2 were quantified by flow cytometry. (E) U937-DC-SIGN cell-based flow cytometric assay of ZIKV neutralizing activity (NT50) of mouse sera collected on day 49. Data are presented as the mean or mean ± SEM from two independent experiments. The nonparametric Kruskal-Wallis and Mann-Whitney tests were used to compare three groups (B and E) and two groups (C and D), respectively.
Next, we evaluated the function of vaccine-elicited T cells in peripheral blood cells and splenocytes prepared on days 35 and 49 after vaccination, respectively. The cells were stimulated in vitro with a pool of NS3 or prM-E peptides, and the responding cytokine-secreting cells were quantified by flow cytometry. As shown in Fig. 3C, peripheral blood cells from NS3-vaccinated mice contained a significantly higher frequency of IFNγ-, IFNγ/tumor necrosis factor (TNF)–, and IFNγ/TNF/interleukin-2 (IL-2)–producing CD8+ T cells compared with cells from control mice. Similarly, the abundance of CD8+ T cells producing IFNγ-, IFNγ/TNF-, and IFNγ/TNF/IL-2 in the spleen was significantly higher in NS3-vaccinated mice compared with control mice (Fig. 3D). In comparison, relatively few cytokine-producing CD8+ T cells were induced by prM-E vaccination (Fig. 3, C and D).
The pattern of nAb production in vaccinated HLA-B*0702 Ifnar1−/− mice was similar to that in C57BL/6 mice. Thus, markedly higher ZIKV neutralizing activity (Fig. 3E) and ZIKV E-reactive IgG level (fig. S2A) were detected in sera from prM-E–vaccinated mice compared with NS3-vaccinated mice. Together, these data indicate that NS3 and prM-E vaccines are both immunogenic in HLA-B*0702 Ifnar1−/− mice, but whereas the NS3 vaccine stimulated a predominantly CD8+ T cell response without induction of nAbs, essentially the opposite response pattern was observed in prM-E–vaccinated mice.
NS3 vaccine–induced control of ZIKV burden and lethality in HLA-B*0702 Ifnar1−/− mice
Having established that the NS3 and prM-E vaccines could elicit T and B cell immunity in HLA-B*0702 Ifnar1−/− mice, we next evaluated the protective effect of vaccine-elicited immunity on ZIKV infection. Mice were vaccinated and boosted as described above, infected retro-orbitally with 103 focus-forming units (FFU) of ZIKV SD001 on day 49, and evaluated on day 52 (Fig. 4A). A notable reduction in the level of ZIKV RNA in the serum, brain, spleen, and liver of vaccinated mice compared with control mice (>100-fold reduction) was observed 3 days after infection, with the levels being nearly undetectable in most vaccinated mice (Fig. 4B). These results demonstrate that the ZIKV NS3 and prM-E vaccines could both induce robust immunity against ZIKV.
Fig. 4. Control of ZIKV infection in HLA-B*0702 mice by NS3 and prM-E vaccines.
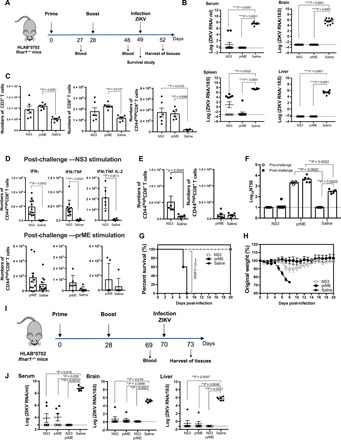
(A) HLA-B*0702 Ifnar1−/− mice were intramuscularly injected with 10 μg of NS3 or prM-E vaccines or with saline and boosted on day 28. (B) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of ZIKV RNA levels in the serum, brain, spleen, and liver of vaccinated mice with NS3 or prM-E (n = 11) or saline (n = 12) on day 52. (C) Number of CD3+, CD8+, CD44highCD8+ T cells and (D) CD44highCD8+ T cells producing IFNγ, IFNγ/TNF, or IFNγ/TNF/IL-2 or (E) granzyme B were reported. (F) Neutralizing activity (NT50) against ZIKV SD001 on days 49 (pre-challenge) and 52 (post-challenge). On day 49, vaccinated mice (NS3, n = 6; prM-E, n = 6; saline, n = 5) were lethally challenged retro-orbitally and survival (G) and weight loss (H) were monitored. (I) HLA-B*0702 Ifnar1−/− mice were intramuscularly immunized with 20 μg of NS3, prM-E (n = 6), or NS3 + prM-E (n = 5) vaccines or with saline (n = 6) as represented. (J) qRT-PCR analysis of ZIKV RNA levels in the serum, brain, and liver after infection (day 73). Data are presented as the mean or mean ± SEM from two independent experiments. The nonparametric Kruskal-Wallis and Mann-Whitney tests were used to compare more than two groups (B, C, F, and J) or two groups (D and E), and the Gehan-Breslow-Wilcoxon test was used to compare survival data (G).
To assess the effect of vaccination on the immune response to ZIKV infection, we isolated splenocytes on day 52, 3 days after infection, and examined the abundance and function of T cells after in vitro stimulation with NS3 or prM-E peptides. Compared with the control mice, both groups of vaccinated mice showed an expansion of total CD3+, CD8+, and activated CD8+ T cells (CD44highCD62LnegCD8+; referred to here as CD44highCD8+ T cells) at 3 days after ZIKV infection, although it was statistically significant in NS3-vaccinated mice only for the CD44highCD8+ T cells due to the high variability in response between mice (Fig. 4C). Effector CD8+ T cell function, as assessed by enumeration of activated CD8+ T cells producing cytokines, was also increased in the NS3- and prM-E–vaccinated mice compared with control mice after ZIKV infection (Fig. 4D). However, vaccination with NS3 induced a considerably more robust cytokine response by CD8+ effector T cells compared with prM-E vaccination, which was only slightly increased relative to the response in control mice (Fig. 4D). Moreover, NS3-vaccinated mice showed a notable increase in IFNγ/TNF/IL-2–producing CD8+ T cells, which are thought to respond more vigorously than IFNγ/TNF–producing cells to secondary viral infections (Fig. 4D) (40). To evaluate the cytotoxic T cell response, we also measured the number of CD8+ T cells producing the lytic protein granzyme B in the mice at 3 days after ZIKV infection (Fig. 4E). Here, too, we detected a significantly greater abundance of activated granzyme B–secreting CD8+ T cells in NS3-vaccinated mice compared with either the control or prM-E–vaccinated mice (Fig. 4E). Thus, the NS3 vaccine induced a polyfunctional CD8+ T cell response that included cells secreting IFNγ, TNF, IL-2, and granzyme B in response to ZIKV infection.
Notably, while we detected a robust nAb response in HLA-B*0702 Ifnar1−/− mice vaccinated and boosted with prM-E, the response did not increase 3 days after ZIKV infection, which contrasted with the significant increase in neutralizing activity observed after infection of control, unvaccinated mice (Fig. 4F). As expected, NS3-vaccinated mice did not display a detectable nAb response before or after ZIKV infection (Fig. 4F). The inability of ZIKV infection to further increase neutralizing activity in prM-E–vaccinated mice suggests that the prM-E vaccine induces sterilizing humoral immunity (Fig. 4B).
To determine whether vaccination with the NS3 and prM-E replicons could protect against lethal ZIKV infection, HLA-B*0702 Ifnar1−/− mice were vaccinated and boosted, infected on day 49 with a lethal dose of ZIKV SD001 (104 FFU), and monitored for weight loss, clinical signs of disease, and survival. All of the control unvaccinated mice succumbed within 8 days of ZIKV infection; however, all vaccinated mice survived until the end of the experiment (day 20), although some transient weight loss was observed in NS3-vaccinated mice (Fig. 4, G and H). The clinical disease scores followed a similar pattern (fig. S3). Thus, the NS3 vaccine efficiently protects against lethal ZIKV infection in the absence of nAb response. To examine the long-term efficacy of the vaccines, HLA-B*0702 Ifnar1−/− mice were vaccinated with NS3, prM-E, or NS3 + prM-E vaccines on day 0, boosted on day 28, and challenged with ZIKV SD001 on day 70. Tissues were harvested for analysis 3 days after challenge (Fig. 4I). Compared with the control mice, most of the NS3- and prM-E–vaccinated mice showed good control of viral growth, while the combination of NS3 + prM-E vaccines resulted in virtually complete viral control (Fig. 4J). Collectively, these data demonstrate that NS3 and prM-E vaccines alone can confer long-term protection against ZIKV infection, and the combination of both vaccines elicits even stronger long-lasting protection.
Efficacy of ZIKV NS3 and prM-E vaccines in pregnant HLA-B*0702 Ifnar1−/− mice
Because the consequences of maternal ZIKV transmission can be devastating, we next asked whether vaccination of dams with NS3 or prM-E can protect the fetuses from maternal ZIKV infection. For these experiments, female HLA-B*0702 Ifnar1−/− mice were vaccinated, boosted, and mated with BALB/c (H-2d) sires (Fig. 5A). To simulate ZIKV infection in humans during the first trimester, when the risk of congenital ZIKV syndrome is high (30), dams were infected on E7.5 with ZIKV (103 FFU) and then sacrificed at E14.5 for collection of the placenta, serum, and brain. The uteri (placenta, decidua, and fetuses) from NS3- and prM-E–vaccinated mice were larger than those from saline-injected mice and contained more viable fetuses (Fig. 5B), which were significantly heavier and larger (Fig. 5, C and D). Approximately 90% of fetuses from NS3- and prM-E–vaccinated dams were viable on E14.5, with only about 10% resorption (Fig. 5, C and D). In contrast, 56% of fetuses from control mice showed resorption and 44% showed growth restriction (Fig. 5, C and D). Data show the mean of viable fetuses (blue), growth-restricted fetuses (green), and resorbed fetuses (red; weight and size were based on the residual placenta). ZIKV RNA was either undetectable or several orders of magnitude lower (<100-fold) in the placentas of NS3- and prM-E–vaccinated mice compared with the control mice (Fig. 5E). Similarly, ZIKV RNA was undetectable in the serum and brain of all vaccinated pregnant mice compared with the control mice (Fig. 5F). Collectively, these data demonstrate that ZIKV NS3 and prM-E replicon-based vaccines markedly reduce transplacental transmission of ZIKV.
Fig. 5. Effects of NS3 and prM-E vaccines on the phenotypes of ZIKV-infected pregnant mice and fetuses.
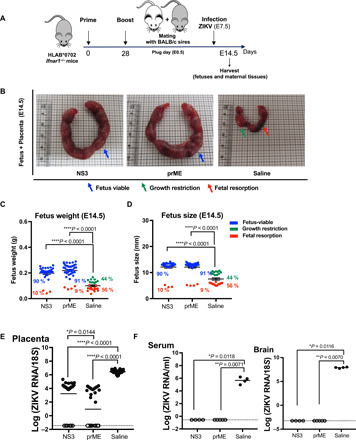
(A) Experimental protocol. HLA-B*0702 Ifnar1−/− dams (all n = 12) were intramuscularly immunized with 10 μg of NS3 or prM-E vaccines or with saline and boosted in the same manner on day 28. Dams were then mated with BALB/c sires. On embryonic day 7.5 (E7.5), pregnant mice were infected retro-orbitally with 103 FFU of ZIKV SD001. Seven days later (E14.5), mice were sacrificed, and fetuses and maternal tissues were harvested for analysis. (B) Representative placenta and fetuses from dams in the NS3, prM-E, and saline groups at E14.5. Color-coded arrows indicate viable fetus (blue), growth restriction (green), and fetal resorption (red). (C and D) Fetal body weight (C) and size (D) on E14.5. (E and F) qRT-PCR analysis of ZIKV RNA levels in the placentas (E) and maternal serum and brain (F) on E14.5. Data were pooled from independent experiments and represent a total of 32 fetuses from 4 mothers for the NS3 group, 40 fetuses from 5 mothers for the prM-E group, and 25 fetuses from 4 mothers for the saline group. Data are presented as the mean ± SEM. The nonparametric Kruskal-Wallis test was used to compare three groups. (B) Photo credit: J. A. Regla-Nava, La Jolla Institute for Immunology, CA.
Mechanism of NS3-mediated protection against lethal ZIKV infection in HLA-B*0702 Ifnar1−/− mice
To probe the mechanism of the NS3 vaccine–induced protection in more detail, we examined the effects of depletion or adoptive transfer of CD8+ T cells on viral clearance in vaccinated mice. For the depletion experiments, HLA-B*0702 Ifnar1−/− mice were vaccinated, boosted, and then injected with a CD8+ cell–depleting Ab or isotype control Ab on days −3 and −1 before challenge with ZIKV (Fig. 6A). Spleens analyzed 3 days after infection showed more than 90% depletion of CD8+ T cells (fig. S4A). These experiments revealed a notable effect of CD8+ T cell depletion on viral clearance. As expected, vaccinated mice treated with the isotype control Ab were fully protected against ZIKV infection, as revealed by the near undetectable levels of ZIKV RNA in serum, brain, spleen, and liver in NS3-vaccinated mice compared with control mice (Fig. 6B). In marked contrast, the levels of ZIKV RNA in tissues from CD8+ T cell–depleted NS3-vaccinated mice were comparable to those of the control infected mice (Fig. 6B).
Fig. 6. Contribution of CD8T cells to NS3 vaccine protection against ZIKV infection.
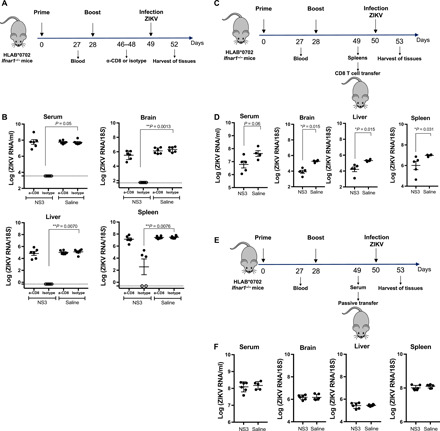
(A) HLA-B*0702 Ifnar1−/− mice were vaccinated with 10 μg of NS3 vaccine (n = 11) or saline (n = 12), boosted, and then intraperitoneally treated with CD8+ cell–depleting Ab (2.43) or isotype control Ab before ZIKV SD001 infection. (B) Serum, brain, liver, and spleen ZIKV RNA levels were analyzed by qRT-PCR (CD8- or isotype-treated mice). (C) HLA-B*0702 Ifnar1−/− mice were vaccinated as described for (A). On day 49, CD8+ T cells were isolated and transferred into naïve HLA-B*0702 Ifnar1−/− mice (1 × 107 cells). One day later, all mice were infected retro-orbitally with 103 FFU of ZIKV SD001. (D) ZIKV RNA levels in serum, brain, liver, and spleen were analyzed by qRT-PCR (NS3, n = 5; saline, n = 4). (E) HLA-B*0702 Ifnar1−/− mice were immunized as described for (A) (NS3, n = 12; saline, n = 12), and pooled sera from day 49 were passively transferred. (F) qRT-PCR analysis of ZIKV RNA in the serum, brain, liver, and spleen of mice transferred with serum from NS3-vaccinated mice (n = 6) or mice injected with saline (n = 5) 3 days after infection. Data are pooled from two independent experiments and are presented as the mean ± SEM. The nonparametric Kruskal-Wallis and Mann-Whitney tests were used to compare three groups and two groups, respectively.
Next, we performed adoptive transfer experiments to investigate whether CD8+ T cells were sufficient for viral clearance. HLA-B*0702 Ifnar1−/− mice were vaccinated and boosted with NS3 vaccine on days 0 and 28 (Fig. 6C). On day 49, CD8+ T cells were purified (>90 to 95% purity; fig. S4B) from pooled spleens and transferred into naïve HLA-B*0702 Ifnar1−/−mice (107 cells per mouse). One day later, the recipient mice were infected with 103 FFU ZIKV SD001. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of serum, brain, liver, and spleen at 3 days after infection revealed significantly lower ZIKV RNA loads (~10-fold) in the tissues of mice transferred with CD8+ T cells purified from NS3-vaccinated mice compared with control mice (Fig. 6D), indicating that NS3-elicited CD8+ T cells could protect naïve mice from ZIKV infection. Although we showed earlier that the NS3 vaccine does not elicit ZIKV nAbs (Fig. 3E), we wished to confirm this by performing passive transfer of sera from NS3-vaccinated mice. For these experiments, HLA-B*0702 Ifnar1−/− mice were vaccinated and boosted with NS3 on days 0 and 28, and serum was harvested on day 49. The sera were pooled and transferred to naïve HLA-B*0702 Ifnar1−/− mice (300 μl per mouse), and 1 day later, the recipient mice were infected with 103 FFU ZIKV SD001(Fig. 6E). As expected from the earlier observations, we observed no difference in tissue levels of ZIKV RNA in mice that received serum from NS3-vaccinated mice compared with those that received serum from control mice (Fig. 6F).
Together, these data demonstrate unequivocally that CD8+ T cells are necessary and sufficient to mediate protective immunity to ZIKV induced by vaccination with NS3. Moreover, the NS3 vaccine does not induce nAbs.
DISCUSSION
The first vaccine candidates reported to protect against ZIKV infection in mice and monkeys were DNA vaccines expressing ZIKV prM-E, and the nAb titers elicited by the vaccines were shown to correlate with protection against ZIKV viremia (17, 41). Since then, ZIKV vaccine development has focused on induction of a protective nAb response using the same antigen with multiple delivery platforms (17, 36, 42). All ZIKV vaccine candidates that have completed phase 1 trials in humans are E protein centric and are focused on eliciting nAb responses (43). As mentioned, however, a suboptimal Ab response to one flavivirus renders the host susceptible to exacerbation of disease upon infection with heterologous flaviviruses or serotypes through ADE (39). The clinical relevance of this observation has been supported by results from clinical trials with Dengvaxia, the only licensed DENV vaccine. The vaccine was found to elicit suboptimal Ab responses with varying neutralizing activity against the four DENV serotypes, and vaccinated individuals carried the risk of manifesting severe dengue disease upon primary DENV infection (19). Dengvaxia is thus currently recommended only for DENV-immune individuals aged 9 years or older. The failure of Dengvaxia to fully protect may be due to the absence of a robust vaccine-elicited CD8+ T cell response against DENV, as suggested by the finding that, in DENV-naïve individuals, Dengvaxia induces a strong anti–yellow fever virus CD8+ T cell response but a weak anti-DENV CD8+ T cell response (44). The potential for vaccine-induced ADE, together with the fact that no ZIKV vaccines have yet been approved for use in humans, prompted us to take a different approach to anti-flaviviral vaccine development by exploiting the known ability of CD8+ T cells to protect against infection with DENV (45–47) and ZIKV (11, 15, 16, 29, 30) in multiple mouse models. Here, we constructed and tested alphavirus-based mRNA replicon vaccines expressing ZIKV NS3 and prM-E proteins, and the results provide proof of concept that the NS3 vaccine not only is a potent immunogen but also elicits a human-relevant T cell response that protects against lethal infection, reduces transplacental transmission, and prevents fetal damage. In our study, prM-E induced nAbs with high neutralization titers, comparable to those induced by other prM-E vaccines (36). While NS3 and prM-E vaccines were both protective and elicited a T cell response, the NS3 vaccine did not induce a nAb response, reducing the risk of ADE mediated by an inefficient or waning vaccine Ab response.
The rationale for our approach was based on accumulating evidence from human studies that both CD8+ and CD4+ effector T cells protect against DENV (48–50) and ZIKV (8–10, 34) and on our previous work establishing NS3 as the major target of T cells in anti-DENV and anti-ZIKV immunity in humans (8–10, 14). To provide a robust comparator for the efficacy of the NS3 vaccine, we also designed and tested a replicon vaccine expressing prM-E using the same approach. As noted, flaviviral E proteins are the major targets of the nAb response, but their similarity, particularly in the EDII FL, where the immunodominant epitope for subneutralizing Abs is located, provides the potential for pathogenic ADE (17). Thus, we constructed the prM-E vaccine with a mutant EDII protein that reduces cross-reactivity (36). The alphavirus mRNA replicon approach was selected as the vaccine platform because it allows high antigen expression, is safer than DNA vaccines because replication is confined to the cytosol, and has previously been used to induce humoral and cellular immunity against several viruses, including influenza and cytomegalovirus, in animal models (51, 52).
To ensure the relevance of our findings to vaccine development for humans, we tested the NS3 and prM-E replicon vaccines in HLA-B*0702 Ifnar1−/− mice, which have been used extensively to identify and investigate the role of DENV and ZIKV CD8+ T cell epitopes restricted by an HLA allele that is common to many ethnic and geographic populations. In particular, HLA-B*0702 Ifnar1−/− have been used in studies investigating epitopes of relevance to human responses during primary DENV infection (38), secondary heterotypic DENV infection (46), primary ZIKV infection, and sequential heterologous DENV-ZIKV infections (16). These studies not only identified human-relevant CD8+ T cells specificities but also revealed that preexisting T cell immunity to DENV shaped the immunodominance pattern during subsequent infection with ZIKV or heterotypic DENV, and these observations were confirmed using T cells from DENV-exposed individuals (8, 53). Although the absence of IFN receptors in HLA-B*0702 Ifnar1−/− mice does not recapitulate the natural system, it is necessary to allow ZIKV replication and the development of anti-ZIKV immunity (which is dependent on antigen load) for vaccine testing. Immunocompetent C57BL/6 and HLA-B*0702 Ifnar1−/− mouse models both provide insights into vaccine efficacy and mechanisms. In the present study, HLA-B*0702 Ifnar1−/− mice were used as a stringent ZIKV challenge system to enable assessment of the translatability of the anti-NS3 T cell response to humans and the potential role of the T cell response in disease.
The NS3 vaccine–induced CD8+ T cell response in the present study was polyfunctional with respect to cytokine secretion and cytotoxicity and elicited memory responses 3 days after ZIKV challenge. Notably, the NS3 vaccine stimulated a robust IFNγ/TNF/IL-2 triple-positive CD8+ T cell response, which is thought to play a key role in protection during secondary antiviral responses (40). The triple-positive cells were detected in the periphery after boosting and were rapidly expanded in the spleen after ZIKV infection. Although robust activation of T cells was observed after ZIKV infection of prM-E–vaccinated mice, the cytokine response in vitro was low and we could not detect evidence of polyfunctionality 3 days after infection. One potential explanation is that ZIKV E protein contains relatively few immunodominant HLA-B7–restricted T cell epitopes compared with NS3 protein (16). In addition, it is possible that the T cell response could be delayed or diminished when a strong nAb response is concomitantly elicited by infection.
The NS3 sequence, which is conserved among the DENV serotypes, has been used in DNA vaccines to elicit protection against DENV. Costa and colleagues (54) showed that 90% of BALB/c mice immunized with a plasmid encoding full-length DENV2 NS3 were protected against a lethal dose of DENV2. These authors also showed that a DNA vaccine encoding the NS3 helicase sequence induced partial protection against lethal challenge (70% survival), whereas one encoding the NS3 protease sequence was ineffective. CD8+ T cells from the full-length NS3-vaccinated mice produced IFNγ, as detected by IFNγ-ELISpot; however, whether CD8+ T cells were crucial to the mechanism of the vaccine-induced protection was not examined (54). Similarly, although many ZIKV vaccines have shown efficacy in animal models, very few have examined the mechanisms of protection. A recent study demonstrated that Abs, but not CD8+ T cells, were responsible for protection mediated by a gorilla adenovirus–based ZIKV prM-E vaccine against ZIKV infection in mice (55). A 2017 study by Brault et al. (56) described the development of a modified vaccinia Ankara vector expressing ZIKV NS1 that was able to protect against lethal intracranial challenge with ZIKV. Although the vaccine induced robust nAb and CD8+ T cell responses, the authors did not investigate which response(s) mediated protection (56). Bullard et al. (57) developed an adenovirus type 4 vector–based vaccine expressing ZIKV prM-E that protected mice against lethal ZIKV challenge without inducing anti-ZIKV nAbs. However, the authors did not investigate whether CD4+ and/or CD8+ T cells mediated the vaccine-induced protection. More recently, Grubor-Bauk et al. (58) developed a DNA vaccine expressing ZIKV NS1 protein that failed to protect Ifnar1−/− mice but did confer protection against viremia in BALB/c mice, which are highly resistant to ZIKV infection and do not develop disease. IgG transfer and T cell depletion studies using the BALB/c mouse model revealed that NS1 DNA vaccine–induced Abs decreased viremia and that both CD4+ and CD8+ T cells were required for the viral control (58). In comparison to these studies, our CD8+ T cell depletion and adoptive transfer experiments showed that CD8+ T cells were necessary and sufficient for protective immunity in NS3-vaccinated HLA-B*0702 Ifnar1−/− mice. Our data thus establish the crucial role of CD8+ T cells in vaccine-mediated protection against ZIKV in a highly susceptible mouse model with human-relevant T cell responses, suggesting that an NS3-based vaccine might afford protection in vulnerable pregnant individuals.
Accumulating evidence supports a role for interplay between Ab and T cell responses in determining the outcome of DENV and ZIKV infections. In this context, our results suggest that the mechanism of protection induced by ZIKV vaccines will differ depending on the vaccine platform. Overall, more studies on the mechanisms of vaccine-induced protection will be needed to understand the implications of Ab versus T cell responses in the development of optimal ZIKV vaccines. The present study establishes proof of concept for the efficacy of an NS3-based, T cell–centric ZIKV vaccine and demonstrates that a vaccine targeting both T cell and Ab responses could be the superior option for eliciting long-term protection. Although epidemiologic evidence in support of ZIKV-ADE in humans is currently sparse (26), DENV-ADE is considered a major challenge in flavivirus vaccine development. Animal model studies recapitulating key epidemiologic scenarios suggest that ZIKV vaccine–induced Ab responses may precipitate DENV-ADE (27, 28). In contrast, studies in mice have demonstrated that the presence of anti-DENV CD8+ T cells can abrogate DENV-ADE (47), and that DENV/ZIKV cross-reactive CD8+ T cells can protect against ZIKV and DENV infections (16, 29, 30). Last, recent studies in humans have revealed that DENV immunity cross-protects against ZIKV infection, although whether this is mediated by humoral or cellular immunity is unclear (31–33). Thus, an NS3-based, T cell–centric ZIKV vaccine, in combination with an Ab-centric ZIKV vaccine, may afford long-term protection against both DENV and ZIKV while avoiding ADE of infection to either virus.
MATERIALS AND METHODS
Production of vaccine
SMARRT was derived from the Venezuelan equine encephalitis virus (strain TC-83), whose structural proteins have been deleted and replaced with a gene of interest, NS3 (amino acids 1520 to 2021) and prM-E (amino acids 107 to 195) in our study (accession number KU321639.1). ZIKV NS3 and prM-E were synthesized at IDT and cloned downstream of the replicon 26S promoter using Gibson assembly. Single clones were screened for the correct insert. The entire replicon and plasmid were sequenced-verified either by Sanger sequencing or next-generation sequencing on Illumina’s miSeq platform. Lipid nanoparticle formulations encapsulating replicon RNA were prepared by mixing an ethanolic solution of proprietary lipids with an aqueous solution of replicon RNA using a Nanoassemblr microfluidic device. The particles thus obtained were diluted with phosphate buffer and dialyzed against Hepes buffer (pH 7.4) using regenerated cellulose membranes (Spectra/Por Tube-A-Lyzers, Spectrum Laboratories). Purified formulations were concentrated using ultra-spin centrifugal filter units (Amicon Ultra Centrifugal Filter Units, EMD Millipore) and then filtered through a 0.2-μm polyethersulfone filter. Analytical characterization of the formulation included measurement of particle size and polydispersity by dynamic light scattering (ZEN3600, Malvern Instruments), and replicon content and encapsulation efficiency by a fluorometric assay using RiboGreen RNA reagent (Molecular Probes, Invitrogen). In this study, the constructs are termed NS3 and prM-E vaccines.
Vaccine electroporation and dsRNA detection
BHK-21 cells [American Type Culture Collection (ATCC), Manassas, VA, CCL10] were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gemini, #100-106), MEM nonessential amino acids (Gibco, #11140050), and penicillin/streptomycin/glutamine (Gibco, #10378016). BHK-21 cells were electroporated in strip cuvettes with 3 μg of RNA per 106 cells using SF buffer (Lonza) and 4D-Nucleofector. At 20 hours after electroporation, cells were removed from wells by incubation with trypsin-EDTA solution and washed in PBS containing 5% bovine serum albumin (BSA). The cells were stained for dsRNA using the J2 anti-dsRNA Ab (Scicons, #10010500) conjugated to R-PE using a Lightning-Link R-PE conjugation kit (Innova Biosciences). After staining, cells were evaluated on a ZE5 Cell analyzer (Bio-Rad) and the data were analyzed using FlowJo 10 (Tree Star, Ashland, OR).
Western blotting
BHK-21 cell pellets were lysed at 4°C in radioimmunoprecipitation assay buffer containing complete protease inhibitor cocktail (Roche) and then centrifuged. Protein in the supernatant was quantified using a micro-BCA protein assay kit. Aliquots of 35-μg protein were separated on NuPAGE 4 to 12% bis-tris gels and transferred to a nitrocellulose membrane. The membrane was blocked with 5% nonfat dry milk and then incubated with mouse anti-ZIKV E Ab (MyBiosource) or rabbit anti-ZIKV NS3 Ab (GeneTex). The membrane was then washed and incubated with the appropriate horseradish peroxidase (HRP)–conjugated secondary Ab. After washing, the membrane was incubated with SuperSignal West Pico Plus solution and then imaged using a Bio-Rad ChemiDoc system.
Virus collection and titration
ZIKV SD001 was isolated in 2016 from a woman who had traveled to Caracas, Venezuela (59). DENV2 S221 strain is a previously described mouse-adapted strain (45). Both viruses were cultured in C6/36 Aedes albopictus mosquito cells (CRL 1660, ATCC) as described previously (29) and were concentrated by ultracentrifugation.
Viral titers were measured using a BHK-21 cell–based focus-forming assay. BHK-21 cells were plated at 2 × 105 cells per well in 24-well plates in MEM-α medium containing 10% FBS, 1% penicillin, and 1% Hepes and incubated overnight at 37°C in a 5% CO2 atmosphere. Cells were infected with serial dilutions of virus or tissue homogenates for 1.5 hours with gentle rocking. The medium was then aspirated, fresh medium supplemented with 1% carboxymethyl cellulose (Sigma-Aldrich) was added, and the plates were incubated for an additional 2 to 3 days. The cells were then fixed with 4% formalin (Fisher), permeabilized with 1% Triton X-100 (Sigma-Aldrich), and blocked by incubation with 10% FBS in PBS. Cells were then incubated for 1 hour with the pan-flavivirus E protein–specific monoclonal Ab 4G2 (Bio X Cell), washed with PBS, and incubated for 1.5 hours with HRP-conjugated goat anti-mouse IgG (Sigma-Aldrich). Cell foci were detected by incubation for 30 min with True Blue substrate (KPL) and counted manually.
Mouse experiments
All experiments were performed in strict accordance with recommendations set forth in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at the La Jolla Institute for Immunology (protocol number AP00001029). HLA-B*0702 Ifnar1−/− transgenic mice and AG129 (Ifnα/β/γR−/−) mice were bred under pathogen-free conditions at La Jolla Institute for Immunology. Wild-type C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All groups were matched for age and sex (males and females, 4 to 8 weeks of age). None of the experiments were performed blinded.
Mice were intramuscularly vaccinated with 1, 10, or 20 μg of NS3 or prM-E vaccine or saline and boosted 28 days later in the same manner. Three or 6 weeks after the boost, mice were infected retro-orbitally with 103 or 104 (lethal dose) FFU of ZIKV SD001. Mice were weighed and observed for clinical signs of disease daily. Clinical scores were based on mouse appearance and mobility on a seven-point scale. Animals with ≥20% body weight loss were humanely euthanized. Blood samples were collected into serum collection tubes (Sarstedt) or EDTA-containing tubes (Sarstedt) from a facial vein on days 27 and 49 after vaccination. For tissue collection, mice were euthanized by CO2 inhalation 3 days after infection, and organs were collected and stored in RNAlater (Invitrogen) at 4°C until analysis. For experiments with pregnant mice, dams were mated with BALB/c males and checked daily for the appearance of vaginal plugs. Seven days after the detection of vaginal plugs (E7.5), dams were infected retro-orbitally with 103 FFU of ZIKV SD001. Dams were euthanized on E14.5, blood was collected by cardiac puncture, and tissues were harvested for analysis.
Viral RNA quantification
RNA was isolated from mouse sera using the QIAmp Viral RNA Mini Kit (Qiagen) and from tissues using the RNeasy Mini Kit (Qiagen). qRT-PCR was performed using the qScript One-Step qRT-PCR Kit (Quanta Bioscience) with a CFX96 Touch real-time PCR detection system (Bio-Rad, CFX Manager 3.1). ZIKV-specific primers have been previously described (2). Cycling conditions were as follows: 45°C for 15 min, 95°C for 15 min, followed by 40 cycles of 95°C for 15 s and 60°C for 15 s, and a final extension of 72°C for 30 min. Viral RNA concentration was calculated using a standard curve composed of four 100-fold serial dilutions of in vitro–transcribed RNA from ZIKV strain FSS13025.
Ex vivo IFNγ-ELISpot assay
Spleens were harvested from each group of mice and pooled, and a single-cell suspension of splenocytes was prepared. Aliquots of 2 × 105 cells per well were incubated for 20 hours with 10 μg/ml of a pool of NS3 or prM-E ZIKV-derived peptides in 96-well flat-bottom plates (Immobilon-P, Millipore, MA) precoated with anti-mouse IFNγ Ab (clone AN18, Mabtech, Sweden). The plates were developed as previously detailed (11), and SFCs were counted. Data are presented as the mean SFC per 106 cells. All epitopes are listed in Table 1.
In vivo ADE assay
On day 49, sera were prepared from blood samples of C57BL/6 mice that were primed and boosted with NS3 or prM-E or injected with saline. Samples of 1- or 10-μl sera diluted in PBS, 15 μg of 2H2 Ab (37), or PBS were injected into 5- to 6-week-old AG129 mice 1 day before intravenous injection of 105 FFU of DENV2 S221. Weight loss and survival were monitored daily.
Enzyme-linked immunosorbent assay
ELISA plates (96-well; Costar) were coated with ZIKV E protein (1 μg/ml; ZIKVSU-ENV, Native Antigen) in coating buffer (0.1 M NaHCO3) overnight at 4°C and then blocked for 1 hour at room temperature with 5% Blocker Casein in PBS (Thermo Fisher Scientific). Mouse serum samples were diluted threefold (from 1:30 to 1:65,610) in 1% BSA/PBS, added to the coated wells, and incubated for 1.5 hours at room temperature. Plates were then washed with wash buffer [0.05% Tween 20 (Promega) in PBS], and HRP-conjugated goat anti-mouse IgG Fc (1:5000 in 1% BSA/PBS) was added to each well for 1.5 hours at room temperature. TMB chromogen solution (eBioscience) was added to the wells, the reaction was stopped by addition of sulfuric acid (2N), and the absorbance at 450 nm was read on a SpectraMax M2E microplate reader (Molecular Devices). ZIKV-specific end point titers were calculated as the reciprocal of the highest serum dilution that gave a reading twice the cutoff absorbance of the negative control (1% BSA/PBS).
Intracellular cytokine staining (ICS)
Splenocytes were resuspended in 10% FBS/RPMI medium, plated at 2 × 106 cells per well in 96-well plates, and incubated for 6 hours with 10 μg/ml of pooled NS3 or prM-E ZIKV-derived peptides as previously detailed (11). Positive [cell stimulation cocktail, phorbol 12-myristate 13-acetate (PMA), and ionomycin at 500×; eBioscience] and negative (medium) controls were included in all experiments. Cells were surface-labeled with anti-CD3 (clone 145-2C11), anti-CD8 (clone 53-67), anti-CD44 (clone IM7), and anti-CD62L (clone Mel-14) and then fixed, permeabilized, and stained with anti–granzyme B (clone NGZB), anti-IFNγ (clone XMG 1.2), anti-TNFα (clone MP6-XT22), and anti–IL-2 (clone JES6-5H4). Cells were analyzed on an LSR II flow cytometer (BD Biosciences), and data were analyzed using FlowJo software X 10.0.7.
Adoptive transfer and depletion of CD8+ T cells
Four- to 5-week-old male and female HLA-B*0702 Ifnar1−/− mice were vaccinated and boosted with NS3 vaccine or saline as described above. Spleens were harvested on day 49, and CD8+ T cells were purified from the spleens by negative selection using the EasySep CD8+ T Cell Isolation Kit (StemCell). Purified CD8+ T cells (107 per mouse) were intravenously injected into 7- to 8-week-old HLA-B*0702 Ifnar1−/− recipient mice. One day later, the recipient mice were infected retro-orbitally with ZIKV SD001. For the depletion experiments, vaccinated and boosted mice were intravenously injected with 300 μg of a CD8-depleting monoclonal antibody (mAb) (2.43, Bio X Cell) or an isotype control mAb (rat IgG2b, Bio X Cell) on days −3 and −1 before infection retro-orbitally with ZIKV SD001. In both sets of experiments, blood and organs were harvested 3 days later and ZIKV RNA was quantified.
Serum transfer experiments
Four- to 5-week-old male and female HLA-B*0702 Ifnar1−/− mice were vaccinated and boosted with NS3 vaccine or injected with saline as described above. Blood was harvested on day 49, and sera from each group were prepared and pooled. Pooled sera (300 μl per mouse) were intraperitoneally injected into 7- to 8-week-old HLA-B*0702 Ifnar1−/− recipient mice. One day later, the recipient mice were infected retro-orbitally with ZIKV SD001. Blood and organs were harvested 3 days later, and ZIKV RNA was quantified.
Neutralization assays
Sera from vaccinated mice were heat-inactivated for 30 min at 56°C and then serially diluted 1:5 in RPMI medium containing 1% penicillin/streptomycin and 1% Hepes in 96-well flat-bottom plates. A sufficient amount of ZIKV SD001 to induce 7 to 20% infection of U937-DC-SIGN cells (predetermined by titration of virus) was mixed with the sera and incubated for 1 hour at 37°C. The mixture was then added to a 96-well round-bottom plate containing 105 U937-DC-SIGN cells per well and incubated for 2 hours at 37°C with gentle rocking every 15 min. Cells and virus incubated in the absence of serum served as the positive control. Plates were then centrifuged at 1500 rpm for 5 min at 4°C, the supernatants were aspirated, fresh RPMI medium supplemented with 10% FBS was added, and the cells were incubated for 20 hours at 37°C. Last, the cells were harvested, stained with phycoerythrin (PE)–conjugated anti-CD209 (DC-SIGN; clone DNC246), incubated with Cytofix/Cytoperm solution (BD Biosciences), and stained intracellularly with Alexa Fluor 647–conjugated 4G2 (anti-ZIKV E protein). The cells were analyzed on a FACSCanto II flow cytometer (BD Biosciences), and the percentage of infected cells was determined using FlowJo 10.5.0 software. Percentage neutralization was calculated as (% infected cells in the absence of serum − % infected cells in the presence of serum)/(% infected cells in the absence of serum) × 100%. Data are presented as the log serum dilution giving 50% neutralization (log NT50).
Statistical analysis
Data were analyzed with Prism software v7.0 (GraphPad Software, La Jolla, CA, USA) and are presented as the mean ± SEM. Differences between group means were analyzed by the Kruskal-Wallis test to compare more than two groups or the nonparametric Mann-Whitney test to compare two groups. A P value of <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank the Department of Laboratory Animal Care and the Flow Cytometry facility at the La Jolla Institute for Immunology for their assistance with husbandry and experiments. Funding: This study was funded by NIH grants AI116813, AI140063, and NS106387 to S.S. and a grant from the Chiba-UCSD Center for Mucosal Immunology, Allergy, and Vaccine Development to S.S. Author contributions: A.E.N. and S.S. designed the study. A.E.N. designed, performed, and analyzed experiments and wrote the manuscript. T.S., A.-V.N., J.A.R.-N., M.S., D.S., A.A., M.W., J.S., A.G., E.B., and J.L.D. performed and analyzed experiments. J.B.V., P.P.K., and P.C. performed and analyzed formulation of materials used for studies. A.E.N., K.K., P.A., N.W., and S.S. interpreted the data and edited the manuscript. S.S. supervised the study. Competing interests: D.S., A.A., M.W., J.S., J.L.D., K.K., P.A., and N.W. were employees of Synthetic Genomics Inc. at the time the studies were conducted. J.B.V., P.P.K., and P.C. are employees of Arcturus Therapeutics. All authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions of this paper are present in this manuscript and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/45/eabb2154/DC1
REFERENCES AND NOTES
- 1.Mansuy J. M., Suberbielle E., Chapuy-Regaud S., Mengelle C., Bujan L., Marchou B., Delobel P., Gonzalez-Dunia D., Malnou C. E., Izopet J., Martin-Blondel G., Zika virus in semen and spermatozoa. Lancet Infect. Dis. 16, 1106–1107 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Tang W. W., Young M. P., Mamidi A., Regla-Nava J. A., Kim K., Shresta S., A mouse model of Zika virus sexual transmission and vaginal viral replication. Cell Rep. 17, 3091–3098 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brasil P., Pereira J. P. Jr., Moreira M. E., Ribeiro Nogueira R. M., Damasceno L., Wakimoto M., Rabello R. S., Valderramos S. G., Halai U.-A., Salles T. S., Zin A. A., Horovitz D., Daltro P., Boechat M., Raja Gabaglia C., Carvalho de Sequeira P., Pilotto J. H., Medialdea-Carrera R., Cotrim da Cunha D., Abreu de Carvalho L. M., Pone M., Machado Siqueira A., Calvet G. A., Rodrigues Baião A. E., Neves E. S., Nassar de Carvalho P. R., Hasue R. H., Marschik P. B., Einspieler C., Janzen C., Cherry J. D., Bispo de Filippis A. M., Nielsen-Saines K., Zika virus infection in pregnant women in Rio de Janeiro. N. Engl. J. Med. 375, 2321–2334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calvet G., Aguiar R. S., Melo A. S. O., Sampaio S. A., de Filippis I., Fabri A., Araujo E. S. M., de Sequeira P. C., de Mendonça M. C. L., de Oliveira L., Tschoeke D. A., Schrago C. G., Thompson F. L., Brasil P., dos Santos F. B., Nogueira R. M. R., Tanuri A., de Filippis A. M. B., Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 16, 653–660 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Martines R. B., Bhatnagar J., Keating M. K., Silva-Flannery L., Muehlenbachs A., Gary J., Goldsmith C., Hale G., Ritter J., Rollin D., Shieh W.-J., Luz K. G., de Oliveira Ramos A. M., Davi H. P. F., Kleber de Oliveria W., Lanciotti R., Lambert A., Zaki S., Notes from the field: Evidence of Zika virus infection in brain and placental tissues from two congenitally infected newborns and two fetal losses—Brazil, 2015. MMWR Morb. Mortal. Wkly Rep. 65, 159–160 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Krauer F., Riesen M., Reveiz L., Oladapo O. T., Martínez-Vega R., Porgo T. V., Haefliger A., Broutet N. J., Low N.; WHO Zika causality working group , Zika virus infection as a cause of congenital brain abnormalities and Guillain–Barré syndrome: Systematic review. PLOS Med. 14, e1002203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Figueiredo C. P., Barros-Aragão F. G. Q., Neris R. L. S., Frost P. S., Soares C., Souza I. N. O., Zeidler J. D., Zamberlan D. C., de Sousa V. L., Souza A. S., Guimarães A. L. A., Bellio M., Marcondes de Souza J., Alves-Leon S. V., Neves G. A., Paula-Neto H. A., Castro N. G., De Felice F. G., Assunção-Miranda I., Clarke J. R., Da Poian A. T., Ferreira S. T., Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun. 10, 3890 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grifoni A., Pham J., Sidney J., O’Rourke P. H., Paul S., Peters B., Martini S. R., de Silva A. D., Ricciardi M. J., Magnani D. M., Silveira C. G. T., Maestri A., Costa P. R., de-Oliveira-Pinto L. M., de Azeredo E. L., Damasco P. V., Phillips E., Mallal S., de Silva A. M., Collins M., Durbin A., Diehl S. A., Cerpas C., Balmaseda A., Kuan G., Coloma J., Harris E., Crowe J. E. Jr., Stone M., Norris P. J., Busch M., Vivanco-Cid H., Cox J., Graham B. S., Ledgerwood J. E., Turtle L., Solomon T., Kallas E. G., Watkins D. I., Weiskopf D., Sette A., Prior dengue virus exposure shapes T cell immunity to Zika virus in humans. J. Virol. 91, e01469-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herrera B. B., Tsai W.-Y., Chang C. A., Hamel D. J., Wang W.-K., Lu Y., Mboup S., Kanki P. J., Sustained specific and cross-reactive T cell responses to Zika and dengue virus NS3 in West Africa. J. Virol. 92, e01992-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lim M. Q., Kumaran E. A. P., Tan H. C., Lye D. C., Leo Y. S., Ooi E. E., MacAry P. A., Bertoletti A., Rivino L., Cross-reactivity and anti-viral function of dengue capsid and NS3-specific memory T cells toward Zika virus. Front. Immunol. 9, 2225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elong Ngono A., Vizcarra E. A., Tang W. W., Sheets N., Joo Y., Kim K., Gorman M. J., Diamond M. S., Shresta S., Mapping and role of the CD8+ T cell response during primary Zika virus infection in mice. Cell Host Microbe 21, 35–46 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pardy R. D., Rajah M. M., Condotta S. A., Taylor N. G., Sagan S. M., Richer M. J., Analysis of the T Cell response to Zika virus and identification of a novel CD8+ T cell epitope in immunocompetent mice. PLOS Pathog. 13, e1006184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elong Ngono A., Young M. P., Bunz M., Xu Z., Hattakam S., Vizcarra E., Regla-Nava J. A., Tang W. W., Yamabhai M., Wen J., Shresta S., CD4+ T cells promote humoral immunity and viral control during Zika virus infection. PLOS Pathog. 15, e1007474 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elong Ngono A., Shresta S., Cross-reactive T cell immunity to dengue and Zika viruses: New insights into vaccine development. Front. Immunol. 10, 1316 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hassert M., Harris M. G., Brien J. D., Pinto A. K., Identification of protective CD8 T cell responses in a mouse model of Zika virus infection. Front. Immunol. 10, 1678 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen J., Tang W. W., Sheets N., Ellison J., Sette A., Kim K., Shresta S., Identification of Zika virus epitopes reveals immunodominant and protective roles for dengue virus cross-reactive CD8+ T cells. Nat. Microbiol. 2, 17036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diamond M. S., Ledgerwood J. E., Pierson T. C., Zika virus vaccine development: Progress in the face of new challenges. Annu. Rev. Med. 70, 121–135 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Katzelnick L. C., Gresh L., Halloran M. E., Mercado J. C., Kuan G., Gordon A., Balmaseda A., Harris E., Antibody-dependent enhancement of severe dengue disease in humans. Science 358, 929–932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sridhar S., Luedtke A., Langevin E., Zhu M., Bonaparte M., Machabert T., Savarino S., Zambrano B., Moureau A., Khromava A., Moodie Z., Westling T., Mascareñas C., Frago C., Cortés M., Chansinghakul D., Noriega F., Bouckenooghe A., Chen J., Ng S.-P., Gilbert P. B., Gurunathan S., DiazGranados C. A., Effect of dengue serostatus on dengue vaccine safety and efficacy. N. Engl. J. Med. 379, 327–340 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Salje H., Cummings D. A. T., Rodriguez-Barraquer I., Katzelnick L. C., Lessler J., Klungthong C., Thaisomboonsuk B., Nisalak A., Weg A., Ellison D., Macareo L., Yoon I.-K., Jarman R., Thomas S., Rothman A. L., Endy T., Cauchemez S., Reconstruction of antibody dynamics and infection histories to evaluate dengue risk. Nature 557, 719–723 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bardina S. V., Bunduc P., Tripathi S., Duehr J., Frere J. J., Brown J. A., Nachbagauer R., Foster G. A., Krysztof D., Tortorella D., Stramer S. L., García-Sastre A., Krammer F., Lim J. K., Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 356, 175–180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stettler K., Beltramello M., Espinosa D. A., Graham V., Cassotta A., Bianchi S., Vanzetta F., Minola A., Jaconi S., Mele F., Foglierini M., Pedotti M., Simonelli L., Dowall S., Atkinson B., Percivalle E., Simmons C. P., Varani L., Blum J., Baldanti F., Cameroni E., Hewson R., Harris E., Lanzavecchia A., Sallusto F., Corti D., Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353, 823–826 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Rathore A. P. S., Saron W. A. A., Lim T., Jahan N., St. John A. L., Maternal immunity and antibodies to dengue virus promote infection and Zika virus-induced microcephaly in fetuses. Sci. Adv. 5, eaav3208 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown J. A., Singh G., Acklin J. A., Lee S., Duehr J. E., Chokola A. N., Frere J. J., Hoffman K. W., Foster G. A., Krysztof D., Cadagan R., Jacobs A. R., Stramer S. L., Krammer F., García-Sastre A., Lim J. K., Dengue virus immunity increases Zika virus-induced damage during pregnancy. Immunity 50, 751–762.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmerman M. G., Quicke K. M., O’Neal J. T., Arora N., Machiah D., Priyamvada L., Kauffman R. C., Register E., Adekunle O., Swieboda D., Johnson E. L., Cordes S., Haddad L., Chakraborty R., Coyne C. B., Wrammert J., Suthar M. S., Cross-reactive dengue virus antibodies augment Zika virus infection of human placental macrophages. Cell Host Microbe 24, 731–742.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robbiani D. F., Olsen P. C., Costa F., Wang Q., Oliveira T. Y., Nery N. Jr., Aromolaran A., do Rosário M. S., Sacramento G. A., Cruz J. S., Khouri R., Wunder E. A. Jr., Mattos A., de Paula Freitas B., Sarno M., Archanjo G., Daltro D., Carvalho G. B. S., Pimentel K., de Siqueira I. C., de Almeida J. R. M., Henriques D. F., Lima J. A., Vasconcelos P. F. C., Schaefer-Babajew D., Azzopardi S. A., Bozzacco L., Gazumyan A., Belfort R. Jr., Alcântara A. P., Carvalho G., Moreira L., Araujo K., Reis M. G., Keesler R. I., Coffey L. L., Tisoncik-Go J., Gale M. Jr., Rajagopal L., Adams Waldorf K. M., Dudley D. M., Simmons H. A., Mejia A., O’Connor D. H., Steinbach R. J., Haese N., Smith J., Lewis A., Colgin L., Roberts V., Frias A., Kelleher M., Hirsch A., Streblow D. N., Rice C. M., MacDonald M. R., de Almeida A. R. P., van Rompay K. K. A., Ko A. I., Nussenzweig M. C., Risk of Zika microcephaly correlates with features of maternal antibodies. J. Exp. Med. 216, 2302–2315 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.George J., Valiant W. G., Mattapallil M. J., Walker M., Huang Y.-J. S., Vanlandingham D. L., Misamore J., Greenhouse J., Weiss D. E., Verthelyi D., Higgs S., Andersen H., Lewis M. G., Mattapallil J. J., Prior exposure to Zika virus significantly enhances peak Dengue-2 viremia in rhesus macaques. Sci. Rep. 7, 10498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fowler A. M., Tang W. W., Young M. P., Mamidi A., Viramontes K. M., McCauley M. D., Carlin A. F., Schooley R. T., Swanstrom J., Baric R. S., Govero J., Diamond M. S., Shresta S., Maternally acquired Zika antibodies enhance dengue disease severity in mice. Cell Host Microbe 24, 743–750.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wen J., Elong Ngono A., Regla-Nava J. A., Kim K., Gorman M. J., Diamond M. S., Shresta S., Dengue virus-reactive CD8+ T cells mediate cross-protection against subsequent Zika virus challenge. Nat. Commun. 8, 1459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Regla-Nava J. A., Elong Ngono A., Viramontes K. M., Huynh A.-T., Wang Y.-T., Nguyen A.-V. T., Salgado R., Mamidi A., Kim K., Diamond M. S., Shresta S., Cross-reactive dengue virus-specific CD8+ T cells protect against Zika virus during pregnancy. Nat. Commun. 9, 3042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gordon A., Gresh L., Ojeda S., Katzelnick L. C., Sanchez N., Mercado J. C., Chowell G., Lopez B., Elizondo D., Coloma J., Burger-Calderon R., Kuan G., Balmaseda A., Harris E., Prior dengue virus infection and risk of Zika: A pediatric cohort in Nicaragua. PLOS Med. 16, e1002726 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez-Barraquer I., Costa F., Nascimento E. J. M., Júnior N. N., Castanha P. M. S., Sacramento G. A., Cruz J., Carvalho M., De Olivera D., Hagan J. E., Adhikarla H., Wunder E. A. Jr., Coêlho D. F., Azar S. R., Rossi S. L., Vasilakis N., Weaver S. C., Ribeiro G. S., Balmaseda A., Harris E., Nogueira M. L., Reis M. G., Marques E. T. A., Cummings D. A. T., Ko A. I., Impact of preexisting dengue immunity on Zika virus emergence in a dengue endemic region. Science 363, 607–610 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pedroso C., Fischer C., Feldmann M., Sarno M., Luz E., Moreira-Soto A., Cabral R., Netto E. M., Brites C., Kümmerer B. M., Drexler J. F., Cross-protection of dengue virus infection against congenital Zika syndrome, northeastern Brazil. Emerg. Infect. Dis. 25, 1485–1493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paquin-Proulx D., Leal F. E., Terrassani Silveira C. G., Maestri A., Brockmeyer C., Kitchen S. M., Cabido V. D., Kallas E. G., Nixon D. F., T-cell responses in individuals infected with Zika virus and in those vaccinated against dengue virus. Pathog. Immun. 2, 274–292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knudsen M. L., Ljungberg K., Kakoulidou M., Kostic L., Hallengärd D., García-Arriaza J., Merits A., Esteban M., Liljeström P., Kinetic and phenotypic analysis of CD8+ T cell responses after priming with alphavirus replicons and homologous or heterologous booster immunizations. J. Virol. 88, 12438–12451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richner J. M., Himansu S., Dowd K. A., Butler S. L., Salazar V., Fox J. M., Julander J. G., Tang W. W., Shresta S., Pierson T. C., Ciaramella G., Diamond M. S., Modified mRNA vaccines protect against Zika virus infection. Cell 168, 1114–1125.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zellweger R. M., Prestwood T. R., Shresta S., Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe 7, 128–139 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiskopf D., Yauch L. E., Angelo M. A., John D. V., Greenbaum J. A., Sidney J., Kolla R. V., De Silva A. D., de Silva A. M., Grey H., Peters B., Shresta S., Sette A., Insights into HLA-restricted T cell responses in a novel mouse model of dengue virus infection point toward new implications for vaccine design. J. Immunol. 187, 4268–4279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elong Ngono A., Shresta S., Immune response to dengue and Zika. Annu. Rev. Immunol. 36, 279–308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sa Q., Woodward J., Suzuki Y., IL-2 produced by CD8+ immune T cells can augment their IFN-γ production independently from their proliferation in the secondary response to an intracellular pathogen. J. Immunol. 190, 2199–2207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbink P., Larocca R. A., De La Barrera R. A., Bricault C. A., Moseley E. T., Boyd M., Kirilova M., Li Z., Ng’ang’a D., Nanayakkara O., Nityanandam R., Mercado N. B., Borducchi E. N., Agarwal A., Brinkman A. L., Cabral C., Chandrashekar A., Giglio P. B., Jetton D., Jimenez J., Lee B. C., Mojta S., Molloy K., Shetty M., Neubauer G. H., Stephenson K. E., Peron J. P. S., de Andrade Zanotto P. M., Misamore J., Finneyfrock B., Lewis M. G., Alter G., Modjarrad K., Jarman R. G., Eckels K. H., Michael N. L., Thomas S. J., Barouch D. H., Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 353, 1129–1132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nürnberger C., Bodmer B. S., Fiedler A. H., Gabriel G., Mühlebach M. D., A measles virus-based vaccine candidate mediates protection against Zika virus in an allogeneic mouse pregnancy model. J. Virol. 93, e01485-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrett A. D. T., Current status of Zika vaccine development: Zika vaccines advance into clinical evaluation. NPJ Vaccines 3, 24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harenberg A., Begue S., Mamessier A., Gimenez-Fourage S., Seah C. C., Liang A. W., Ng J. L., Toh X. Y., Archuleta S., Wilder-Smith A., Shek L. P., Wartel-Tram A., Bouckenooghe A., Lang J., Crevat D., Caillet C., Guy B., Persistence of Th1/Tc1 responses one year after tetravalent dengue vaccination in adults and adolescents in Singapore. Hum. Vaccin. Immunother. 9, 2317–2325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yauch L. E., Zellweger R. M., Kotturi M. F., Qutubuddin A., Sidney J., Peters B., Prestwood T. R., Sette A., Shresta S., A protective role for dengue virus-specific CD8+ cells. J. Immunol. 182, 4865–4873 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elong Ngono A., Chen H.-W., Tang W. W., Joo Y., King K., Weiskopf D., Sidney J., Sette A., Shresta S., Protective role of cross-reactive CD8 T cells against dengue virus infection. EBioMedicine 13, 284–293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zellweger R. M., Eddy W. E., Tang W. W., Miller R., Shresta S., CD8+ T cells prevent antigen-induced antibody-dependent enhancement of dengue disease in mice. J. Immunol. 193, 4117–4124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiskopf D., Angelo M. A., de Azeredo E. L., Sidney J., Greenbaum J. A., Fernando A. N., Broadwater A., Kolla R. V., De Silva A. D., de Silva A. M., Mattia K. A., Doranz B. J., Grey H. M., Shresta S., Peters B., Sette A., Comprehensive analysis of dengue virus-specific responses supports an HLA-linked protective role for CD8+ T cells. Proc. Natl. Acad. Sci. U.S.A. 110, E2046–E2053 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simon-Lorière E., Duong V., Tawfik A., Ung S., Ly S., Casadémont I., Prot M., Courtejoie N., Bleakley K., Buchy P., Tarantola A., Dussart P., Cantaert T., Sakuntabhai A., Increased adaptive immune responses and proper feedback regulation protect against clinical dengue. Sci. Transl. Med. 9, eaal5088 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Wijeratne D. T., Fernando S., Gomes L., Jeewandara C., Ginneliya A., Samarasekara S., Wijewickrama A., Hardman C. S., Ogg G. S., Malavige G. N., Quantification of dengue virus specific T cell responses and correlation with viral load and clinical disease severity in acute dengue infection. PLOS Negl. Trop. Dis. 12, e0006540 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Geall A. J., Verma A., Otten G. R., Shaw C. A., Hekele A., Banerjee K., Cu Y., Beard C. W., Brito L. A., Krucker T., O’Hagan D. T., Singh M., Mason P. W., Valiante N. M., Dormitzer P. R., Barnett S. W., Rappuoli R., Ulmer J. B., Mandl C. W., Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. U.S.A. 109, 14604–14609 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hekele A., Bertholet S., Archer J., Gibson D. G., Palladino G., Brito L. A., Otten G. R., Brazzoli M., Buccato S., Bonci A., Casini D., Maione D., Qi Z.-Q., Gill J. E., Caiazza N. C., Urano J., Hubby B., Gao G. F., Shu Y., De Gregorio E., Mandl C. W., Mason P. W., Settembre E. C., Ulmer J. B., Craig Venter J., Dormitzer P. R., Rappuoli R., Geall A. J., Rapidly produced SAM(®) vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2, e52 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weiskopf D., Cerpas C., Angelo M. A., Bangs D. J., Sidney J., Paul S., Peters B., Sanches F. P., Silvera C. G. T., Costa P. R., Kallas E. G., Gresh L., de Silva A. D., Balmaseda A., Harris E., Sette A., Human CD8+ T-cell responses against the 4 dengue virus serotypes are associated with distinct patterns of protein targets. J. Infect. Dis. 212, 1743–1751 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Costa S. M., Yorio A. P., Gonçalves A. J. S., Vidale M. M., Costa E. C. B., Mohana-Borges R., Motta M. A., Freire M. S., Alves A. M. B., Induction of a protective response in mice by the dengue virus NS3 protein using DNA vaccines. PLOS ONE 6, e25685 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hassan A. O., Dmitriev I. P., Kashentseva E. A., Zhao H., Brough D. E., Fremont D. H., Curiel D. T., Diamond M. S., A gorilla adenovirus-based vaccine against Zika virus induces durable immunity and confers protection in pregnancy. Cell Rep. 28, 2634–2646.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brault A. C., Domi A., McDonald E. M., Talmi-Frank D., McCurley N., Basu R., Robinson H. L., Hellerstein M., Duggal N. K., Bowen R. A., Guirakhoo F., A Zika vaccine targeting NS1 protein protects immunocompetent adult mice in a lethal challenge model. Sci. Rep. 7, 14769 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bullard B. L., Corder B. N., Gorman M. J., Diamond M. S., Weaver E. A., Efficacy of a T cell-biased adenovirus vector as a Zika virus vaccine. Sci. Rep. 8, 18017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grubor-Bauk B., Wijesundara D. K., Masavuli M., Abbink P., Peterson R. L., Prow N. A., Larocca R. A., Mekonnen Z. A., Shrestha A., Eyre N. S., Beard M. R., Gummow J., Carr J., Robertson S. A., Hayball J. D., Barouch D. H., Gowans E. J., NS1 DNA vaccination protects against Zika infection through T cell-mediated immunity in immunocompetent mice. Sci. Adv. 5, eaax2388 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carlin A. F., Wen J., Vizcarra E. A., McCauley M., Chaillon A., Akrami K., Kim C., Elong Ngono A., Lara-Marquez M. L., Smith D. M., Glass C. K., Schooley R. T., Benner C., Shresta S., A longitudinal systems immunologic investigation of acute Zika virus infection in an individual infected while traveling to Caracas Venezuela. PLOS Negl. Trop. Dis. 12, e0007053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/45/eabb2154/DC1