

Mitochondrial complex I inhibition in the presence of a mito-antioxidant induces reductive stress, ISR, and cancer cell death.
Abstract
Mitochondria-derived reactive oxygen species (mROS) are required for the survival, proliferation, and metastasis of cancer cells. The mechanism by which mitochondrial metabolism regulates mROS levels to support cancer cells is not fully understood. To address this, we conducted a metabolism-focused CRISPR-Cas9 genetic screen and uncovered that loss of genes encoding subunits of mitochondrial complex I was deleterious in the presence of the mitochondria-targeted antioxidant mito-vitamin E (MVE). Genetic or pharmacologic inhibition of mitochondrial complex I in combination with the mitochondria-targeted antioxidants, MVE or MitoTEMPO, induced a robust integrated stress response (ISR) and markedly diminished cell survival and proliferation in vitro. This was not observed following inhibition of mitochondrial complex III. Administration of MitoTEMPO in combination with the mitochondrial complex I inhibitor phenformin decreased the leukemic burden in a mouse model of T cell acute lymphoblastic leukemia. Thus, mitochondrial complex I is a dominant metabolic determinant of mROS-dependent cellular fitness.
INTRODUCTION
Reactive oxygen species (ROS) can activate signaling pathways that support cancer cell survival and proliferation as well as metastasis and drug resistance (1). Given the oncogenic role of ROS, antioxidants that scavenge ROS have long been hypothesized to be prophylactic or therapeutic. Although early data supported this theory by showing that certain dietary antioxidants diminish oxidative damage markers associated with cancer, many clinical studies have failed to prevent or treat cancer using dietary antioxidants (2). Instead, antioxidants have been shown to exacerbate the risk and progression of the disease (3, 4). These contradictory findings highlight the need for a more complete understanding of ROS biology in cancer.
Cancer cells exhibit a unique state of redox homeostasis compared to nontransformed cells. Oncogene activation and tumor suppressor loss increase mitochondria- and NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidase (NOX)–dependent superoxide (O2−) generation (5). O2− is quickly converted to hydrogen peroxide (H2O2) that oxidizes specific cysteine residues on target proteins. This modulation can alter the protein’s enzymatic activity, which supports the downstream protumorigenic and metastatic signaling pathways (1). Excess H2O2, however, can be converted to the hydroxyl radical (•OH), which can react with polyunsaturated fatty acids (PUFAs) to produce lipid hydroperoxides. Lipid peroxidation can substantially damage cellular membranes and induce cell death (6). Therefore, surviving cancer cells rely on elevated antioxidant response to scavenge lipid hydroperoxides. This process is driven, in part, by activation of the transcription factor nuclear factor erythroid-2–related factor 2 (NRF2), which increases glutathione (GSH) synthesis and glutathione peroxidase 4 (GPX4) activity (7–14). Thus, redox biology and metabolism are important contributors to tumorigenesis and metastasis (15). The specific type of ROS that is scavenged, either H2O2 or lipid hydroperoxides, can suppress or exacerbate cancer, respectively.
Targeted antioxidant molecules that regulate the localized pools of signaling H2O2 generated by the mitochondria or NOXs may be efficacious anticancer agents (16). Various mitochondria-targeted antioxidants (mito-antioxidants) have demonstrated capacities to suppress different types of cancer in vitro and in vivo (17). A mitochondria-targeted O2− scavenger, MitoTEMPO, has been shown to inhibit metastasis of various cancer cells (18). In addition, transgenic introduction of mitochondria-targeted catalase (mCatalase), which converts mitochondrial H2O2 to water (H2O), reduces the spontaneous formation of colon cancer in a mouse model of adenomatous polyposis coli multiple intestinal neoplasia (19). Despite the well-established necessity of mitochondria-derived ROS (mROS) for cancer, the mechanism by which intracellular metabolism controls the production of mROS to support cancer cell survival and proliferation is not fully understood. In this study, we addressed this question by performing a negative selection CRISPR screen of metabolic genes using the mitochondria-targeted antioxidant mito-vitamin E (MVE) (20). Our unbiased functional genomic screen revealed the metabolic determinants of mROS-dependent cancer cell fitness.
RESULTS
A negative selection CRISPR screen using MVE uncovers the selective requirement of mitochondrial complex I
To investigate how intracellular metabolism regulates mROS generation to promote cancer cell fitness, we performed a CRISPR-based negative selection screen for metabolic genes whose loss was deleterious in the presence of a mild concentration of the mitochondria-targeted antioxidant MVE. The genes that are required for cancer cells to adapt to diminished levels of mROS were uncovered by this screen, elucidating the critical metabolic processes for mROS-dependent cancer cell survival and proliferation. Human Jurkat T-cell acute lymphoblastic leukemia (T-ALL) cells were transduced with a lentiviral single-guide RNA (sgRNA) library consisting of ~30,000 unique sgRNAs that target ~3000 metabolism-related genes. The knockout pool of Jurkat cells was subsequently cultured in the presence or absence of 50 nM MVE (Fig. 1A), a concentration that slightly suppressed the rate of population doubling (~17% after 4 days) of wild-type Jurkat cells (Fig. 1B). Following ~15 population doublings, genomic DNA was prepared from the initial, untreated, and MVE-treated cell populations and deep sequencing was performed to measure the frequency of each sgRNA. The change in abundance of each sgRNA was assessed as the log2 fold change of its frequency within the untreated or MVE-treated cell population compared to that in the initial population. For each gene, a score was defined as the median of the changes in abundance across all sgRNAs targeting a particular gene (~10 sgRNAs per gene). If a gene is essential for Jurkat cells to survive and/or proliferate amid a mild reduction of mROS, then sgRNAs targeting that gene will be underrepresented in the final MVE-treated surviving population of cells, yielding a negative differential gene score when compared to the untreated population.
Fig. 1. Loss of mitochondrial complex I is deleterious in the presence of mito-antioxidants.
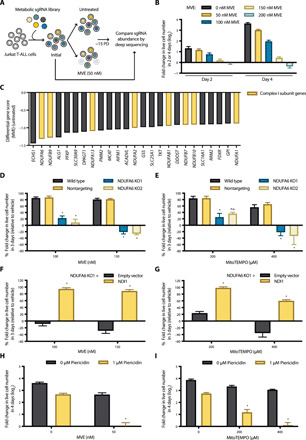
(A) Schematic representation of the CRISPR-based negative selection screen. PD, population doublings. (B) Concentration-dependent effects of MVE on the population doubling of Jurkat cells. n = 5; mean + SEM. (C) Differential gene scores of the top 25 genes whose sgRNAs were underrepresented in the MVE-treated population. (D and E) Wild-type, nontargeting control, and NDUFA6-null (NDUFA6 KO1 and KO2) Jurkat cell lines were treated with MVE (D) or MitoTEMPO (E) for 7 days. Population doubling of the cells during the last 3 days of each treatment was assessed and normalized to the vehicle treatment. It should be noted that our MVE had lost its potency since the screen was conducted. n = 4; mean + SEM; *P < 0.0001 (D), *P = 0.0118 (E; 200 μM; KO1), *P = 0.0002 (E; 400 μM; KO1), *P < 0.0001 (E; 400 μM; KO2) compared to nontargeting. (F and G) Empty vector or NDI1 was ectopically expressed in the NDUFA6 KO1 cell line, and the sensitivity to MVE (F) or MitoTEMPO (G) was measured as described above. n = 4; mean + SEM; *P < 0.0001 compared to empty vector. (H and I) Wild-type Jurkat cells were treated with piericidin ± MVE (H) or MitoTEMPO (I) for 4 days, and the population doublings were assessed. n = 5; mean + SEM; *P < 0.0001 compared to piericidin or MVE alone (H) and piericidin or MitoTEMPO alone (I).
Among the top 25 genes whose loss sensitizes Jurkat cells to a low concentration of MVE, we observed many genes encoding subunits of mitochondrial complex I within the electron transport chain (ETC) (Fig. 1C). These include NDUFA6, NDUFB9, NDUFA13, NDUFA2, NDUFAB1, NDUFB7, NDUFB10, and NDUFA1 (Fig. 1C and fig. S1). The top-scoring gene ECHS1 encodes short-chain enoyl-CoA (coenzyme A) hydratase (ECHS1), which catalyzes the second step of fatty acid oxidation, where 2-trans-enoyl-CoA is hydrated to l-3-hydroxyacyl-CoA. Most ECHS1-deficient patients present with Leigh syndrome, a neurometabolic disorder traditionally associated with defects in mitochondrial complex I activity. Various degrees of complex I dysfunction were detected in ECHS1-deficient patients (21). Collectively, the data from our MVE negative selection CRISPR screen strongly suggest an essential role of mitochondrial complex I in mROS-dependent cellular fitness.
Inhibition of mitochondrial complex I sensitizes cells to mito-antioxidants
To validate our screen results, we generated two clonal CRISPR-mediated NDUFA6-knockout Jurkat cell lines (NDUFA6 KO1 and KO2) using two independent sgRNAs targeting NDUFA6, sgNDUFA6_3 and sgNDUFA6_7, respectively (figs. S1A and S2A). In addition, we generated a clonal control cell line using a nontargeting sgRNA. NDUFA6, the top-scoring mitochondrial complex I subunit hit from our screen, is an accessory subunit required for the catalytic activity of complex I. Our NDUFA6-null cell lines exhibited significantly reduced proliferation in glucose-free medium with galactose, indicating that loss of NDUFA6 was sufficient to impair mitochondrial oxidative phosphorylation (fig. S2B). Moreover, in agreement with the screen result, our NDUFA6-null cell lines were markedly more sensitive to MVE compared to wild-type or nontargeting control cells (Fig. 1D). To ensure that these results were not specific to MVE, we assessed the sensitivity of NDUFA6-null Jurkat cells to another mito-antioxidant, MitoTEMPO. As observed with MVE, NDUFA6-null cell lines were hypersensitive to MitoTEMPO compared to control cell lines (Fig. 1E). Furthermore, we ectopically expressed the Saccharomyces cerevisiae alternative NADH (reduced form of nicotinamide adenine dinucleotide) dehydrogenase (NDI1) gene in NDUFA6-null cells (fig. S2C). NDI1 can oxidize NADH while reducing ubiquinone to ubiquinol, thus restoring the electron transport activity of mitochondrial complex I. As shown in fig. S2D, NDUFA6-null cells expressing NDI1 were capable of proliferating in galactose-containing medium. Moreover, NDI1 expression was sufficient to restore NDUFA6-null Jurkat cell resistance to both MVE and MitoTEMPO (Fig. 1, F and G).
Next, we tested whether pharmacologic inhibition of mitochondrial complex I would have a similar effect on mito-antioxidant sensitivities as genetic inhibition of complex I. Jurkat cells treated with the mitochondrial complex I inhibitor piericidin in combination with either MVE or MitoTEMPO had a significantly reduced proliferation compared to cells treated with piericidin or the mito-antioxidant alone (Fig. 1, H and I). These findings validate the screen result and show that mitochondrial complex I is required for maintaining cellular fitness in the presence of a mito-antioxidant.
Inhibition of mitochondrial complex I, but not mitochondrial complex III, synergizes with a mito-antioxidant to impair cellular fitness
Mammalian mitochondrial complex I is a 46-subunit enzyme within the ETC with multiple metabolic roles. One key function of complex I is to oxidize NADH to NAD+, which can fuel the oxidative tricarboxylic acid (TCA) cycle, allowing the efficient generation of metabolites to support macromolecule synthesis. To examine whether this anabolic function of complex I is required for cellular fitness in the presence of a mito-antioxidant, we supplemented cells with pyruvate and uridine that provide biosynthetic precursors amid ETC inhibition (22–24). In addition, we tested whether inhibition of mitochondrial complex III, which also abolishes the oxidative TCA cycle, would have a similar effect on mito-antioxidant sensitivities as complex I inhibition. In the presence of pyruvate and uridine, the complex I inhibitor piericidin continued to sensitize Jurkat cells to the mito-antioxidants MVE and MitoTEMPO (Fig. 2, A and B, respectively). Moreover, the complex III inhibitor antimycin failed to increase mito-antioxidant sensitivities. The rate of population doubling following treatment with antimycin and either MVE or MitoTEMPO was comparable to the rate following treatment with antimycin alone (Fig. 2, A and B).
Fig. 2. Combination of mitochondrial complex I inhibition and a mito-antioxidant induces a unique profile of metabolites.
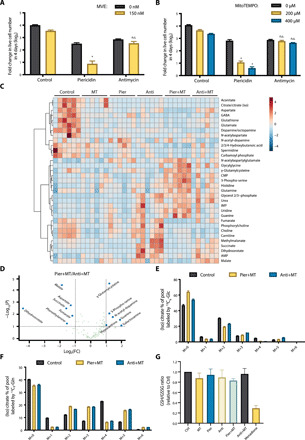
(A and B) Jurkat cells supplemented with pyruvate and uridine were treated with 1 μM piericidin or 1 μM antimycin ± MVE (A) or MitoTEMPO (B) for 4 days, and the population doublings were assessed. n = 5; mean + SEM; *P < 0.0001 compared to piericidin alone; n.s.P > 0.9999 (A), n.s.P > 0.9999 (B; 200 μM), n.s.P = 0.9053 (B; 400 μM) compared to antimycin alone. (C) Heatmap of the metabolites whose abundances were significantly different among Jurkat cells treated with vehicle (Control), MitoTEMPO (MT), piericidin (Pier), antimycin (Anti), piericidin and MitoTEMPO (Pier+MT), or antimycin and MitoTEMPO (Anti+MT) for 24 hours. The relative abundance of each metabolite is depicted as z score across rows (red, high; blue, low) (n = 6, FDR ≤ 0.1). (D) Volcano plot of the metabolites whose abundances were significantly different between Jurkat cells treated with Pier+MT and Anti+MT for 24 hours (dashed line: fold change threshold = 2 and P value threshold = 0.1, n = 6). (E and F) Jurkat cells treated with vehicle (Control), Pier+MT, or Anti+MT for 24 hours were labeled for 8 hours with [U-13C6]glucose (E) or [U-13C5]glutamine (F), and the percentage of labeled (iso)citrate pools was assessed (n = 5, mean + SEM). (G) GSH/GSSG ratio in Jurkat cells treated with vehicle (Control), MT, Pier, Anti, Pier+MT, Anti+MT, or menadione for 24 hours (n = 4, mean + SEM).
Furthermore, bromodeoxyuridine (BrdU) and annexin V staining was performed to assess proliferation and apoptosis, respectively, in Jurkat cells supplemented with pyruvate and uridine. Consistent with the rate of population doubling data described in Fig. 2B, piericidin and MitoTEMPO significantly reduced the percentage of proliferating cells compared to piericidin or MitoTEMPO alone, while cells treated with antimycin and MitoTEMPO had a similar ratio of BrdU incorporation as cells treated with antimycin alone (fig. S3, A and B). Moreover, treatment with piericidin and MitoTEMPO for 4 days markedly increased the annexin V+ apoptotic population of cells, while either drug alone had little to no effect on cell viability (fig. S3, C and D). Therefore, inhibition of mitochondrial complex I, but not mitochondrial complex III, synergizes with mito-antioxidants to impair both cell proliferation and survival.
Mitochondrial complex I inhibition in combination with a mito-antioxidant induces a unique profile of metabolites
To further investigate why mitochondrial complex I is essential for cellular fitness in the presence of a mito-antioxidant, we examined the molecular signatures of Jurkat cells treated with either piericidin and MitoTEMPO or antimycin and MitoTEMPO. First, we assessed the metabolite contents in cells treated with vehicle (Control), MitoTEMPO (MT), piericidin (Pier), antimycin (Anti), piericidin and MitoTEMPO (Pier+MT), or antimycin and MitoTEMPO (Anti+MT). Pier+MT induced a metabolite profile distinct from the other treatment groups (Fig. 2C). Closer examination of the differential metabolite contents revealed that Pier+MT-treated cells had a reduction in dihydroorotate compared to Anti+MT-treated cells (Fig. 2D). This difference is attributed to inhibition of dihydroorotate dehydrogenase during complex III inhibition (24). In addition, Pier+MT-treated cells had decreased levels of TCA cycle–associated metabolites, including malate, aspartate, succinate, and fumarate, compared to Anti+MT-treated cells (Fig. 2D). However, both Pier+MT- and Anti+MT-treated cells showed reduced flux of oxidative TCA cycle, identified by the levels of M+2 mass isotopomers of (iso)citrate, α-ketoglutarate, succinate, and aspartate from [U-13C6]glucose, and M+4 mass isotopomers of (iso)citrate, fumarate, malate, and aspartate from [U-13C5]glutamine (Fig. 2, E and F, and fig. S4). Moreover, Pier+MT- and Anti+MT-treated cells showed similar elevation in reductive TCA cycle flux, as the levels of M+5 isotopomer of (iso)citrate and M+3 isotopomer of malate from [U-13C5]glutamine were comparably increased (Fig. 2F and fig. S4, E and G). These data suggest that cells treated with Pier+MT, as well as Anti+MT, generate biosynthetic precursors mainly through reductive TCA cycle.
The differential metabolite contents data also revealed increased levels of 3-phospho-serine, guanine, and hypoxanthine in cells treated with Pier+MT compared to those treated with Anti+MT (Fig. 2D). Pier+MT-treated cells had elevated levels of M+3 mass isotopomers of 3-phospho-serine and serine from [U-13C6]glucose (fig. S5, A to C). However, the cells showed reduced level of M+2 mass isotopomer of glycine, possibly leading to decreased levels of M+7 isotopomers of inosine monophosphate (IMP) and guanosine monophosphate (GMP) from [U-13C6]glucose (fig. S5, D to F).
γ-Glutamyl cysteine was another metabolite whose level was elevated in Pier+MT-treated cells compared to Anti+MT-treated cells (Fig. 2D). γ-Glutamyl cysteine is a critical precursor of GSH synthesis, an essential regulator of cellular redox homeostasis. Pier+MT- and Anti+MT-treated cells showed similar flux of de novo GSH synthesis, identified by M+5 or M+2 mass isotopomer of GSH from [U-13C5]glutamine or [U-13C6]glucose, respectively (fig. S6, A to D). Pier+MT-treated cells had decreased level of GSH compared to Anti+MT-treated cells (fig. S6E). However, there was no significant difference in GSH/oxidized glutathione (GSSG) ratios, which reflects the state of whole-cell redox homeostasis (Fig. 2G).
Mitochondrial complex I inhibition in combination with a mito-antioxidant suppresses generation of mitochondrial H2O2
We next examined mROS generation in cells treated with Pier+MT or Anti+MT. Specifically, we exposed saponin-permeabilized Jurkat cells to either vehicle (Control), MT, Pier, Pier+MT, Anti, or Anti+MT and measured the levels of mitochondria-derived H2O2 using Amplex red and superoxide dismutase 1 (SOD1). SOD1 instantaneously dismutates O2− generated by the mitochondria to H2O2. Amplex red, a membrane impermeable substrate, gets oxidized by H2O2 at a 1:1 ratio to become a highly fluorescent resorufin. As expected, compared to Control cells, cells treated with MT alone had reduced levels of mitochondria-derived H2O2, while cells treated with Pier or Anti alone had increased levels. Cells treated with Pier+MT had a markedly lower level of mitochondrial H2O2 compared to cells treated with Anti+MT (Fig. 3A). This result may be attributed to differing levels of mitochondrial intermembrane O2−, resulting from distinct interactions of piericidin with MitoTEMPO versus antimycin with MitoTEMPO. Piericidin augments electron leakage from mitochondrial complex I, inducing elevated formation of O2− in the mitochondrial matrix. Antimycin augments electron leakage from both mitochondrial complex I and III, thereby increasing the levels of O2− in the mitochondrial intermembrane space as well as the matrix. MitoTEMPO, which is localized to the matrix, can readily access mitochondrial matrix O2− induced by piericidin or antimycin. Antimycin, however, also releases O2− into the intermembrane space, which cannot be as efficiently attenuated by MitoTEMPO compared to the matrix O2− (Fig. 3B). Consequently, the intermembrane O2− induced by antimycin can cross the mitochondrial outer membrane via voltage-dependent anion channels to enter the cytosol where it is quickly converted into H2O2 by SOD1.
Fig. 3. Combination of mitochondrial complex I inhibition and a mito-antioxidant suppresses generation of mitochondrial H2O2.
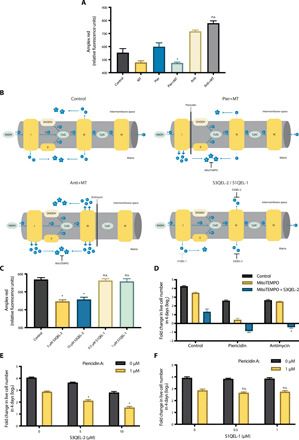
(A) Levels of mitochondria-derived H2O2 following treatment of permeabilized Jurkat cells with vehicle (Control), MT, Pier, Anti, Pier+MT, or Anti+MT for 1 hour. n = 4; mean + SEM; *P = 0.0051 compared to Pier; n.s.P = 0.6009 compared to Anti. (B) Schematic representation of mitochondrial electron leakage and superoxide (O2−) formation in the presence of Pier+MT, Anti+MT, or S3QEL-2, and S1QEL-1. (C) Levels of mitochondria-derived H2O2 following treatment of permeabilized Jurkat cells with S3QEL-2 and S1QEL-1 of indicated concentrations for 1 hour. n = 4; mean + SEM; *P < 0.0001 and n.s.P > 0.9999 compared to Control. (D) Jurkat cells supplemented with pyruvate and uridine were treated with 1 μM piericidin or 1 μM antimycin ± 400 μM MitoTEMPO ± 10 μM S3QEL-2 for 4 days, and the population doublings were assessed. n = 4; mean + SEM; *P < 0.0001 compared to Anti+MT. (E and F) Jurkat cells were treated with piericidin ± S3QEL-2 (E) or S1QEL-1 (F) for 4 days, and the population doublings were assessed. n = 5; mean + SEM; *P < 0.0001 and n.s.P > 0.9999 compared to piericidin alone.
The importance of intermembrane O2− was further investigated using S3QEL-2. S3QEL-2 specifically inhibits the generation of intermembrane O2− from complex III by suppressing its electron leakage without affecting the electron flow through ETC (Fig. 3B) (25). S3QEL-2 by itself reduced the level of mitochondria-derived H2O2 (Fig. 3C). Addition of S3QEL-2 to Jurkat cells in the presence of antimycin and MitoTEMPO significantly reduced the rate of population doubling (Fig. 3D). The result highlights the necessity of mitochondrial complex III–generated intermembrane O2− for cellular fitness and suggests that the deleterious effect of the combination of complex I inhibition and a mito-antioxidant is potentially attributed to diminished intermembrane O2−.
In addition, we used piericidin in combination with S3QEL-2. Because piericidin inhibits mitochondrial complex I, the electron flow through complex III is reduced; thus, piericidin alone decreases the levels of mitochondrial intermembrane O2− derived from complex III. However, addition of S3QEL-2 to piericidin-treated cells would further eliminate all complex III–generated intermembrane O2−. As shown in Fig. 3E, cells treated with piericidin and S3QEL-2 had reduced rate of population doubling compared to either drug treatment alone. This data indicates that inhibition of complex III–derived intermembrane O2− is sufficient to impair cellular fitness when combined with complex I inhibition. Moreover, as a control experiment, we treated cells with piericidin and S1QEL-1, a specific inhibitor of mitochondrial complex I generation of O2− during reverse electron transport (RET) (Fig. 3B) (26). Under normal culture conditions, wild-type Jurkat cells conduct forward electron transport (FET), and thus, their level of mitochondrial H2O2 is refractory to S1QEL-1 (Fig. 3C). We observed that S1QEL-1 alone or in combination with piericidin had no effect on cellular fitness (Fig. 3F).
Mitochondrial complex I inhibition in combination with a mito-antioxidant induces a robust integrated stress response
Given that mitochondria-derived H2O2 is a signaling molecule necessary for cell survival and proliferation, we hypothesized that the differential generation of mitochondrial H2O2 resulting from piericidin and MitoTEMPO versus antimycin and MitoTEMPO may lead to distinct effects on redox signaling. To test this hypothesis, we performed RNA sequencing (RNA-seq) on cells treated with either MT, Pier, Anti, Pier+MT, or Anti+MT. Treatment of Jurkat cells with Pier+MT induced the most distinct transcriptomic profile (fig. S7A). Differential pathway expression analysis of cells treated with Pier+MT compared to Anti+MT revealed an elevated expression of genes down-regulated by KRAS signaling (KRAS signaling DN) and a reduced expression of genes involved in phosphatidylinositol 3-kinase (PI3K)–AKT–mammalian target of rapamycin (mTOR) signaling in the Pier+MT condition (Fig. 4A). This data suggests that Pier+MT treatment dysregulates the KRAS and PI3K-AKT-mTOR signaling pathways that have previously been reported to require mitochondria-derived H2O2 (5, 27).
Fig. 4. Combination of mitochondrial complex I inhibition and a mito-antioxidant induces a robust ISR.
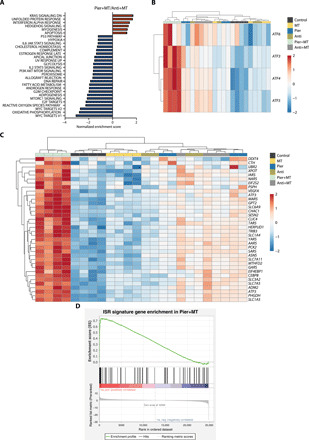
(A) Gene set enrichment analysis of top gene signatures that are up-regulated (red) or down-regulated (blue) in Jurkat cells treated with Pier+MT compared to Anti+MT for 24 hours. n = 4; FDR ≤ 0.05. (B) Heatmap of unsupervised clustering, representing ATF transcripts in Jurkat cells treated with vehicle (Control), MT, Pier, Anti, Pier+MT, or Anti+MT for 24 hours. The relative abundance of each transcript is depicted as z score across rows (red, high; blue, low) (n = 4). (C) Heatmap of unsupervised clustering, representing ISR signature genes whose abundances were significantly different between Jurkat cells treated with Pier+MT and Anti+MT for 24 hours. The relative abundance of each transcript is depicted as z score across rows (red, high; blue, low) (n = 4; FDR ≤ 0.05). (D) Gene set enrichment analysis of the ISR signature genes in Jurkat cells treated with Pier+MT for 24 hours (n = 4).
The unfolded protein response (UPR) was among the pathways up-regulated in cells treated with Pier+MT (Fig. 4A). UPR has overlapping target genes with the integrated stress response (ISR), a cytoprotective signaling pathway activated following diverse stress stimuli such as mitochondrial dysfunction. Defects in mitochondrial oxidative phosphorylation, redox balance, or proteostasis activate the general control nonderepressible 2 (GCN2) kinase to phosphorylate eukaryotic translation initiation factor 2A (EIF2A). EIF2A subsequently induces activating transcription factors (ATFs) that up-regulate genes involved in cellular adaptation (28). As shown in Fig. 4B, the transcript levels of the main ISR effector ATF4, as well as ATF3, ATF5, and ATF6, were up-regulated in the cells treated with Pier+MT compared to the other treatment groups. Further query of the RNA-seq data with a subset of genes associated with ISR activation revealed a robust enrichment of ISR-related genes in cells treated with Pier+MT (Fig. 4, C and D; fig. S8A; and table S1) (29).
Among the genes that were up-regulated in Pier+MT-treated cells were phosphoglycerate dehydrogenase (PHGDH), as well as phosphoserine aminotransferase 1 (PSAT1) (fig. S8B). PHGDH and PSAT1 are regulatory enzymes of de novo serine biosynthesis that were previously reported to be activated during ISR (30). Serine hydroxymethyltransferase 1 (SHMT1), on the other hand, was down-regulated in cells treated with Pier+MT (fig. S8B). Such dysregulation in one-carbon metabolism is consistent with our previous observation where Pier+MT-treated cells had elevated glucose-derived synthesis of 3-phosphoserine and serine, yet reduced generation of glycine compared to Anti+MT-treated cells (fig. S5, A to D). In addition, the signatures of canonical mitochondrial UPR (UPRmt) genes, including DNA-damage inducible transcript 3 (DDIT3), mitochondrial heat shock proteins (HSPs), and proteases, were not induced following treatment with Pier+MT (fig. S8C) (30).
Combination of a mitochondrial complex I inhibitor and a mito-antioxidant attenuates the leukemic burden of mice in vivo
Next, we investigated the anticancer potential of using a complex I inhibitor in combination with a mito-antioxidant in a syngeneic mouse cancer model. Specifically, we used a mouse model of T-ALL, the potent in vivo mitochondrial complex I inhibitor phenformin (31), and the mito-antioxidant MitoTEMPO. The combination of phenformin and MitoTEMPO, compared to each drug alone, significantly reduced the rate of population doubling of Jurkat and MOLT-4 T-ALL cell lines in vitro (Fig. 5, A and B). For the in vivo experiments, we generated mice with Notch1 mutation (Notch1dE)–induced T-ALL, which recapitulates most of the features of human T-ALL. Hematopoietic stem cells (HSCs) from healthy donors were transformed with a Notch1dE-GFP (green fluorescent protein) construct and adoptively transferred to primary recipients. Upon T-ALL establishment, T-ALL cells were isolated from the spleen and bone marrow of the primary recipients and subsequently transplanted into immunocompetent secondary recipients. These animals were then segregated into four different treatment groups, receiving either vehicle [phosphate-buffered saline (PBS)], phenformin alone, MitoTEMPO alone, or a combination of phenformin and MitoTEMPO (Fig. 5C). The mice that received either phenformin alone or MitoTEMPO alone had GFP+ T-ALL cell contents comparable to control mice (Fig. 5, D to F, and fig. S9). However, the mice that received the combination therapy (phenformin and MitoTEMPO) showed significantly reduced T-ALL in the spleen (Fig. 5D and fig. S9, A and B), bone marrow (Fig. 5E and fig. S9C), and peripheral lymph nodes (Fig. 5F and fig. S9D) compared to control mice. These data are consistent with our observed in vitro effects using a complex I inhibitor and a mito-antioxidant, and it suggests a potential combination therapy to significantly suppress cancer burden in vivo.
Fig. 5. Combination of a mitochondrial complex I inhibitor and a mito-antioxidant attenuates the leukemic burden of mice in vivo.
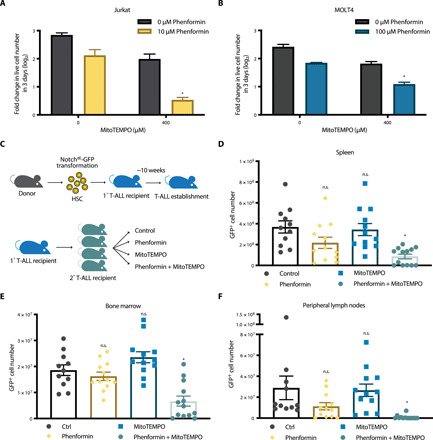
(A and B) Jurkat (A) or MOLT-4 (B) T-ALL cell lines were treated with phenformin ± MitoTEMPO for 7 days. Population doubling of the cells during the last 3 days of each treatment was assessed. n = 5; mean + SEM; *P < 0.0001 compared to phenformin or MitoTEMPO alone. (C) Schematic representation of a T-ALL adoptive transfer in syngeneic, immunocompetent mouse recipients. (D to F) Number of GFP+ T-ALL cells in the spleen (D), bone marrow (E), or peripheral lymph nodes (F) from T-ALL recipients treated with vehicle (Control), phenformin, MitoTEMPO, or Phenformin+MitoTempo for 15 days [Control: n = 11; phenformin: n = 12; MitoTEMPO: n = 12; Phenformin+MitoTEMPO: n = 13; mean ±SEM; *P = 0.0002 (D), *P = 0.0020 (E), *P < 0.0001 (F) compared to control; n.s.P values are reported in Material and Methods].
DISCUSSION
In this study, we conducted a CRISPR-based functional genomic screen to identify the metabolic determinants of mROS-dependent cell proliferation and survival. A negative selection CRISPR screen using a mild concentration of the mROS scavenger MVE uncovered subunits of mitochondrial complex I to be selectively required for cellular fitness in the presence of a mito-antioxidant. Cells lacking the mitochondrial complex I subunit NDUFA6 were hypersensitive to low concentrations of the mito-antioxidants MVE and MitoTEMPO, validating our screen results. Moreover, cellular fitness was impaired when wild-type Jurkat cells were treated with a pharmacological inhibitor of mitochondrial complex I in combination with a low concentration of a mito-antioxidant. In contrast to inhibition of mitochondrial complex I, we found that inhibition of mitochondrial complex III did not synergize with mito-antioxidants to impair cell survival. Furthermore, we showed that this unique cytotoxic effect of complex I inhibition in the presence of a mito-antioxidant was associated with its capacity to suppress the levels of mitochondria-derived H2O2. Collectively, our data strongly suggest that mitochondrial complex I is the dominant metabolic determinant facilitating mROS generation to support cellular fitness.
As a consequence, cells treated with a mitochondrial complex I inhibitor and a mito-antioxidant exhibited multiple signs of reductive stress. Mitochondria-derived H2O2 can oxidize specific cysteine residues on target proteins to modulate downstream signaling pathways. We observed that the combination of piericidin and MitoTEMPO led to a down-regulation of several redox-sensitive signaling pathways, including KRAS and PI3K-AKT-mTOR (5, 27). Mitochondrial H2O2 is also essential for formations of disulfide bonds that determine the orderly structure and trafficking of many proteins. Therefore, it is possible that piericidin and MitoTEMPO disrupt intracellular proteostasis (32). Consistent with this hypothesis, we found that the combination of piericidin and MitoTEMPO up-regulated genes associated with UPR activation.
The combination of mitochondrial complex I inhibition and a mito-antioxidant also induced a robust ISR. Cells treated with piericidin and MitoTEMPO showed up-regulation of the main ISR effector ATF4, along with a plethora of ISR signature genes. ATF4 signaling promotes metabolic remodeling, such as increased de novo serine biosynthesis (30, 33, 34). The metabolite profile of cells treated with piericidin and MitoTEMPO showed elevated levels of total 3-phospho-serine, as well as glucose-derived 3-phospho-serine and serine. In addition, it is important to note that ISR activation is widely studied in the context of oxidative stress rather than reductive stress (35–37); thus, we report a potential association of mitochondrial reductive stress with the ISR. Chronic reductive stress, in which most electron acceptors are reduced, can force proteins to donate their electrons to oxygen instead, resulting in an increase in intracellular ROS levels (38). It will be of interest to examine whether this feedback loop between mitochondrial reductive and oxidative stress induces the ISR and cell death.
In summary, our screening approach identified mitochondrial complex I as the dominant metabolic determinant of mROS generation. Inhibition of mitochondrial complex I in the presence of a mild concentration of a mito-antioxidant impaired cancer cell fitness both in vitro and in vivo. Our findings suggest that inducing reductive stress by limiting mROS formation may be an effective strategy to target cancer cells. This potential anticancer therapy is exciting given that reductive stress is a largely unexplored subject in the fields of ROS biology and cancer. Supporting this, hyperactivation of NRF2, a master regulator of cellular antioxidant responses, was recently reported to induce tumor suppression in vivo (39, 40). However, given the abundance of evidence that NRF2 is also required for tumorigenesis (7–14), the tumor suppressive effect of reductive stress is likely to be highly context dependent: Are certain types of cancer more vulnerable to reductive stress? Do cancers in different stages (e.g., initiation, growth, or metastasis) have varying sensitivities to reductive stress? Does the molecular context, including cellular localization, type of ROS, or degree, of the reductive stress determine its biological outcome? Future studies will examine the context-dependent effects of reductive stress on cancer biology.
MATERIALS AND METHODS
CRISPR-based negative selection screen
Negative selection CRISPR screen using metabolism-focused sgRNA library was performed as previously described (22). At 2 days after transduction of Jurkat cells with lentiviral sgRNA library, infected cells were selected with puromycin (0.5 μg/ml) for 3 days. Subsequently, the negative selection MVE screen was initiated as follows. An initial population of 50 million cells were collected and stored at −80°C. One hundred million cells were seeded in two spinner flasks with 50 million cells per flask at 100,000 cells/ml. The cells were passaged for approximately 15 population doublings in the presence or absence of 50 nM MVE (a gift from B. Kalyanaraman at the Medical College of Wisconsin). Genomic DNA was then extracted from the MVE-treated and untreated populations, as well as the initial population. sgRNA inserts were polymerase chain reaction (PCR)–amplified while adding index and Illumina adapter (P5 and P7) sequences. The libraries were sequenced by HiSeq 2500 (Illumina), using previously reported sequencing primers (22). The abundance of sgRNAs was analyzed and compared among the initial, MVE-treated, and untreated populations. For each sgRNA, its frequency was first assessed by dividing its number of reads with the total sgRNA reads after adding a pseudocount of one. The changes in abundance of each sgRNA were then calculated as log2 fold change of its frequency within MVE-treated or untreated population compared to that in the initial population. Median of these changes in abundance across all sgRNAs targeting a particular gene (~10 sgRNAs per gene) was termed its gene score. The difference between gene scores in the MVE-treated and untreated populations is the differential gene score.
Cell culture
Jurkat and MOLT-4 cells were cultured in RPMI 1640 medium without l-glutamine (Corning, #15040CM) with addition of 10% Nu-Serum IV Growth Medium Supplement (Corning), 1% GlutaMAX (Gibco), and 1% antibiotic-antimycotic solution (Gibco). Whenever inhibiting mitochondrial complex III, the medium was additionally supplemented with 1 mM methyl-pyruvate (Sigma-Aldrich) and 400 μM uridine (Sigma-Aldrich). For galactose sensitivity assays, Jurkat cells were also cultured in RPMI 1640 medium without glucose (Gibco, #11879020) with addition of 11.1 mM galactose (Sigma-Aldrich), 10% dialyzed fetal bovine serum (FBS) (PEAK Serum), 1% GlutaMAX (Gibco), and 1% antibiotic-antimycotic solution (Gibco). Cells were kept at 37°C, 5% CO2, and 95% humidity. All cell lines used were periodically tested for mycoplasma contamination using a PCR-based detection kit (American Type Culture Collection).
Generation of cell lines with knockouts and ectopic expressions
sgRNA oligonucleotides targeting NDUFA6, as well as a nontargeting control, were cloned into pSpCas9(BB)-2A-GFP (PX458) plasmid (a gift from F. Zhang at the Massachusetts Institute of Technology; Addgene, #48138), according to the provider’s instructions. Sequences of the oligonucleotides were as follows: sgNDUFA6_3: 5′-ACTGAAAATGGGCTTCACGA-3′ (targeting + strand in exon 1); sgNDUFA6_7: 5′-GATGCTTTGGCAAGATGGCG-3′ (targeting − strand in exon 1); nontargeting: 5′-GTAGCGAACGTGTCCGGCGT-3′. The sgRNA-Cas9-2A-GFP vectors were then transfected into Jurkat cells, using jetPRIME transfection reagent (Polyplus). At 2 days after transfection, GFP-positive cells were single cell–sorted into 96-well plates using BD FACSAria SORP systems. The clones were cultured for 2 to 3 weeks, and established clonal cell lines were expanded. NDUFA6 knockouts were verified by Simple Western analysis (see below). To ectopically express NDI1 in the NDUFA6-null cell line, NDI1 coding sequence from pWPI-NDI1-BFP construct (a gift from E. Dufour at the University of Tampere) was subcloned into the pLV-EF1-RFP (red fluorescent protein) vector (VectorBuilder). The NDI1-EF1-RFP vector or empty vector control, along with pMD2.G and psPAX2 lentiviral packaging vectors, was then transfected into 293T cells, using jetPRIME (Polyplus). Resultant supernatants containing NDI1 or control lentivirus were collected and stored at −80°C. For transduction, 1 million cells of NDUFA6 KO1 cell line were seeded per well in a six-well plate with 1 ml of the NDI1 or control virus containing polybrene (8 μg/ml). The cells were spin-infected by centrifugation at 1500 rcf in room temperature for 90 min, followed by an overnight incubation at 37°C. At approximately 24 hours after transduction, virus was removed from the cells. On the following day, RFP+ cells were sorted on BD FACSAria SORP systems. The cells were periodically sorted to maintain high RFP expressions. NDI1 expression was confirmed by Simple Western analysis (see below).
Immunoblotting with Simple Western assay
To assess NDUFA6 and NDI1 protein expressions, whole-cell lysates were extracted from indicated cell lines by resuspending the cells in 1× cell lysis buffer (Cell Signaling Technology) with Halt protease inhibitor cocktail (Thermo Fisher Scientific). The cell suspension was then exposed to a complete freeze-thaw cycle between −80°C and room temperature. Protein concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Immunoblotting was performed on Wes platform (ProteinSimple), according to the manufacturer’s protocol. For analysis of NDUFA6 protein, 20 μg of the lysates (at 4 μg/μl) was loaded per well in a 12- to 230-kDa Wes Separation Module (ProteinSimple). A rabbit polyclonal antibody against NDUFA6 (1:10 dilution; Santa Cruz Biotechnology, #86755) and mouse monoclonal antibody against GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (1:20,000 dilution; Santa Cruz Biotechnology, #32233) were used, in combination with the Anti-Rabbit or Anti-Mouse Detection Modules for Wes (ProteinSimple), respectively. For analysis of NDI1 protein, 2.5 μg of the lysates (at 0.5 μg/μl) was loaded per well. A rabbit antibody against NDI1 (1:500 dilution; a gift from E. Dufour at the University of Tampere) and rabbit monoclonal antibody against vinculin (1:100 dilution; Cell Signaling Technology, #13901) were used. The chemiluminescence of all proteins was quantified on Compass software (version 4.0; ProteinSimple).
Drug treatments, followed by assessments of cell sensitivity, proliferation, and viability
Two hundred thousand cells of indicated Jurkat or MOLT-4 cell lines were seeded per well in 12-well plates at 100,000 cells/ml. Where noted, cells were treated with galactose medium (described above), vehicle [1× Dulbecco’s PBS (DPBS; Corning), dimethyl sulfoxide (DMSO; Sigma-Aldrich, #D2650), or alcohol (J.T.Baker, #9229-01)], MVE (a gift from B. Kalyanaraman at the Medical College of Wisconsin), MitoTEMPO (Sigma-Aldrich, #SML0737), piericidin (Cayman Chemical, #15379), antimycin (Sigma-Aldrich, #A8674), S3QEL-2 (Sigma-Aldrich, SML1554), S1QEL-1 (Sigma-Aldrich, SML1948), or phenformin (Sigma-Aldrich, P7045) of indicated concentrations for specified durations. For treatments exceeding 4 days in glucose-containing medium, cells were collected on day 4 and reseeded at 200,000 cells/well and 100,000 cells/ml with a fresh concentration of drug. To assess cell sensitivities to treatments, cells were collected and resuspended in fluorescence-activated cell sorting (FACS) buffer [1× DPBS (Corning) with 10% Nu-Serum IV Growth Medium Supplement (Corning)]. Live cells were counted using ViaStain PI Stain Solution on a Cellometer K2 platform (Nexcelom). Last, the rates of population doubling were calculated by taking log2 of the live cell number divided by the initial cell number (200,000). When comparing the treatment sensitivities between multiple cell lines that proliferate at different rates, the rates of population doubling under treatments were normalized with those under control (glucose medium or vehicle treatments). To assess cell proliferation, 10 μM BrdU was added to cells 1 hour before the specified endpoint of a treatment. Cells were labeled with BrdU for 2 hours, after which they were collected, resuspended in FACS buffer, and counted on Cellometer K2 (Nexcelom). One million (or less, if necessary) cells were permeabilized, fixed, and stained for BrdU with the FITC BrdU Flow Kit (BD Biosciences), according to the manufacturer’s protocol. The percentage of proliferating population [fluorescein isothiocyanate (FITC) BrdU+] was analyzed on a BD FACSymphony cell analyzer and FlowJo software (version 10.4.2; BD Life Sciences). To assess cell viability, cells were collected, resuspended in FACS buffer, and counted on Cellometer K2 (Nexcelom). One hundred thousand cells were stained for annexin V with the Annexin V-FITC Apoptosis Detection Kit (eBioscience), according to the manufacturer’s instructions. The percentage of live (annexin V−, PI−), necrotic (annexin V−, PI+), early apoptotic (annexin V+, PI−), and late apoptotic (annexin V+, PI+) populations were measured on a BD FACSymphony cell analyzer and FlowJo software (version 10.4.2; BD Life Sciences).
H2O2 measurement
To measure the mitochondria-derived H2O2, 300,000 Jurkat cells were plated per well in a 96-well black polystyrene plate with clear bottom (Corning, #3603). The cells were permeabilized in 50 μl of KHEB buffer [120 mM KCl (Sigma-Aldrich), 5 mM Hepes (Corning, #25060), 1 mM EGTA (Sigma-Aldrich), 2% bovine serum albumin (Thermo Fisher Scientific, #BP9704100)] with saponin (100 μg/ml; Sigma-Aldrich) for 15 min at room temperature. Permeabilized cells were immediately treated with vehicle [DMSO (Sigma-Aldrich, #D2650) and alcohol (J.T.Baker, #9229-01)], 400 μM MitoTEMPO, 1 μM piericidin, 1 μM antimycin, 1 μM piericidin + 400 μM MitoTEMPO, 1 μM antimycin + 400 μM MitoTEMPO, 5 or 10 μM S3QEL-2, or 0.5 or 1 μM S1QEL-1 in the presence of 12.5 μM Amplex red (Molecular Probe), SOD1 (12.5 U/ml; Sigma-Aldrich), and horseradish peroxidase (2.5 U/ml; Thermo Fisher Scientific) in KHEB of final volume up to 200 μl per well. Resorufin fluorescence with excitation of 544 nm and emission of 590 nm was measured every minute for 1 hour using SpectraMax M2 (Molecular Devices) set at 37°C.
Metabolomics
One million Jurkat cells were seeded per plate in 10-cm plates at 100,000 cells/ml. The cells were treated with vehicle (DMSO and alcohol), 400 μM MitoTEMPO, 1 μM piericidin, 1 μM antimycin, 400 μM MitoTEMPO + 1 μM piericidin, or 400 μM MitoTEMPO + 1 μM antimycin for 24 hours. For carbon tracing experiments, isotopic labeling was performed for 8 hours, starting at 16 hours after beginning of the drug treatments. Cell medium was switched to labeling medium containing RPMI 1640 medium without glucose (Gibco, #11879020) or glutamine (Corning, #15040CM) with addition of [U-13C6]glucose (2000 mg/liter; Cambridge Isotope Laboratories) or 2 mM [U-13C5]glutamine (Cambridge Isotope Laboratories), respectively, as well as 10% dialyzed FBS (PEAK serum), 1% antibiotic-antimycotic solution (Gibco), 1 mM methyl-pyruvate (Sigma-Aldrich), 400 μM uridine (Sigma-Aldrich), and fresh concentrations of the drugs. After the treatment and/or labeling, cells were washed once with 0.9% NaCl at 4°C. The cell pellets were flash-frozen in liquid nitrogen and stored at −80°C until ready for metabolite extractions. To extract metabolites, the pellets were briefly thawed on ice and resuspended in 225 μl/1 million cells of high-performance liquid chromatography (HPLC)–grade methanol in water (80/20, v/v) that was previously cooled to −80°C. The cell suspensions were exposed to three complete freeze-thaw cycles between liquid nitrogen and 37°C water bath, followed by centrifugation at 18,000 rcf at 4°C for 15 min. The supernatants containing metabolites were collected and evaporated to dryness in a SpeedVac concentrator (Thermo Savant). The metabolites were reconstituted in acetonitrile in analytical-grade water (50/50, v/v), centrifuged to remove debris, and then analyzed by high-resolution HPLC–tandem mass spectrometry. The system consisted of a Q-Exactive (Thermo Fisher Scientific) in line with an electrospray source and an UltiMate 3000 (Thermo Fisher Scientific) series HPLC consisting of a binary pump, degasser, and autosampler outfitted with a XBridge Amide column (Waters; dimensions of 4.6 mm by 100 mm and a 3.5-μm particle size). Mobile phase A contained 95% (v/v) water, 5% (v/v) acetonitrile, 10 mM ammonium hydroxide, and 10 mM ammonium acetate (pH 9.0), and mobile phase B contained 100% acetonitrile. The gradient was set to 0 min, 15% A; 2.5 min, 30% A; 7 min, 43% A; 16 min, 62% A; 16.1 to 18 min, 75% A; 18 to 25 min, 15% A, with a flow rate of 400 μl/min. The capillary of the ESI (electrospray ionization) source was set to 275°C, with sheath gas at 45 arbitrary units, auxiliary gas at 5 arbitrary units, and the spray voltage at 4.0 kV. In positive/negative polarity switching mode, a mass/charge ratio scan ranging from 70 to 850 was chosen, and MS1 data were collected at a resolution of 70,000. The automatic gain control (AGC) target was set at 1 × 106, and the maximum injection time was 200 ms. The top five precursor ions were subsequently fragmented, using the higher-energy collisional dissociation (HCD) cell with normalized collision energy (NCE) of 30% in MS2 at a resolution of 17,500. Sample volumes of 10 μl were injected. Data acquisition was performed using Xcalibur software (version 4.1; Thermo Fisher Scientific). The peak area of each detected metabolite was normalized by the total ion current, an integration of all of the recorded peaks within the window of acquisition. Data analysis was subsequently carried out by MetaboAnalyst (version 4.0). Features with more than 50% missing values were removed. Leftover missing values were replaced by half the minimum positive values. For the heatmap representation of metabolites in multiple groups (Control, MT, Pier, Anti, Pier+MT, and Anti+MT), multiple one-way analyses of variance (ANOVAs) with Fisher’s least significant difference post hoc analysis and false discovery rate (FDR) threshold of 0.1 were performed to generate a list of metabolites whose levels are significantly different among the groups. The list was then plotted as a heatmap, using Euclidean distance measures and Ward’s clustering algorithm of metabolites/rows. Each row represents z scores of each metabolite. For the volcano plot representation of metabolites between Pier+MT and Anti+MT, unpaired t test with a fold change threshold of 2 and P value threshold of 0.1 was used to identify significantly changed metabolites between the two groups. The data from carbon tracing experiments were analyzed by Excel. Normalized peak areas of differentially labeled metabolites (e.g., M+0, M+1, and M+2) were added up to calculate the total normalized peak area of each metabolite. Subsequently, percentage of pool for each labeled metabolite was determined by dividing its normalized peak area by the total normalized peak area.
GSH/GSSG ratio measurement
Two hundred thousand Jurkat cells were seeded per well in 12-well plates at 100,000 cells/ml. The cells were treated with vehicle (DMSO and alcohol), 400 μM MitoTEMPO, 1 μM piericidin, 1 μM antimycin, 400 μM MitoTEMPO + 1 μM piericidin, 400 μM MitoTEMPO + 1 μM antimycin, or 5 μM Menadione for 24 hours. Cells were harvested and diluted into 400,000 cells/ml in Hanks’ balanced salt solution (HBSS) (Gibco, #14025092) with fresh concentrations of the drugs. Twenty-five microliters of the cell suspension (or 10,000 cells) was added per well of a 96-well white polystyrene plate with clear bottom (Corning, #3610). GSH/GSSG ratios were measured using GSH/GSSG-Glo Assay (Promega, #V6611), according to the manufacturer’s protocol. Luminescence was measured using SpectraMax M2 (Molecular Devices).
RNA sequencing
Five hundred thousand Jurkat cells were seeded per plate in 10-cm plates at 100,000 cells/ml. The cells were treated with vehicle (DMSO and alcohol), 400 μM MitoTEMPO, 1 μM piericidin, 1 μM antimycin, 400 μM MitoTEMPO + 1 μM piericidin, or 400 μM MitoTEMPO + 1 μM antimycin for 24 hours. After the treatment, cells were collected and resuspended in 350 μl of RLT Plus buffer (Qiagen) with 0.01% 2-mercaptoethanol (Sigma-Aldrich). The cell suspensions were homogenized by 29-guage needles and QIAshredder Spin Columns (Qiagen). RNA was extracted with the RNeasy Plus Mini Kit (Qiagen), following the manufacturer’s protocol with an additional on-column deoxyribonuclease (DNase) treatment using RNase-Free DNase Set (Qiagen). The quality and quantity of the RNA were assessed by Agilent 4200 TapeStation, using the RNA ScreenTape System (Agilent Technologies). mRNA libraries were prepared with NEBNext Ultra RNA Library Prep Kit for Illumina (NEB). Pooled library (2.5 pM) was sequenced on Illumina NextSeq 500 system, using 1% PhiX control and NextSeq 500/550 High Output Kit v2 (75 cycles) (Illumina). Raw BCL read files were demultiplexed and converted to FASTQ files using bcl2fastq (Illumina) and trimmed using Trimmomatic (version 0.39). The reads were then aligned to the human hg38 reference genome using STAR software to generate BAM files. HTSeq framework was used to count reads within the exons. Likelihood ratio tests and pairwise differential gene expression analyses were carried out using the R package DESeq2. Gene set enrichment analysis (GSEA) was performed using the GSEA software (version 4.0.0). The pairwise differential gene expression output from DESeq2 was converted via Wald test to a preranked list of genes from significantly up-regulated to significantly down-regulated. The list was submitted to the GSEA software, where GSEA was performed based on Hallmark Gene Sets from Molecular Signatures Database (MSigDB) (version 7.0) or a curated list of ISR genes (table S1; a gift from C. Sidrauski at the Calico Life Sciences) (29).
T-ALL adoptive transfer
Notch1dE-GFP retrovirus was prepared by transfecting the MIGR1-Notch1dE-GFP vector (a gift from P. Ntziachristos in the Northwestern University) into Platinum-E retroviral packaging cells (a gift from P. Ntziachristos in the Northwestern University) using jetPRIME (Polyplus). The virus was freshly produced before every transduction. Eight-week-old C57Bl/6J mice were euthanized to extract bone marrow cells, from which HSCs were isolated using the EasySep Mouse CD117 Positive Selection Kit (StemCell). In the following steps, HSCs were kept in Opti-MEM (Gibco) supplemented with interleukin-3 (IL-3) (10 ng/ml; PeproTech, #213-13), interleukin-7 (IL-7) (10 ng/ml; PeproTech, #217-17), stem cell factor (SCF) (50 ng/ml; PeproTech, #250-03), Fms-related tyrosine kinase 3 ligand (Flt3L) (50 ng/ml; PeproTech, #250-31L), and interleukin-6 (IL-6) (20 ng/ml; PeproTech, #216-16). To transform HSCs, approximately 1 million cells were plated per well in six-well plates with 5 ml of Notch1dE-GFP retrovirus containing polybrene (4 μg/ml). The cells were spin-infected by centrifugation at 1500 rcf in room temperature for 90 min, followed by an incubation with the virus overnight at 37°C. At 18 hours after transduction, the virus was removed and the cells were allowed to rest for 2 days. On the day of adoptive transfer, lineage (CD4, CD8a, B220, CD11b, Gr-1, NK1.1, Ter-119)–negative and GFP-positive cells were sorted on BD FACSAria SORP systems. T-ALL cells (50,000 to 100,000), along with 500,000 support bone marrow cells, were injected intravenously into 8- to 12-week-old C57Bl/6J recipients, which were irradiated at 1000 cGy within 24 hours before the transfer. Upon T-ALL establishment in the primary recipients, splenocytes and bone marrow cells were harvested. The percentage of GFP+ cells was then analyzed on a BD FACSymphony cell analyzer and FlowJo software (version 10.4.2; BD Life Sciences). The total number of live cells was counted using ViaStain AOPI Stain Solution on Cellometer K2 (Nexcelom). Fifteen thousand of the live GFP+ T-ALL cells were injected intravenously into 8- to 12-week-old C57Bl/6J recipients, which were irradiated at 450 cGy within 24 hours before the transfer. The T-ALL cells were allowed to engraft for 4 days, after which the secondary recipients were allocated into four treatment groups, where they received daily intraperitoneal injections of vehicle (PBS; Amresco, #K813), phenformin (75 mg/kg; Sigma-Aldrich, #P7045), MitoTEMPO (5 mg/kg; Sigma-Aldrich, #SML0737), or phenformin (75 mg/kg) + MitoTEMPO (5 mg/kg) for 15 days. At the end of the treatment, cells were harvested from the spleen, one set of femur and tibia, and peripheral lymph nodes of each recipient. The cells were stained with Ghost Dye Red 780 (Tonbo Biosciences) and resuspended in FACS buffer with PKH26 Reference Microbeads (Sigma-Aldrich). The numbers and percentages of GFP-positive T-ALL cells were analyzed on a BD FACSymphony cell analyzer and FlowJo software (version 10.4.2; BD Life Sciences). Statistical significance was determined by Kruskal-Wallis nonparametric test, using GraphPad Prism (version 8; GraphPad Software). Specific P values of statistical significance (*) are noted in the figure legend. P values of nonsignificance (n.s.) are as follows: n.s.P = 0.0612 (Fig. 5D; Phenformin); n.s.P = 0.8723 (Fig. 5D; MitoTEMPO); n.s.P = 0.4904 (Fig. 5E; Phenformin); n.s.P = 0.1818 (Fig. 5E; MitoTEMPO); n.s.P = 0.1303 (Fig. 5F; Phenformin); n.s.P = 0.8095 (Fig. 5F; MitoTEMPO); n.s.P = 0.9256 (fig. S9A; MitoTEMPO); n.s.P = 0.4074 (fig. S9B; Phenformin); n.s.P = 0.2305 (fig. S9B; MitoTEMPO); n.s.P = 0.5719 (fig. S9C; MitoTEMPO); n.s.P = 0.0746 (fig. S9D; Phenformin); and n.s.P = 0.9767 (fig. S9D; MitoTEMPO) compared to control. All animal care and experiments were conducted in accordance with the Northwestern University Institutional Animal Care and Use Committee (IACUC) guidelines.
Statistical analysis
Data are presented as mean + or ± standard error of the mean (SEM). Minimum of n = 3 independent experiments were performed, and the numbers of replicates are noted in the figure legends. No samples were excluded from the analysis. Experiments were neither randomized nor blinded. Statistical significance (P values) was determined by one-way ANOVA (with single variable) or two-way ANOVA (with two variables) with Bonferroni’s multiple comparisons test, using GraphPad Prism (version 8; GraphPad software). Statistical significance (*) was defined as P < 0.05. Particular P values between groups of interest are noted in the figure legends. P values are reported as four digits after the decimal point: P values less than 0.0001 are shown as <0.0001; P values higher than 0.9999 are shown as >0.9999. Detailed methods of statistical analysis of metabolomics, RNA-seq, and in vivo T-ALL data are described above.
Supplementary Material
Acknowledgments
We are grateful to K. Birsoy (The Rockefeller University) for providing advice and reagents for our CRISPR screen. We thank the Genome Technology Core of the Whitehead Institute for deep sequencing the sgRNA inserts. We are grateful to B. Kalyanaraman (Medical College of Wisconsin) for providing us MVE. We are grateful to G.R. Scott Budinger (Northwestern University) for providing us with the Cellometer K2 instrument to count live cells. We would also like to thank the Metabolomics Core of Northwestern University for the quantification of metabolites. We thank the Flow Cytometry Core of Northwestern University for providing us with the BD FACSymphony cell analyzer and cell sorting services using FACSAria SORP systems. We are grateful to H. Abdala-Valencia (Northwestern University) for deep sequencing the RNA transcripts. We are also grateful to E. T. Bartom (Northwestern University) for providing us the Ceto pipeline for RNA-seq analysis. Last, we thank L. Diebold Petrovich (Northwestern University) and J. S. Stoolman (Northwestern University) for their help in harvesting organs from T-ALL recipient animals. Funding: This work was supported by an NIH grant (5R35CA197532) to N.S.C., an NCI grant (T32 CA009560) and an NIH grant (2T32HL076139-16) to H.K., an NCI grant (T32 CA009560) to G.S.M., and a Cancer Research Institute Irvington postdoctoral fellowship to E.M.S. D.M.S. is an investigator of the Howard Hughes Medical Institute and an American Cancer Society Research professor. Author contributions: H.K., C.R.R., D.M.S., and N.S.C. initiated the project and designed the research plan. H.K. carried out most of the experiments in the paper. C.R.R. conducted the CRISPR screen. G.S.M. analyzed metabolomics and RNA-seq data. E.M.S. provided technical expertise with animal experiments. T.W. performed the bioinformatics analysis of the screen. H.K. and N.S.C. wrote the manuscript, with input from all authors. Competing interests: N.S.C. is a scientific advisor of the Raphael Pharmaceuticals Inc. D.M.S. and T.W. are cofounders of and D.M.S. is a consultant to KSQ Therapeutics Inc. D.M.S. and T.W. are inventors on a patent for functional genomics using CRISPR-Cas (US 15/141,348). The rest of the authors declare that they have no financial or nonfinancial competing interests. Data and materials availability: All data needed to evaluate the conclusions of the paper are present in the paper and/or the Supplementary Materials. RNA-seq data related to this paper is available on the GEO repository (accession number GSE157912). Please contact N.S.C. for additional data and material requests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/45/eabb7272/DC1
REFERENCES AND NOTES
- 1.Reczek C. R., Chandel N. S., The two faces of reactive oxygen species in cancer. Annu. Rev. Cancer Biol. 1, 79–98 (2017). [Google Scholar]
- 2.Chandel N. S., Tuveson D. A., The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 371, 177–178 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Klein E. A., Thompson I. M., Tangen C. M., Crowley J. J., Lucia M. S., Goodman P. J., Minasian L. M., Ford L. G., Parnes H. L., Gaziano J. M., Karp D. D., Lieber M. M., Walther P. J., Klotz L., Parsons J. K., Chin J. L., Darke A. K., Lippman S. M., Goodman G. E., Meyskens F. L., Baker L. H., Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 306, 1549–1556 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiel C., Le Gal K., Ibrahim M. X., Jahangir C. A., Kashif M., Yao H., Ziegler D. V., Xu X., Ghosh T., Mondal T., Kanduri C., Lindahl P., Sayin V. I., Bergo M. O., BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell 178, 330–345.e22 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Liou G.-Y., Döppler H., DelGiorno K. E., Zhang L., Leitges M., Crawford H. C., Murphy M. P., Storz P., Mutant Kras-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 14, 2325–2336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conrad M., Kagan V. E., Bayir H., Pagnussat G. C., Head B., Traber M. G., Stockwell B. R., Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 32, 602–619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schafer Z. T., Grassian A. R., Song L., Jiang Z., Gerhart-Hines Z., Irie H. Y., Gao S., Puigserver P., Brugge J. S., Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris I. S., Treloar A. E., Inoue S., Sasaki M., Gorrini C., Lee K. C., Yung K. Y., Brenner D., Knobbe-Thomsen C. B., Cox M. A., Elia A., Berger T., Cescon D. W., Adeoye A., Brüstle A., Molyneux S. D., Mason J. M., Li W. Y., Yamamoto K., Wakeham A., Berman H. K., Khokha R., Done S. J., Kavanagh T. J., Lam C.-W., Mak T. W., Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 (2015). [DOI] [PubMed] [Google Scholar]
- 9.DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K. H., Yeo C. J., Calhoun E. S., Scrimieri F., Winter J. M., Hruban R. H., Iacobuzio-Donahue C., Kern S. E., Blair I. A., Tuveson D. A., Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero R., Sayin V. I., Davidson S. M., Bauer M. R., Singh S. X., LeBoeuf S. E., Karakousi T. R., Ellis D. C., Bhutkar A., Sánchez-Rivera F. J., Subbaraj L., Martinez B., Bronson R. T., Prigge J. R., Schmidt E. E., Thomas C. J., Goparaju C., Davies A., Dolgalev I., Heguy A., Allaj V., Poirier J. T., Moreira A. L., Rudin C. M., Pass H. I., Heiden M. G. V., Jacks T., Papagiannakopoulos T., Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 23, 1362–1368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bar-Peled L., Kemper E. K., Suciu R. M., Vinogradova E. V., Backus K. M., Horning B. D., Paul T. A., Ichu T.-A., Svensson R. U., Olucha J., Chang M. W., Kok B. P., Zhu Z., Ihle N. T., Dix M. M., Jiang P., Hayward M. M., Saez E., Shaw R. J., Cravatt B. F., Chemical proteomics identifies druggable vulnerabilities in a genetically defined cancer. Cell 171, 696–709.e23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Viswanathan V. S., Ryan M. J., Dhruv H. D., Gill S., Eichhoff O. M., Seashore-Ludlow B., Kaffenberger S. D., Eaton J. K., Shimada K., Aguirre A. J., Viswanathan S. R., Chattopadhyay S., Tamayo P., Yang W. S., Rees M. G., Chen S., Boskovic Z. V., Javaid S., Huang C., Wu X., Tseng Y.-Y., Roider E. M., Gao D., Cleary J. M., Wolpin B. M., Mesirov J. P., Haber D. A., Engelman J. A., Boehm J. S., Kotz J. D., Hon C. S., Chen Y., Hahn W. C., Levesque M. P., Doench J. G., Berens M. E., Shamji A. F., Clemons P. A., Stockwell B. R., Schreiber S. L., Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hangauer M. J., Viswanathan V. S., Ryan M. J., Bole D., Eaton J. K., Matov A., Galeas J., Dhruv H. D., Berens M. E., Schreiber S. L., McCormick F., McManus M. T., Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao J. Y., Poddar A., Magtanong L., Lumb J. H., Mileur T. R., Reid M. A., Dovey C. M., Wang J., Locasale J. W., Stone E., Cole S. P. C., Carette J. E., Dixon S. J., A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep. 26, 1544–1556.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung E. C., DeNicola G. M., Nixon C., Blyth K., Labuschagne C. F., Tuveson D. A., Vousden K. H., Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 37, 168–182.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung E. C., Lee P., Ceteci F., Nixon C., Blyth K., Sansom O. J., Vousden K. H., Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev. 30, 52–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zielonka J., Joseph J., Sikora A., Hardy M., Ouari O., Vasquez-Vivar J., Cheng G., Lopez M., Kalyanaraman B., Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 117, 10043–10120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Porporato P. E., Payen V. L., Pérez-Escuredo J., De Saedeleer C. J., Danhier P., Copetti T., Dhup S., Tardy M., Vazeille T., Bouzin C., Feron O., Michiels C., Gallez B., Sonveaux P., A mitochondrial switch promotes tumor metastasis. Cell Rep. 8, 754–766 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Woo D. K., Green P. D., Santos J. H., D’Souza A. D., Walther Z., Martin W. D., Christian B. E., Chandel N. S., Shadel G. S., Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APCMin/+ mice. Am. J. Pathol. 180, 24–31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhanasekaran A., Kotamraju S., Kalivendi S. V., Matsunaga T., Shang T., Keszler A., Joseph J., Kalyanaraman B., Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J. Biol. Chem. 279, 37575–37587 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Sharpe A., McKenzie M., Mitochondrial fatty acid oxidation disorders associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) deficiency. Cell 7, 46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birsoy K., Wang T., Chen W. W., Freinkman E., Abu-Remaileh M., Sabatini D. M., An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan L. B., Gui D. Y., Hosios A. M., Bush L. N., Freinkman E., Vander Heiden M. G., Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Löffler M., Jöckel J., Schuster G., Becker C., Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides. Mol. Cell. Biochem. 174, 125–129 (1997). [PubMed] [Google Scholar]
- 25.Orr A. L., Vargas L., Turk C. N., Baaten J. E., Matzen J. T., Dardov V. J., Attle S. J., Li J., Quackenbush D. C., Goncalves R. L. S., Perevoshchikova I. V., Petrassi H. M., Meeusen S. L., Ainscow E. K., Brand M. D., Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 11, 834–836 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brand M. D., Goncalves R. L. S., Orr A. L., Vargas L., Gerencser A. A., Borch Jensen M., Wang Y. T., Melov S., Turk C. N., Matzen J. T., Dardov V. J., Petrassi H. M., Meeusen S. L., Perevoshchikova I. V., Jasper H., Brookes P. S., Ainscow E. K., Suppressors of superoxide-H2O2 production at site IQ of mitochondrial complex I protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab. 24, 582–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J.-H., Choi T. G., Park S., Yun H. R., Nguyen N. N. Y., Jo Y. H., Jang M., Kim J., Kim J., Kang I., Ha J., Murphy M. P., Tang D. G., Kim S. S., Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ. 25, 1921–1937 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pakos-Zebrucka K., Koryga I., Mnich K., Ljujic M., Samali A., Gorman A. M., The integrated stress response. EMBO Rep. 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong Y. L., LeBon L., Basso A. M., Kohlhaas K. L., Nikkel A. L., Robb H. M., Donnelly-Roberts D. L., Prakash J., Swensen A. M., Rubinstein N. D., Krishnan S., McAllister F. E., Haste N. V., O’Brien J. J., Roy M., Ireland A., Frost J. M., Shi L., Riedmaier S., Martin K., Dart M. J., Sidrauski C., eIF2B activator prevents neurological defects caused by a chronic integrated stress response. eLife 8, e42940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forsström S., Jackson C. B., Carroll C. J., Kuronen M., Pirinen E., Pradhan S., Marmyleva A., Auranen M., Kleine I.-M., Khan N. A., Roivainen A., Marjamäki P., Liljenbäck H., Wang L., Battersby B. J., Richter U., Velagapudi V., Nikkanen J., Euro L., Suomalainen A., Fibroblast growth factor 21 drives dynamics of local and systemic stress responses in mitochondrial myopathy with mtDNA deletions. Cell Metab. 30, 1040–1054.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Birsoy K., Possemato R., Lorbeer F. K., Bayraktar E. C., Thiru P., Yucel B., Wang T., Chen W. W., Clish C. B., Sabatini D. M., Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang Y., Song Y., Loscalzo J., Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 104, 10813–10817 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bao X. R., Ong S.-E., Goldberger O., Peng J., Sharma R., Thompson D. A., Vafai S. B., Cox A. G., Marutani E., Ichinose F., Goessling W., Regev A., Carr S. A., Clish C. B., Mootha V. K., Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 5, e10575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nikkanen J., Forsström S., Euro L., Paetau I., Kohnz R. A., Wang L., Chilov D., Viinamäki J., Roivainen A., Marjamäki P., Liljenbäck H., Ahola S., Buzkova J., Terzioglu M., Khan N. A., Pirnes-Karhu S., Paetau A., Lönnqvist T., Sajantila A., Isohanni P., Tyynismaa H., Nomura D. K., Battersby B. J., Velagapudi V., Carroll C. J., Suomalainen A., Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 23, 635–648 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D. F., Bell J. C., Hettmann T., Leiden J. M., Ron D., An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Liu L., Wise D. R., Diehl J. A., Simon M. C., Hypoxic reactive oxygen species regulate the integrated stress response and cell survival. J. Biol. Chem. 283, 31153–31162 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suragani R. N. V. S., Zachariah R. S., Velazquez J. G., Liu S., Sun C.-W., Townes T. M., Chen J.-J., Heme-regulated eIF2α kinase activated Atf4 signaling pathway in oxidative stress and erythropoiesis. Blood 119, 5276–5284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Korge P., Calmettes G., Weiss J. N., Increased reactive oxygen species production during reductive stress: The roles of mitochondrial glutathione and thioredoxin reductases. Biochim. Biophys. Acta 1847, 514–525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang Y. P., Torrente L., Falzone A., Elkins C. M., Liu M., Asara J. M., Dibble C. C., DeNicola G. M., Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. eLife 8, e45572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamada S., Shimosegawa T., Taguchi K., Nabeshima T., Yamamoto M., Masamune A., Simultaneous K-ras activation and Keap1 deletion cause atrophy of pancreatic parenchyma. Am. J. Physiol. Gastrointest. Liver Physiol. 314, G65–G74 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/45/eabb7272/DC1